
Comparative Metabolomics in Single Ventricle Patients after Fontan Palliation: A Strong Case for a Targeted Metabolic Therapy


Abstract
:1. Introduction
2. Cardiac Energy Metabolism
2.1. Energy Metabolism in the Healthy Heart
2.2. Energy Metabolism in Biventricular Patients with Congestive Heart Failure
2.3. Right Ventricular Failure: The Case of Pulmonary Arterial Hypertension
2.4. Energy Metabolism and the Single Ventricle after Fontan Palliation
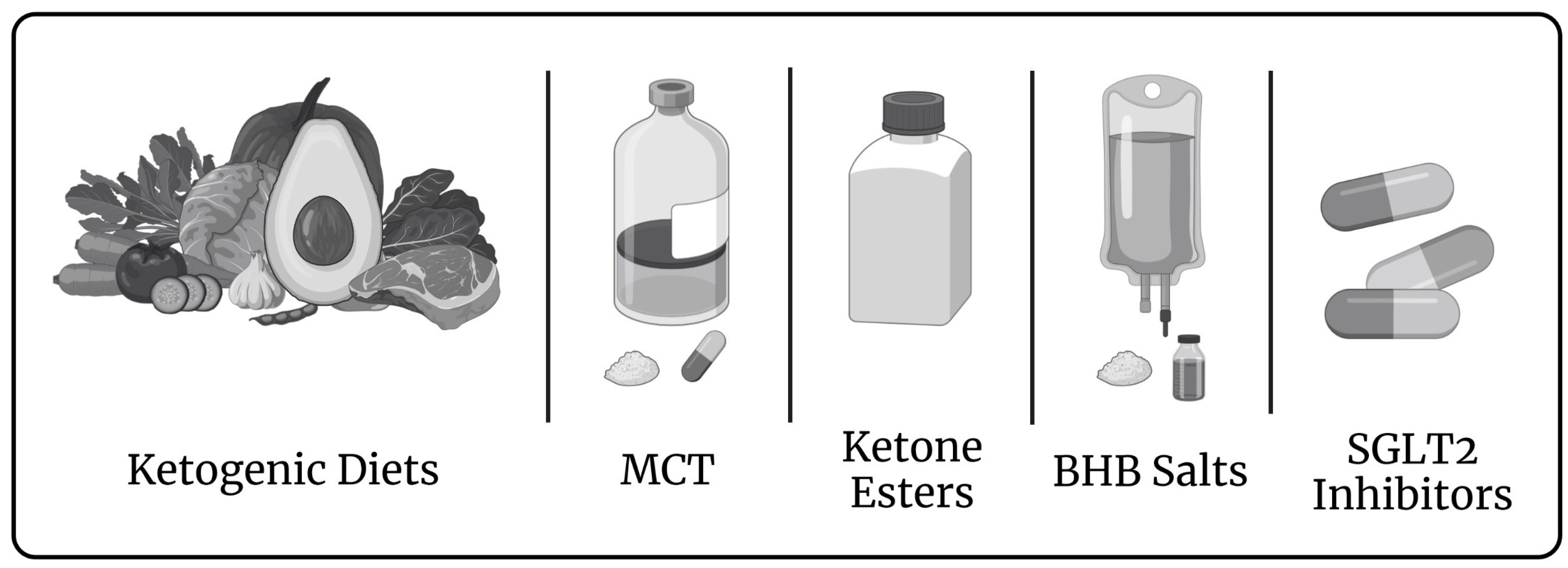
3. Induced Ketosis in Patients with a Failing Biventricular Heart
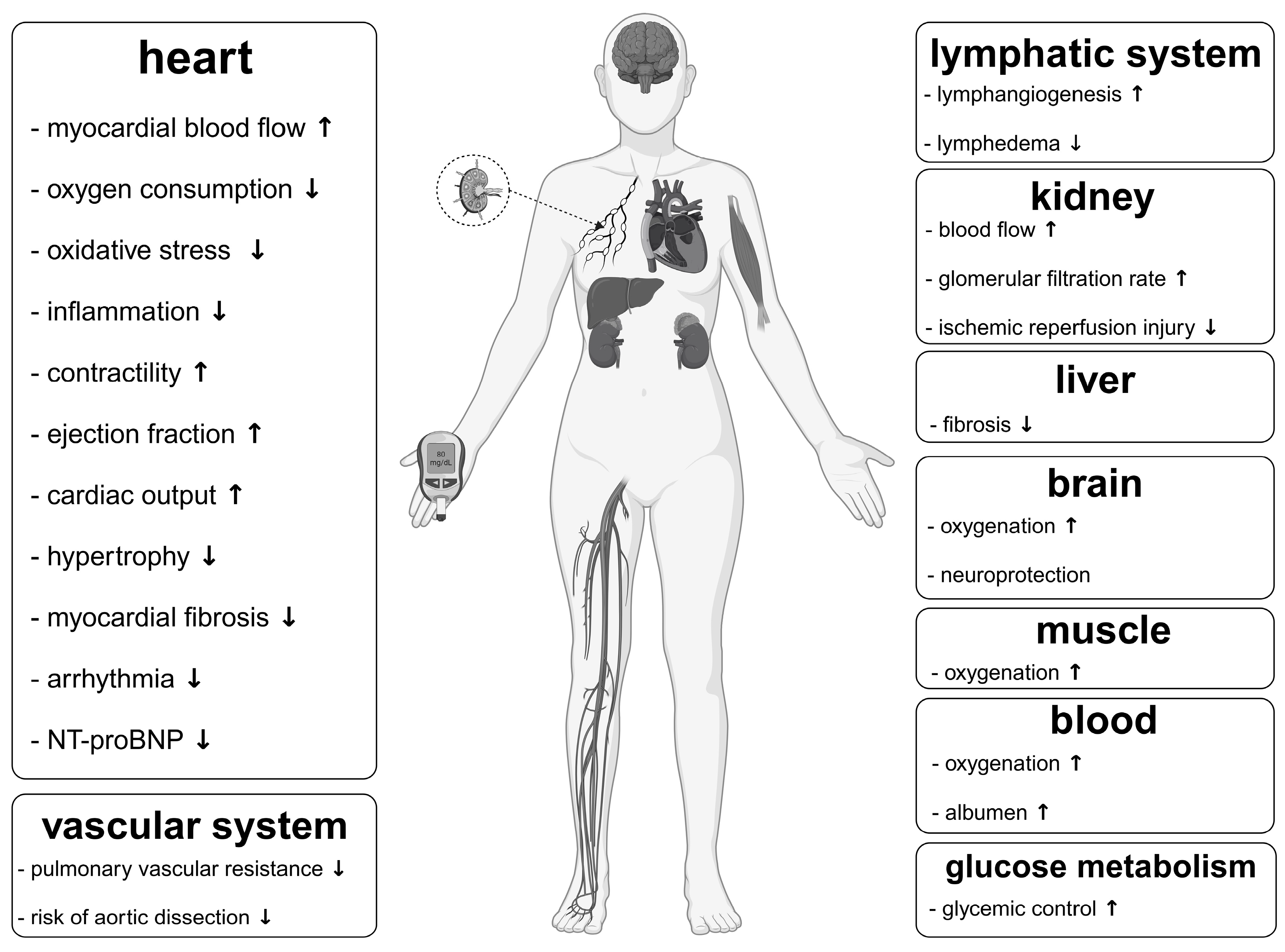
4. Impact of Ketones Apart from That on the Cardiovascular System Relevant to Fontan Circulation
5. Rationale of a Targeted Metabolic Therapy in Fontan Patients
6. Limitations
7. Methods
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rychik, J.; Atz, A.M.; Celermajer, D.S.; Deal, B.J.; Gatzoulis, M.A.; Gewillig, M.H.; Hsia, T.-Y.; Hsu, D.T.; Kovacs, A.H.; McCrindle, B.W.; et al. Evaluation and Management of the Child and Adult with Fontan Circulation: A Scientific Statement from the American Heart Association. Circulation 2019, 140, e234–e284. [Google Scholar] [CrossRef]
- Fontan, F.; Baudet, E. Surgical Repair of Tricuspid Atresia. Thorax 1971, 26, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Kreutzer, G.; Galíndez, E.; Bono, H.; De Palma, C.; Laura, J.P. An Operation for the Correction of Tricuspid Atresia. J. Thorac. Cardiovasc. Surg. 1973, 66, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Gewillig, M.; Brown, S.C. The Fontan Circulation after 45 Years: Update in Physiology. Heart 2016, 102, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Alsoufi, B.; Gillespie, S.; Kim, D.; Shashidharan, S.; Kanter, K.; Maher, K.; Kogon, B. The Impact of Dominant Ventricle Morphology on Palliation Outcomes of Single Ventricle Anomalies. Ann. Thorac. Surg. 2016, 102, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Kutty, S.; Jacobs, M.L.; Thompson, W.R.; Danford, D.A. Fontan Circulation of the Next Generation: Why It’s Necessary, What It Might Look Like. J. Am. Heart Assoc. 2020, 9, e013691. [Google Scholar] [CrossRef] [PubMed]
- Rychik, J. Path Taken in a Fontan Circulation: Room for Optimism in the Face of Uncertainty. Heart 2021, 107, 521–522. [Google Scholar] [CrossRef]
- Zhu, A.; Meza, J.M.; Prabhu, N.K.; McCrary, A.W.; Allareddy, V.; Turek, J.W.; Andersen, N.D. Survival After Intervention for Single-Ventricle Heart Disease Over 15 Years at a Single Institution. Ann. Thorac. Surg. 2022, 114, 2303–2312. [Google Scholar] [CrossRef]
- Lewis, M.; Rosenbaum, M. The Miracle Baby Grows Up: Hypoplastic Left Heart Syndrome in the Adult. Curr. Cardiol. Rep. 2017, 19, 74. [Google Scholar] [CrossRef]
- Julsrud, P.R.; Weigel, T.J.; Van Son, J.A.; Edwards, W.D.; Mair, D.D.; Driscoll, D.J.; Danielson, G.K.; Puga, F.J.; Offord, K.P. Influence of Ventricular Morphology on Outcome after the Fontan Procedure. Am. J. Cardiol. 2000, 86, 319–323. [Google Scholar] [CrossRef]
- McGuirk, S. The Impact of Ventricular Morphology on Midterm Outcome Following Completion Total Cavopulmonary Connection. Eur. J. Cardiothorac. Surg. 2003, 24, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, P.A.W.; Sleeper, L.A.; Mahony, L.; Colan, S.D.; Atz, A.M.; Breitbart, R.E.; Gersony, W.M.; Gallagher, D.; Geva, T.; Margossian, R.; et al. Contemporary Outcomes after the Fontan Procedure. J. Am. Coll. Cardiol. 2008, 52, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Backer, C.L. The Functionally Univentricular Heart. J. Am. Coll. Cardiol. 2012, 59, 1186–1187. [Google Scholar] [CrossRef] [PubMed]
- d’Udekem, Y.; Xu, M.Y.; Galati, J.C.; Lu, S.; Iyengar, A.J.; Konstantinov, I.E.; Wheaton, G.R.; Ramsay, J.M.; Grigg, L.E.; Millar, J.; et al. Predictors of Survival After Single-Ventricle Palliation. J. Am. Coll. Cardiol. 2012, 59, 1178–1185. [Google Scholar] [CrossRef] [Green Version]
- King, G.; Buratto, E.; Daley, M.; Iyengar, A.; Alphonso, N.; Grigg, L.; Cordina, R.; d’Udekem, Y.; Konstantinov, I.E. Impact of Aortic Atresia After Fontan Operation in Patients With Hypoplastic Left Heart Syndrome. Ann. Thorac. Surg. 2022, 116, 95–102. [Google Scholar] [CrossRef]
- Iyengar, A.J.; Winlaw, D.S.; Galati, J.C.; Wheaton, G.R.; Gentles, T.L.; Grigg, L.E.; Justo, R.N.; Radford, D.J.; Weintraub, R.G.; Bullock, A.; et al. The Extracardiac Conduit Fontan Procedure in Australia and New Zealand: Hypoplastic Left Heart Syndrome Predicts Worse Early and Late Outcomes. Eur. J. Cardiothorac. Surg. 2014, 46, 465–473. [Google Scholar] [CrossRef]
- Book, W.M.; Gerardin, J.; Saraf, A.; Marie Valente, A.; Rodriguez, F. Clinical Phenotypes of Fontan Failure: Implications for Management: Fontan Phenotypes. Congenit. Heart Dis. 2016, 11, 296–308. [Google Scholar] [CrossRef]
- Sable, C.; Foster, E.; Uzark, K.; Bjornsen, K.; Canobbio, M.M.; Connolly, H.M.; Graham, T.P.; Gurvitz, M.Z.; Kovacs, A.; Meadows, A.K.; et al. Best Practices in Managing Transition to Adulthood for Adolescents with Congenital Heart Disease: The Transition Process and Medical and Psychosocial Issues: A Scientific Statement from the American Heart Association. Circulation 2011, 123, 1454–1485. [Google Scholar] [CrossRef] [Green Version]
- Michel, M.; Zlamy, M.; Entenmann, A.; Pichler, K.; Scholl-Bürgi, S.; Karall, D.; Geiger, R.; Salvador, C.; Niederwanger, C.; Ohuchi, H. Impact of the Fontan Operation on Organ Systems. Cardiovasc. Hematol. Disord. Drug Targets 2019, 19, 205–214. [Google Scholar] [CrossRef]
- Harteveld, L.M.; Blom, N.A.; Terol Espinosa de Los Monteros, C.; Kuipers, I.M.; Rammeloo, L.A.J.; Hazekamp, M.G.; van Dijk, J.G.; ten Harkel, A.D.J. 3-Month Enalapril Treatment in Pediatric Fontan Patients with Moderate to Good Systolic Ventricular Function. Am. J. Cardiol. 2022, 163, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Shaddy, R.E.; Boucek, M.M.; Hsu, D.T.; Boucek, R.J.; Canter, C.E.; Mahony, L.; Ross, R.D.; Pahl, E.; Blume, E.D.; Dodd, D.A.; et al. Carvedilol for Children and Adolescents With Heart Failure: A Randomized Controlled Trial. JAMA 2007, 298, 1171. [Google Scholar] [CrossRef] [PubMed]
- Schranz, D.; Voelkel, N.F. “Nihilism” of Chronic Heart Failure Therapy in Children and Why Effective Therapy Is Withheld. Eur. J. Pediatr. 2016, 175, 445–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, P.A.W.; Breitbart, R.E.; McCrindle, B.W.; Sleeper, L.A.; Atz, A.M.; Hsu, D.T.; Lu, M.; Margossian, R.; Williams, R.V. The Fontan Patient: Inconsistencies in Medication Therapy Across Seven Pediatric Heart Network Centers. Pediatr. Cardiol. 2010, 31, 1219–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanayem, N.S.; Berger, S.; Tweddell, J.S. Medical Management of the Failing Fontan. Pediatr. Cardiol. 2007, 28, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Taegtmeyer, H. Metabolism—The Lost Child of Cardiology. J. Am. Coll. Cardiol. 2000, 36, 1386–1388. [Google Scholar] [CrossRef] [Green Version]
- Taegtmeyer, H. Cardiac Metabolism as a Target for the Treatment of Heart Failure. Circulation 2004, 110, 894–896. [Google Scholar] [CrossRef] [Green Version]
- Ashrafian, H.; Frenneaux, M.P.; Opie, L.H. Metabolic Mechanisms in Heart Failure. Circulation 2007, 116, 434–448. [Google Scholar] [CrossRef]
- Kimball, T.H.; Vondriska, T.M. Metabolism, Epigenetics, and Causal Inference in Heart Failure. Trends Endocrinol. Metab. 2020, 31, 181–191. [Google Scholar] [CrossRef]
- Selvaraj, S.; Kelly, D.P.; Margulies, K.B. Implications of Altered Ketone Metabolism and Therapeutic Ketosis in Heart Failure. Circulation 2020, 141, 1800–1812. [Google Scholar] [CrossRef]
- Taegtmeyer, H. Energy Metabolism of the Heart: From Basic Concepts to Clinical Applications Applications. Curr. Probl. Cardiol. 1994, 19, 61–86. [Google Scholar] [CrossRef]
- Christensen, K.H. Treatment with the Ketone Body 3-Hydroxybutyrate in Patients with Acute Heart Failure. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04442555 (accessed on 20 July 2022).
- Yurista, S.R.; Chong, C.-R.; Badimon, J.J.; Kelly, D.P.; de Boer, R.A.; Westenbrink, B.D. Therapeutic Potential of Ketone Bodies for Patients with Cardiovascular Disease. J. Am. Coll. Cardiol. 2021, 77, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Takahara, S.; Soni, S.; Phaterpekar, K.; Kim, T.T.; Maayah, Z.H.; Levasseur, J.L.; Silver, H.L.; Freed, D.H.; Ferdaoussi, M.; Dyck, J.R.B. Chronic Exogenous Ketone Supplementation Blunts the Decline of Cardiac Function in the Failing Heart. ESC Heart Fail. 2021, 8, 5606–5612. [Google Scholar] [CrossRef] [PubMed]
- Takahara, S.; Soni, S.; Maayah, Z.H.; Ferdaoussi, M.; Dyck, J.R.B. Ketone Therapy for Heart Failure: Current Evidence for Clinical Use. Cardiovasc. Res. 2022, 118, 977–987. [Google Scholar] [CrossRef]
- Monzo, L.; Sedlacek, K.; Hromanikova, K.; Tomanova, L.; Borlaug, B.A.; Jabor, A.; Kautzner, J.; Melenovsky, V. Myocardial Ketone Body Utilization in Patients with Heart Failure: The Impact of Oral Ketone Ester. Metabolism 2021, 115, 154452. [Google Scholar] [CrossRef] [PubMed]
- Papazafiropoulou, A.; Georgopoulos, M.; Katsilambros, N. Ketone Bodies and the Heart. Arch. Med. Sci. Atheroscler. Dis. 2021, 6, 209–214. [Google Scholar] [CrossRef]
- Schulze, P.C.; Wu, J.M.F. Ketone Bodies for the Starving Heart. Nat. Metab. 2020, 2, 1183–1185. [Google Scholar] [CrossRef]
- Horton, J.L.; Davidson, M.T.; Kurishima, C.; Vega, R.B.; Powers, J.C.; Matsuura, T.R.; Petucci, C.; Lewandowski, E.D.; Crawford, P.A.; Muoio, D.M.; et al. The Failing Heart Utilizes 3-Hydroxybutyrate as a Metabolic Stress Defense. JCI Insight 2019, 4, e124079. [Google Scholar] [CrossRef] [Green Version]
- Uchihashi, M.; Hoshino, A.; Okawa, Y.; Ariyoshi, M.; Kaimoto, S.; Tateishi, S.; Ono, K.; Yamanaka, R.; Hato, D.; Fushimura, Y.; et al. Cardiac-Specific Bdh1 Overexpression Ameliorates Oxidative Stress and Cardiac Remodeling in Pressure Overload–Induced Heart Failure. Circ. Heart Fail. 2017, 10, e004417. [Google Scholar] [CrossRef]
- Schugar, R.C.; Moll, A.R.; André d’Avignon, D.; Weinheimer, C.J.; Kovacs, A.; Crawford, P.A. Cardiomyocyte-Specific Deficiency of Ketone Body Metabolism Promotes Accelerated Pathological Remodeling. Mol. Metab. 2014, 3, 754–769. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Röhling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone Bodies: From Enemy to Friend and Guardian Angel. BMC Med. 2021, 19, 313. [Google Scholar] [CrossRef]
- Michel, M.; Dubowy, K.-O.; Entenmann, A.; Karall, D.; Adam, M.G.; Zlamy, M.; Odri Komazec, I.; Geiger, R.; Niederwanger, C.; Salvador, C.; et al. Targeted Metabolomic Analysis of Serum Amino Acids in the Adult Fontan Patient with a Dominant Left Ventricle. Sci. Rep. 2020, 10, 8930. [Google Scholar] [CrossRef]
- Michel, M.; Dubowy, K.-O.; Zlamy, M.; Karall, D.; Adam, M.G.; Entenmann, A.; Keller, M.A.; Koch, J.; Odri Komazec, I.; Geiger, R.; et al. Targeted Metabolomic Analysis of Serum Phospholipid and Acylcarnitine in the Adult Fontan Patient with a Dominant Left Ventricle. Ther. Adv. Chronic Dis. 2020, 11, 204062232091603. [Google Scholar] [CrossRef] [PubMed]
- Opie, L.H. Heart Physiology: From Cell to Circulation, 4th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2004. [Google Scholar]
- Barth, E. Ultrastructural Quantitation of Mitochondria and Myofilaments in Cardiac Muscle from 10 Different Animal Species Including Man. J. Mol. Cell. Cardiol. 1992, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Ingwall, J.S.; Weiss, R.G. Is the Failing Heart Energy Starved?: On Using Chemical Energy to Support Cardiac Function. Circ. Res. 2004, 95, 135–145. [Google Scholar] [CrossRef]
- Herrmann, G.; Decherd, G. The chemical nature of heart failure. Ann. Intern. Med. 1939, 12, 1233. [Google Scholar] [CrossRef]
- Neubauer, S. The Failing Heart—An Engine Out of Fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [Green Version]
- Sack, M.N.; Rader, T.A.; Park, S.; Bastin, J.; McCune, S.A.; Kelly, D.P. Fatty Acid Oxidation Enzyme Gene Expression Is Downregulated in the Failing Heart. Circulation 1996, 94, 2837–2842. [Google Scholar] [CrossRef]
- Katz, A.M. Energetics and the Failing Heart. Hosp. Pract. 1991, 26, 78–90. [Google Scholar] [CrossRef]
- Taegtmeyer, H. Failing Heart and Starving Brain. Circulation 2016, 134, 265–266. [Google Scholar] [CrossRef] [PubMed]
- Giussani, D.A.; Bennet, L.; Sferruzzi-Perri, A.N.; Vaughan, O.R.; Fowden, A.L. Hypoxia, fetal and neonatal physiology: 100 years on from Sir Joseph Barcroft. J. Physiol. 2016, 594, 1105–1111. [Google Scholar] [CrossRef] [Green Version]
- Girard, J.; Ferre, P.; Pegorier, J.P.; Duee, P.H. Adaptations of Glucose and Fatty Acid Metabolism during Perinatal Period and Suckling-Weaning Transition. Physiol. Rev. 1992, 72, 507–562. [Google Scholar] [CrossRef]
- Ascuitto, R.J.; Ross-Ascuitto, N.T. Substrate Metabolism in the Developing Heart. Semin. Perinatol. 1996, 20, 542–563. [Google Scholar] [CrossRef]
- Itoi, T.; Lopaschuk, G.D. The Contribution of Glycolysis, Glucose Oxidation, Lactate Oxidation, and Fatty Acid Oxidation to ATP Production in Isolated Biventricular Working Hearts from 2-Week-Old Rabbits. Pediatr. Res. 1993, 34, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Dimasi, C.G.; Darby, J.R.T.; Morrison, J.L. A change of heart: Understanding the mechanisms regulating cardiac proliferation and metabolism before and after birth. J. Physiol. 2023, 601, 1319–1341. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Jaswal, J.S. Energy Metabolic Phenotype of the Cardiomyocyte During Development, Differentiation, and Postnatal Maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Ritterhoff, J.; Tian, R. Metabolism in Cardiomyopathy: Every Substrate Matters. Cardiovasc. Res. 2017, 113, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Taegtmeyer, H.; Sen, S.; Vela, D. Return to the Fetal Gene Program: A Suggested Metabolic Link to Gene Expression in the Heart. Ann. N. Y. Acad. Sci. 2010, 1188, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.; Shannon, R.P. Insulin Resistance in Dilated Cardiomyopathy. Rev. Cardiovasc. Med. 2003, 4, S50–S57. [Google Scholar] [PubMed]
- Nikolaidis, L. The Development of Myocardial Insulin Resistance in Conscious Dogs with Advanced Dilated Cardiomyopathy. Cardiovasc. Res. 2004, 61, 297–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taegtmeyer, H.; Golfman, L.; Sharma, S.; Razeghi, P.; Arsdall, M. Linking Gene Expression to Function: Metabolic Flexibility in the Normal and Diseased Heart. Ann. N. Y. Acad. Sci. 2004, 1015, 202–213. [Google Scholar] [CrossRef]
- Schulze, P.C.; Drosatos, K.; Goldberg, I.J. Lipid Use and Misuse by the Heart. Circ. Res. 2016, 118, 1736–1751. [Google Scholar] [CrossRef]
- Kolwicz, S.C.; Airhart, S.; Tian, R. Ketones Step to the Plate: A Game Changer for Metabolic Remodeling in Heart Failure? Circulation 2016, 133, 689–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedi, K.C.; Snyder, N.W.; Brandimarto, J.; Aziz, M.; Mesaros, C.; Worth, A.J.; Wang, L.L.; Javaheri, A.; Blair, I.A.; Margulies, K.B.; et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 2016, 133, 706–716. [Google Scholar] [CrossRef] [Green Version]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Krüger, M.; Hoppel, C.L.; et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 2016, 133, 698–705. [Google Scholar] [CrossRef] [Green Version]
- Huynh, K. Ketone Bodies as Fuel in Heart Failure. Nat. Rev. Cardiol. 2016, 13, 123. [Google Scholar] [CrossRef]
- Manolis, A.S.; Manolis, T.A.; Manolis, A.A. Ketone Bodies and Cardiovascular Disease: An Alternate Fuel Source to the Rescue. Int. J. Mol. Sci. 2023, 24, 3534. [Google Scholar] [CrossRef]
- Voros, G.; Ector, J.; Garweg, C.; Droogne, W.; Van Cleemput, J.; Peersman, N.; Vermeersch, P.; Janssens, S. Increased Cardiac Uptake of Ketone Bodies and Free Fatty Acids in Human Heart Failure and Hypertrophic Left Ventricular Remodeling. Circ. Heart Fail. 2018, 11, e004953. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Ketone Body Can Be a Fuel Substrate for Failing Heart. Cardiovasc. Res. 2019, 115, 1567–1569. [Google Scholar] [CrossRef]
- Liao, S.; Tang, Y.; Yue, X.; Gao, R.; Yao, W.; Zhou, Y.; Zhang, H. β-Hydroxybutyrate Mitigated Heart Failure with Preserved Ejection Fraction by Increasing Treg Cells via Nox2/GSK-3β. J. Inflamm. Res. 2021, 14, 4697–4706. [Google Scholar] [CrossRef]
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting Mitochondria-Inflammation Circuit by β-Hydroxybutyrate Mitigates HFpEF. Circ. Res. 2021, 128, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative Stress and Heart Failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [Green Version]
- Beer, M.; Seyfarth, T.; Sandstede, J.; Landschütz, W.; Lipke, C.; Köstler, H.; von Kienlin, M.; Harre, K.; Hahn, D.; Neubauer, S. Absolute Concentrations of High-Energy Phosphate Metabolites in Normal, Hypertrophied, and Failing Human Myocardium Measured Noninvasively with 31P-SLOOP Magnetic Resonance Spectroscopy. J. Am. Coll. Cardiol. 2002, 40, 1267–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, R.G.; Gerstenblith, G.; Bottomley, P.A. ATP Flux through Creatine Kinase in the Normal, Stressed, and Failing Human Heart. Proc. Natl. Acad. Sci. USA 2005, 102, 808–813. [Google Scholar] [CrossRef]
- Smith, C.S.; Bottomley, P.A.; Schulman, S.P.; Gerstenblith, G.; Weiss, R.G. Altered Creatine Kinase Adenosine Triphosphate Kinetics in Failing Hypertrophied Human Myocardium. Circulation 2006, 114, 1151–1158. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R.; Møller, N.; Gormsen, L.C.; Tolbod, L.P.; Hansson, N.H.; Sorensen, J.; Harms, H.J.; Frøkiær, J.; Eiskjaer, H.; Jespersen, N.R.; et al. Cardiovascular Effects of Treatment with the Ketone Body 3-Hydroxybutyrate in Chronic Heart Failure Patients. Circulation 2019, 139, 2129–2141. [Google Scholar] [CrossRef]
- Brittain, E.L.; Talati, M.; Fessel, J.P.; Zhu, H.; Penner, N.; Calcutt, M.W.; West, J.D.; Funke, M.; Lewis, G.D.; Gerszten, R.E.; et al. Fatty Acid Metabolic Defects and Right Ventricular Lipotoxicity in Human Pulmonary Arterial Hypertension. Circulation 2016, 133, 1936–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugh, M.E.; Robbins, I.M.; Rice, T.W.; West, J.; Newman, J.H.; Hemnes, A.R. Unrecognized Glucose Intolerance Is Common in Pulmonary Arterial Hypertension. J. Heart Lung Transplant. 2011, 30, 904–911. [Google Scholar] [CrossRef]
- West, J.; Niswender, K.D.; Johnson, J.A.; Pugh, M.E.; Gleaves, L.; Fessel, J.P.; Hemnes, A.R. A Potential Role for Insulin Resistance in Experimental Pulmonary Hypertension. Eur. Respir. J. 2013, 41, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Zamanian, R.T.; Hansmann, G.; Snook, S.; Lilienfeld, D.; Rappaport, K.M.; Reaven, G.M.; Rabinovitch, M.; Doyle, R.L. Insulin Resistance in Pulmonary Arterial Hypertension. Eur. Respir. J. 2008, 33, 318–324. [Google Scholar] [CrossRef]
- Zare, E.; Kafshbani, P.; Chenaghlou, M.; Noori, M.; Ghaemmaghami, Z.; Amin, A.; Taghavi, S.; Naderi, N. Prognostic Significance of Insulin Resistance in Pulmonary Hypertension. ESC Heart Fail. 2022, 9, 318–326. [Google Scholar] [CrossRef]
- University of Aarhus. Ketones for Pulmonary Hypertension—Effects on Hemodynamics (KEPAH). 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04615754 (accessed on 22 March 2023).
- Nielsen, R.; Christensen, K.H.; Gopalasingam, N.; Berg-Hansen, K.; Seefeldt, J.; Homilius, C.; Boedtkjer, E.; Andersen, M.J.; Wiggers, H.; Møller, N.; et al. Hemodynamic Effects of Ketone Bodies in Patients with Pulmonary Hypertension. J. Am. Heart Assoc. 2023, 12, e028232. [Google Scholar] [CrossRef]
- Blake, M.; Puchalska, P.; Kazmirczak, F.; Thenappan, T.; Crawford, P.A.; Prins, K.W. Ketone Bodies in Right Ventricular Failure: A Unique Therapeutic Opportunity. bioRxiv 2023. [Google Scholar] [CrossRef]
- McCullough, D.J.; Kue, N.; Mancini, T.; Vang, A.; Clements, R.T.; Choudhary, G. Endurance Exercise Training in Pulmonary Hypertension Increases Skeletal Muscle Electron Transport Chain Supercomplex Assembly. Pulm. Circ. 2020, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cawthon, D.; Beers, K.; Bottje, W.G. Electron Transport Chain Defect and Inefficient Respiration May Underlie Pulmonary Hypertension Syndrome (Ascites)-Associated Mitochondrial Dysfunction in Broilers. Poult. Sci. 2001, 80, 474–484. [Google Scholar] [CrossRef]
- Xu, W.; Comhair, S.A.A.; Chen, R.; Hu, B.; Hou, Y.; Zhou, Y.; Mavrakis, L.A.; Janocha, A.J.; Li, L.; Zhang, D.; et al. Integrative Proteomics and Phosphoproteomics in Pulmonary Arterial Hypertension. Sci. Rep. 2019, 9, 18623. [Google Scholar] [CrossRef] [Green Version]
- Huertas, A.; Tu, L.; Humbert, M.; Guignabert, C. Chronic Inflammation within the Vascular Wall in Pulmonary Arterial Hypertension: More than a Spectator. Cardiovasc. Res. 2020, 116, 885–893. [Google Scholar] [CrossRef]
- Fowler, E.D.; Hauton, D.; Boyle, J.; Egginton, S.; Steele, D.S.; White, E. Energy Metabolism in the Failing Right Ventricle: Limitations of Oxygen Delivery and the Creatine Kinase System. Int. J. Mol. Sci. 2019, 20, 1805. [Google Scholar] [CrossRef] [Green Version]
- Garcia, A.M.; Toni, L.S.; Miyano, C.A.; Sparagna, G.C.; Jonscher, R.; Phillips, E.K.; Karimpour-Fard, A.; Chapman, H.L.; Baybayon-Grandgeorge, A.N.; Pietra, A.E.; et al. Cardiac Transcriptome Remodeling and Impaired Bioenergetics in Single-Ventricle Congenital Heart Disease. JACC Basic Transl. Sci. 2023, 8, 258–279. [Google Scholar] [CrossRef]
- Pires da Silva, J.; Pietra, A.E.; Baybayon-Grandgeorge, A.N.; Garcia, A.M. Serum Metabolic Profiling Identifies Key Differences between Patients with Single-Ventricle Heart Disease and Healthy Controls. Int. J. Transl. Med. 2022, 2, 78–96. [Google Scholar] [CrossRef]
- Xu, X.; Lin, J.-H.I.; Bais, A.S.; Reynolds, M.J.; Tan, T.; Gabriel, G.C.; Kondos, Z.; Liu, X.; Shiva, S.S.; Lo, C.W. Mitochondrial Respiration Defects in Single-Ventricle Congenital Heart Disease. Front. Cardiovasc. Med. 2021, 8, 734388. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA Damage and Dysfunction Associated with Oxidative Stress in Failing Hearts After Myocardial Infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef]
- Tsutsui, H. Mitochondrial Oxidative Stress and Heart Failure. Intern. Med. 2006, 45, 809–813. [Google Scholar] [CrossRef] [Green Version]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 281, 785–789. [Google Scholar] [CrossRef]
- Koeslag, J.H.; Noakes, T.D.; Sloan, A.W. Post-Exercise Ketosis. J. Physiol. 1980, 301, 79–90. [Google Scholar] [CrossRef]
- Robinson, A.M.; Williamson, D.H. Physiological Roles of Ketone Bodies as Substrates and Signals in Mammalian Tissues. Physiol. Rev. 1980, 60, 143–187. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Ho, K.L.; Pherwani, S.; Ketema, E.B. Ketone Metabolism in the Failing Heart. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2020, 1865, 158813. [Google Scholar] [CrossRef]
- Hue, L.; Taegtmeyer, H. The Randle Cycle Revisited: A New Head for an Old Hat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E578–E591. [Google Scholar] [CrossRef] [Green Version]
- Barger, P.M.; Kelly, D.P. PPAR Signaling in the Control of Cardiac Energy Metabolism. Trends Cardiovasc. Med. 2000, 10, 238–245. [Google Scholar] [CrossRef]
- Taegtmeyer, H.; Wilson, C.R.; Razeghi, P.; Sharma, S. Metabolic Energetics and Genetics in the Heart. Ann. N. Y. Acad. Sci. 2005, 1047, 208–218. [Google Scholar] [CrossRef]
- Kelly, D.P. PPARs of the Heart: Three Is a Crowd. Circ. Res. 2003, 92, 482–484. [Google Scholar] [CrossRef]
- Lehman, J.J.; Kelly, D.P. Gene Regulatory Mechanisms Governing Energy Metabolism during Cardiac Hypertrophic Growth. Heart Fail. Rev. 2002, 7, 175–185. [Google Scholar] [CrossRef]
- Razeghi, P.; Young, M.E.; Abbasi, S.; Taegtmeyer, H. Hypoxia in Vivo Decreases Peroxisome Proliferator-Activated Receptor α-Regulated Gene Expression in Rat Heart. Biochem. Biophys. Res. Commun. 2001, 287, 5–10. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, Y.; Ren, J. Acetylation in Cardiovascular Diseases: Molecular Mechanisms and Clinical Implications. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165836. [Google Scholar] [CrossRef]
- Shogren-Knaak, M.; Ishii, H.; Sun, J.-M.; Pazin, M.J.; Davie, J.R.; Peterson, C.L. Histone H4-K16 Acetylation Controls Chromatin Structure and Protein Interactions. Science 2006, 311, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Peleg, S.; Feller, C.; Ladurner, A.G.; Imhof, A. The Metabolic Impact on Histone Acetylation and Transcription in Ageing. Trends Biochem. Sci. 2016, 41, 700–711. [Google Scholar] [CrossRef] [Green Version]
- Verdin, E.; Dequiedt, F.; Kasler, H.G. Class II Histone Deacetylases: Versatile Regulators. Trends Genet. 2003, 19, 286–293. [Google Scholar] [CrossRef] [Green Version]
- McKinsey, T.A.; Olson, E.N. Cardiac Histone Acetylation—Therapeutic Opportunities Abound. Trends Genet. 2004, 20, 206–213. [Google Scholar] [CrossRef]
- Backs, J.; Olson, E.N. Control of Cardiac Growth by Histone Acetylation/Deacetylation. Circ. Res. 2006, 98, 15–24. [Google Scholar] [CrossRef]
- Allard, M.F.; Schonekess, B.O.; Henning, S.L.; English, D.R.; Lopaschuk, G.D. Contribution of Oxidative Metabolism and Glycolysis to ATP Production in Hypertrophied Hearts. Am. J. Physiol. Heart Circ. Physiol. 1994, 267, H742–H750. [Google Scholar] [CrossRef]
- Diakos, N.A.; Navankasattusas, S.; Abel, E.D.; Rutter, J.; McCreath, L.; Ferrin, P.; McKellar, S.H.; Miller, D.V.; Park, S.Y.; Richardson, R.S.; et al. Evidence of Glycolysis Up-Regulation and Pyruvate Mitochondrial Oxidation Mismatch During Mechanical Unloading of the Failing Human Heart. JACC Basic Transl. Sci. 2016, 1, 432–444. [Google Scholar] [CrossRef] [Green Version]
- Bottomley, P.A.; Panjrath, G.S.; Lai, S.; Hirsch, G.A.; Wu, K.; Najjar, S.S.; Steinberg, A.; Gerstenblith, G.; Weiss, R.G. Metabolic Rates of ATP Transfer Through Creatine Kinase (CK Flux) Predict Clinical Heart Failure Events and Death. Sci. Transl. Med. 2013, 5, 215re3. [Google Scholar] [CrossRef] [Green Version]
- Mey, J.T.; Hari, A.; Axelrod, C.L.; Fealy, C.E.; Erickson, M.L.; Kirwan, J.P.; Dweik, R.A.; Heresi, G.A. Lipids and Ketones Dominate Metabolism at the Expense of Glucose Control in Pulmonary Arterial Hypertension: A Hyperglycaemic Clamp and Metabolomics Study. Eur. Respir. J. 2020, 55, 1901700. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial Dysfunction in Pathophysiology of Heart Failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [Green Version]
- Takimoto, E.; Kass, D.A. Role of Oxidative Stress in Cardiac Hypertrophy and Remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating Oxidative Stress in Heart Failure: Past, Present and Future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Nascimben, L.; Ingwall, J.S.; Pauletto, P.; Friedrich, J.; Gwathmey, J.K.; Saks, V.; Pessina, A.C.; Allen, P.D. Creatine Kinase System in Failing and Nonfailing Human Myocardium. Circulation 1996, 94, 1894–1901. [Google Scholar] [CrossRef]
- Keceli, G.; Gupta, A.; Sourdon, J.; Gabr, R.; Schär, M.; Dey, S.; Tocchetti, C.G.; Stuber, A.; Agrimi, J.; Zhang, Y.; et al. Mitochondrial Creatine Kinase Attenuates Pathologic Remodeling in Heart Failure. Circ. Res. 2022, 130, 741–759. [Google Scholar] [CrossRef]
- Olson, E.N.; Backs, J.; McKinsey, T.A. Control of Cardiac Hypertrophy and Heart Failure by Histone Acetylation/Deacetylation. In Novartis Foundation Symposia; Bock, G., Goode, J., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2008; pp. 3–19. [Google Scholar] [CrossRef]
- Fukushima, A.; Zhang, L.; Huqi, A.; Lam, V.H.; Rawat, S.; Altamimi, T.; Wagg, C.S.; Dhaliwal, K.K.; Hornberger, L.K.; Kantor, P.F.; et al. Acetylation Contributes to Hypertrophy-Caused Maturational Delay of Cardiac Energy Metabolism. JCI Insight 2018, 3, e99239. [Google Scholar] [CrossRef] [Green Version]
- Castillo, E.C.; Morales, J.A.; Chapoy-Villanueva, H.; Silva-Platas, C.; Treviño-Saldaña, N.; Guerrero-Beltrán, C.E.; Bernal-Ramírez, J.; Torres-Quintanilla, A.; García, N.; Youker, K.; et al. Mitochondrial Hyperacetylation in the Failing Hearts of Obese Patients Mediated Partly by a Reduction in SIRT3: The Involvement of the Mitochondrial Permeability Transition Pore. Cell Physiol. Biochem. 2019, 53, 465–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, J.Y.Y.; Tuano, N.K.; Rafehi, H.; Gao, X.-M.; Ziemann, M.; Du, X.-J.; El-Osta, A. HDAC Inhibition Attenuates Cardiac Hypertrophy by Acetylation and Deacetylation of Target Genes. Epigenetics 2015, 10, 418–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.-H.; Seok, Y.M.; Song, M.; Lee, H.-A.; Kurz, T.; Kim, I. Histone Deacetylase Inhibition Attenuates Cardiac Hypertrophy and Fibrosis through Acetylation of Mineralocorticoid Receptor in Spontaneously Hypertensive Rats. Mol. Pharmacol. 2015, 87, 782–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, B.; Schulte, J.S.; Hamer, S.; Himmler, K.; Pluteanu, F.; Seidl, M.D.; Stein, J.; Wardelmann, E.; Hammer, E.; Völker, U.; et al. HDAC (Histone Deacetylase) Inhibitor Valproic Acid Attenuates Atrial Remodeling and Delays the Onset of Atrial Fibrillation in Mice. Circ. Arrhythm. Electrophysiol. 2019, 12, e007071. [Google Scholar] [CrossRef]
- Colussi, C.; Berni, R.; Rosati, J.; Straino, S.; Vitale, S.; Spallotta, F.; Baruffi, S.; Bocchi, L.; Delucchi, F.; Rossi, S.; et al. The Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid Reduces Cardiac Arrhythmias in Dystrophic Mice. Cardiovasc. Res. 2010, 87, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Bertsch, T.; Volke, J.; Schmid, A.; Klingbeil, R.; Metodiev, Y.; Karaca, B.; Kim, S.-H.; Lindner, S.; Schupp, T.; et al. Narrative Review of Metabolomics in Cardiovascular Disease. J. Thorac. Dis. 2021, 13, 2532–2550. [Google Scholar] [CrossRef]
- Bassareo, P.P.; McMahon, C.J. Metabolomics: A New Tool in Our Understanding of Congenital Heart Disease. Children 2022, 9, 1803. [Google Scholar] [CrossRef]
- Michel, M.; Laser, K.T.; Dubowy, K.-O.; Scholl-Bürgi, S.; Michel, E. Metabolomics and Random Forests in Patients with Complex Congenital Heart Disease. Front. Cardiovasc. Med. 2022, 9, 994068. [Google Scholar] [CrossRef]
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive Quantification of Fuel Use by the Failing and Nonfailing Human Heart. Science 2020, 370, 364–368. [Google Scholar] [CrossRef]
- Smith, E.; Fernandez, C.; Melander, O.; Ottosson, F. Altered Acylcarnitine Metabolism Is Associated With an Increased Risk of Atrial Fibrillation. J. Am. Heart Assoc. 2020, 9, e016737. [Google Scholar] [CrossRef]
- Ruiz, M.; Labarthe, F.; Fortier, A.; Bouchard, B.; Thompson Legault, J.; Bolduc, V.; Rigal, O.; Chen, J.; Ducharme, A.; Crawford, P.A.; et al. Circulating Acylcarnitine Profile in Human Heart Failure: A Surrogate of Fatty Acid Metabolic Dysregulation in Mitochondria and Beyond. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H768–H781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penny, D.J.; Redington, A.N. Function of the Left and Right Ventricles and the Interactions Between Them. Pediatr. Crit. Care Med. 2016, 17, S112–S118. [Google Scholar] [CrossRef] [PubMed]
- Sanz, J.; Sánchez-Quintana, D.; Bossone, E.; Bogaard, H.J.; Naeije, R. Anatomy, Function, and Dysfunction of the Right Ventricle. J. Am. Coll. Cardiol. 2019, 73, 1463–1482. [Google Scholar] [CrossRef] [PubMed]
- Kondo, R.P.; Dederko, D.A.; Teutsch, C.; Chrast, J.; Catalucci, D.; Chien, K.R.; Giles, W.R. Comparison of Contraction and Calcium Handling between Right and Left Ventricular Myocytes from Adult Mouse Heart: A Role for Repolarization Waveform: Interventricular Heterogeneity of Cardiac Myocyte Contractions. J. Physiol. 2006, 571, 131–146. [Google Scholar] [CrossRef] [Green Version]
- Sedmera, D. Form Follows Function: Developmental and Physiological View on Ventricular Myocardial Architecture. Eur. J. Cardiothorac. Surg. 2005, 28, 526–528. [Google Scholar] [CrossRef] [Green Version]
- Garcia, A.M.; Beatty, J.-T.; Nakano, S.J. Heart Failure in Single Right Ventricle Congenital Heart Disease: Physiological and Molecular Considerations. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H947–H965. [Google Scholar] [CrossRef] [PubMed]
- Friehs, I.; Cowan, D.B.; Choi, Y.-H.; Black, K.M.; Barnett, R.; Bhasin, M.K.; Daly, C.; Dillon, S.J.; Libermann, T.A.; McGowan, F.X.; et al. Pressure-Overload Hypertrophy of the Developing Heart Reveals Activation of Divergent Gene and Protein Pathways in the Left and Right Ventricular Myocardium. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H697–H708. [Google Scholar] [CrossRef] [Green Version]
- Schreckenberg, R.; Rebelo, M.; Deten, A.; Weber, M.; Rohrbach, S.; Pipicz, M.; Csonka, C.; Ferdinandy, P.; Schulz, R.; Schlüter, K.-D. Specific Mechanisms Underlying Right Heart Failure: The Missing Upregulation of Superoxide Dismutase-2 and Its Decisive Role in Antioxidative Defense. Antioxid. Redox Signal. 2015, 23, 1220–1232. [Google Scholar] [CrossRef]
- Nagendran, J.; Gurtu, V.; Fu, D.Z.; Dyck, J.R.B.; Haromy, A.; Ross, D.B.; Rebeyka, I.M.; Michelakis, E.D. A Dynamic and Chamber-Specific Mitochondrial Remodeling in Right Ventricular Hypertrophy Can Be Therapeutically Targeted. J. Thorac. Cardiovasc. Surg. 2008, 136, 168–178.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokhari, S.; Raina, A.; Berman Rosenweig, E.; Schulze, P.C.; Bokhari, J.; Einstein, A.J.; Barst, R.J.; Johnson, L.L. PET Imaging May Provide a Novel Biomarker and Understanding of Right Ventricular Dysfunction in Patients With Idiopathic Pulmonary Arterial Hypertension. Circ. Cardiovasc. Imaging 2011, 4, 641–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Arroyo, J.; Mizuno, S.; Szczepanek, K.; Van Tassell, B.; Natarajan, R.; dos Remedios, C.G.; Drake, J.I.; Farkas, L.; Kraskauskas, D.; Wijesinghe, D.S.; et al. Metabolic Gene Remodeling and Mitochondrial Dysfunction in Failing Right Ventricular Hypertrophy Secondary to Pulmonary Arterial Hypertension. Circ. Heart Fail. 2013, 6, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piao, L.; Marsboom, G.; Archer, S.L. Mitochondrial Metabolic Adaptation in Right Ventricular Hypertrophy and Failure. J. Mol. Med. 2010, 88, 1011–1020. [Google Scholar] [CrossRef] [Green Version]
- Fessel, J.P.; Hamid, R.; Wittmann, B.M.; Robinson, L.J.; Blackwell, T.; Tada, Y.; Tanabe, N.; Tatsumi, K.; Hemnes, A.R.; West, J.D. Metabolomic Analysis of Bone Morphogenetic Protein Receptor Type 2 Mutations in Human Pulmonary Endothelium Reveals Widespread Metabolic Reprogramming. Pulm. Circ. 2012, 2, 201–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schooneman, M.G.; Vaz, F.M.; Houten, S.M.; Soeters, M.R. Acylcarnitines. Diabetes 2013, 62, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalim, S.; Clish, C.B.; Wenger, J.; Elmariah, S.; Yeh, R.W.; Deferio, J.J.; Pierce, K.; Deik, A.; Gerszten, R.E.; Thadhani, R.; et al. A Plasma Long-Chain Acylcarnitine Predicts Cardiovascular Mortality in Incident Dialysis Patients. J. Am. Heart Assoc. 2013, 2, e000542. [Google Scholar] [CrossRef] [Green Version]
- Aitken-Buck, H.M.; Krause, J.; Zeller, T.; Jones, P.P.; Lamberts, R.R. Long-Chain Acylcarnitines and Cardiac Excitation-Contraction Coupling: Links to Arrhythmias. Front. Physiol. 2020, 11, 577856. [Google Scholar] [CrossRef]
- Brunner, N.W.; Skhiri, M.; Fortenko, O.; Hsi, A.; Haddad, F.; Khazeni, N.; Zamanian, R.T. Impact of Insulin Resistance on Ventricular Function in Pulmonary Arterial Hypertension. J. Heart Lung Transplant. 2014, 33, 721–726. [Google Scholar] [CrossRef]
- Assad, T.R.; Hemnes, A.R. Metabolic Dysfunction in Pulmonary Arterial Hypertension. Curr. Hypertens. Rep. 2015, 17, 20. [Google Scholar] [CrossRef] [Green Version]
- Graham, B.B.; Kumar, R.; Mickael, C.; Sanders, L.; Gebreab, L.; Huber, K.M.; Perez, M.; Smith-Jones, P.; Serkova, N.J.; Tuder, R.M. Severe Pulmonary Hypertension Is Associated with Altered Right Ventricle Metabolic Substrate Uptake. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L435–L440. [Google Scholar] [CrossRef] [Green Version]
- Ohuchi, H.; Negishi, J.; Hayama, Y.; Miike, H.; Suzuki, D.; Nakajima, K.; Konagai, N.; Iwasa, T.; Sakaguchi, H.; Kurosaki, K.; et al. Abnormal Glucose Metabolism in Patients with Fontan Circulation: Unique Characteristics and Associations with Fontan Pathophysiology. Am. Heart J. 2019, 216, 125–135. [Google Scholar] [CrossRef]
- Sharma, S.; Sud, N.; Wiseman, D.A.; Carter, A.L.; Kumar, S.; Hou, Y.; Rau, T.; Wilham, J.; Harmon, C.; Oishi, P.; et al. Altered Carnitine Homeostasis Is Associated with Decreased Mitochondrial Function and Altered Nitric Oxide Signaling in Lambs with Pulmonary Hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L46–L56. [Google Scholar] [CrossRef]
- Fowler, E.D.; Benoist, D.; Drinkhill, M.J.; Stones, R.; Helmes, M.; Wüst, R.C.I.; Stienen, G.J.M.; Steele, D.S.; White, E. Decreased Creatine Kinase Is Linked to Diastolic Dysfunction in Rats with Right Heart Failure Induced by Pulmonary Artery Hypertension. J. Mol. Cell. Cardiol. 2015, 86, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Bernal-Ramirez, J.; Díaz-Vesga, M.C.; Talamilla, M.; Méndez, A.; Quiroga, C.; Garza-Cervantes, J.A.; Lázaro-Alfaro, A.; Jerjes-Sanchez, C.; Henríquez, M.; García-Rivas, G.; et al. Exploring Functional Differences between the Right and Left Ventricles to Better Understand Right Ventricular Dysfunction. Oxidative Med. Cell. Longev. 2021, 2021, 9993060. [Google Scholar] [CrossRef]
- Bogaard, H.J.; Mizuno, S.; Hussaini, A.A.A.; Toldo, S.; Abbate, A.; Kraskauskas, D.; Kasper, M.; Natarajan, R.; Voelkel, N.F. Suppression of Histone Deacetylases Worsens Right Ventricular Dysfunction after Pulmonary Artery Banding in Rats. Am. J. Respir. Crit. Care Med. 2011, 183, 1402–1410. [Google Scholar] [CrossRef]
- Chelladurai, P.; Boucherat, O.; Stenmark, K.; Kracht, M.; Seeger, W.; Bauer, U.; Bonnet, S.; Pullamsetti, S.S. Targeting Histone Acetylation in Pulmonary Hypertension and Right Ventricular Hypertrophy. Br. J. Pharmacol. 2021, 178, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Luo, F.; Wu, P.; Huang, Y.; Das, A.; Chen, S.; Chen, J.; Hu, X.; Li, F.; Fang, Z.; et al. Metabolomics Reveals Metabolite Changes of Patients with Pulmonary Arterial Hypertension in China. J. Cell. Mol. Med. 2020, 24, 2484–2496. [Google Scholar] [CrossRef]
- Rawat, S.; Fukushima, A.; Zhang, L.; Hugi, A.; Lam, V.; Altamimi, T.; Wagg, C.; Petinelli, R.; Dhaliwal, K.; Hornberger, L.; et al. Control of cardiac fatty acid metabolism in infants with hypoplastic left heart syndrome. J. Mol. Cell. Cardiol. 2018, 124, 91–92. [Google Scholar] [CrossRef]
- Motoki, N.; Motoki, H.; Utsumi, M.; Yamazaki, S.; Obinata, H.; Takei, K.; Yasukochi, S. Identification of metabolomic profile related to adult Fontan pathophysiology. Int. J. Cardiol. Heart Vasc. 2021, 37, 100921. [Google Scholar] [CrossRef]
- Li, M.; Zhou, S.; Chen, C.; Ma, L.; Luo, D.; Tian, X.; Dong, X.; Zhou, Y.; Yang, Y.; Cui, Y. Therapeutic Potential of Pyruvate Therapy for Patients with Mitochondrial Diseases: A Systematic Review. Ther. Adv. Endocrinol. Metab. 2020, 11, 204201882093824. [Google Scholar] [CrossRef]
- Des Rosiers, C.; Labarthe, F.; Lloyd, S.G.; Chatham, J.C. Cardiac Anaplerosis in Health and Disease: Food for Thought. Cardiovasc. Res. 2011, 90, 210–219. [Google Scholar] [CrossRef] [Green Version]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Verma, S. Mechanisms of Cardiovascular Benefits of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors. JACC Basic Transl. Sci. 2020, 5, 632–644. [Google Scholar] [CrossRef]
- Maejima, Y. SGLT2 Inhibitors Play a Salutary Role in Heart Failure via Modulation of the Mitochondrial Function. Front. Cardiovasc. Med. 2020, 6, 186. [Google Scholar] [CrossRef]
- Pietschner, R.; Kolwelter, J.; Bosch, A.; Striepe, K.; Jung, S.; Kannenkeril, D.; Ott, C.; Schiffer, M.; Achenbach, S.; Schmieder, R.E. Effect of Empagliflozin on Ketone Bodies in Patients with Stable Chronic Heart Failure. Cardiovasc. Diabetol. 2021, 20, 219. [Google Scholar] [CrossRef]
- Takada, S.; Sabe, H.; Kinugawa, S. Treatments for Skeletal Muscle Abnormalities in Heart Failure: Sodium-Glucose Transporter 2 and Ketone Bodies. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H117–H128. [Google Scholar] [CrossRef]
- Muneuchi, J.; Sugitani, Y.; Kobayashi, M.; Ezaki, H.; Yamada, H.; Watanabe, M. Feasibility and Safety of Sodium Glucose Cotransporter-2 Inhibitors in Adults with Heart Failure after the Fontan Procedure. Case Rep. Cardiol. 2022, 2022, 5243594. [Google Scholar] [CrossRef]
- Ghelani, S.J.; Opotowsky, A.R.; Harrild, D.M.; Powell, A.J.; Azcue, N.; Ahmad, S.; Clair, N.S.; Bradwin, G.; Rathod, R.H. Characterization of Circulating and Urinary Biomarkers in the Fontan Circulation and Their Correlation with Cardiac Imaging. Am. J. Cardiol. 2022, 162, 177–183. [Google Scholar] [CrossRef]
- van den Bosch, E.; Bossers, S.S.M.; Kamphuis, V.P.; Boersma, E.; Roos-Hesselink, J.W.; Breur, J.M.P.J.; Ten Harkel, A.D.J.; Kapusta, L.; Bartelds, B.; Roest, A.A.W.; et al. Associations Between Blood Biomarkers, Cardiac Function, and Adverse Outcome in a Young Fontan Cohort. J. Am. Heart Assoc. 2021, 10, e015022. [Google Scholar] [CrossRef]
- Gom, R.C.; Bhatt, D.; Villa, B.R.; George, A.G.; Lohman, A.W.; Mychasiuk, R.; Rho, J.M.; Teskey, G.C. The Ketogenic Diet Raises Brain Oxygen Levels, Attenuates Postictal Hypoxia, and Protects against Learning Impairments. Neurobiol. Dis. 2021, 154, 105335. [Google Scholar] [CrossRef]
- Trevisan, R.; Nosadini, R.; Fioretto, P.; Avogaro, A.; Duner, E.; Jori, E.; Valerio, A.; Doria, A.; Crepaldi, G. Ketone Bodies Increase Glomerular Filtration Rate in Normal Man and in Patients with Type 1 (Insulin-Dependent) Diabetes Mellitus. Diabetologia 1987, 30, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Wang, D.D.-H.; Qiu, Y.; Airhart, S.; Liu, Y.; Stempien-Otero, A.; O’Brien, K.D.; Tian, R. Boosting NAD Level Suppresses Inflammatory Activation of PBMCs in Heart Failure. J. Clin. Investig. 2020, 130, 6054–6063. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Persad, K.L. Failure to Launch. JACC Basic Transl. Sci. 2023, 8, 280–282. [Google Scholar] [CrossRef]
- Frank, B.S.; Khailova, L.; Silveira, L.; Mitchell, M.B.; Morgan, G.J.; Hsieh, E.W.Y.; DiMaria, M.V.; Twite, M.; Klawitter, J.; Davidson, J.A. Proteomic profiling identifies key differences between inter-stage infants with single ventricle heart disease and healthy controls. Transl. Res. 2021, 229, 24–37. [Google Scholar] [CrossRef]
- Ohuchi, H.; Miyamoto, Y.; Yamamoto, M.; Ishihara, H.; Takata, H.; Miyazaki, A.; Yamada, O.; Yagihara, T. High Prevalence of Abnormal Glucose Metabolism in Young Adult Patients with Complex Congenital Heart Disease. Am. Heart J. 2009, 158, 30–39. [Google Scholar] [CrossRef]
- Whiteside, W.; Tan, M.; Ostlund, R.E.; Yu, S.; Ma, L.; Rocchini, A. Altered Cholesterol Metabolism and Hypocholesterolemia in Patients with Single Ventricle Following Fontan Palliation. J. Pediatr. 2016, 171, 73–77. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, W.; Tan, M.; Yu, S.; Rocchini, A. Low Total, Low-Density Lipoprotein, High-Density Lipoprotein, and Non–High-Density Lipoprotein Cholesterol Levels in Patients with Complex Congenital Heart Disease after Fontan Palliation. J. Pediatr. 2013, 162, 1199–1204. [Google Scholar] [CrossRef]
- Lubert, A.M.; Alsaied, T.; Palermo, J.J.; Anwar, N.; Urbina, E.M.; Brown, N.M.; Alexander, C.; Almeneisi, H.; Wu, F.; Leventhal, A.R.; et al. Fontan-Associated Dyslipidemia. J. Am. Heart Assoc. 2021, 10, e019578. [Google Scholar] [CrossRef]
- Zyblewski, S.C.; Argraves, W.S.; Graham, E.M.; Slate, E.H.; Atz, A.M.; Bradley, S.M.; McQuinn, T.C.; Wilkerson, B.A.; Wing, S.B.; Argraves, K.M. Reduction in postoperative high-density lipoprotein cholesterol levels in children undergoing the Fontan operation. Pediatr. Cardiol. 2012, 33, 1154–1159. [Google Scholar] [CrossRef] [Green Version]
- Saraf, A.; De Staercke, C.; Everitt, I.; Haouzi, A.; Ko, Y.A.; Jennings, S.; Kim, J.H.; Rodriguez, F.H.; Kalogeropoulos, A.P.; Quyyumi, A.; et al. Biomarker profile in stable Fontan patients. Int. J. Cardiol. 2020, 305, 56–62. [Google Scholar] [CrossRef]
- Fillmore, N.; Lopaschuk, G.D. Targeting Mitochondrial Oxidative Metabolism as an Approach to Treat Heart Failure. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Karwi, Q.G.; Biswas, D.; Pulinilkunnil, T.; Lopaschuk, G.D. Myocardial Ketones Metabolism in Heart Failure. J. Card. Fail. 2020, 26, 998–1005. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Dyck, J.R.B. Ketones and the Cardiovascular System. Nat. Cardiovasc. Res. 2023, 2, 425–437. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Sato, K.; Tsuchiya, N.; Thomas, S.; Fell, D.A.; Veech, R.L.; Passonneau, J.V. Control of Glucose Utilization in Working Perfused Rat Heart. J. Biol. Chem. 1994, 269, 25502–25514. [Google Scholar] [CrossRef]
- Cahill, G.F.; Veech, R.L. Ketoacids? Good Medicine? Trans. Am. Clin. Climatol. Assoc. 2003, 114, 149–163. [Google Scholar]
- Veech, R.L.; Chance, B.; Kashiwaya, Y.; Lardy, H.A.; Cahill, G.F., Jr. Ketone Bodies, Potential Therapeutic Uses. IUBMB Life Int. Union Biochem. Mol. Biol. Life 2001, 51, 241–247. [Google Scholar] [CrossRef]
- Veech, R.L. The Therapeutic Implications of Ketone Bodies: The Effects of Ketone Bodies in Pathological Conditions: Ketosis, Ketogenic Diet, Redox States, Insulin Resistance, and Mitochondrial Metabolism. Prostaglandins Leukot. Essent. Fatty Acids 2004, 70, 309–319. [Google Scholar] [CrossRef]
- Sato, K.; Kashiwaya, Y.; Keon, C.A.; Tsuchiya, N.; King, M.T.; Radda, G.K.; Chance, B.; Clarke, K.; Veech, R.L. Insulin, Ketone Bodies, and Mitochondrial Energy Transduction. FASEB J. 1995, 9, 651–658. [Google Scholar] [CrossRef]
- Mudaliar, S.; Alloju, S.; Henry, R.R. Can a Shift in Fuel Energetics Explain the Beneficial Cardiorenal Outcomes in the EMPA-REG OUTCOME Study? A Unifying Hypothesis. Diabetes Care 2016, 39, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Ho, K.L.; Karwi, Q.G.; Wagg, C.; Zhang, L.; Vo, K.; Altamimi, T.; Uddin, G.M.; Ussher, J.R.; Lopaschuk, G.D. Ketones Can Become the Major Fuel Source for the Heart but Do Not Increase Cardiac Efficiency. Cardiovasc. Res. 2021, 117, 1178–1187. [Google Scholar] [CrossRef]
- Berg-Hansen, K.; Christensen, K.H.; Gopalasingam, N.; Nielsen, R.; Eiskjær, H.; Møller, N.; Birkelund, T.; Christensen, S.; Wiggers, H. Beneficial Effects of Ketone Ester in Patients with Cardiogenic Shock: A Randomized, Controlled, Double-Blind Trial. JACC Heart Fail. 2023; ahead of print. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G. Jump Starting the Heart: Ketone Esters Improve Cardiac Function in Patients with Cardiogenic Shock. JACC Heart Fail. 2023, in press. [CrossRef]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef]
- Grieve, D. Oxidative Stress in Heart Failure More than Just Damage. Eur. Heart J. 2003, 24, 2161–2163. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of Oxidative Stress by β-Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Zhang, C.; Shang, F.-F.; Luo, M.; You, Y.; Zhai, Q.; Xia, Y.; Suxin, L. Ketogenic Diet Ameliorates Cardiac Dysfunction via Balancing Mitochondrial Dynamics and Inhibiting Apoptosis in Type 2 Diabetic Mice. Aging Dis. 2020, 11, 229. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.P.; Kakkar, R.; McCarthy, C.P.; Januzzi, J.L. Inflammation in Heart Failure. J. Am. Coll. Cardiol. 2020, 75, 1324–1340. [Google Scholar] [CrossRef]
- Shirazi, L.F.; Bissett, J.; Romeo, F.; Mehta, J.L. Role of Inflammation in Heart Failure. Curr. Atheroscler. Rep. 2017, 19, 27. [Google Scholar] [CrossRef]
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the Role of Inflammation in Heart Failure. Nat. Rev. Cardiol. 2020, 17, 269–285. [Google Scholar] [CrossRef]
- Suetomi, T.; Willeford, A.; Brand, C.S.; Cho, Y.; Ross, R.S.; Miyamoto, S.; Brown, J.H. Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca2+/Calmodulin-Dependent Protein Kinase II δ Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation 2018, 138, 2530–2544. [Google Scholar] [CrossRef] [Green Version]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The Ketone Metabolite β-Hydroxybutyrate Blocks NLRP3 Inflammasome–Mediated Inflammatory Disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones Inhibit Mitochondrial Production of Reactive Oxygen Species Production Following Glutamate Excitotoxicity by Increasing NADH Oxidation. Neuroscience 2007, 145, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Forsythe, C.E.; Phinney, S.D.; Fernandez, M.L.; Quann, E.E.; Wood, R.J.; Bibus, D.M.; Kraemer, W.J.; Feinman, R.D.; Volek, J.S. Comparison of Low Fat and Low Carbohydrate Diets on Circulating Fatty Acid Composition and Markers of Inflammation. Lipids 2008, 43, 65–77. [Google Scholar] [CrossRef]
- Greco, T.; Glenn, T.C.; Hovda, D.A.; Prins, M.L. Ketogenic Diet Decreases Oxidative Stress and Improves Mitochondrial Respiratory Complex Activity. J. Cereb. Blood Flow Metab. 2016, 36, 1603–1613. [Google Scholar] [CrossRef]
- Yang, D.; Liu, H.-Q.; Liu, F.-Y.; Guo, Z.; An, P.; Wang, M.-Y.; Yang, Z.; Fan, D.; Tang, Q.-Z. Mitochondria in Pathological Cardiac Hypertrophy Research and Therapy. Front. Cardiovasc. Med. 2022, 8, 822969. [Google Scholar] [CrossRef]
- Abel, E.D.; Doenst, T. Mitochondrial Adaptations to Physiological vs. Pathological Cardiac Hypertrophy. Cardiovasc. Res. 2011, 90, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Kolwicz, S.C.; Tian, R. Glucose Metabolism and Cardiac Hypertrophy. Cardiovasc. Res. 2011, 90, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial Fatty Acid Metabolism in Health and Disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Kee, H.J.; Kook, H. Roles and Targets of Class I and IIa Histone Deacetylases in Cardiac Hypertrophy. J. Biomed. Biotechnol. 2011, 2011, 928326. [Google Scholar] [CrossRef] [Green Version]
- Hewitson, R.; Dargan, J.; Collis, D.; Green, A.; Moorjani, N.; Ohri, S.; Townsend, P.A. Heart Failure: The Pivotal Role of Histone Deacetylases. Int. J. Biochem. Cell Biol. 2013, 45, 448–453. [Google Scholar] [CrossRef]
- Yurista, S.R.; Matsuura, T.R.; Silljé, H.H.W.; Nijholt, K.T.; McDaid, K.S.; Shewale, S.V.; Leone, T.C.; Newman, J.C.; Verdin, E.; van Veldhuisen, D.J.; et al. Ketone Ester Treatment Improves Cardiac Function and Reduces Pathologic Remodeling in Preclinical Models of Heart Failure. Circ. Heart Fail. 2021, 14, e007684. [Google Scholar] [CrossRef]
- Nakamura, M.; Odanovic, N.; Nakada, Y.; Dohi, S.; Zhai, P.; Ivessa, A.; Yang, Z.; Abdellatif, M.; Sadoshima, J. Dietary Carbohydrates Restriction Inhibits the Development of Cardiac Hypertrophy and Heart Failure. Cardiovasc. Res. 2021, 117, 2365–2376. [Google Scholar] [CrossRef]
- Okere, I.C.; Young, M.E.; McElfresh, T.A.; Chess, D.J.; Sharov, V.G.; Sabbah, H.N.; Hoit, B.D.; Ernsberger, P.; Chandler, M.P.; Stanley, W.C. Low Carbohydrate/High-Fat Diet Attenuates Cardiac Hypertrophy, Remodeling, and Altered Gene Expression in Hypertension. Hypertension 2006, 48, 1116–1123. [Google Scholar] [CrossRef] [Green Version]
- Egbe, A.C.; Connolly, H.M.; Miranda, W.R.; Ammash, N.M.; Hagler, D.J.; Veldtman, G.R.; Borlaug, B.A. Hemodynamics of Fontan Failure: The Role of Pulmonary Vascular Disease. Circ. Heart Fail. 2017, 10, e004515. [Google Scholar] [CrossRef]
- Castaldi, B.; Bordin, G.; Padalino, M.; Cuppini, E.; Vida, V.; Milanesi, O. Hemodynamic Impact of Pulmonary Vasodilators on Single Ventricle Physiology. Cardiovasc. Ther. 2018, 36, e12314. [Google Scholar] [CrossRef] [Green Version]
- Zuchi, C.; Tritto, I.; Carluccio, E.; Mattei, C.; Cattadori, G.; Ambrosio, G. Role of Endothelial Dysfunction in Heart Failure. Heart Fail. Rev. 2020, 25, 21–30. [Google Scholar] [CrossRef]
- Giannitsi, S.; Maria, B.; Bechlioulis, A.; Naka, K. Endothelial Dysfunction and Heart Failure: A Review of the Existing Bibliography with Emphasis on Flow Mediated Dilation. JRSM Cardiovasc. Dis. 2019, 8, 204800401984304. [Google Scholar] [CrossRef]
- McCarthy, C.G.; Chakraborty, S.; Singh, G.; Yeoh, B.S.; Schreckenberger, Z.J.; Singh, A.; Mell, B.; Bearss, N.R.; Yang, T.; Cheng, X.; et al. Ketone Body β-Hydroxybutyrate Is an Autophagy-Dependent Vasodilator. JCI Insight 2021, 6, e149037. [Google Scholar] [CrossRef]
- Gormsen, L.C.; Svart, M.; Thomsen, H.H.; Søndergaard, E.; Vendelbo, M.H.; Christensen, N.; Tolbod, L.P.; Harms, H.J.; Nielsen, R.; Wiggers, H.; et al. Ketone Body Infusion With 3-Hydroxybutyrate Reduces Myocardial Glucose Uptake and Increases Blood Flow in Humans: A Positron Emission Tomography Study. J. Am. Heart Assoc. 2017, 6, e005066. [Google Scholar] [CrossRef]
- Ibrahim, A. The Effect of Ketone on β-Aminopropionitrile-Induced Vascular Remodeling. Master’s Thesis, Georgia State University, Atlanta, GA, USA, 2022. [Google Scholar] [CrossRef]
- Coleman, K.; Phillips, J.; Sciarini, M.; Stubbs, B.; Jackson, O.; Kernagis, D. A Metabolic Intervention for Improving Human Cognitive Performance During Hypoxia. Aerosp. Med. Hum. Perform. 2021, 92, 556–562. [Google Scholar] [CrossRef]
- Prins, P.J.; Buxton, J.D.; McClure, T.S.; D’Agostino, D.P.; Ault, D.L.; Welton, G.L.; Jones, D.W.; Atwell, A.D.; Slack, M.A.; Slack, M.L.; et al. Ketone Bodies Impact on Hypoxic CO2 Retention Protocol During Exercise. Front. Physiol. 2021, 12, 780755. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Pawlosky, R.; Markis, W.; King, M.T.; Bergman, C.; Srivastava, S.; Murray, A.; Clarke, K.; Veech, R.L. A Ketone Ester Diet Increases Brain Malonyl-CoA and Uncoupling Proteins 4 and 5 While Decreasing Food Intake in the Normal Wistar Rat. J. Biol. Chem. 2010, 285, 25950–25956. [Google Scholar] [CrossRef] [Green Version]
- Puchalska, P.; Crawford, P.A. Multi-Dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Roberts, C.; McKenzie, A.; George, M.P. Nutritional Ketosis to Treat Pulmonary Hypertension Associated with Obesity and Metabolic Syndrome: A Case Report. Pulm. Circ. 2021, 11, 2045894021991426. [Google Scholar] [CrossRef]
- Chowdhury, B.; Luu, A.Z.; Luu, V.Z.; Kabir, M.G.; Pan, Y.; Teoh, H.; Quan, A.; Sabongui, S.; Al-Omran, M.; Bhatt, D.L.; et al. The SGLT2 Inhibitor Empagliflozin Reduces Mortality and Prevents Progression in Experimental Pulmonary Hypertension. Biochem. Biophys. Res. Commun. 2020, 524, 50–56. [Google Scholar] [CrossRef]
- Rychik, J. The Relentless Effects of the Fontan Paradox. Semin. Thorac. Cardiovasc. Surg. Pediatr. Card. Surg. Annu. 2016, 19, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Mooli, R.G.R.; Ramakrishnan, S.K. Emerging Role of Hepatic Ketogenesis in Fatty Liver Disease. Front. Physiol. 2022, 13, 946474. [Google Scholar] [CrossRef]
- Liao, Y.-J.; Wang, Y.-H.; Wu, C.-Y.; Hsu, F.-Y.; Chien, C.-Y.; Lee, Y.-C. Ketogenic Diet Enhances the Cholesterol Accumulation in Liver and Augments the Severity of CCl4 and TAA-Induced Liver Fibrosis in Mice. Int. J. Mol. Sci. 2021, 22, 2934. [Google Scholar] [CrossRef]
- Moore, M.P.; Cunningham, R.P.; Davis, R.A.H.; Deemer, S.E.; Roberts, B.M.; Plaisance, E.P.; Rector, R.S. A Dietary Ketone Ester Mitigates Histological Outcomes of NAFLD and Markers of Fibrosis in High-Fat Diet Fed Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G564–G572. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Dufour, S.; Lyu, K.; Zhang, X.-M.; Hakkarainen, A.; Lehtimäki, T.E.; Cline, G.W.; Petersen, K.F.; Shulman, G.I.; Yki-Järvinen, H. Effect of a Ketogenic Diet on Hepatic Steatosis and Hepatic Mitochondrial Metabolism in Nonalcoholic Fatty Liver Disease. Proc. Natl. Acad. Sci. USA 2020, 117, 7347–7354. [Google Scholar] [CrossRef] [Green Version]
- Sripongpun, P.; Churuangsuk, C.; Bunchorntavakul, C. Current Evidence Concerning Effects of Ketogenic Diet and Intermittent Fasting in Patients with Nonalcoholic Fatty Liver. J. Clin. Transl. Hepatol. 2022, 10, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Khuong, J.N.; Wilson, T.G.; Grigg, L.E.; Bullock, A.; Celermajer, D.; Disney, P.; Wijesekera, V.A.; Hornung, T.; Zannino, D.; Iyengar, A.J.; et al. Fontan-Associated Nephropathy: Predictors and Outcomes. Int. J. Cardiol. 2020, 306, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Levin, A.; Kiess, M.; Sexsmith, G.; Chakrabarti, S.; Barlow, A.; Human, D.; Grewal, J. Chronic Kidney Damage in the Adult Fontan Population. Int. J. Cardiol. 2018, 257, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Binotto, M.A. Renal Function and Fontan Patients: What Is the Real Impact in the Long-Term Outcomes? Int. J. Cardiol. 2020, 306, 86–87. [Google Scholar] [CrossRef]
- Niaz, T.; Stephens, E.H.; Gleich, S.J.; Dearani, J.A.; Johnson, J.N.; Sas, D.J.; Bly, S.; Driscoll, D.J.; Cetta, F. Acute Kidney Injury and Renal Replacement Therapy After Fontan Operation. Am. J. Cardiol. 2021, 161, 84–94. [Google Scholar] [CrossRef]
- Zafar, F.; Lubert, A.M.; Katz, D.A.; Hill, G.D.; Opotowsky, A.R.; Alten, J.A.; Goldstein, S.L.; Alsaied, T. Long-Term Kidney Function After the Fontan Operation. J. Am. Coll. Cardiol. 2020, 76, 334–341. [Google Scholar] [CrossRef]
- Hems, D.A.; Brosnan, J.T. Effects of Ischaemia on Content of Metabolites in Rat Liver and Kidney in Vivo. Biochem. J. 1970, 120, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1α Drives NAD Biosynthesis Linking Oxidative Metabolism to Renal Protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef] [Green Version]
- Ritmeester, E.; Veger, V.A.; van der Ven, J.P.G.; van Tussenbroek, G.M.J.W.; van Capelle, C.I.; Udink ten Cate, F.E.A.; Helbing, W.A. Fontan Circulation Associated Organ Abnormalities Beyond the Heart, Lungs, Liver, and Gut: A Systematic Review. Front. Cardiovasc. Med. 2022, 9, 826096. [Google Scholar] [CrossRef]
- Puchowicz, M.A.; Emancipator, D.S.; Xu, K.; Magness, D.L.; Ndubuizu, O.I.; Lust, W.D.; LaManna, J.C. Adaptation to Chronic Hypoxia During Diet-Induced Ketosis. In Oxygen Transport to Tissue XXVI; Okunieff, P., Williams, J., Chen, Y., Eds.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2005; Volume 566, pp. 51–57. [Google Scholar] [CrossRef]
- Poffé, C.; Robberechts, R.; Podlogar, T.; Kusters, M.; Debevec, T.; Hespel, P. Exogenous Ketosis Increases Blood and Muscle Oxygenation but Not Performance during Exercise in Hypoxia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2021, 321, R844–R857. [Google Scholar] [CrossRef]
- García-Caballero, M.; Zecchin, A.; Souffreau, J.; Truong, A.-C.K.; Teuwen, L.-A.; Vermaelen, W.; Martín-Pérez, R.; de Zeeuw, P.; Bouché, A.; Vinckier, S.; et al. Role and Therapeutic Potential of Dietary Ketone Bodies in Lymph Vessel Growth. Nat. Metab. 2019, 1, 666–675. [Google Scholar] [CrossRef]
- Universitaire Ziekenhuizen KU Leuven. Ketogenic Diet: A Novel Metabolic Strategy to Treat Lymphedema Patients? 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03991897 (accessed on 4 April 2023).
- Puchalska, P.; Crawford, P.A. Ketogenic Therapies for Lymphedema? Nat. Metab. 2019, 1, 656–657. [Google Scholar] [CrossRef]
- Dodeja, A.; Urbina, F.; Moore-Padilla, M.; Mah, M.L.; Bradley, D.; Bradley, E. Fontan-Associated Liver Disease: Is Insulin Sensitivity Important? J. Am. Coll. Cardiol. 2020, 75, 549. [Google Scholar] [CrossRef]
- Emamaullee, J.; Zaidi, A.N.; Schiano, T.; Kahn, J.; Valentino, P.L.; Hofer, R.E.; Taner, T.; Wald, J.W.; Olthoff, K.M.; Bucuvalas, J.; et al. Fontan-Associated Liver Disease: Screening, Management, and Transplant Considerations. Circulation 2020, 142, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, J.Z.; Day, A.; Brinkworth, G.D.; Sato, J.; Yamada, S.; Jönsson, T.; Beardsley, J.; Johnson, J.A.; Thabane, L.; Johnston, B.C. Efficacy and Safety of Low and Very Low Carbohydrate Diets for Type 2 Diabetes Remission: Systematic Review and Meta-Analysis of Published and Unpublished Randomized Trial Data. BMJ 2021, 372, m4743. [Google Scholar] [CrossRef]
- Tommerdahl, K.L.; Nelson, R.G.; Bjornstad, P. Dapagliflozin in young people with type 2 diabetes. Lancet Diabetes Endocrinol. 2022, 10, 303–304. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Liao, J.; Zhou, D.; Mu, J. Ketogenic Diet Therapy for Epilepsy: Past 100 Years of Practice. Acta Epileptol. 2022, 4, 15. [Google Scholar] [CrossRef]
- Suo, C.; Liao, J.; Lu, X.; Fang, K.; Hu, Y.; Chen, L.; Cao, D.; Huang, T.; Li, B.; Li, C. Efficacy and Safety of the Ketogenic Diet in Chinese Children. Seizure 2013, 22, 174–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, J.; Swaminathan, A.; Paseka, J.; Hanson, C. Efficacy and Safety of a Ketogenic Diet in Children and Adolescents with Refractory Epilepsy—A Review. Nutrients 2020, 12, 1809. [Google Scholar] [CrossRef]
- Dressler, A.; Trimmel-Schwahofer, P. The Ketogenic Diet for Infants: How Long Can You Go? Epilepsy Res. 2020, 164, 106339. [Google Scholar] [CrossRef]
- Mady, M.A.; Kossoff, E.H.; McGregor, A.L.; Wheless, J.W.; Pyzik, P.L.; Freeman, J.M. The Ketogenic Diet: Adolescents Can Do It, Too. Epilepsia 2003, 44, 847–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholl-Bürgi, S.; Höller, A.; Pichler, K.; Michel, M.; Haberlandt, E.; Karall, D. Ketogenic Diets in Patients with Inherited Metabolic Disorders. J. Inherit. Metab. Dis. 2015, 38, 765–773. [Google Scholar] [CrossRef]
- Lin, K.-L.; Lin, J.-J.; Wang, H.-S. Application of Ketogenic Diets for Pediatric Neurocritical Care. Biomed. J. 2020, 43, 218–225. [Google Scholar] [CrossRef]
- Li, J.; Zhang, H.; Dai, Z. Cancer Treatment with the Ketogenic Diet: A Systematic Review and Meta-Analysis of Animal Studies. Front. Nutr. 2021, 8, 594408. [Google Scholar] [CrossRef] [PubMed]
- Lauzier, B.; Vaillant, F.; Merlen, C.; Gélinas, R.; Bouchard, B.; Rivard, M.-E.; Labarthe, F.; Dolinsky, V.W.; Dyck, J.R.B.; Allen, B.G.; et al. Metabolic Effects of Glutamine on the Heart: Anaplerosis versus the Hexosamine Biosynthetic Pathway. J. Mol. Cell. Cardiol. 2013, 55, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Saavedra, D.; Sanders, L.; Freeman, S.; Reisz, J.A.; Lee, M.H.; Mickael, C.; Kumar, R.; Kassa, B.; Gu, S.; D’ Alessandro, A.; et al. Stable Isotope Metabolomics of Pulmonary Artery Smooth Muscle and Endothelial Cells in Pulmonary Hypertension and with TGF-Beta Treatment. Sci. Rep. 2020, 10, 413. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Olson, K.C.; Gao, C.; Prosdocimo, D.A.; Zhou, M.; Wang, Z.; Jeyaraj, D.; Youn, J.-Y.; Ren, S.; Liu, Y.; et al. Catabolic Defect of Branched-Chain Amino Acids Promotes Heart Failure. Circulation 2016, 133, 2038–2049. [Google Scholar] [CrossRef]
- Vockley, J.; Charrow, J.; Ganesh, J.; Eswara, M.; Diaz, G.A.; McCracken, E.; Conway, R.; Enns, G.M.; Starr, J.; Wang, R.; et al. Triheptanoin Treatment in Patients with Pediatric Cardiomyopathy Associated with Long Chain-Fatty Acid Oxidation Disorders. Mol. Genet. Metab. 2016, 119, 223–231. [Google Scholar] [CrossRef]
- Zöggeler, T.; Stock, K.; Jörg-Streller, M.; Spenger, J.; Konstantopoulou, V.; Hufgard-Leitner, M.; Scholl-Bürgi, S.; Karall, D. Long-Term Experience with Triheptanoin in 12 Austrian Patients with Long-Chain Fatty Acid Oxidation Disorders. Orphanet J. Rare Dis. 2021, 16, 28. [Google Scholar] [CrossRef]
- Lei, I.; Tian, S.; Gao, W.; Liu, L.; Guo, Y.; Tang, P.; Chen, E.; Wang, Z. Acetyl-CoA Production by Specific Metabolites Promotes Cardiac Repair after Myocardial Infarction via Histone Acetylation. eLife 2021, 10, e60311. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.N.; Driscoll, D.J.; O’Leary, P.W. Protein-Losing Enteropathy and the Fontan Operation. Nutr. Clin. Pract. 2012, 27, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Violante, S.; IJlst, L.; te Brinke, H.; Almeida, I.T.; Wanders, R.J.A.; Ventura, F.V.; Houten, S.M. Carnitine Palmitoyltransferase 2 and Carnitine/Acylcarnitine Translocase Are Involved in the Mitochondrial Synthesis and Export of Acylcarnitines. FASEB J. 2013, 27, 2039–2044. [Google Scholar] [CrossRef] [PubMed]
- Pereyra, A.S.; Harris, K.L.; Soepriatna, A.H.; Waterbury, Q.A.; Bharathi, S.S.; Zhang, Y.; Fisher-Wellman, K.H.; Goergen, C.J.; Goetzman, E.S.; Ellis, J.M. Octanoate Is Differentially Metabolized in Liver and Muscle and Fails to Rescue Cardiomyopathy in CPT2 Deficiency. J. Lipid Res. 2021, 62, 100069. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Metabolic State | ||||||
|---|---|---|---|---|---|---|
| Substrate Consumption | ETC | CK | Therapeutic Ketosis | |||
| Fatty acids | Glucose | Ketone bodies | ||||
| Healthy heart | 60–70% [57,58] | Remaining [57,58] | Remaining [57,58] | balanced | ||
| HfrEF | Early HF: no change [48,59,60] Late HF: ↓ [48,59,60] | Early HF: ↑ [48,59,60] Late HF: ↓ [60,61,62,63,64,65,66] | Progressing HF: ↑ [35,65,67,68,69,70,71,72,73,74,75] | Loss of electrons [76] Accumulation of ROS [76] | early HF: ↓ [48,77,78,79] | CO +40% [80] EF +8% [80] |
| BV-HF | ↓ [81] | ↑ [82,83,84,85] | Progressing HF: ↑ [69,86,87,88] | Loss of electrons [89,90] Accumulation of ROS [91,92] | ↓ [89,93] | CO +27% [87] PVR—18% [87] |
| SV-HF | ↓ [94] | ↓ [95] | ? | Loss of electrons [96] Accumulation of ROS [76,97,98] | ? | ? |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renaud, D.; Scholl-Bürgi, S.; Karall, D.; Michel, M. Comparative Metabolomics in Single Ventricle Patients after Fontan Palliation: A Strong Case for a Targeted Metabolic Therapy. Metabolites 2023, 13, 932. https://doi.org/10.3390/metabo13080932
Renaud D, Scholl-Bürgi S, Karall D, Michel M. Comparative Metabolomics in Single Ventricle Patients after Fontan Palliation: A Strong Case for a Targeted Metabolic Therapy. Metabolites. 2023; 13(8):932. https://doi.org/10.3390/metabo13080932
Chicago/Turabian StyleRenaud, David, Sabine Scholl-Bürgi, Daniela Karall, and Miriam Michel. 2023. "Comparative Metabolomics in Single Ventricle Patients after Fontan Palliation: A Strong Case for a Targeted Metabolic Therapy" Metabolites 13, no. 8: 932. https://doi.org/10.3390/metabo13080932
APA StyleRenaud, D., Scholl-Bürgi, S., Karall, D., & Michel, M. (2023). Comparative Metabolomics in Single Ventricle Patients after Fontan Palliation: A Strong Case for a Targeted Metabolic Therapy. Metabolites, 13(8), 932. https://doi.org/10.3390/metabo13080932