
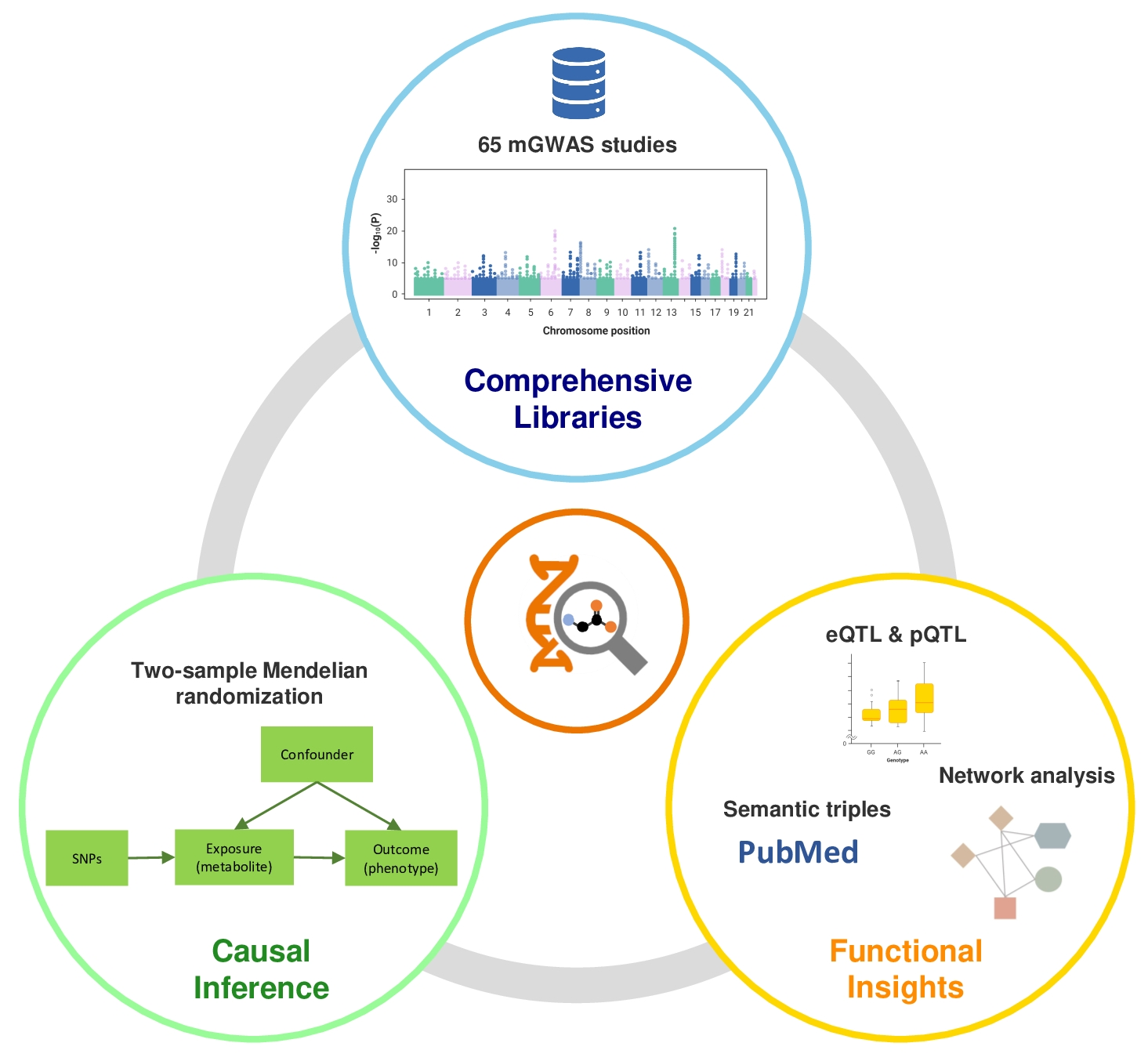
mGWAS-Explorer 2.0: Causal Analysis and Interpretation of Metabolite–Phenotype Associations



Abstract
:
1. Introduction
- Implemented a two-sample MR strategy to allow the investigation of causal relationships between >4000 metabolites and various phenotypes;
- Integration of semantic triples with eQTL and pQTL data to support functional annotation and mechanistic insights from MR results;
- Added a new “Joint Search” module that allows users to flexibly enter and search different molecules of interest;
- Enhanced data harmonization workflow and released the underlying mGWASR package to support reproducible analysis.
2. Materials and Methods
2.1. Knowledgebase Curation
2.2. Methods for MR Analysis
2.3. Pre-Computed Phenome-Wide MR
2.4. Semantic Triples
2.5. R Package
3. Results
3.1. Two-Sample Mendelian Randomization (2SMR)
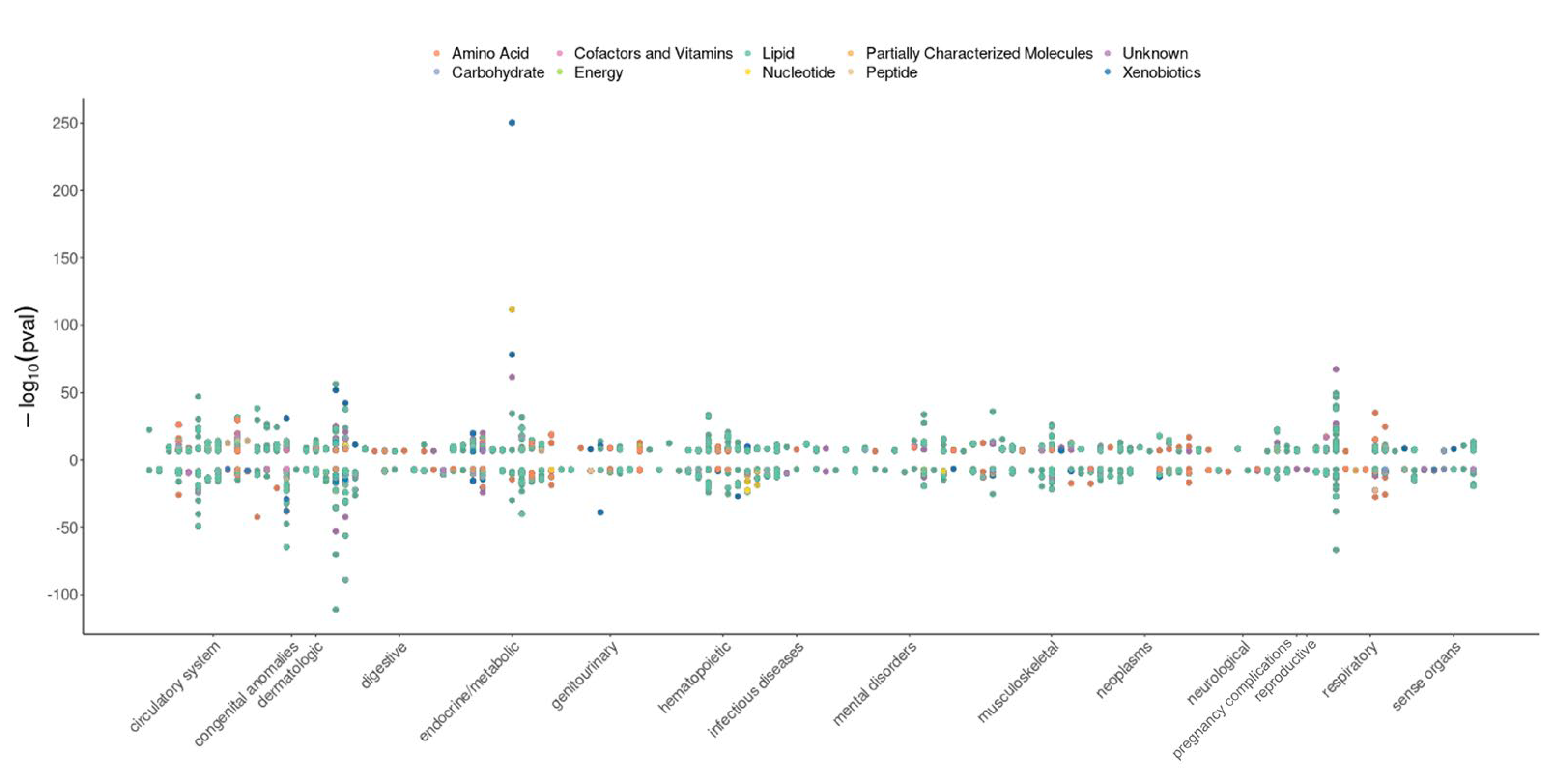
3.2. Pre-Computed Phenome-Wide MR
3.3. Triangulating Evidence from Semantic Triples
3.4. Enabling Joint SNP/Metabolite Analysis
3.5. Improving Transparency/Reproducibility through Releasing mGWASR Package
3.6. Case Studies
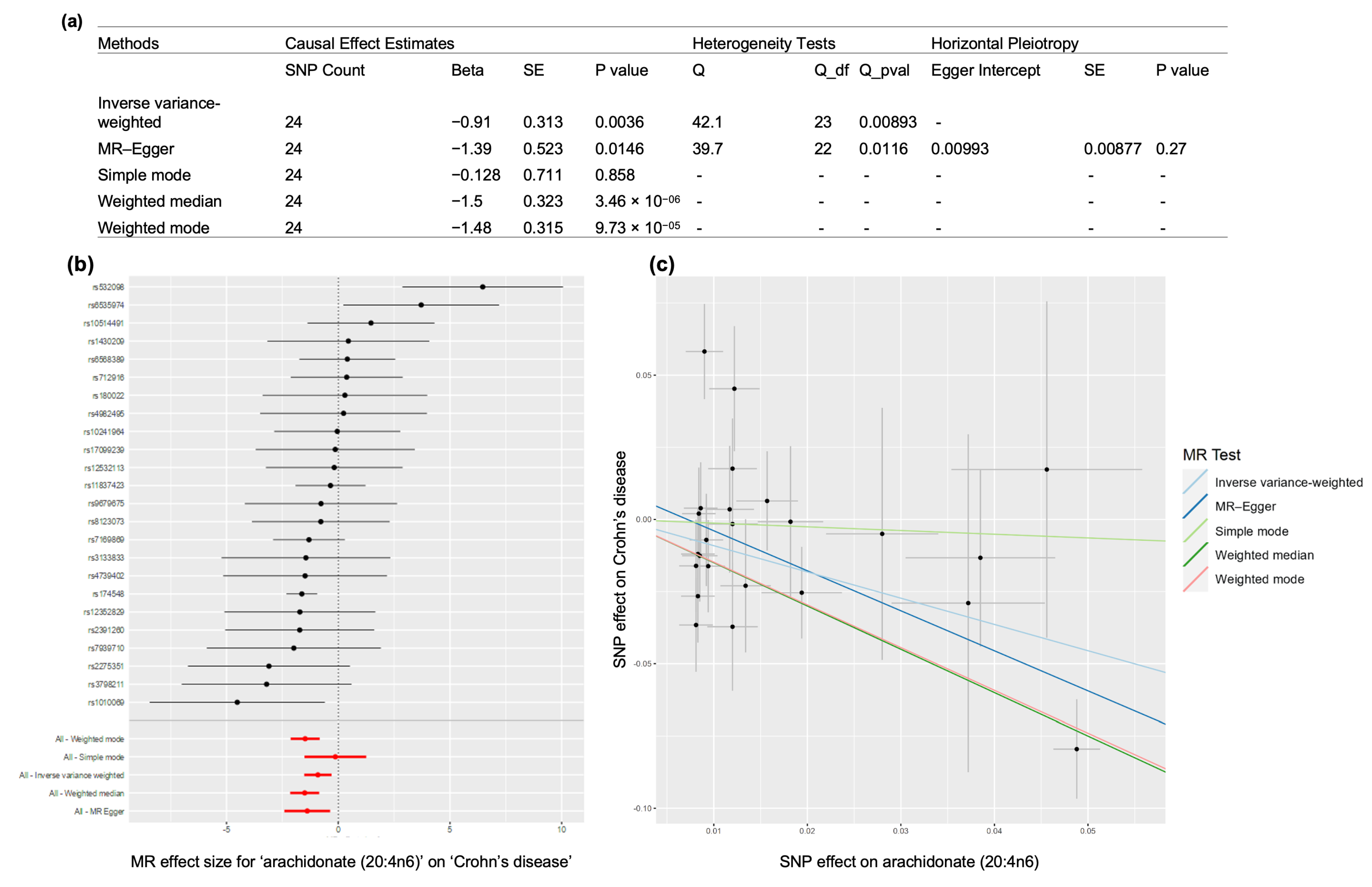
3.6.1. Crohn’s Disease Case Study
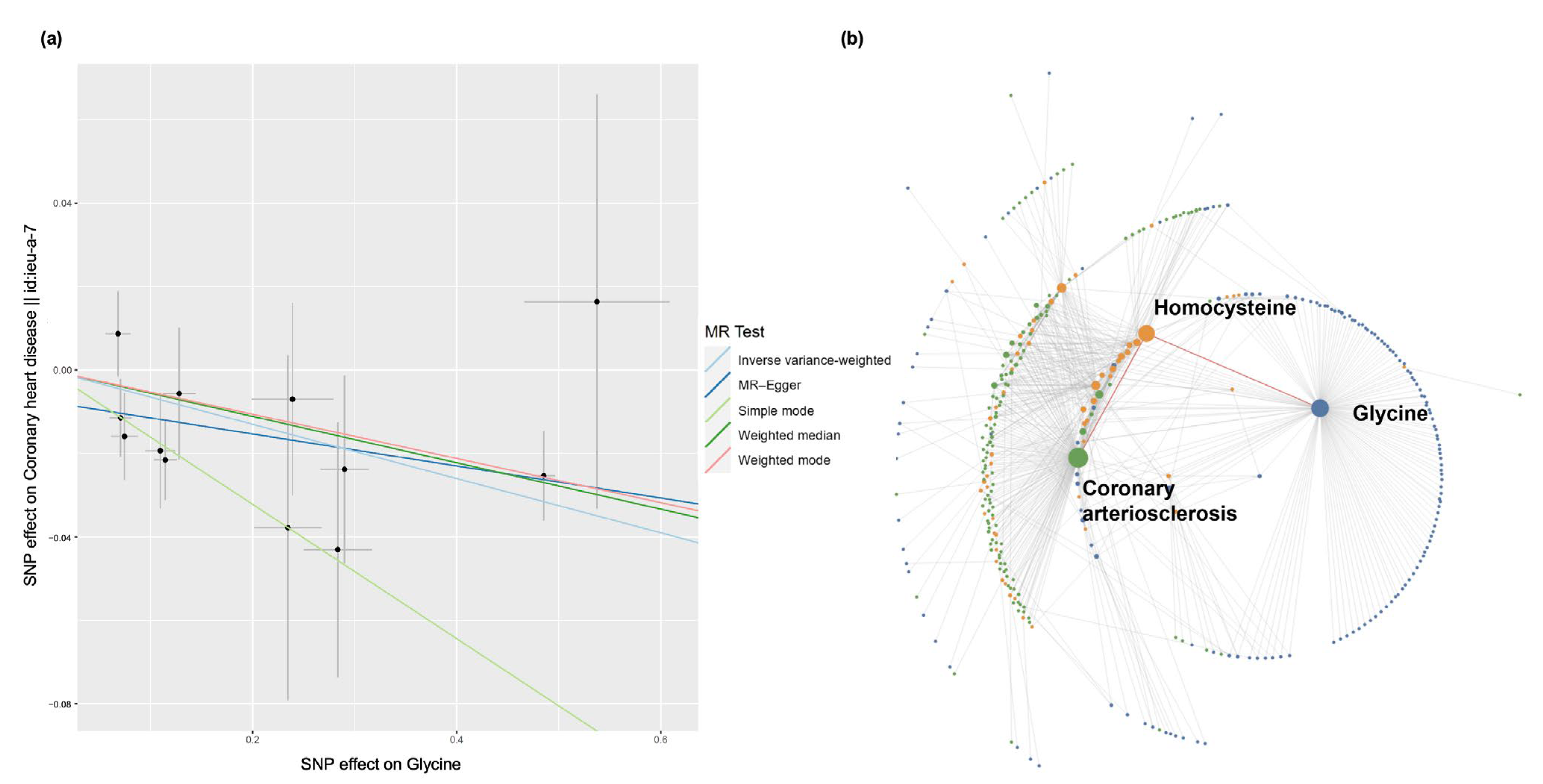
3.6.2. Coronary Heart Disease Case Study
3.7. Comparison with Other Tools
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotta, L.A.; Pietzner, M.; Stewart, I.D.; Wittemans, L.B.L.; Li, C.; Bonelli, R.; Raffler, J.; Biggs, E.K.; Oliver-Williams, C.; Auyeung, V.P.W.; et al. A cross-platform approach identifies genetic regulators of human metabolism and health. Nat. Genet. 2021, 53, 54–64. [Google Scholar] [CrossRef]
- Surendran, P.; Stewart, I.D.; Au Yeung, V.P.W.; Pietzner, M.; Raffler, J.; Wörheide, M.A.; Li, C.; Smith, R.F.; Wittemans, L.B.L.; Bomba, L.; et al. Rare and common genetic determinants of metabolic individuality and their effects on human health. Nat. Med. 2022, 28, 2321–2332. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.-Y.; Fauman, E.B.; Petersen, A.-K.; Krumsiek, J.; Santos, R.; Huang, J.; Arnold, M.; Erte, I.; Forgetta, V.; Yang, T.-P. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014, 46, 543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieger, C.; Geistlinger, L.; Altmaier, E.; De Angelis, M.H.; Kronenberg, F.; Meitinger, T.; Mewes, H.-W.; Wichmann, H.-E.; Weinberger, K.M.; Adamski, J.; et al. Genetics meets metabolomics: A genome-wide association study of metabolite profiles in human serum. PLoS Genet. 2008, 4, e1000282. [Google Scholar] [CrossRef] [Green Version]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Evangelou, E.; Ioannidis, J.P. Meta-analysis methods for genome-wide association studies and beyond. Nat. Rev. Genet. 2013, 14, 379–389. [Google Scholar] [CrossRef]
- Kastenmüller, G.; Raffler, J.; Gieger, C.; Suhre, K. Genetics of human metabolism: An update. Hum. Mol. Genet. 2015, 24, R93–R101. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Zhou, G.; Ou, H.; Xia, J. mGWAS-Explorer: Linking SNPs, Genes, Metabolites, and Diseases for Functional Insights. Metabolites 2022, 12, 526. [Google Scholar] [CrossRef]
- Lord, J.; Jermy, B.; Green, R.; Wong, A.; Xu, J.; Legido-Quigley, C.; Dobson, R.; Richards, M.; Proitsi, P. Mendelian randomization identifies blood metabolites previously linked to midlife cognition as causal candidates in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2009808118. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Méric, G.; Long, T.; Watrous, J.D.; Burgess, S.; Havulinna, A.S.; Ritchie, S.C.; Brożyńska, M.; Jousilahti, P.; Perola, M.; et al. Genome-wide association and Mendelian randomization analysis prioritizes bioactive metabolites with putative causal effects on common diseases. medRxiv, 2020; preprint. [Google Scholar] [CrossRef]
- Tanha, H.M.; Sathyanarayanan, A.; Nyholt, D.R. Genetic overlap and causality between blood metabolites and migraine. Am. J. Hum. Genet. 2021, 108, 2086–2098. [Google Scholar] [CrossRef]
- Zheng, J.; Haberland, V.; Baird, D.; Walker, V.; Haycock, P.C.; Hurle, M.R.; Gutteridge, A.; Erola, P.; Liu, Y.; Luo, S.; et al. Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat. Genet. 2020, 52, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Kolber, P.; Krüger, R. Gene-environment interaction and Mendelian randomisation. Rev. Neurol 2019, 175, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Ebrahim, S. ‘Mendelian randomization’: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef] [Green Version]
- de Leeuw, C.; Savage, J.; Bucur, I.G.; Heskes, T.; Posthuma, D. Understanding the assumptions underlying Mendelian randomization. Eur. J. Hum. Genet. 2022, 30, 653–660. [Google Scholar] [CrossRef]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 2018, 7, e34408. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef] [Green Version]
- Verbanck, M.; Chen, C.-Y.; Neale, B.; Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Scott, R.A.; Timpson, N.J.; Davey Smith, G.; Thompson, S.G.; Consortium, E.-I. Using published data in Mendelian randomization: A blueprint for efficient identification of causal risk factors. Eur. J. Epidemiol. 2015, 30, 543–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsworth, B.; Lyon, M.; Alexander, T.; Liu, Y.; Matthews, P.; Hallett, J.; Bates, P.; Palmer, T.; Haberland, V.; Smith, G.D.; et al. The MRC IEU OpenGWAS data infrastructure. bioRxiv, 2020; preprint. [Google Scholar] [CrossRef]
- Porcu, E.; Sjaarda, J.; Lepik, K.; Carmeli, C.; Darrous, L.; Sulc, J.; Mounier, N.; Kutalik, Z. Causal Inference Methods to Integrate Omics and Complex Traits. Cold Spring Harb. Perspect. Med. 2021, 11, a040493. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Yoo, W.; Alesh, I.; Mahajan, N.; Mirowska, K.K.; Mewada, A.; Kahn, J.; Afonso, L.; Williams, K.A., Sr.; Flack, J.M. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: A Mendelian randomization analysis. J. Am. Coll. Cardiol. 2012, 60, 2631–2639. [Google Scholar] [CrossRef] [Green Version]
- Holmes, M.V.; Simon, T.; Exeter, H.J.; Folkersen, L.; Asselbergs, F.W.; Guardiola, M.; Cooper, J.A.; Palmen, J.; Hubacek, J.A.; Carruthers, K.F.; et al. Secretory phospholipase A(2)-IIA and cardiovascular disease: A mendelian randomization study. J. Am. Coll. Cardiol. 2013, 62, 1966–1976. [Google Scholar] [CrossRef]
- Julian, T.H.; Cooper-Knock, J.; MacGregor, S.; Guo, H.; Aslam, T.; Sanderson, E.; Black, G.C.M.; Sergouniotis, P.I. Phenome-wide Mendelian randomisation analysis identifies causal factors for age-related macular degeneration. eLife 2023, 12, e82546. [Google Scholar] [CrossRef]
- Dong, S.S.; Zhang, K.; Guo, Y.; Ding, J.M.; Rong, Y.; Feng, J.C.; Yao, S.; Hao, R.H.; Jiang, F.; Chen, J.B.; et al. Phenome-wide investigation of the causal associations between childhood BMI and adult trait outcomes: A two-sample Mendelian randomization study. Genome Med. 2021, 13, 48. [Google Scholar] [CrossRef]
- Liu, Y.; Elsworth, B.; Erola, P.; Haberland, V.; Hemani, G.; Lyon, M.; Zheng, J.; Lloyd, O.; Vabistsevits, M.; Gaunt, T.R. EpiGraphDB: A database and data mining platform for health data science. Bioinformatics 2021, 37, 1304–1311. [Google Scholar] [CrossRef]
- Lawlor, D.A.; Tilling, K.; Davey Smith, G. Triangulation in aetiological epidemiology. Int. J. Epidemiol. 2017, 45, 1866–1886. [Google Scholar] [CrossRef] [Green Version]
- Kilicoglu, H.; Shin, D.; Fiszman, M.; Rosemblat, G.; Rindflesch, T.C. SemMedDB: A PubMed-scale repository of biomedical semantic predications. Bioinformatics 2012, 28, 3158–3160. [Google Scholar] [CrossRef] [Green Version]
- Elsworth, B.; Gaunt, T.R. MELODI Presto: A fast and agile tool to explore semantic triples derived from biomedical literature. Bioinformatics 2021, 37, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Bose, D.; Kwon, A.; Hanks, S.C.; Jackson, A.U.; Stringham, H.M.; Welch, R.; Oravilahti, A.; Fernandes Silva, L.; Locke, A.E.; et al. Integrating transcriptomics, metabolomics, and GWAS helps reveal molecular mechanisms for metabolite levels and disease risk. Am. J. Hum. Genet. 2022, 109, 1727–1741. [Google Scholar] [CrossRef]
- Pietzner, M.; Wheeler, E.; Carrasco-Zanini, J.; Cortes, A.; Koprulu, M.; Wörheide, M.A.; Oerton, E.; Cook, J.; Stewart, I.D.; Kerrison, N.D.; et al. Mapping the proteo-genomic convergence of human diseases. Science 2021, 374, eabj1541. [Google Scholar] [CrossRef] [PubMed]
- Võsa, U.; Claringbould, A.; Westra, H.-J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Yazar, S.; et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Zhang, Z.; Liu, Y.; Diao, L.; Han, L. A Multi-Omics Perspective of Quantitative Trait Loci in Precision Medicine. Trends Genet 2020, 36, 318–336. [Google Scholar] [CrossRef] [PubMed]
- Shariatipour, N.; Heidari, B.; Richards, C. Meta-QTL for morphological traits and pharmaceutical alkaloids in periwinkle (Catharanthus roseus (L.)‘G. Don’). J. Hortic. Sci. Biotechnol. 2022, 98, 87–98. [Google Scholar] [CrossRef]
- Shariatipour, N.; Heidari, B.; Richards, C.M. Meta-analysis of QTLome for grain zinc and iron contents in wheat (Triticum aestivum L.). Euphytica 2021, 217, 86. [Google Scholar] [CrossRef]
- The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [CrossRef]
- Zheng, Z.; Huang, D.; Wang, J.; Zhao, K.; Zhou, Y.; Guo, Z.; Zhai, S.; Xu, H.; Cui, H.; Yao, H.; et al. QTLbase: An integrative resource for quantitative trait loci across multiple human molecular phenotypes. Nucleic Acids Res. 2019, 48, D983–D991. [Google Scholar] [CrossRef] [Green Version]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Lu, T.; Pettersson-Kymmer, U.; Stewart, I.D.; Butler-Laporte, G.; Nakanishi, T.; Cerani, A.; Liang, K.Y.H.; Yoshiji, S.; Willett, J.D.S.; et al. Genomic atlas of the plasma metabolome prioritizes metabolites implicated in human diseases. Nat. Genet. 2023, 55, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Hysi, P.G.; Mangino, M.; Christofidou, P.; Falchi, M.; Karoly, E.D.; NIHR Bioresource Investigators; Mohney, R.P.; Valdes, A.M.; Spector, T.D.; Menni, C. Metabolome Genome-Wide Association Study Identifies 74 Novel Genomic Regions Influencing Plasma Metabolites Levels. Metabolites 2022, 12, 61. [Google Scholar] [CrossRef]
- Long, T.; Hicks, M.; Yu, H.-C.; Biggs, W.H.; Kirkness, E.F.; Menni, C.; Zierer, J.; Small, K.S.; Mangino, M.; Messier, H.; et al. Whole-genome sequencing identifies common-to-rare variants associated with human blood metabolites. Nat. Genet. 2017, 49, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Hemani, G.; Tilling, K.; Davey Smith, G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017, 13, e1007081. [Google Scholar] [CrossRef] [Green Version]
- Bowden, J.; Del Greco, M.F.; Minelli, C.; Davey Smith, G.; Sheehan, N.; Thompson, J. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat. Med. 2017, 36, 1783–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, T.; Jung, Y.; Moon, M.K.; Kwon, O.; Hwang, G.S.; Park, T. Integrative Pathway Analysis of SNP and Metabolite Data Using a Hierarchical Structural Component Model. Front. Genet. 2022, 13, 814412. [Google Scholar] [CrossRef]
- Chang, H.Y.; Colby, S.M.; Du, X.; Gomez, J.D.; Helf, M.J.; Kechris, K.; Kirkpatrick, C.R.; Li, S.; Patti, G.J.; Renslow, R.S.; et al. A Practical Guide to Metabolomics Software Development. Anal. Chem. 2021, 93, 1912–1923. [Google Scholar] [CrossRef]
- Chu, X.; Jaeger, M.; Beumer, J.; Bakker, O.B.; Aguirre-Gamboa, R.; Oosting, M.; Smeekens, S.P.; Moorlag, S.; Mourits, V.P.; Koeken, V.A.C.M.; et al. Integration of metabolomics, genomics, and immune phenotypes reveals the causal roles of metabolites in disease. Genome Biol. 2021, 22, 198. [Google Scholar] [CrossRef]
- de Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.G.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef] [Green Version]
- McCarty, M.F.; O'Keefe, J.H.; DiNicolantonio, J.J. Interleukin-1beta may act on hepatocytes to boost plasma homocysteine—The increased cardiovascular risk associated with elevated homocysteine may be mediated by this cytokine. Med. Hypotheses 2017, 102, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Nian, S.; Zhang, S.; Xu, W.; Zhang, X.; Ye, D.; Zheng, L. The associations between serum biomarkers and stenosis of the coronary arteries. Oncotarget 2016, 7, 39231–39240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, W.C.; Han, I.; Lee, W.; Choi, Y.J.; Lee, K.Y.; Kim, D.G.; Jung, S.H.; Oh, S.H.; Lee, B.H. Inhibition of homocysteine-induced endoplasmic reticulum stress and endothelial cell damage by l-serine and glycine. Toxicol Vitr. 2016, 34, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Movva, R.; Figueredo, V.M. Alcohol and the heart: To abstain or not to abstain? Int. J. Cardiol. 2013, 164, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Gallegos, S.; Muñoz, B.; Araya, A.; Aguayo, L.G. High ethanol sensitive glycine receptors regulate firing in D1 medium spiny neurons in the nucleus accumbens. Neuropharmacology 2019, 160, 107773. [Google Scholar] [CrossRef]
- Grira, N.; Lahidheb, D.; Lamine, O.; Ayoub, M.; Wassaifi, S.; Aouni, Z.; Fehri, W.; Mazigh, C. The Association of IL-6, TNFα and CRP Gene Polymorphisms with Coronary Artery Disease in a Tunisian Population: A Case-Control study. Biochem. Genet. 2021, 59, 751–766. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Wu, H.; Chen, S.; Zhu, H.; Zhang, J.; Hou, Y.; Hu, C.A.; Zhang, G. Glycine enhances muscle protein mass associated with maintaining Akt-mTOR-FOXO1 signaling and suppressing TLR4 and NOD2 signaling in piglets challenged with LPS. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R365–R373. [Google Scholar] [CrossRef] [Green Version]
- Halama, A.; Zaghlool, S.; Thareja, G.; Kader, S.; Muftha, W.A.; Mook-Kanamori, M.; Sarwath, H.; Mohamoud, Y.A.; Ameling, S.; Baković, M.P.; et al. The Molecular Human—A Roadmap of Molecular Interactions Linking Multiomics Networks with Disease Endpoints. medRxiv, 2022; preprint. [Google Scholar] [CrossRef]
- Pingault, J.-B.; O’Reilly, P.F.; Schoeler, T.; Ploubidis, G.B.; Rijsdijk, F.; Dudbridge, F. Using genetic data to strengthen causal inference in observational research. Nat. Rev. Genet. 2018, 19, 566–580. [Google Scholar] [CrossRef] [Green Version]
- Pingault, J.B.; Richmond, R.; Davey Smith, G. Causal Inference with Genetic Data: Past, Present, and Future. Cold Spring Harb. Perspect. Med. 2022, 12, a041271. [Google Scholar] [CrossRef]
- Davies, N.M.; Holmes, M.V.; Davey Smith, G. Reading Mendelian randomisation studies: A guide, glossary, and checklist for clinicians. Bmj 2018, 362, k601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallima, H.; El Ridi, R. Arachidonic acid: Physiological roles and potential health benefits—A review. J. Adv. Res. 2018, 11, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Trebble, T.M.; Arden, N.K.; Wootton, S.A.; Mullee, M.A.; Calder, P.C.; Burdge, G.C.; Fine, D.R.; Stroud, M.A. Peripheral blood mononuclear cell fatty acid composition and inflammatory mediator production in adult Crohn’s disease. Clin. Nutr. 2004, 23, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Esteve-Comas, M.; Ramírez, M.; Fernández-Bañares, F.; Abad-Lacruz, A.; Gil, A.; Cabré, E.; González-Huix, F.; Moreno, J.; Humbert, P.; Guilera, M. Plasma polyunsaturated fatty acid pattern in active inflammatory bowel disease. Gut 1992, 33, 1365–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittemans, L.B.L.; Lotta, L.A.; Oliver-Williams, C.; Stewart, I.D.; Surendran, P.; Karthikeyan, S.; Day, F.R.; Koulman, A.; Imamura, F.; Zeng, L.; et al. Assessing the causal association of glycine with risk of cardio-metabolic diseases. Nat. Commun. 2019, 10, 1060. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, P.; Alam, S.F. Role of homocysteine in the development of cardiovascular disease. Nutr. J. 2015, 14, 6. [Google Scholar] [CrossRef] [Green Version]
- Su, J.; Xu, K.; Li, Z.; Hu, Y.; Hu, Z.; Zheng, X.; Song, S.; Tang, Z.; Li, L. Genome-wide association study and Mendelian randomization analysis provide insights for improving rice yield potential. Sci. Rep. 2021, 11, 6894. [Google Scholar] [CrossRef]
- Gallagher, M.D.; Chen-Plotkin, A.S. The Post-GWAS Era: From Association to Function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tool Name | mGWAS-Explorer | EpiGraphDB | The Molecular Human | MR-Base | |
|---|---|---|---|---|---|
| 2.0 | 1.0 | ||||
| Data input and processing | |||||
| Metabolite | √ | √ | √ | √ | √ |
| SNP | √ | √ | √ | √ | − |
| Gene | √ | √ | √ | √ | − |
| MR exposure | √ | − | √ | − | √ |
| MR outcome | √ | − | √ | − | √ |
| Output | |||||
| Data table | √ | √ | √ | √ | √ |
| Interactive network | +++ | +++ | ++ | ++ | − |
| Forest plot | √ | − | − | − | √ |
| Scatter plot | √ | − | − | − | √ |
| Funnel plot | √ | − | − | − | √ |
| Functions and resources | |||||
| Mendelian randomization | √ | − | * √ | − | √ |
| Exposure (metabolite) | ** 4238 metabolic traits, 65 studies | − | 123 metabolic traits, 1 study | − | 123 metabolic traits, 1 study |
| Enrichment analysis | √ | √ | − | − | − |
| Pre-computed phenome-wide MR | √ | − | √ | − | − |
| Semantic triples evidence | √ | − | √ | − | − |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, L.; Zhou, G.; Xia, J. mGWAS-Explorer 2.0: Causal Analysis and Interpretation of Metabolite–Phenotype Associations. Metabolites 2023, 13, 826. https://doi.org/10.3390/metabo13070826
Chang L, Zhou G, Xia J. mGWAS-Explorer 2.0: Causal Analysis and Interpretation of Metabolite–Phenotype Associations. Metabolites. 2023; 13(7):826. https://doi.org/10.3390/metabo13070826
Chicago/Turabian StyleChang, Le, Guangyan Zhou, and Jianguo Xia. 2023. "mGWAS-Explorer 2.0: Causal Analysis and Interpretation of Metabolite–Phenotype Associations" Metabolites 13, no. 7: 826. https://doi.org/10.3390/metabo13070826
APA StyleChang, L., Zhou, G., & Xia, J. (2023). mGWAS-Explorer 2.0: Causal Analysis and Interpretation of Metabolite–Phenotype Associations. Metabolites, 13(7), 826. https://doi.org/10.3390/metabo13070826