
Metabolic Profile of Alzheimer’s Disease: Is 10-Hydroxy-2-decenoic Acid a Pertinent Metabolic Adjuster?


Abstract
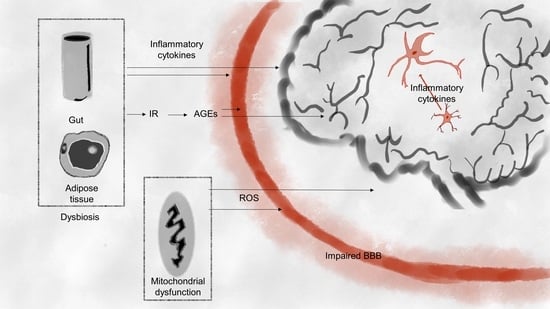
:
1. Introduction
2. Metabolic Syndrome and Alzheimer’s Disease
2.1. Type II Diabetes Mellitus
2.1.1. Insulin Resistance
2.1.2. Inflammation and Vascular Impairment
2.1.3. Glucose Metabolism Dysfunction and Mitochondrial Oxidative Stress
2.1.4. Hyperglycemia and Advanced Glycation End Products
2.2. Obesity
2.2.1. Inflammation
2.2.2. Leptin
2.2.3. Endoplasmic Reticulum Stress
2.2.4. Mitochondrial Oxidative Stress and Impaired Blood–Brain Barrier
2.2.5. Gut Microbiota
3. Biomarkers
4. Properties of 10-Hydroxy-2-decenoic Acid
4.1. Anti-Neurodegeneration and Immunomodulation
4.2. Antitumor
4.3. Metabolic Adjusting Properties
5. Summary and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| MetS | Metabolic syndromes |
| DM | Diabetes mellitus |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| 10-HDA | 10-Hydroxy-2-decenoic acid |
| RJ | Royal Jelly |
| ER | Endoplasmic reticulum |
| IR | Insulin resistance |
| MCI | Mild cognitive impairment |
| GLP-1R | Glucagon-like peptide-1 receptor |
| PPARs | Peroxisome proliferator-activated receptors |
| GSK-3 | Glycogen synthase kinase-3 |
| TREM2 | Triggering receptor expressed on myeloid cells 2 |
| BMI | Body mass index |
| BBB | Blood–brain barrier |
| AGEs | Advanced glycation end products |
| RMSD | Root mean square deviation |
References
- Darenskaya, M.A.; Kolesnikova, L.I.; Kolesnikov, S.I. Oxidative Stress: Pathogenetic Role in Diabetes Mellitus and Its Complications and Therapeutic Approaches to Correction. Bull Exp. Biol. Med. 2021, 171, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.C.W. Epidemiology of diabetes and diabetic complications in China. Diabetologia 2018, 61, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Chung, C.M.; Leu, H.B.; Lin, L.Y.; Chiu, C.C.; Hsu, C.Y.; Chiang, C.H.; Huang, P.H.; Chen, T.J.; Lin, S.J.; et al. Diabetes mellitus and the risk of Alzheimer’s disease: A nationwide population-based study. PLoS ONE 2014, 9, e87095. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Zhou, F.; Mei, J.; Yang, H.; Li, H. The Effect of Type 2 Diabetes Mellitus on Neuropsychological Symptoms in Chinese Early Alzheimer’s Disease Population. Neuropsychiatr. Dis. Treat. 2020, 16, 829–836. [Google Scholar] [CrossRef]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Et. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef]
- Piché, M.E.; Tchernof, A.; Després, J.P. Obesity Phenotypes, Diabetes, and Cardiovascular Diseases. Circ. Res. 2020, 126, 1477–1500. [Google Scholar] [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2021, 17, 327–406. [CrossRef]
- Kim, B.; Backus, C.; Oh, S.; Feldman, E.L. Hyperglycemia-induced tau cleavage in vitro and in vivo: A possible link between diabetes and Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2013, 34, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Cooper, M.E.; Del Prato, S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet 2014, 383, 1068–1083. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.V.F.; Loures, C.M.G.; Alves, L.C.V.; de Souza, L.C.; Borges, K.B.G.; Carvalho, M.D.G. Alzheimer’s disease: Risk factors and potentially protective measures. J. Biomed. Sci. 2019, 26, 33. [Google Scholar] [CrossRef] [PubMed]
- Singh-Manoux, A.; Czernichow, S.; Elbaz, A.; Dugravot, A.; Sabia, S.; Hagger-Johnson, G.; Kaffashian, S.; Zins, M.; Brunner, E.J.; Nabi, H.; et al. Obesity phenotypes in midlife and cognition in early old age: The Whitehall II cohort study. Neurology 2012, 79, 755–762. [Google Scholar] [CrossRef]
- De Felice, F.G.; Ferreira, S.T. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef]
- Morabito, M.V.; Berman, D.E.; Schneider, R.T.; Zhang, Y.; Leibel, R.L.; Small, S.A. Hyperleucinemia causes hippocampal retromer deficiency linking diabetes to Alzheimer’s disease. Neurobiol. Dis. 2014, 65, 188–192. [Google Scholar] [CrossRef]
- Wu, M.; Liao, M.; Huang, R.; Chen, C.; Tian, T.; Wang, H.; Li, J.; Li, J.; Sun, Y.; Wu, C.; et al. Hippocampal overexpression of TREM2 ameliorates high fat diet induced cognitive impairment and modulates phenotypic polarization of the microglia. Genes Dis. 2022, 9, 401–414. [Google Scholar] [CrossRef]
- Sonar, S.A.; Lal, G. Blood-brain barrier and its function during inflammation and autoimmunity. J. Leukoc. Biol. 2018, 103, 839–853. [Google Scholar] [CrossRef]
- Galea, I. The blood-brain barrier in systemic infection and inflammation. Cell Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; Kunugi, H. Royal Jelly as an Intelligent Anti-Aging Agent-A Focus on Cognitive Aging and Alzheimer’s Disease: A Review. Antioxidants 2020, 9, 937. [Google Scholar] [CrossRef]
- Cornara, L.; Biagi, M.; Xiao, J.; Burlando, B. Therapeutic Properties of Bioactive Compounds from Different Honeybee Products. Front Pharm. 2017, 8, 412. [Google Scholar] [CrossRef]
- Guardia de Souza, E.S.T.; do Val de Paulo, M.E.F.; da Silva, J.R.M.; da Silva Alves, A.; Britto, L.R.G.; Xavier, G.F.; Lopes Sandoval, M.R. Oral treatment with royal jelly improves memory and presents neuroprotective effects on icv-STZ rat model of sporadic Alzheimer’s disease. Heliyon 2020, 6, e03281. [Google Scholar] [CrossRef]
- Guo, J.; Wang, Z.; Chen, Y.; Cao, J.; Tian, W.; Ma, B.; Dong, Y. Active components and biological functions of royal jelly. J. Funct. Foods 2021, 82, 104514. [Google Scholar] [CrossRef]
- Fratini, F.; Cilia, G.; Mancini, S.; Felicioli, A. Royal Jelly: An ancient remedy with remarkable antibacterial properties. Microbiol. Res. 2016, 192, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Sakai, H. Anti-Cancer and Protective Effects of Royal Jelly for Therapy-Induced Toxicities in Malignancies. Int. J. Mol. Sci. 2018, 19, 3270. [Google Scholar] [CrossRef]
- Botezan, S.; Baci, G.-M.; Bagameri, L.; Pașca, C.; Dezmirean, D.S. Current Status of the Bioactive Properties of Royal Jelly: A Comprehensive Review with a Focus on Its Anticancer, Anti-Inflammatory, and Antioxidant Effects. Molecules 2023, 28, 1510. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Montine, T.J.; Sonnen, J.A.; Montine, K.S.; Crane, P.K.; Larson, E.B. Adult Changes in Thought study: Dementia is an individually varying convergent syndrome with prevalent clinically silent diseases that may be modified by some commonly used therapeutics. Curr. Alzheimer Res. 2012, 9, 718–723. [Google Scholar] [CrossRef]
- Ryu, J.C.; Zimmer, E.R.; Rosa-Neto, P.; Yoon, S.O. Consequences of Metabolic Disruption in Alzheimer’s Disease Pathology. Neurother. J. Am. Soc. Exp. NeuroTherapeutics 2019, 16, 600–610. [Google Scholar] [CrossRef]
- Li, W.; Risacher, S.L.; Gao, S.; Boehm, S.L., 2nd; Elmendorf, J.S.; Saykin, A.J. Type 2 diabetes mellitus and cerebrospinal fluid Alzheimer’s disease biomarker amyloid β1-42 in Alzheimer’s Disease Neuroimaging Initiative participants. Alzheimer’s Dement. 2018, 10, 94–98. [Google Scholar] [CrossRef]
- Motta, C.; Assogna, M.; Bonomi, C.G.; Mascolo, A.P.; De Lucia, V.; Semprini, R.; Mercuri, N.B.; Koch, G.; Martorana, A. Diabetes mellitus contributes to higher cerebrospinal fluid tau levels selectively in Alzheimer’s disease patients with the APOE4 genotype. Eur. J. Neurol. 2021, 28, 3965–3971. [Google Scholar] [CrossRef]
- Karki, R.; Madan, S.; Gadiya, Y.; Domingo-Fernández, D.; Kodamullil, A.T.; Hofmann-Apitius, M. Data-Driven Modeling of Knowledge Assemblies in Understanding Comorbidity Between Type 2 Diabetes Mellitus and Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2020, 78, 87–95. [Google Scholar] [CrossRef]
- Huang, C.; Luo, J.; Wen, X.; Li, K. Linking Diabetes Mellitus with Alzheimer’s Disease: Bioinformatics Analysis for the Potential Pathways and Characteristic Genes. Biochem. Genet. 2022, 60, 1049–1075. [Google Scholar] [CrossRef]
- Ahmed, A.S.; Elgharabawy, R.M.; Al-Najjar, A.H. Ameliorating effect of anti-Alzheimer’s drugs on the bidirectional association between type 2 diabetes mellitus and Alzheimer’s disease. Exp. Biol. Med. 2017, 242, 1335–1344. [Google Scholar] [CrossRef]
- Michailidis, M.; Moraitou, D.; Tata, D.A.; Kalinderi, K.; Papamitsou, T.; Papaliagkas, V. Alzheimer’s Disease as Type 3 Diabetes: Common Pathophysiological Mechanisms between Alzheimer’s Disease and Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 2687. [Google Scholar] [CrossRef]
- Milstein, J.L.; Ferris, H.A. The brain as an insulin-sensitive metabolic organ. Mol. Metab. 2021, 52, 101234. [Google Scholar] [CrossRef]
- Hopkins, D.F.; Williams, G. Insulin receptors are widely distributed in human brain and bind human and porcine insulin with equal affinity. Diabet. Med. A J. Br. Diabet. Assoc. 1997, 14, 1044–1050. [Google Scholar] [CrossRef]
- Ebrahimpour, S.; Zakeri, M.; Esmaeili, A. Crosstalk between obesity, diabetes, and alzheimer’s disease: Introducing quercetin as an effective triple herbal medicine. Ageing Res. Rev. 2020, 62, 101095. [Google Scholar] [CrossRef]
- Adam, A.P. A Potential New Mechanism Linking Type II Diabetes Mellitus and Alzheimer’s Disease. BioEssays News Rev. Mol. Cell. Dev. Biol. 2018, 40, e1800061. [Google Scholar] [CrossRef]
- Ahmed, B.; Sultana, R.; Greene, M.W. Adipose tissue and insulin resistance in obese. Biomed Pharm. 2021, 137, 111315. [Google Scholar] [CrossRef]
- Minton, K. B1 B cells link gut dysbiosis and insulin resistance. Nat. Rev. Immunol. 2019, 19, 1. [Google Scholar] [CrossRef]
- Sędzikowska, A.; Szablewski, L. Insulin and Insulin Resistance in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9987. [Google Scholar] [CrossRef]
- Gannon, O.J.; Robison, L.S.; Salinero, A.E.; Abi-Ghanem, C.; Mansour, F.M.; Kelly, R.D.; Tyagi, A.; Brawley, R.R.; Ogg, J.D.; Zuloaga, K.L. High-fat diet exacerbates cognitive decline in mouse models of Alzheimer’s disease and mixed dementia in a sex-dependent manner. J. Neuroinflamm. 2022, 19, 110. [Google Scholar] [CrossRef]
- Folch, J.; Olloquequi, J.; Ettcheto, M.; Busquets, O.; Sánchez-López, E.; Cano, A.; Espinosa-Jiménez, T.; García, M.L.; Beas-Zarate, C.; Casadesús, G.; et al. The Involvement of Peripheral and Brain Insulin Resistance in Late Onset Alzheimer’s Dementia. Front. Aging Neurosci. 2019, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Liu, C.C.; Van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129.e115. [Google Scholar] [CrossRef]
- Velazquez, R.; Tran, A.; Ishimwe, E.; Denner, L.; Dave, N.; Oddo, S.; Dineley, K.T. Central insulin dysregulation and energy dyshomeostasis in two mouse models of Alzheimer’s disease. Neurobiol. Aging 2017, 58, 1–13. [Google Scholar] [CrossRef]
- Litwiniuk, A.; Bik, W.; Kalisz, M.; Baranowska-Bik, A. Inflammasome NLRP3 Potentially Links Obesity-Associated Low-Grade Systemic Inflammation and Insulin Resistance with Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 5603. [Google Scholar] [CrossRef]
- Kamal, M.A.; Priyamvada, S.; Anbazhagan, A.N.; Jabir, N.R.; Tabrez, S.; Greig, N.H. Linking Alzheimer’s disease and type 2 diabetes mellitus via aberrant insulin signaling and inflammation. CNS Neurol. Disord. Drug Targets 2014, 13, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Bozluolcay, M.; Andican, G.; Fırtına, S.; Erkol, G.; Konukoglu, D. Inflammatory hypothesis as a link between Alzheimer’s disease and diabetes mellitus. Geriatr. Gerontol. Int. 2016, 16, 1161–1166. [Google Scholar] [CrossRef]
- Nowell, J.; Blunt, E.; Gupta, D.; Edison, P. Antidiabetic agents as a novel treatment for Alzheimer’s and Parkinson’s disease. Ageing Res. Rev. 2023, 89, 101979. [Google Scholar] [CrossRef]
- Van Dyken, P.; Lacoste, B. Impact of Metabolic Syndrome on Neuroinflammation and the Blood-Brain Barrier. Front. Neurosci. 2018, 12, 930. [Google Scholar] [CrossRef]
- Miao, J.; Ma, H.; Yang, Y.; Liao, Y.; Lin, C.; Zheng, J.; Yu, M.; Lan, J. Microglia in Alzheimer’s disease: Pathogenesis, mechanisms, and therapeutic potentials. Front. Aging Neurosci. 2023, 15, 1201982. [Google Scholar] [CrossRef] [PubMed]
- Song, G.J.; Suk, K. Pharmacological Modulation of Functional Phenotypes of Microglia in Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, W.; Chen, H.L.; Zhang, H.Y.; Zhang, N.X. Interplay between Alzheimer’s disease and global glucose metabolism revealed by the metabolic profile alterations of pancreatic tissue and serum in APP/PS1 transgenic mice. Acta Pharmacol. Sin. 2019, 40, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef]
- Kyrtata, N.; Emsley, H.C.A.; Sparasci, O.; Parkes, L.M.; Dickie, B.R. A Systematic Review of Glucose Transport Alterations in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 626636. [Google Scholar] [CrossRef]
- Domingues, R.; Pereira, C.; Cruz, M.T.; Silva, A. Therapies for Alzheimer’s disease: A metabolic perspective. Mol. Genet. Metab. 2021, 132, 162–172. [Google Scholar] [CrossRef]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; Espindola de Freitas, A.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef]
- Theurey, P.; Connolly, N.M.C.; Fortunati, I.; Basso, E.; Lauwen, S.; Ferrante, C.; Moreira Pinho, C.; Joselin, A.; Gioran, A.; Bano, D.; et al. Systems biology identifies preserved integrity but impaired metabolism of mitochondria due to a glycolytic defect in Alzheimer’s disease neurons. Aging Cell 2019, 18, e12924. [Google Scholar] [CrossRef]
- Connolly, N.M.C.; Theurey, P.; Pizzo, P. Glucose dysregulation in pre-clinical Alzheimer’s disease. Aging 2019, 11, 5296–5297. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, N.; Leon, J.; Sheng, Z.; Oka, S.; Hamasaki, H.; Iwaki, T.; Nakabeppu, Y. Molecular pathophysiology of impaired glucose metabolism, mitochondrial dysfunction, and oxidative DNA damage in Alzheimer’s disease brain. Mech. Ageing Dev. 2017, 161, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Zhang, M.; Jeong, Y.Y.; Margolis, D.J.; Cai, Q. The role of mitophagy in the regulation of mitochondrial energetic status in neurons. Autophagy 2021, 17, 4182–4201. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Bacskai, B.J. Mitochondria and Calcium in Alzheimer’s Disease: From Cell Signaling to Neuronal Cell Death. Trends Neurosci. 2021, 44, 136–151. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Han, E.; Han, K.D.; Lee, B.W.; Kang, E.S.; Cha, B.S.; Ko, S.H.; Lee, Y.H. Severe Hypoglycemia Increases Dementia Risk and Related Mortality: A Nationwide, Population-based Cohort Study. J. Clin. Endocrinol. Metab. 2022, 107, e1976–e1986. [Google Scholar] [CrossRef]
- Rawlings, A.M.; Sharrett, A.R.; Albert, M.S.; Coresh, J.; Windham, B.G.; Power, M.C.; Knopman, D.S.; Walker, K.; Burgard, S.; Mosley, T.H.; et al. The Association of Late-Life Diabetes Status and Hyperglycemia With Incident Mild Cognitive Impairment and Dementia: The ARIC Study. Diabetes. Care 2019, 42, 1248–1254. [Google Scholar] [CrossRef]
- Ferreiro, E.; Lanzillo, M.; Canhoto, D.; Carvalho da Silva, A.M.; Mota, S.I.; Dias, I.S.; Ferreira, I.L.; Fontes, A.R.; Mastrella, G.; Pinheiro, P.; et al. Chronic hyperglycemia impairs hippocampal neurogenesis and memory in an Alzheimer’s disease mouse model. Neurobiol. Aging 2020, 92, 98–113. [Google Scholar] [CrossRef]
- Cherbuin, N.; Sachdev, P.; Anstey, K.J. Higher normal fasting plasma glucose is associated with hippocampal atrophy: The PATH Study. Neurology 2012, 79, 1019–1026. [Google Scholar] [CrossRef]
- Pappas, C.; Small, B.J.; Andel, R.; Laczó, J.; Parizkova, M.; Ondrej, L.; Hort, J. Blood Glucose Levels May Exacerbate Executive Function Deficits in Older Adults with Cognitive Impairment. J. Alzheimer’s Dis. JAD 2019, 67, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Ogama, N.; Sakurai, T.; Kawashima, S.; Tanikawa, T.; Tokuda, H.; Satake, S.; Miura, H.; Shimizu, A.; Kokubo, M.; Niida, S.; et al. Postprandial Hyperglycemia Is Associated With White Matter Hyperintensity and Brain Atrophy in Older Patients With Type 2 Diabetes Mellitus. Front. Aging Neurosci. 2018, 10, 273. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, S.; Hong, Z.; Chen, X.; Shan, X.; Fischbach, S.; Xiao, X. Chronic hyperglycemia regulates microglia polarization through ERK5. Aging 2019, 11, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Vittal Rao, H.; Bihaqi, S.W.; Iannucci, J.; Sen, A.; Grammas, P. Thrombin Signaling Contributes to High Glucose-Induced Injury of Human Brain Microvascular Endothelial Cells. J. Alzheimer’s Dis. JAD 2021, 79, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Kong, Y.; Wang, F.; Wang, J.; Liu, C.; Zhou, Y.; Xu, Z.; Zhang, C.; Sun, B.; Guan, Y. Pathological Mechanisms Linking Diabetes Mellitus and Alzheimer’s Disease: The Receptor for Advanced Glycation End Products (RAGE). Front. Aging Neurosci. 2020, 12, 217. [Google Scholar] [CrossRef]
- He, C.; Li, Q.; Cui, Y.; Gao, P.; Shu, W.; Zhou, Q.; Wang, L.; Li, L.; Lu, Z.; Zhao, Y.; et al. Recurrent moderate hypoglycemia accelerates the progression of Alzheimer’s disease through impairment of the TRPC6/GLUT3 pathway. JCI Insight. 2022, 7, e154595. [Google Scholar] [CrossRef]
- Nilsson, M.; Jensen, N.; Gejl, M.; Bergmann, M.L.; Storgaard, H.; Zander, M.; Miskowiak, K.; Rungby, J. Experimental non-severe hypoglycaemia substantially impairs cognitive function in type 2 diabetes: A randomised crossover trial. Diabetologia 2019, 62, 1948–1958. [Google Scholar] [CrossRef]
- Puente, E.C.; Silverstein, J.; Bree, A.J.; Musikantow, D.R.; Wozniak, D.F.; Maloney, S.; Daphna-Iken, D.; Fisher, S.J. Recurrent moderate hypoglycemia ameliorates brain damage and cognitive dysfunction induced by severe hypoglycemia. Diabetes 2010, 59, 1055–1062. [Google Scholar] [CrossRef]
- Walker, J.M.; Harrison, F.E. Shared Neuropathological Characteristics of Obesity, Type 2 Diabetes and Alzheimer’s Disease: Impacts on Cognitive Decline. Nutrients 2015, 7, 7332–7357. [Google Scholar] [CrossRef]
- Picone, P.; Di Carlo, M.; Nuzzo, D. Obesity and Alzheimer’s disease: Molecular bases. Eur. J. Neurosci. 2020, 52, 3944–3950. [Google Scholar] [CrossRef] [PubMed]
- Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Mitschelen, M.; Koller, A.; Szalai, G.; Sonntag, W.E.; Ungvari, Z.; Csiszar, A. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: Effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 1212–1226. [Google Scholar] [CrossRef]
- Alford, S.; Patel, D.; Perakakis, N.; Mantzoros, C.S. Obesity as a risk factor for Alzheimer’s disease: Weighing the evidence. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2018, 19, 269–280. [Google Scholar] [CrossRef]
- Verdile, G.; Keane, K.N.; Cruzat, V.F.; Medic, S.; Sabale, M.; Rowles, J.; Wijesekara, N.; Martins, R.N.; Fraser, P.E.; Newsholme, P. Inflammation and Oxidative Stress: The Molecular Connectivity between Insulin Resistance, Obesity, and Alzheimer’s Disease. Mediat. Inflamm. 2015, 2015, 105828. [Google Scholar] [CrossRef]
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Nakandakari, S.; Muñoz, V.R.; Kuga, G.K.; Gaspar, R.C.; Sant’Ana, M.R.; Pavan, I.C.B.; da Silva, L.G.S.; Morelli, A.P.; Simabuco, F.M.; da Silva, A.S.R.; et al. Short-term high-fat diet modulates several inflammatory, ER stress, and apoptosis markers in the hippocampus of young mice. Brain Behav. Immun. 2019, 79, 284–293. [Google Scholar] [CrossRef]
- Kacířová, M.; Železná, B.; Blažková, M.; Holubová, M.; Popelová, A.; Kuneš, J.; Šedivá, B.; Maletínská, L. Aging and high-fat diet feeding lead to peripheral insulin resistance and sex-dependent changes in brain of mouse model of tau pathology THY-Tau22. J. Neuroinflamm. 2021, 18, 141. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Huang, N.; Liu, J.; Huang, J.; Shi, J.; Jin, F. AMPK: A bridge between diabetes mellitus and Alzheimer’s disease. Behav. Brain Res. 2021, 400, 113043. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Z.H.; Kang, S.S.; Liu, X.; Xia, Y.; Chan, C.B.; Ye, K. High-fat diet-induced diabetes couples to Alzheimer’s disease through inflammation-activated C/EBPβ/AEP pathway. Mol. Psychiatry 2022, 27, 3396–3409. [Google Scholar] [CrossRef]
- Duquenne, M.; Folgueira, C.; Bourouh, C.; Millet, M.; Silva, A.; Clasadonte, J.; Imbernon, M.; Fernandois, D.; Martinez-Corral, I.; Kusumakshi, S.; et al. Leptin brain entry via a tanycytic LepR–EGFR shuttle controls lipid metabolism and pancreas function. Nat. Metab. 2021, 3, 1071–1090. [Google Scholar] [CrossRef]
- Hamilton, K.; Harvey, J. The Neuronal Actions of Leptin and the Implications for Treating Alzheimer’s Disease. Pharmaceuticals 2021, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- Letra, L.; Matafome, P.; Rodrigues, T.; Duro, D.; Lemos, R.; Baldeiras, I.; Patrício, M.; Castelo-Branco, M.; Caetano, G.; Seiça, R.; et al. Association between Adipokines and Biomarkers of Alzheimer’s Disease: A Cross-Sectional Study. J. Alzheimer’s Dis. JAD 2019, 67, 725–735. [Google Scholar] [CrossRef]
- Khemka, V.K.; Bagchi, D.; Bandyopadhyay, K.; Bir, A.; Chattopadhyay, M.; Biswas, A.; Basu, D.; Chakrabarti, S. Altered serum levels of adipokines and insulin in probable Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2014, 41, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, C.E.; van der Flier, W.M.; Scheltens, P.; Duits, A.; Wijnstok, N.; Nijpels, G.; Dekker, J.M.; Blankenstein, R.M.; Heijboer, A.C. Serum leptin is not altered nor related to cognitive decline in Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2015, 44, 809–813. [Google Scholar] [CrossRef]
- Maioli, S.; Lodeiro, M.; Merino-Serrais, P.; Falahati, F.; Khan, W.; Puerta, E.; Codita, A.; Rimondini, R.; Ramirez, M.J.; Simmons, A.; et al. Alterations in brain leptin signalling in spite of unchanged CSF leptin levels in Alzheimer’s disease. Aging Cell 2015, 14, 122–129. [Google Scholar] [CrossRef]
- Bonda, D.J.; Stone, J.G.; Torres, S.L.; Siedlak, S.L.; Perry, G.; Kryscio, R.; Jicha, G.; Casadesus, G.; Smith, M.A.; Zhu, X.; et al. Dysregulation of leptin signaling in Alzheimer disease: Evidence for neuronal leptin resistance. J. Neurochem. 2014, 128, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Buchan, M.; Vitanova, K.; Aitken, L.; Gunn-Moore, F.J.; Ramsay, R.R.; Doherty, G. Neuroprotective actions of leptin facilitated through balancing mitochondrial morphology and improving mitochondrial function. J. Neurochem. 2020, 155, 191–206. [Google Scholar] [CrossRef]
- Uddin, M.S.; Yu, W.S.; Lim, L.W. Exploring ER stress response in cellular aging and neuroinflammation in Alzheimer’s disease. Ageing Res. Rev. 2021, 70, 101417. [Google Scholar] [CrossRef]
- Nakagawa, K.; Islam, S.; Ueda, M.; Nakagawa, T. Endoplasmic reticulum stress contributes to the decline in doublecortin expression in the immature neurons of mice with long-term obesity. Sci. Rep. 2022, 12, 1022. [Google Scholar] [CrossRef]
- Roh, H.T.; Cho, S.Y.; So, W.Y. Obesity promotes oxidative stress and exacerbates blood-brain barrier disruption after high-intensity exercise. J. Sport Health. Sci. 2017, 6, 225–230. [Google Scholar] [CrossRef]
- Nagano, H.; Ito, S.; Masuda, T.; Ohtsuki, S. Effect of Insulin Receptor-Knockdown on the Expression Levels of Blood-Brain Barrier Functional Proteins in Human Brain Microvascular Endothelial Cells. Pharm. Res. 2022, 39, 1561–1574. [Google Scholar] [CrossRef] [PubMed]
- Martin-Jiménez, C.A.; Gaitán-Vaca, D.M.; Echeverria, V.; González, J.; Barreto, G.E. Relationship Between Obesity, Alzheimer’s Disease, and Parkinson’s Disease: An Astrocentric View. Mol. Neurobiol. 2017, 54, 7096–7115. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2017, 58, 1–15. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Gut microbiota and pro/prebiotics in Alzheimer’s disease. Aging 2020, 12, 5539–5550. [Google Scholar] [CrossRef]
- Carranza-Naval, M.J.; Vargas-Soria, M.; Hierro-Bujalance, C.; Baena-Nieto, G.; Garcia-Alloza, M.; Infante-Garcia, C.; Del Marco, A. Alzheimer’s Disease and Diabetes: Role of Diet, Microbiota and Inflammation in Preclinical Models. Biomolecules 2021, 11, 262. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, T.; Cao, M.; Yuan, C.; Reiter, R.J.; Zhao, Z.; Zhao, Y.; Chen, L.; Fan, W.; Wang, X.; et al. Gut Microbiota Dysbiosis Induced by Decreasing Endogenous Melatonin Mediates the Pathogenesis of Alzheimer’s Disease and Obesity. Front. Immunol. 2022, 13, 900132. [Google Scholar] [CrossRef]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef]
- Shen, H.; Guan, Q.; Zhang, X.; Yuan, C.; Tan, Z.; Zhai, L.; Hao, Y.; Gu, Y.; Han, C. New mechanism of neuroinflammation in Alzheimer’s disease: The activation of NLRP3 inflammasome mediated by gut microbiota. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 100, 109884. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Gates, E.J.; Ranger, A.L.; Klegeris, A. Short-chain fatty acids (SCFAs) alone or in combination regulate select immune functions of microglia-like cells. Mol. Cell. Neurosci. 2020, 105, 103493. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, K.; Li, X.; Xu, L.; Yang, Z. Sodium butyrate ameliorates the impairment of synaptic plasticity by inhibiting the neuroinflammation in 5XFAD mice. Chem. Biol. Interact. 2021, 341, 109452. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, L.; Ettorre, E.; Zicari, A.M.; Inghilleri, M.; Nocella, C.; Perri, L.; Spalice, A.; Fossati, C.; De Lucia, M.C.; Pigozzi, F.; et al. Oxidative Stress and Gut-Derived Lipopolysaccharides in Neurodegenerative Disease: Role of NOX2. Oxidative Med. Cell. Longev. 2020, 2020, 8630275. [Google Scholar] [CrossRef] [PubMed]
- Rezaei Asl, Z.; Sepehri, G.; Salami, M. Probiotic treatment improves the impaired spatial cognitive performance and restores synaptic plasticity in an animal model of Alzheimer’s disease. Behav. Brain Res. 2019, 376, 112183. [Google Scholar] [CrossRef]
- Kim, S.Y.; Chae, C.W.; Lee, H.J.; Jung, Y.H.; Choi, G.E.; Kim, J.S.; Lim, J.R.; Lee, J.E.; Cho, J.H.; Park, H.; et al. Sodium butyrate inhibits high cholesterol-induced neuronal amyloidogenesis by modulating NRF2 stabilization-mediated ROS levels: Involvement of NOX2 and SOD1. Cell Death Dis. 2020, 11, 469. [Google Scholar] [CrossRef] [PubMed]
- Bonfili, L.; Cuccioloni, M.; Gong, C.; Cecarini, V.; Spina, M.; Zheng, Y.; Angeletti, M.; Eleuteri, A.M. Gut microbiota modulation in Alzheimer’s disease: Focus on lipid metabolism. Clin. Nutr. 2022, 41, 698–708. [Google Scholar] [CrossRef]
- Bonfili, L.; Cecarini, V.; Gogoi, O.; Berardi, S.; Scarpona, S.; Angeletti, M.; Rossi, G.; Eleuteri, A.M. Gut microbiota manipulation through probiotics oral administration restores glucose homeostasis in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2020, 87, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Govindarajulu, M.; Pinky, P.D.; Steinke, I.; Bloemer, J.; Ramesh, S.; Kariharan, T.; Rella, R.T.; Bhattacharya, S.; Dhanasekaran, M.; Suppiramaniam, V.; et al. Gut Metabolite TMAO Induces Synaptic Plasticity Deficits by Promoting Endoplasmic Reticulum Stress. Front. Mol. Neurosci. 2020, 13, 138. [Google Scholar] [CrossRef]
- Ouwens, D.M.; van Duinkerken, E.; Schoonenboom, S.N.; Herzfeld de Wiza, D.; Klein, M.; van Golen, L.; Pouwels, P.J.; Barkhof, F.; Moll, A.C.; Snoek, F.J.; et al. Cerebrospinal fluid levels of Alzheimer’s disease biomarkers in middle-aged patients with type 1 diabetes. Diabetologia 2014, 57, 2208–2214. [Google Scholar] [CrossRef]
- McLimans, K.E.; Willette, A.A. Autotaxin is Related to Metabolic Dysfunction and Predicts Alzheimer’s Disease Outcomes. J. Alzheimer’s Dis. JAD 2017, 56, 403–413. [Google Scholar] [CrossRef]
- Hong, S.H.; Han, K.; Park, S.; Kim, S.M.; Kim, N.H.; Choi, K.M.; Baik, S.H.; Park, Y.G.; Yoo, H.J. Gamma-Glutamyl Transferase Variability and Risk of Dementia in Diabetes Mellitus: A Nationwide Population-Based Study. J. Clin. Endocrinol. Metab. 2020, 105, e119–e129. [Google Scholar] [CrossRef]
- Ha, J.; Moon, M.K.; Kim, H.; Park, M.; Cho, S.Y.; Lee, J.; Lee, J.Y.; Kim, E. Plasma Clusterin as a Potential Link Between Diabetes and Alzheimer Disease. J. Clin. Endocrinol. Metab. 2020, 105, 3058–3068. [Google Scholar] [CrossRef]
- Ciudin, A.; Simó-Servat, O.; Hernández, C.; Arcos, G.; Diego, S.; Sanabria, Á.; Sotolongo, Ó.; Hernández, I.; Boada, M.; Simó, R. Retinal Microperimetry: A New Tool for Identifying Patients With Type 2 Diabetes at Risk for Developing Alzheimer Disease. Diabetes 2017, 66, 3098–3104. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Liu, Y.; He, T.; Zhang, Y.; He, J.; Li, M.; Jiang, B.; Gao, Y.; Chen, C.; Ke, D.; et al. Platelet biomarkers identifying mild cognitive impairment in type 2 diabetes patients. Aging Cell 2021, 20, e13469. [Google Scholar] [CrossRef] [PubMed]
- Marutani, N.; Akamine, S.; Kanayama, D.; Gotoh, S.; Yanagida, K.; Maruyama, R.; Mori, K.; Miyamoto, T.; Adachi, H.; Sakagami, Y.; et al. Plasma NfL is associated with mild cognitive decline in patients with diabetes. Psychogeriatr. Off. J. Jpn. Psychogeriatr. Soc. 2022, 22, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Yao, R.; Sun, C.; Leng, B.; Shen, T.; Wang, T.; Zhang, S.; Li, M.; Yang, Y.; Sun, H.; et al. Blood Neuroexosomal Mitochondrial Proteins Predict Alzheimer Disease in Diabetes. Diabetes 2022, 71, 1313–1323. [Google Scholar] [CrossRef]
- Wang, C.; Huang, X.; Tian, S.; Huang, R.; Guo, D.; Lin, H.; Wang, J.; Wang, S. High Plasma Resistin Levels Portend the Insulin Resistance-Associated Susceptibility to Early Cognitive Decline in Patients with Type 2 Diabetes Mellitus. J. Alzheimer’s Dis. JAD 2020, 75, 807–815. [Google Scholar] [CrossRef]
- Wang, J.; Yuan, Y.; Cai, R.; Huang, R.; Tian, S.; Lin, H.; Guo, D.; Wang, S. Association between Plasma Levels of PAI-1, tPA/PAI-1 Molar Ratio, and Mild Cognitive Impairment in Chinese Patients with Type 2 Diabetes Mellitus. J. Alzheimer’s Dis. JAD 2018, 63, 835–845. [Google Scholar] [CrossRef]
- Xu, Z.P.; Yang, S.L.; Zhao, S.; Zheng, C.H.; Li, H.H.; Zhang, Y.; Huang, R.X.; Li, M.Z.; Gao, Y.; Zhang, S.J.; et al. Biomarkers for Early Diagnostic of Mild Cognitive Impairment in Type-2 Diabetes Patients: A Multicentre, Retrospective, Nested Case-Control Study. EBioMedicine 2016, 5, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Graff-Radford, J.; Yong, K.X.X.; Apostolova, L.G.; Bouwman, F.H.; Carrillo, M.; Dickerson, B.C.; Rabinovici, G.D.; Schott, J.M.; Jones, D.T.; Murray, M.E. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol 2021, 20, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef]
- Shinohara, M.; Tachibana, M.; Kanekiyo, T.; Bu, G. Role of LRP1 in the pathogenesis of Alzheimer’s disease: Evidence from clinical and preclinical studies. J. Lipid. Res. 2017, 58, 1267–1281. [Google Scholar] [CrossRef] [PubMed]
- Goding, J.W.; Grobben, B.; Slegers, H. Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim. Biophys. Acta 2003, 1638, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Gasecka, A.; Siwik, D.; Gajewska, M.; Jaguszewski, M.J.; Mazurek, T.; Filipiak, K.J.; Postuła, M.; Eyileten, C. Early Biomarkers of Neurodegenerative and Neurovascular Disorders in Diabetes. J. Clin. Med. 2020, 9, 2807. [Google Scholar] [CrossRef]
- Chen, F.; Swartzlander, D.B.; Ghosh, A.; Fryer, J.D.; Wang, B.; Zheng, H. Clusterin secreted from astrocyte promotes excitatory synaptic transmission and ameliorates Alzheimer’s disease neuropathology. Mol. Neurodegener. 2021, 16, 5. [Google Scholar] [CrossRef]
- Chrem Mendez, P.; Surace, E.; Bérgamo, Y.; Calandri, I.; Vázquez, S.; Sevlever, G.; Allegri, R.F. Biomarkers for Alzheimer’s disease. Where we stand and where we are headed. Med. B Aires 2019, 79, 546–551. [Google Scholar]
- Mooldijk, S.S.; Ikram, M.K.; Ikram, M.A. Adiponectin, Leptin, and Resistin and the Risk of Dementia. J Gerontol A Biol. Sci. Med. Sci. 2022, 77, 1245–1249. [Google Scholar] [CrossRef]
- Liu, R.M. Aging, Cellular Senescence, and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 1989. [Google Scholar] [CrossRef]
- Huang, S.; Tao, R.; Zhou, J.; Qian, L.; Wu, J. Trans-10-Hydroxy-2-decenoic Acid Alleviates Dextran Sulfate Sodium-Induced Colitis in Mice via Regulating the Inflammasome-Mediated Pyroptotic Pathway and Enhancing Colonic Barrier Function. Mol. Nutr. Food Res. 2022, 66, e2100821. [Google Scholar] [CrossRef]
- You, M.; Miao, Z.; Tian, J.; Hu, F. Trans-10-hydroxy-2-decenoic acid protects against LPS-induced neuroinflammation through FOXO1-mediated activation of autophagy. Eur. J. Nutr. 2020, 59, 2875–2892. [Google Scholar] [CrossRef]
- You, M.; Miao, Z.; Pan, Y.; Hu, F. Trans-10-hydroxy-2-decenoic acid alleviates LPS-induced blood-brain barrier dysfunction by activating the AMPK/PI3K/AKT pathway. Eur. J. Pharm. 2019, 865, 172736. [Google Scholar] [CrossRef]
- Eslami-Kaliji, F.; Sarafbidabad, M.; Kiani-Esfahani, A.; Mirahmadi-Zare, S.Z.; Dormiani, K. 10-hydroxy-2-decenoic acid a bio-immunomodulator in tissue engineering; generates tolerogenic dendritic cells by blocking the toll-like receptor4. J. Biomed. Mater Res. A 2021, 109, 1575–1587. [Google Scholar] [CrossRef]
- Eslami-Kaliji, F.; Mirahmadi-Zare, S.Z.; Nazem, S.; Shafie, N.; Ghaedi, R.; Asadian-Esfahani, M.H. A label-free SPR biosensor for specific detection of TLR4 expression; introducing of 10-HDA as an antagonist. Int. J. Biol. Macromol. 2022, 217, 142–149. [Google Scholar] [CrossRef]
- Saad Al Shehri, Z.; Alanazi, A.D.; Alnomasy, S.F. Anti-Cancer Effects of Queen Bee Acid (10-Hydroxy-2-decenoic Acid) and Its Cellular Mechanisms against Human Hepatoma Cells. Molecules 2023, 28, 1972. [Google Scholar] [CrossRef]
- Albalawi, A.E.; Althobaiti, N.A.; Alrdahe, S.S.; Alhasani, R.H.; Alaryani, F.S.; BinMowyna, M.N. Anti-Tumor Effects of Queen Bee Acid (10-Hydroxy-2-decenoic Acid) Alone and in Combination with Cyclophosphamide and Its Cellular Mechanisms against Ehrlich Solid Tumor in Mice. Molecules 2021, 26, 7021. [Google Scholar] [CrossRef]
- Lin, X.M.; Liu, S.B.; Luo, Y.H.; Xu, W.T.; Zhang, Y.; Zhang, T.; Xue, H.; Zuo, W.B.; Li, Y.N.; Lu, B.X.; et al. 10-HDA Induces ROS-Mediated Apoptosis in A549 Human Lung Cancer Cells by Regulating the MAPK, STAT3, NF-κB, and TGF-β1 Signaling Pathways. BioMed Res. Int. 2020, 2020, 3042636. [Google Scholar] [CrossRef]
- Hu, X.; Liu, Z.; Lu, Y.; Chi, X.; Han, K.; Wang, H.; Wang, Y.; Ma, L.; Xu, B. Glucose metabolism enhancement by 10-hydroxy-2-decenoic acid via the PI3K/AKT signaling pathway in high-fat-diet/streptozotocin induced type 2 diabetic mice. Food Funct. 2022, 13, 9931–9946. [Google Scholar] [CrossRef]
- Zhang, Y.; Geng, S.; Di, Y.; Sun, Y.; Liu, Y.; Li, J.; Zhang, L. 10-Hydroxy-trans-2-decenoic Acid, a New Potential Feed Additive for Broiler Chickens to Improve Growth Performance. Animals 2022, 12, 1846. [Google Scholar] [CrossRef]
- Fan, P.; Sha, F.; Ma, C.; Wei, Q.; Zhou, Y.; Shi, J.; Fu, J.; Zhang, L.; Han, B.; Li, J. 10-Hydroxydec-2-Enoic Acid Reduces Hydroxyl Free Radical-Induced Damage to Vascular Smooth Muscle Cells by Rescuing Protein and Energy Metabolism. Front. Nutr. 2022, 9, 873892. [Google Scholar] [CrossRef]
- Huang, M.; Xiao, M.; Dong, J.; Huang, Y.; Sun, H.; Wang, D. Synergistic anti-inflammatory effects of graphene oxide quantum dots and trans-10-hydroxy-2-decenoic acid on LPS-stimulated RAW 264.7 macrophage cells. Biomater. Adv. 2022, 136, 212774. [Google Scholar] [CrossRef]
- Gao, K.; Su, B.; Dai, J.; Li, P.; Wang, R.; Yang, X. Anti-Biofilm and Anti-Hemolysis Activities of 10-Hydroxy-2-decenoic Acid against Staphylococcus aureus. Molecules 2022, 27, 1485. [Google Scholar] [CrossRef]
- Perminaite, K.; Marksa, M.; Stančiauskaitė, M.; Juknius, T.; Grigonis, A.; Ramanauskiene, K. Formulation of Ocular In Situ Gels with Lithuanian Royal Jelly and Their Biopharmaceutical Evaluation In Vitro. Molecules 2021, 26, 3552. [Google Scholar] [CrossRef]
- Fan, P.; Han, B.; Hu, H.; Wei, Q.; Zhang, X.; Meng, L.; Nie, J.; Tang, X.; Tian, X.; Zhang, L.; et al. Proteome of thymus and spleen reveals that 10-hydroxydec-2-enoic acid could enhance immunity in mice. Expert Opin. Targets 2020, 24, 267–279. [Google Scholar] [CrossRef]
- Pandeya, P.R.; Lamichhane, R.; Lee, K.H.; Kim, S.G.; Lee, D.H.; Lee, H.K.; Jung, H.J. Bioassay-guided isolation of active anti-adipogenic compound from royal jelly and the study of possible mechanisms. BMC Complement Altern. Med. 2019, 19, 33. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Chou, W.M.; Widowati, D.A.; Lin, I.P.; Peng, C.C. 10-hydroxy-2-decenoic acid of royal jelly exhibits bactericide and anti-inflammatory activity in human colon cancer cells. BMC Complement Altern. Med. 2018, 18, 202. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.C.; Sun, H.T.; Lin, I.P.; Kuo, P.C.; Li, J.C. The functional property of royal jelly 10-hydroxy-2-decenoic acid as a melanogenesis inhibitor. BMC Complement Altern. Med. 2017, 17, 392. [Google Scholar] [CrossRef] [PubMed]
- Filipič, B.; Gradišnik, L.; Rihar, K.; Šooš, E.; Pereyra, A.; Potokar, J. The influence of royal jelly and human interferon-alpha (HuIFN-αN3) on proliferation, glutathione level and lipid peroxidation in human colorectal adenocarcinoma cells in vitro. Arh. Hig. Rada. Toksikol. 2015, 66, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Araki, Y.; Hata, T.; Ichihara, K.; Ito, M.; Tanaka, M.; Honda, S. 10-Hydroxy-2-decenoic Acid, the Major Lipid Component of Royal Jelly, Extends the Lifespan of Caenorhabditis elegans through Dietary Restriction and Target of Rapamycin Signaling. J. Aging Res. 2015, 2015, 425261. [Google Scholar] [CrossRef]
- Zheng, J.; Lai, W.; Zhu, G.; Wan, M.; Chen, J.; Tai, Y.; Lu, C. 10-Hydroxy-2-decenoic acid prevents ultraviolet A-induced damage and matrix metalloproteinases expression in human dermal fibroblasts. J. Eur. Acad. Derm. Venereol. 2013, 27, 1269–1277. [Google Scholar] [CrossRef]
- Park, H.M.; Hwang, E.; Lee, K.G.; Han, S.M.; Cho, Y.; Kim, S.Y. Royal jelly protects against ultraviolet B-induced photoaging in human skin fibroblasts via enhancing collagen production. J. Med. Food 2011, 14, 899–906. [Google Scholar] [CrossRef]
- Cheng, D.; Yang, S.; Zhao, X.; Wang, G. The Role of Glucagon-Like Peptide-1 Receptor Agonists (GLP-1 RA) in Diabetes-Related Neurodegenerative Diseases. Drug Des. Dev. Ther. 2022, 16, 665–684. [Google Scholar] [CrossRef]
- Drucker, D.J. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 2018, 27, 740–756. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2012, 8, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Contreras, A.V.; Torres, N.; Tovar, A.R. PPAR-α as a key nutritional and environmental sensor for metabolic adaptation. Adv. Nutr. 2013, 4, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef]
- Lin, C.H.; Nicol, C.J.B.; Cheng, Y.C.; Chen, S.J.; Yen, C.H.; Huang, R.N.; Chiang, M.C. Rosiglitazone rescues human neural stem cells from amyloid-beta induced ER stress via PPARγ dependent signaling. Exp. Cell Res. 2018, 370, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Corbett, G.T.; Gonzalez, F.J.; Pahan, K. Activation of peroxisome proliferator-activated receptor α stimulates ADAM10-mediated proteolysis of APP. Proc. Natl. Acad. Sci. USA 2015, 112, 8445–8450. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Jana, M.; Corbett, G.T.; Ramaswamy, S.; Kordower, J.H.; Gonzalez, F.J.; Pahan, K. Regulation of cyclic AMP response element binding and hippocampal plasticity-related genes by peroxisome proliferator-activated receptor α. Cell Rep. 2013, 4, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Chougule, A.; Baroi, S.; Czernik, P.J.; Crowe, E.; Chang, M.R.; Griffin, P.R.; Lecka-Czernik, B. Osteocytes contribute via nuclear receptor PPAR-alpha to maintenance of bone and systemic energy metabolism. Front. Endocrinol. 2023, 14, 1145467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, N.Q.; Yan, F.; Jin, H.; Zhou, S.Y.; Shi, J.S.; Jin, F. Diabetes mellitus and Alzheimer’s disease: GSK-3β as a potential link. Behav. Brain Res. 2018, 339, 57–65. [Google Scholar] [CrossRef]
- Park, M.; Yi, J.W.; Kim, E.M.; Yoon, I.J.; Lee, E.H.; Lee, H.Y.; Ji, K.Y.; Lee, K.H.; Jang, J.H.; Oh, S.S.; et al. Triggering receptor expressed on myeloid cells 2 (TREM2) promotes adipogenesis and diet-induced obesity. Diabetes 2015, 64, 117–127. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.; Zheng, Y.; Luo, Y.; Du, Y.; Zhao, Y.; Guan, J.; Zhang, X.; Fu, J. TREM-2-p38 MAPK signaling regulates neuroinflammation during chronic cerebral hypoperfusion combined with diabetes mellitus. J. Neuroinflamm. 2020, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Takikawa, M.; Kumagai, A.; Hirata, H.; Soga, M.; Yamashita, Y.; Ueda, M.; Ashida, H.; Tsuda, T. 10-Hydroxy-2-decenoic acid, a unique medium-chain fatty acid, activates 5’-AMP-activated protein kinase in L6 myotubes and mice. Mol. Nutr. Food Res. 2013, 57, 1794–1802. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarker | Fluid Source | Function | Results | References |
|---|---|---|---|---|
| pTau | Cerebrospinal fluid | Correlated with the intensity of neurodegeneration and neurofibrillary-tangle pathology, respectively | The level of total tau was raised in APOE E4+ AD patients with DM | [31,129] |
| Aβ42 | Cerebrospinal fluid | Forms neurotic plaques and causes impaired synaptic plasticity and neuronal cell death | CSF levels of Aβ42 were higher in patients with type 1 diabetes than in controls | [118,130] |
| sLRP1 | Cerebrospinal fluid | A member of the LDL receptor family | CSF levels of LRP1 were higher in patients with type 1 diabetes than in controls | [118,131] |
| autotaxin | Cerebrospinal fluid | Hydrolyzes lysophosphatidylcholine into lysophosphatidic acid | Autotaxin levels were significantly higher in MCI and AD | [119,132] |
| GGT | Serum | Cellular antioxidant, glutathione metabolism | Higher levels of GGT activity were correlated with dementia in patients with DM | [133] |
| GSK-3beta | Serum | A serine/threonine kinase | Serum level of GSK-3β protein was higher in the T2DM-MCI group than the T2DM-nMCI group | [128] |
| Clusterin | Plasma | Participates in several kinds of cellular processes, such as synaptic regulation, lipid transport, extracellular misfolded protein clearance, and complement inhibition | Clusterin increased with disease severity in AD and DM patients | [121,134] |
| NfL | Plasma | An intermediate filament of the neuronal cytoskeleton | Elevated blood levels of NfL can be used to screen for AD | [135] |
| NDUFS3 | Plasma | Subunits of electron transport chain complex | NDUFS3 was lower in patients with T2DM with AD dementia and progressive MCI | [125] |
| SDHB | Plasma | Subunits of electron transport chain complex | SDHB was lower in patients with T2DM with AD dementia and progressive MCI | [125] |
| resistin | Plasma | Play a role in energy homeostasis and regulation of metabolism | Higher plasma levels of resistin were associated with a decreased risk of dementia and AD | [136] |
| PAI-1 | Plasma | A serine protease inhibitor and cell senescence marker | Plasma PAI-1 protein levels were increased in the elderly and in the AD brain | [137] |
| Related Mechanisms | Results | Model | References |
|---|---|---|---|
| Apoptosis | Inhibits apoptosis in human hepatoma cells. | Human hepatoma cell line. | [143] |
| Inflammation Antioxidation | Hypoglycemic effects on diabetic mice, through the PI3K/AKT/GSK3β signaling pathway. | Diabetic C57BL/6J mice. | [146] |
| Inflammation | Blocks TLR4. | HEK293T cells with high TLR4 expression. | [142] |
| Inflammation Antioxidation | Increases serum concentrations of immunoglobulin G at d 21, as well as IgM and interleukin-10 at d 42, while decreasing the levels of tumor necrosis factor-α. | Broiler Chickens. | [147] |
| Inflammation Antioxidation | Inhibits inflammasome-mediated pyroptosis induced by LPS/ATP. | Male C57BL/6 mice. | [138] |
| Antioxidation Energy metabolism Vascular function | Maintains vascular health via scavenging •OH. | Vascular smooth-muscle cells. | [148] |
| Inflammation | Attenuates the secretion of TNF-α, IL-6, and IL-1β. | Macrophages (RAW264.7 cells) | [149] |
| Antimicrobial | Decreases biofilm viability and effectively eradicates mature biofilms. | Staphylococcus aureus. | [150] |
| Antitumor | Decreases tumor volume, tumor markers (AFP and CEA), and TNF-α level. | Female Swiss albino mice. | [144] |
| Immunomodulation | Blocks TLR4. | Dendritic cells | [141] |
| Antimicrobial Antioxidation | Shows antioxidant and antimicrobial activity. | Statens Seruminstitut Rabbit Cornea cell culture line. | [151] |
| Apoptosis Antioxidation | Induces apoptosis through ROS-mediated MAPK, STAT3, NF-κB, and TGF-β1 signaling pathways. | A549 human lung cancer cells. | [145] |
| Autophagy | Protects against neuroinflammation through FOXO1-mediated activation of autophagy. | Microglial BV-2 cells (LPS-induced). | [139] |
| Immunomodulation | Improves immunity in the thymus and spleen | BALB/c mice. | [152] |
| Vascular function | Improves blood–brain barrier dysfunction by activating the AMPK/PI3K/AKT pathway. | C57BL/6 mice (LPS-stimulated). | [140] |
| Insulin signaling Anti-adipogenesis | Inhibits cAMP/PKA pathway and p-Akt- and MAPK-dependent insulin signaling pathway. | 3 T3-L1 adipocyte cell line. | [153] |
| Inflammation Antimicrobial | Modulates interleukin-8, IL-1β, and tumor necrosis factor-alpha. | WiDr cell. | [154] |
| Melanogenesis inhibitor | Inhibits the activity of tyrosinase and the expression of tyrosinase-related protein 1, TRP-2, and microphthalmia-associated transcription factor. | B16F1 melanoma cells. | [155] |
| Antioxidation | Decreases tumorigenic potential of various tumor cells. | Human colorectal adenocarcinoma cells. | [156] |
| Insulin-like signaling | Extends lifespan through dietary restriction signaling. | Caenorhabditis elegans. | [157] |
| Antioxidation | Reduces the UVA-induced activation of the JNK and p38 MAPK pathways. | Human dermal fibroblasts. | [158] |
| Inflammation | Increases procollagen type I and TGF-β1 production. | Human dermal fibroblasts. | [159] |
| Macromolecule | PDB | DeltaG (KJ/mol) | RMSD (Å) | Binding Site (Number) | Hydrogen Bonds |
|---|---|---|---|---|---|
| GLP-1R | 3c5t | −24.27 | 2.193 | Ala28, Ser31, Thr35, and Pro90 | 4 |
| PPAR-gamma | 2q59 | −20.59 | 0.956 | Asn375, Lys230, and Asp381(2) | 4 |
| PPAR-alpha | 3vi8 | −22.47 | 1.598 | Tyr468(2), Met467, Gln445(2), and Lys448 | 6 |
| GSK-3 | 1q5k | −23.81 | 1.556 | Lys292, Lys94, and Arg96 | 4 |
| TREM2 | 6yye | −12.38 | 1.212 | Ser106, Asn109, Asn173(2), and Ala189 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, Y.; Luo, H.; Li, Z.; Feng, Y.; Liu, Z.; Chang, J. Metabolic Profile of Alzheimer’s Disease: Is 10-Hydroxy-2-decenoic Acid a Pertinent Metabolic Adjuster? Metabolites 2023, 13, 954. https://doi.org/10.3390/metabo13080954
Gong Y, Luo H, Li Z, Feng Y, Liu Z, Chang J. Metabolic Profile of Alzheimer’s Disease: Is 10-Hydroxy-2-decenoic Acid a Pertinent Metabolic Adjuster? Metabolites. 2023; 13(8):954. https://doi.org/10.3390/metabo13080954
Chicago/Turabian StyleGong, Yuan, Hongjie Luo, Zeju Li, Yijun Feng, Zhen Liu, and Jie Chang. 2023. "Metabolic Profile of Alzheimer’s Disease: Is 10-Hydroxy-2-decenoic Acid a Pertinent Metabolic Adjuster?" Metabolites 13, no. 8: 954. https://doi.org/10.3390/metabo13080954
APA StyleGong, Y., Luo, H., Li, Z., Feng, Y., Liu, Z., & Chang, J. (2023). Metabolic Profile of Alzheimer’s Disease: Is 10-Hydroxy-2-decenoic Acid a Pertinent Metabolic Adjuster? Metabolites, 13(8), 954. https://doi.org/10.3390/metabo13080954