
SMetaS: A Sample Metadata Standardizer for Metabolomics



Abstract
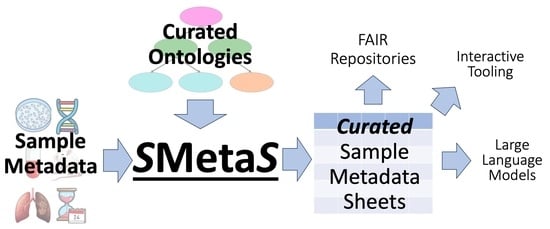
:
1. Introduction
1.1. Motivation
1.2. Sample Metadata in -Omics
1.3. Sample Metadata in Metabolomics—Tool Critiques
1.4. Tool Critique—Metabolomics Workbench
1.5. Tool Critique—ReDU
1.6. Tool Critique—MetaboLights
1.7. SMetaS
2. Materials and Methods
3. Results
3.1. Overview
3.2. Extended Descriptions of Components
3.3. Use Case
3.4. Construction of Vocabularies
4. Discussion
Shortcomings and Future Developments of SMetaS
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, J.; Yu, H.; Xing, S.; Huan, T. Addressing Big Data Challenges in Mass Spectrometry-Based Metabolomics. Chem. Commun. 2022, 58, 9979–9990. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, J.A. Translating Metabolomics into Clinical Practice. Nat. Rev. Bioeng. 2023, 1, 228–229. [Google Scholar] [CrossRef]
- Forcisi, S.; Moritz, F.; Thompson, C.J.; Kanawati, B.; Uhl, J.; Afonso, C.; Bader, C.D.; Barsch, A.; Boughton, B.A.; Chu, R.K.; et al. Large-Scale Interlaboratory DI-FT-ICR MS Comparability Study Employing Various Systems. J. Am. Soc. Mass Spectrom. 2022, 33, 2203–2214. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.A.; Koal, T. Progress in Metabolomics Standardisation and Its Significance in Future Clinical Laboratory Medicine. EJIFCC 2016, 27, 331–343. [Google Scholar]
- Martínez-Reyes, I.; Chandel, N.S. Cancer Metabolism: Looking Forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]
- Goveia, J.; Pircher, A.; Conradi, L.-C.; Kalucka, J.; Lagani, V.; Dewerchin, M.; Eelen, G.; DeBerardinis, R.J.; Wilson, I.D.; Carmeliet, P. Meta-Analysis of Clinical Metabolic Profiling Studies in Cancer: Challenges and Opportunities. EMBO Mol. Med. 2016, 8, 1134–1142. [Google Scholar] [CrossRef]
- Eisenstein, M. Machine Learning Powers Biobank-Driven Drug Discovery. Nat. Biotechnol. 2022, 40, 1303–1305. [Google Scholar] [CrossRef]
- Large Language Models Demonstrate the Potential of Statistical Learning in Language—Contreras Kallens—2023—Cognitive Science—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1111/cogs.13256 (accessed on 11 July 2023).
- Spicer, R.A.; Salek, R.; Steinbeck, C. A Decade after the Metabolomics Standards Initiative It’s Time for a Revision. Sci. Data 2017, 4, 170138. [Google Scholar] [CrossRef] [PubMed]
- Long, N.P.; Nghi, T.D.; Kang, Y.P.; Anh, N.H.; Kim, H.M.; Park, S.K.; Kwon, S.W. Toward a Standardized Strategy of Clinical Metabolomics for the Advancement of Precision Medicine. Metabolites 2020, 10, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, D.; Garrity, G.; Gray, T.; Morrison, N.; Selengut, J.; Sterk, P.; Tatusova, T.; Thomson, N.; Allen, M.J.; Angiuoli, S.V.; et al. The Minimum Information about a Genome Sequence (MIGS) Specification. Nat. Biotechnol. 2008, 26, 541–547. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y. Toward a Sample Metadata Standard in Public Proteomics Repositories. J. Proteome Res. 2020, 19, 3906–3909. [Google Scholar] [CrossRef] [PubMed]
- Specimen and Sample Metadata Standards for Biodiversity Genomics: A Proposal from the Darwin Tree of Life Project. Wellcome Open Research. Available online: https://wellcomeopenresearch.org/articles/7-187/v1?src=rss (accessed on 22 July 2023).
- Sasse, J.; Darms, J.; Fluck, J. Semantic Metadata Annotation Services in the Biomedical Domain—A Literature Review. Appl. Sci. 2022, 12, 796. [Google Scholar] [CrossRef]
- Batista, D.; Gonzalez-Beltran, A.; Sansone, S.-A.; Rocca-Serra, P. Machine Actionable Metadata Models. Sci. Data 2022, 9, 592. [Google Scholar] [CrossRef]
- Moxon, S.; Solbrig, H.; Unni, D.; Jiao, D.; Bruskiewich, R.; Balhoff, J.; Vaidya, G.; Duncan, W.; Hegde, H.; Miller, M.; et al. The Linked Data Modeling Language (LinkML): A General-Purpose Data Modeling Framework Grounded in Machine-Readable Semantics. In Proceedings of the CEUR Workshop Proceedings, CEUR-WS. Bozen-Bolzano, Italy, 16–18 September 2021; Volume 3073, pp. 148–151. [Google Scholar]
- Schriml, L.M.; Chuvochina, M.; Davies, N.; Eloe-Fadrosh, E.A.; Finn, R.D.; Hugenholtz, P.; Hunter, C.I.; Hurwitz, B.L.; Kyrpides, N.C.; Meyer, F.; et al. COVID-19 Pandemic Reveals the Peril of Ignoring Metadata Standards. Sci. Data 2020, 7, 188. [Google Scholar] [CrossRef] [PubMed]
- Nichols, B.N.; Ghosh, S.S.; Auer, T.; Grabowski, T.; Maumet, C.; Keator, D.; Martone, M.E.; Pohl, K.M.; Poline, J.-B. Linked Data in Neuroscience: Applications, Benefits, and Challenges. bioRxiv 2016. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, J.D.; Inácio, B.; Salek, R.M.; Couto, F.M. Assessing Public Metabolomics Metadata, Towards Improving Quality. J. Integr. Bioinform. 2017, 14, 20170054. [Google Scholar] [CrossRef]
- Bremer, P.L.; Wohlgemuth, G.; Fiehn, O. The BinDiscover Database: A Biology-Focused Meta-Analysis Tool for 156,000 GC–TOF MS Metabolome Samples. J. Cheminformatics 2023, 15, 66. [Google Scholar] [CrossRef]
- Hawkins, N.T.; Maldaver, M.; Yannakopoulos, A.; Guare, L.A.; Krishnan, A. Systematic Tissue Annotations of Genomics Samples by Modeling Unstructured Metadata. Nat. Commun. 2022, 13, 6736. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S.; et al. Metabolomics Workbench: An International Repository for Metabolomics Data and Metadata, Metabolite Standards, Protocols, Tutorials and Training, and Analysis Tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef] [Green Version]
- ReDU: A Framework to Find and Reanalyze Public Mass Spectrometry Data. Nature Methods. Available online: https://www.nature.com/articles/s41592-020-0916-7 (accessed on 5 June 2023).
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A Resource Evolving in Response to the Needs of Its Scientific Community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef] [Green Version]
- Mölder, F.; Jablonski, K.P.; Letcher, B.; Hall, M.B.; Tomkins-Tinch, C.H.; Sochat, V.; Forster, J.; Lee, S.; Twardziok, S.O.; Kanitz, A.; et al. Sustainable Data Analysis with Snakemake. F1000Res 2021, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, A.; Swart, P.; Chult, D.S. Exploring Network Structure, Dynamics, and Function Using Networkx; Los Alamos National Lab. (LANL): Los Alamos, NM, USA, 2008. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Sparck Jones, K. A Statistical Interpretation of Term Specificity and Its Application in Retrieval. J. Doc. 1972, 28, 11–21. [Google Scholar] [CrossRef]
- Cover, T.; Hart, P. Nearest Neighbor Pattern Classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Stevens, N.C.; Brown, V.J.; Domanico, M.C.; Edwards, P.C.; Van Winkle, L.S.; Fiehn, O. Alteration of Glycosphingolipid Metabolism by Ozone Is Associated with Exacerbation of Allergic Asthma Characteristics in Mice. Toxicol. Sci. 2023, 191, 79–89. [Google Scholar] [CrossRef]
- Rogers, F.B. Medical Subject Headings. Bull. Med. Libr. Assoc. 1963, 51, 114–116. [Google Scholar]
- GenBank. Nucleic Acids Research. Oxford Academic. Available online: https://academic.oup.com/nar/article/47/D1/D94/5144964 (accessed on 11 July 2023).
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; Mcveigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A Comprehensive Update on Curation, Resources and Tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef]
- Bairoch, A. The Cellosaurus, a Cell-Line Knowledge Resource. J. Biomol. Tech. 2018, 29, 25–38. [Google Scholar] [CrossRef]
- NCI Thesaurus. Available online: https://ncithesaurus.nci.nih.gov/ncitbrowser/ (accessed on 11 July 2023).
- Center for Drug Evaluation and Research. Drugs@FDA Data Files; FDA: Silver Spring, MD, USA, 2023. [Google Scholar]
- Scholz, M.; Fiehn, O. SetupX—A Public Study Design Database for Metabolomic Projects. Pac. Symp. Biocomput. 2007, 169–180. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Design Principle |
|---|---|
| 1 | headers with orthogonal vocabularies |
| 2 | vocabularies with non-redundant terms |
| 3 | inclusion of a synonym set for each “main term” to facilitate the loose expression of a term |
| 4 | vocabularies/models that expand to incorporate new terms easily submitted by users |
| 5 | machine learning models that increase speed-of-use and make the program typo tolerant |
| 6 | a deference for simplicity when possible, because we believe that user apathy/disinterest is as much a problem as any technical challenge |
| Grouping | Metadata Category | Term Count | Initial Vocabulary Description |
|---|---|---|---|
| Core Sample Type | species | 724,962 | NCBI ontology less -rank ‘strain’ -parent node scientific name contained ‘environmental sample’ -parent node scientific name contained ‘unclassified’ -rank ‘no rank’ that contained ‘/’ -rank ‘species’ containing numerical characters -rank ‘species’ containing ‘vector’ |
| organ | 11,494 | MeSH ontology heading ‘A’ and lower | |
| cellLine | 247,365 | Cellosaurus ontology | |
| material | 2056 | MeSH ontology: -heading ‘D20’ and lower -heading ‘G16’ and lower | |
| Sample Description | massUnit | 49 | Unit Ontology: -heading UO0000002 and lower |
| volumeUnit | 79 | Unit Ontology: -heading UO0000095 and lower | |
| sex | 3 | All sexes | |
| heightUnit | 48 | Unit Ontology: -heading UO0000001 and lower | |
| weightUnit | 49 | Unit Ontology: -heading UO0000002 and lower | |
| ageUnit | 22 | Unit Ontology: -heading UO0000003 and lower | |
| ethnicity | 1057 | NCIT Ontology: -header C17049 and lower | |
| geographicalOrigin | 799 | MeSH ontology: -header Z01 and lower -header G16.500.275 and lower | |
| strain | 2282 | NCIT Ontology: -header C14250 and lower except those terms which exist in the NCBI ontology or are descendants of Gene header in NCIT ontology | |
| Study Factors | drugName | 9537 | FDA drug vocabulary |
| drugDoseUnit | 753 | Unit Ontology | |
| geneKnockout | 141,605 | NCBI human gene vocabulary | |
| disease | 36,378 | MeSH ontology: -header C and lower | |
| diet | 1164 | MeSH heading G07.203 and lower | |
| exercise | 569 | MeSH heading I03 and lower, MeSH heading G11.427.410.698 and lower | |
| Time Series | zeroTimeEvent | 69,321 | NCIT: ontology: -header C43431 and lower |
| timeUnit | 22 | All Units | |
| Other | inclusion | 0 | None in initial vocabulary |
| exclusion | 0 | None in initial vocabulary | |
| comment | 0 | None in initial vocabulary |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bremer, P.L.; Fiehn, O. SMetaS: A Sample Metadata Standardizer for Metabolomics. Metabolites 2023, 13, 941. https://doi.org/10.3390/metabo13080941
Bremer PL, Fiehn O. SMetaS: A Sample Metadata Standardizer for Metabolomics. Metabolites. 2023; 13(8):941. https://doi.org/10.3390/metabo13080941
Chicago/Turabian StyleBremer, Parker Ladd, and Oliver Fiehn. 2023. "SMetaS: A Sample Metadata Standardizer for Metabolomics" Metabolites 13, no. 8: 941. https://doi.org/10.3390/metabo13080941
APA StyleBremer, P. L., & Fiehn, O. (2023). SMetaS: A Sample Metadata Standardizer for Metabolomics. Metabolites, 13(8), 941. https://doi.org/10.3390/metabo13080941