
Cancer Cachexia: Underlying Mechanisms and Potential Therapeutic Interventions


Abstract
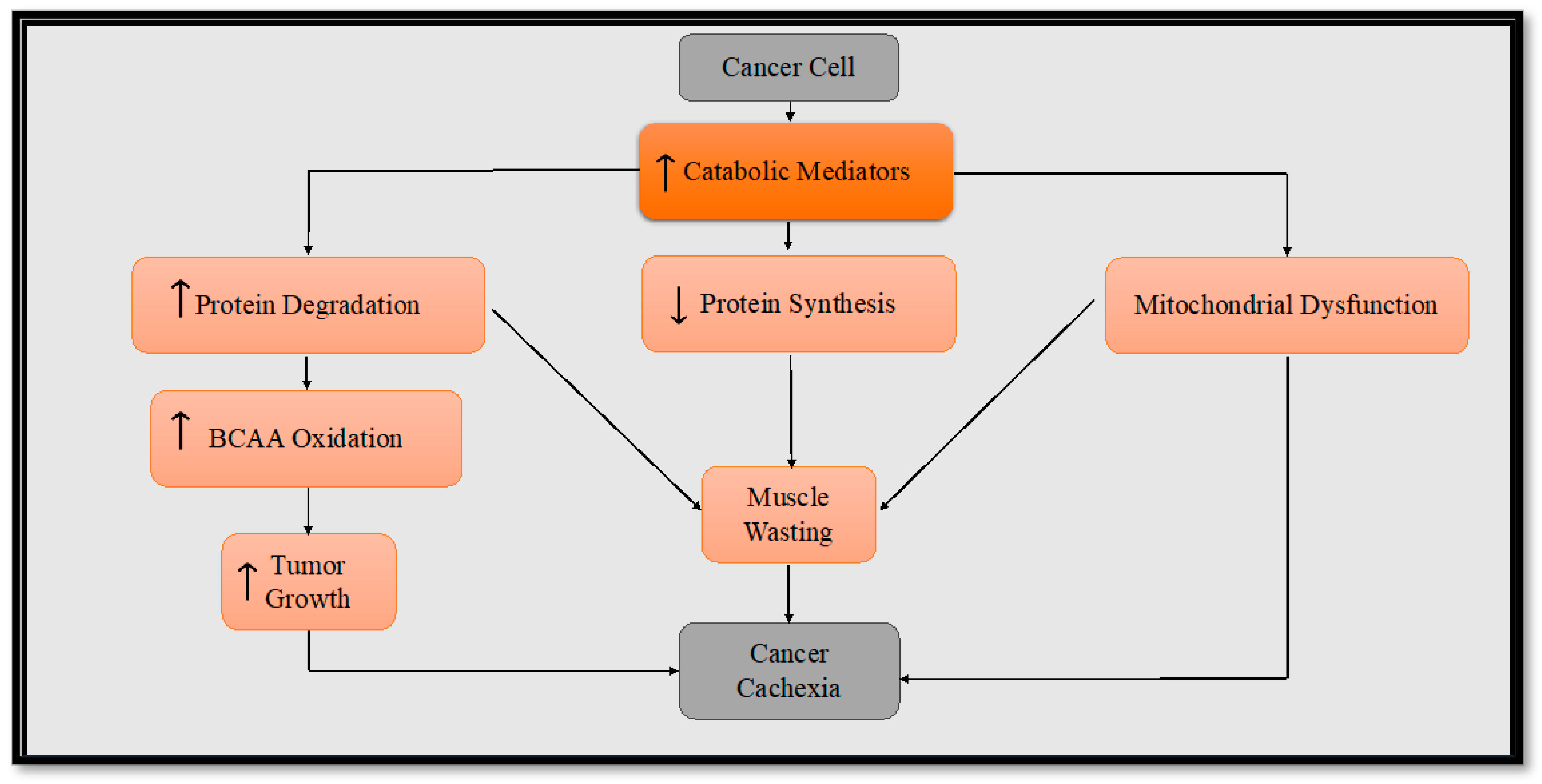
:1. Introduction
2. Preclinical Models of Cancer Cachexia
2.1. APCMin/+ Mouse Model
2.2. C26 Colon Carcinoma Model
2.3. Lewis Lung Carcinoma Model
2.4. Other Genetically Engineered Models

{kind=link}
{kind=link}
{kind=link}
| Model Name | Notable Features | Reference |
|---|---|---|
| APCMin/+ Mouse Model |
| [7,13] |
| Colon-26 Carcinoma Model |
| [14,18,22] |
| Lewis Lung Carcinoma Model |
| [19] |
| Luan and Colleagues Mouse Model |
| [27] |
| KPP Mouse Model |
| [28] |
3. Metabolic Reprogramming in Cancer Cachexia
4. Inflammatory Mediators in Cancer Cachexia
5. Skeletal Muscle Alteration in Cancer Cachexia
5.1. Protein Synthesis
5.2. Muscle Proteolysis
6. Altered Mitochondrial Metabolism in Cancer Cachexia
7. Myokines as Potential Therapeutic Agents for Cancer Cachexia
7.1. Myostatin
7.2. Fibroblast Growth Factor 21
7.3. Interleukin-15
8. Dysfunction of Adipose Tissue in Cancer Cachexia
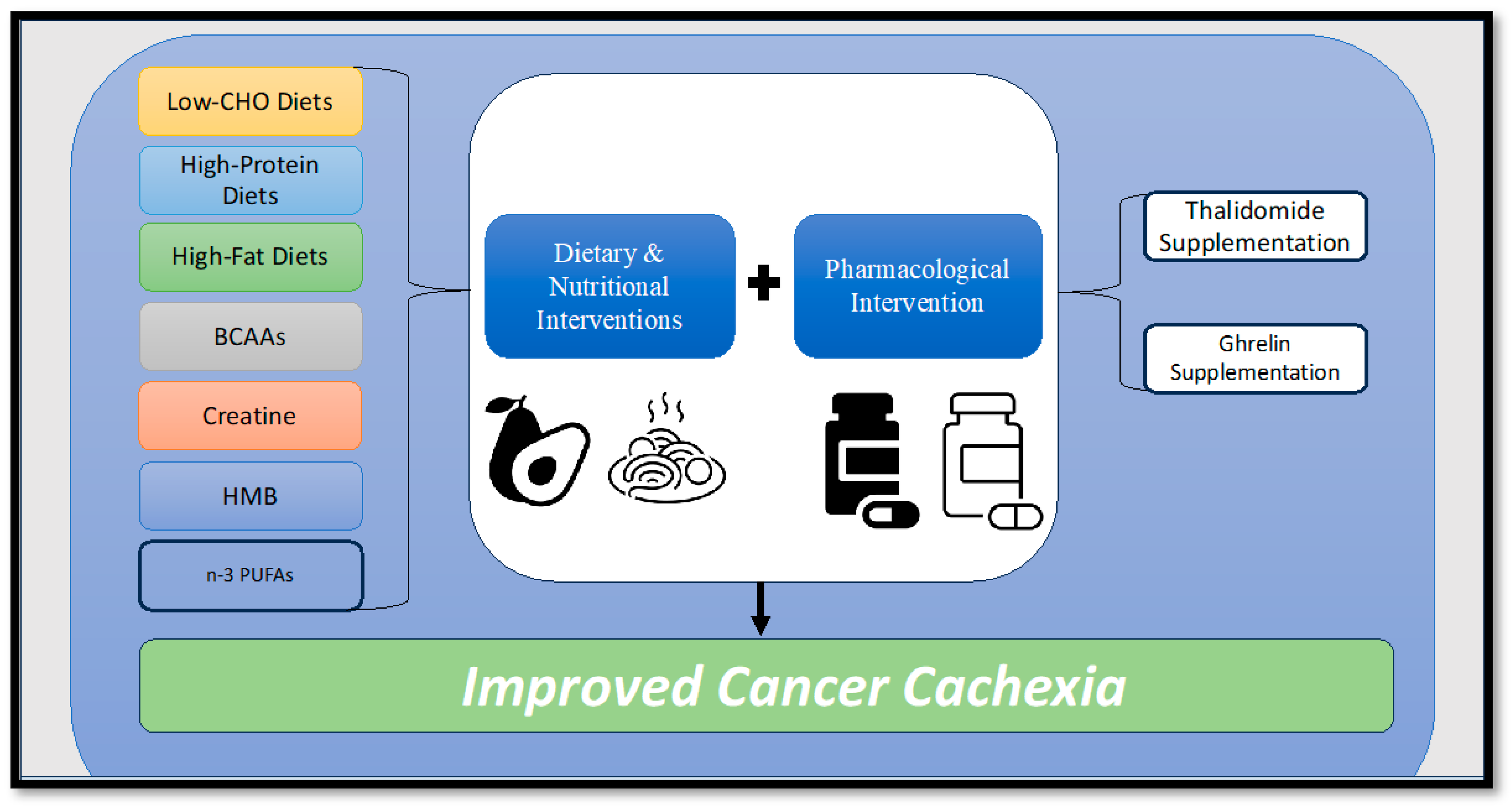
9. Potential Therapeutic Interventions for Cancer Cachexia
9.1. Clinical Care in Cancer Cachexia
9.2. Nutritional Interventions
9.2.1. Omega-3 Polyunsaturated Fatty Acids
9.2.2. Creatine
9.2.3. Branched-chain Amino Acids
9.2.4. Hydroxymethylbuterate
9.3. Dietary Intake Interventions
9.3.1. High-Fat Diets
9.3.2. Carbohydrate Diets
9.3.3. Protein Diets
9.4. Pharmacological Interventions
Ghrelin Supplementation
9.5. Gene Therapy
9.6. Anti-Inflammatory Treatment
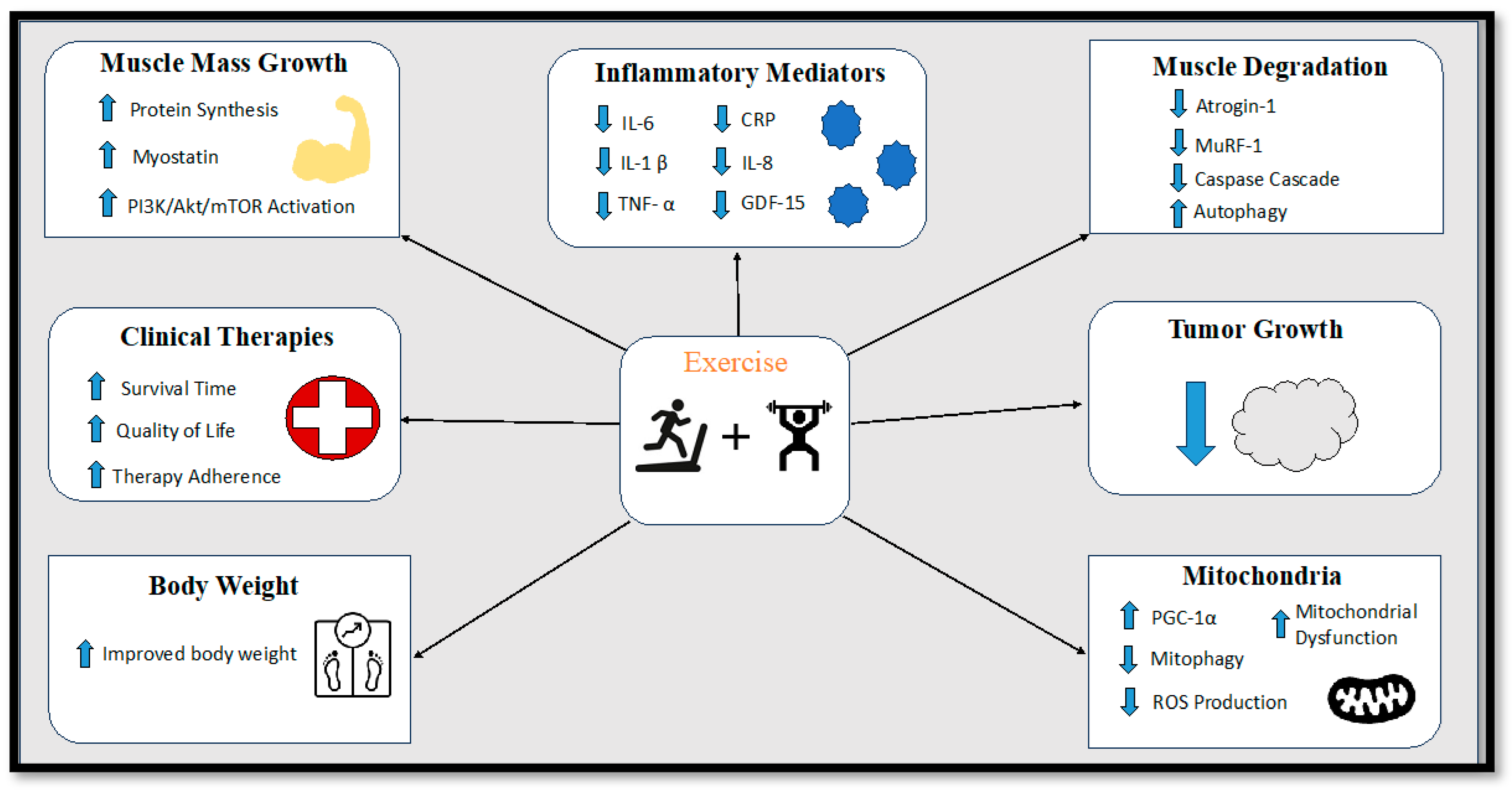
9.7. Exercise Interventions
9.7.1. Endurance Exercise Training
9.7.2. Resistance Exercise Training
9.7.3. Concurrent Exercise Training
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Puppa, M.J.; White, J.P.; Sato, S.; Cairns, M.; Baynes, J.W.; Carson, J.A. Gut barrier dysfunction in the ApcMin/+ mouse model of colon cancer cachexia. Biochim. Biophys. Acta-Mol. Basis Dis. 2011, 1812, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Dodson, S.; Baracos, V.E.; Jatoi, A.; Evans, W.J.; Cella, D.; Dalton, J.T.; Steiner, M.S. Muscle wasting in cancer cachexia: Clinical implications, diagnosis, and emerging treatment strategies. Annu. Rev. Med. 2011, 62, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Skipworth, R.J.; Stewart, G.D.; Dejong, C.H.; Preston, T.; Fearon, K.C. Pathophysiology of cancer cachexia: Much more than host-tumour interaction? Clin. Nutr. 2007, 26, 667–676. [Google Scholar] [CrossRef]
- Dewys, W.D.; Begg, C.; Lavin, P.T.; Band, P.R.; Bennett, J.M.; Bertino, J.R.; Cohen, M.H.; Douglass, H.O.; Engstrom, P.F.; Ezdinli, E.Z.; et al. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am. J. Med. 1980, 69, 491–497. [Google Scholar] [CrossRef]
- Porporato, P.E. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 2016, 5, e200. [Google Scholar] [CrossRef]
- Ren, J.; Sui, H.; Fang, F.; Li, Q.; Li, B. The application of Apc. J. Cancer Res. Clin. Oncol. 2019, 145, 1111–1122. [Google Scholar] [CrossRef]
- White, J.P.; Baynes, J.W.; Welle, S.L.; Kostek, M.C.; Matesic, L.E.; Sato, S.; Carson, J.A. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PLoS ONE 2011, 6, e24650. [Google Scholar] [CrossRef]
- White, J.P.; Puppa, M.J.; Narsale, A.; Carson, J.A. Characterization of the male ApcMin/+ mouse as a hypogonadism model related to cancer cachexia. Biol. Open 2013, 2, 1346–1353. [Google Scholar] [CrossRef]
- VanderVeen, B.N.; Hardee, J.P.; Fix, D.K.; Carson, J.A. Skeletal muscle function during the progression of cancer cachexia in the male Apc. J. Appl. Physiol. 2018, 124, 684–695. [Google Scholar] [CrossRef]
- Baltgalvis, K.A.; Berger, F.G.; Peña, M.M.; Mark Davis, J.; White, J.P.; Carson, J.A. Activity level, apoptosis, and development of cachexia in Apc(Min/+) mice. J. Appl. Physiol. 2010, 109, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Moser, A.R.; Luongo, C.; Gould, K.A.; McNeley, M.K.; Shoemaker, A.R.; Dove, W.F. ApcMin: A mouse model for intestinal and mammary tumorigenesis. Eur. J. Cancer 1995, 31, 1061–1064. [Google Scholar] [CrossRef]
- Parker, T.W.; Neufeld, K.L. APC controls Wnt-induced β-catenin destruction complex recruitment in human colonocytes. Sci. Rep. 2020, 10, 2957. [Google Scholar] [CrossRef] [PubMed]
- Aulino, P.; Berardi, E.; Cardillo, V.M.; Rizzuto, E.; Perniconi, B.; Ramina, C.; Padula, F.; Spugnini, E.P.; Baldi, A.; Faiola, F.; et al. Molecular, cellular and physiological characterization of the cancer cachexia-inducing C26 colon carcinoma in mouse. BMC Cancer 2010, 10, 363. [Google Scholar] [CrossRef] [PubMed]
- Bonetto, A.; Kays, J.K.; Parker, V.A.; Matthews, R.R.; Barreto, R.; Puppa, M.J.; Kang, K.S.; Carson, J.A.; Guise, T.A.; Mohammad, K.S. Differential bone loss in mouse models of colon cancer cachexia. Front. Physiol. 2017, 7, 679. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, A. The Role of Tumor Microenvironment Cells in Colorectal Cancer (CRC) Cachexia. Int. J. Mol. Sci. 2021, 22, 1565. [Google Scholar] [CrossRef]
- Assi, M.; Derbré, F.; Lefeuvre-Orfila, L.; Rébillard, A. Antioxidant supplementation accelerates cachexia development by promoting tumor growth in C26 tumor-bearing mice. Free Radic. Biol. Med. 2016, 91, 204–214. [Google Scholar] [CrossRef]
- Ballarò, R.; Costelli, P.; Penna, F. Animal models for cancer cachexia. Curr. Opin. Support. Palliat. Care 2016, 10, 281–287. [Google Scholar] [CrossRef]
- Penna, F.; Busquets, S.; Argilés, J.M. Experimental cancer cachexia: Evolving strategies for getting closer to the human scenario. Semin. Cell Dev. Biol. 2016, 54, 20–27. [Google Scholar] [CrossRef]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Talbert, E.E.; Metzger, G.A.; He, W.A.; Guttridge, D.C. Modeling human cancer cachexia in colon 26 tumor-bearing adult mice. J. Cachexia Sarcopenia Muscle 2014, 5, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Cosper, P.F.; Leinwand, L.A. Cancer causes cardiac atrophy and autophagy in a sexually dimorphic manner. Cancer Res. 2011, 71, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Brodt, P. Characterization of two highly metastatic variants of Lewis lung carcinoma with different organ specificities. Cancer Res. 1986, 46, 2442–2448. [Google Scholar] [PubMed]
- Yan, L.; Sundaram, S.; Mehus, A.A.; Picklo, M.J. Time-restricted Feeding Attenuates High-fat Diet-enhanced Spontaneous Metastasis of Lewis Lung Carcinoma in Mice. Anticancer. Res. 2019, 39, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.N.; Watari, K.; Fotovati, A.; Hosoi, F.; Yasumoto, K.; Izumi, H.; Kohno, K.; Umezawa, K.; Iguchi, H.; Shirouzu, K.; et al. Inflammatory stimuli from macrophages and cancer cells synergistically promote tumor growth and angiogenesis. Cancer Sci. 2007, 98, 2009–2018. [Google Scholar] [CrossRef]
- Kerr, H.L.; Krumm, K.; Lee, I.I.; Anderson, B.; Christiani, A.; Strait, L.; Breckheimer, B.A.; Irwin, B.; Jiang, A.S.; Rybachok, A.; et al. EXT418, a novel long-acting ghrelin, mitigates Lewis lung carcinoma induced cachexia in mice. J. Cachexia Sarcopenia Muscle 2023, 14, 1337–1348. [Google Scholar] [CrossRef]
- Luan, Y.; Zhang, Y.; Yu, S.Y.; You, M.; Xu, P.C.; Chung, S.; Kurita, T.; Zhu, J.; Kim, S.Y. Development of ovarian tumour causes significant loss of muscle and adipose tissue: A novel mouse model for cancer cachexia study. J. Cachexia Sarcopenia Muscle 2022, 13, 1289–1301. [Google Scholar] [CrossRef]
- Talbert, E.E.; Cuitiño, M.C.; Ladner, K.J.; Rajasekerea, P.V.; Siebert, M.; Shakya, R.; Leone, G.W.; Ostrowski, M.C.; Paleo, B.; Weisleder, N.; et al. Modeling Human Cancer-induced Cachexia. Cell Rep. 2019, 28, 1612–1622.e1614. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Callao, V.; Montoya, E. Toxohormone-like factor from microorganisms with impaired respiration. Science 1961, 134, 2041–2042. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Cantley, L.C. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011, 14, 443–451. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211, Correction to Trends Biochem. Sci. 2016, 41, 287. [Google Scholar] [CrossRef]
- Slavov, N.; Budnik, B.A.; Schwab, D.; Airoldi, E.M.; van Oudenaarden, A. Constant growth rate can be supported by decreasing energy flux and increasing aerobic glycolysis. Cell Rep. 2014, 7, 705–714. [Google Scholar] [CrossRef]
- Epstein, T.; Xu, L.; Gillies, R.J.; Gatenby, R.A. Separation of metabolic supply and demand: Aerobic glycolysis as a normal physiological response to fluctuating energetic demands in the membrane. Cancer Metab. 2014, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 749. [Google Scholar] [CrossRef] [PubMed]
- Lam, V.W.; Poon, R.T. Role of branched-chain amino acids in management of cirrhosis and hepatocellular carcinoma. Hepatol. Res. 2008, 38 (Suppl. S1), S107–S115. [Google Scholar] [CrossRef] [PubMed]
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef]
- Peng, H.; Wang, Y.; Luo, W. Multifaceted role of branched-chain amino acid metabolism in cancer. Oncogene 2020, 39, 6747–6756. [Google Scholar] [CrossRef]
- Ananieva, E.A.; Wilkinson, A.C. Branched-chain amino acid metabolism in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 64. [Google Scholar] [CrossRef]
- Padoan, A.; Plebani, M.; Basso, D. Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity. Int. J. Mol. Sci. 2019, 20, 676. [Google Scholar] [CrossRef] [PubMed]
- Estrov, Z.; Kurzrock, R.; Wetzler, M.; Kantarjian, H.; Blake, M.; Harris, D.; Gutterman, J.U.; Talpaz, M. Suppression of chronic myelogenous leukemia colony growth by interleukin-1 (IL-1) receptor antagonist and soluble IL-1 receptors: A novel application for inhibitors of IL-1 activity. Blood 1991, 78, 1476–1484. [Google Scholar] [CrossRef] [PubMed]
- Berek, J.S.; Chung, C.; Kaldi, K.; Watson, J.M.; Knox, R.M.; Martínez-Maza, O. Serum interleukin-6 levels correlate with disease status in patients with epithelial ovarian cancer. Am. J. Obs. Gynecol. 1991, 164, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef]
- Cao, Z.; Zhao, K.; Jose, I.; Hoogenraad, N.J.; Osellame, L.D. Biomarkers for Cancer Cachexia: A Mini Review. Int. J. Mol. Sci. 2021, 22, 4501. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.; Axelsson, B. The prevalence of deranged C-reactive protein and albumin in patients with incurable cancer approaching death. PLoS ONE 2018, 13, e0193693. [Google Scholar] [CrossRef]
- Han, Y.; Weinman, S.; Boldogh, I.; Walker, R.K.; Brasier, A.R. Tumor necrosis factor-α-inducible IκBα proteolysis mediated by cytosolic m-calpain: A mechanism parallel to the ubiquitin-proteasome pathway for nuclear factor-κb activation. J. Biol. Chem. 1999, 274, 787–794. [Google Scholar] [CrossRef]
- Scheede-Bergdahl, C.; Watt, H.L.; Trutschnigg, B.; Kilgour, R.D.; Haggarty, A.; Lucar, E.; Vigano, A. Is IL-6 the best pro-inflammatory biomarker of clinical outcomes of cancer cachexia? Clin. Nutr. 2012, 31, 85–88. [Google Scholar] [CrossRef]
- Moldawer, L.L.; Rogy, M.A.; Lowry, S.F. The role of cytokines in cancer cachexia. J. Parenter. Enter. Nutr. 1992, 16, 43S–49S. [Google Scholar] [CrossRef]
- Noguchi, Y.; Yoshikawa, T.; Matsumoto, A.; Svaninger, G.; Gelin, J. Are cytokines possible mediators of cancer cachexia? Surg. Today 1996, 26, 467–475. [Google Scholar] [CrossRef]
- Matthys, P.; Billiau, A. Cytokines and cachexia. Nutrition 1997, 13, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.Y.; Choi, Y.S.; Yeom, C.H.; Kwak, S.M.; Yoon, H.M.; Kim, D.G.; Koh, S.J.; Park, J.; Lee, M.A.; Lee, Y.J.; et al. Interleukin-6 but not tumour necrosis factor-alpha predicts survival in patients with advanced cancer. Support. Care Cancer 2013, 21, 3071–3077. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Sun, X.; Fischer, J.E.; Hasselgren, P.O. The expression of genes in the ubiquitin-proteasome proteolytic pathway is increased in skeletal muscle from patients with cancer. Surgery 1999, 126, 744–749; discussion 749–750. [Google Scholar] [CrossRef]
- Doyle, A.; Zhang, G.; Abdel Fattah, E.A.; Eissa, N.T.; Li, Y.P. Toll-like receptor 4 mediates lipopolysaccharide-induced muscle catabolism via coordinate activation of ubiquitin-proteasome and autophagy-lysosome pathways. FASEB J. 2011, 25, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Furuno, K.; Goldberg, A.L. The activation of protein degradation in muscle by Ca2+ or muscle injury does not involve a lysosomal mechanism. Biochem. J. 1986, 237, 859–864. [Google Scholar] [CrossRef]
- Haddad, F.; Zaldivar, F.; Cooper, D.M.; Adams, G.R. IL-6-induced skeletal muscle atrophy. J. Appl. Physiol. 2005, 98, 911–917. [Google Scholar] [CrossRef]
- Washington, T.A.; White, J.P.; Davis, J.M.; Wilson, L.B.; Lowe, L.L.; Sato, S.; Carson, J.A. Skeletal muscle mass recovery from atrophy in IL-6 knockout mice. Acta Physiol. 2011, 202, 657–669. [Google Scholar] [CrossRef]
- Huang, Z.; Zhong, L.; Zhu, J.; Xu, H.; Ma, W.; Zhang, L.; Shen, Y.; Law, B.Y.-K.; Ding, F.; Gu, X. Inhibition of IL-6/JAK/STAT3 pathway rescues denervation-induced skeletal muscle atrophy. Ann. Transl. Med. 2020, 8, 1681. [Google Scholar] [CrossRef]
- White, J.P.; Puppa, M.J.; Gao, S.; Sato, S.; Welle, S.L.; Carson, J.A. Muscle mTORC1 suppression by IL-6 during cancer cachexia: A role for AMPK. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1042–E1052. [Google Scholar] [CrossRef]
- White, J.P.; Baltgalvis, K.A.; Puppa, M.J.; Sato, S.; Baynes, J.W.; Carson, J.A. Muscle oxidative capacity during IL-6-dependent cancer cachexia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R201–R211. [Google Scholar] [CrossRef]
- Liu, M.; Li, H.; Zhang, H.; Zhou, H.; Jiao, T.; Feng, M.; Na, F.; Sun, M.; Zhao, M.; Xue, L.; et al. RBMS1 promotes gastric cancer metastasis through autocrine IL-6/JAK2/STAT3 signaling. Cell Death Dis. 2022, 13, 287. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Seo, Y.; Kwon, J.H.; Shin, Y.; Kim, S.; Park, S.J.; Park, J.J.; Cheon, J.H.; Kim, W.H.; Il Kim, T. IL-6 and IL-8, secreted by myofibroblasts in the tumor microenvironment, activate HES1 to expand the cancer stem cell population in early colorectal tumor. Mol. Carcinog. 2021, 60, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Wahid, I. The role of neuropeptide y in cancer-associated anorexia and its correlation with interleukin-1 beta. Ann. Oncol. 2017, 28, x158. [Google Scholar] [CrossRef]
- Laird, B.J.; McMillan, D.; Skipworth, R.J.; Fallon, M.T.; Paval, D.R.; McNeish, I.; Gallagher, I.J. The emerging role of interleukin 1β (IL-1β) in cancer cachexia. Inflammation 2021, 44, 1223–1228. [Google Scholar] [CrossRef]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef]
- Jung, Y.J.; Isaacs, J.S.; Lee, S.; Trepel, J.; Neckers, L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003, 17, 2115–2117. [Google Scholar] [CrossRef]
- Graziano, F.; Ruzzo, A.; Santini, D.; Humar, B.; Tonini, G.; Catalano, V.; Berardi, R.; Pizzagalli, F.; Arduini, F.; Bearzi, I. Prognostic role of interleukin-1β gene and interleukin-1 receptor antagonist gene polymorphisms in patients with advanced gastric cancer. J. Clin. Oncol. 2005, 23, 2339–2345. [Google Scholar] [CrossRef]
- Zhang, D.; Zheng, H.; Zhou, Y.; Tang, X.; Yu, B.; Li, J. Association of IL-1beta gene polymorphism with cachexia from locally advanced gastric cancer. BMC Cancer 2007, 7, 45. [Google Scholar] [CrossRef]
- McCarthy, H.D.; Dryden, S.; Williams, G. Interleukin-1 beta-induced anorexia and pyrexia in rat: Relationship to hypothalamic neuropeptide Y. Am. J. Physiol.-Endocrinol. Metab. 1995, 269, E852–E857. [Google Scholar] [CrossRef]
- Sugarman, B.J.; Aggarwal, B.B.; Hass, P.E.; Figari, I.S.; Palladino Jr, M.A.; Shepard, H.M. Recombinant human tumor necrosis factor-α: Effects on proliferation of normal and transformed cells in vitro. Science 1985, 230, 943–945. [Google Scholar] [CrossRef]
- Sethi, G.; Sung, B.; Aggarwal, B.B. TNF: A master switch for inflammation to cancer. Front. Biosci.-Landmark 2008, 13, 5094–5107. [Google Scholar] [CrossRef]
- Li, Y.P.; Atkins, C.M.; Sweatt, J.D.; Reid, M.B. Mitochondria mediate tumor necrosis factor-alpha/NF-kappaB signaling in skeletal muscle myotubes. Antioxid. Redox Signal. 1999, 1, 97–104. [Google Scholar] [CrossRef]
- Li, Y.P.; Schwartz, R.J.; Waddell, I.D.; Holloway, B.R.; Reid, M.B. Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-kappaB activation in response to tumor necrosis factor alpha. FASEB J. 1998, 12, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Oliff, A.; Defeo-Jones, D.; Boyer, M.; Martinez, D.; Kiefer, D.; Vuocolo, G.; Wolfe, A.; Socher, S.H. Tumors secreting human TNF/cachectin induce cachexia in mice. Cell 1987, 50, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Peixoto da Silva, S.; Santos, J.M.O.; Costa E Silva, M.P.; Gil da Costa, R.M.; Medeiros, R. Cancer cachexia and its pathophysiology: Links with sarcopenia, anorexia and asthenia. J. Cachexia Sarcopenia Muscle 2020, 11, 619–635. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.J.; Patel, B.M. TNF-α and cancer cachexia: Molecular insights and clinical implications. Life Sci. 2017, 170, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, T.; Ishikawa, T.; Okayama, T.; Oka, K.; Adachi, S.; Mizushima, K.; Kimura, R.; Okajima, M.; Sakai, H.; Sakamoto, N.; et al. Tumor inoculation site affects the development of cancer cachexia and muscle wasting. Int. J. Cancer 2015, 137, 2558–2565. [Google Scholar] [CrossRef]
- Li, Y.-P.; Chen, Y.; John, J.; Moylan, J.; Jin, B.; Mann, D.L.; Reid, M.B. TNF-α acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005, 19, 362. [Google Scholar] [CrossRef]
- Sproston, N.R.; Ashworth, J.J. Role of C-Reactive Protein at Sites of Inflammation and Infection. Front. Immunol. 2018, 9, 754. [Google Scholar] [CrossRef]
- Evans, W.J.; Morley, J.E.; Argilés, J.; Bales, C.; Baracos, V.; Guttridge, D.; Jatoi, A.; Kalantar-Zadeh, K.; Lochs, H.; Mantovani, G.; et al. Cachexia: A new definition. Clin. Nutr. 2008, 27, 793–799. [Google Scholar] [CrossRef]
- Deans, C.; Wigmore, S.J. Systemic inflammation, cachexia and prognosis in patients with cancer. Curr. Opin. Clin. Nutr. Metab. Care 2005, 8, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Amano, K.; Maeda, I.; Morita, T.; Miura, T.; Inoue, S.; Ikenaga, M.; Matsumoto, Y.; Baba, M.; Sekine, R.; Yamaguchi, T.; et al. Clinical Implications of C-Reactive Protein as a Prognostic Marker in Advanced Cancer Patients in Palliative Care Settings. J. Pain. Symptom Manag. 2016, 51, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Sun, M.; Samols, D.; Kushner, I. STAT3 participates in transcriptional activation of the C-reactive protein gene by interleukin-6. J. Biol. Chem. 1996, 271, 9503–9509. [Google Scholar] [CrossRef]
- Ingle, P.V.; Patel, D.M. C-reactive protein in various disease condition-an overview. Asian J. Pharm. Clin. Res. 2011, 4, 9–13. [Google Scholar]
- Hart, P.C.; Rajab, I.M.; Alebraheem, M.; Potempa, L.A. C-Reactive Protein and Cancer-Diagnostic and Therapeutic Insights. Front. Immunol. 2020, 11, 595835. [Google Scholar] [CrossRef] [PubMed]
- Shrotriya, S.; Walsh, D.; Nowacki, A.S.; Lorton, C.; Aktas, A.; Hullihen, B.; Benanni-Baiti, N.; Hauser, K.; Ayvaz, S.; Estfan, B. Serum C-reactive protein is an important and powerful prognostic biomarker in most adult solid tumors. PLoS ONE 2018, 13, e0202555. [Google Scholar] [CrossRef]
- Tavares, P.; Gonçalves, D.M.; Santos, L.L.; Ferreira, R. Revisiting the clinical usefulness of C-reactive protein in the set of cancer cachexia. Porto Biomed. J. 2021, 6, e123. [Google Scholar] [CrossRef]
- Shrotriya, S.; Walsh, D.; Bennani-Baiti, N.; Thomas, S.; Lorton, C. C-Reactive Protein Is an Important Biomarker for Prognosis Tumor Recurrence and Treatment Response in Adult Solid Tumors: A Systematic Review. PLoS ONE 2015, 10, e0143080. [Google Scholar] [CrossRef]
- Hou, Y.C.; Wang, C.J.; Chao, Y.J.; Chen, H.Y.; Wang, H.C.; Tung, H.L.; Lin, J.T.; Shan, Y.S. Elevated Serum Interleukin-8 Level Correlates with Cancer-Related Cachexia and Sarcopenia: An Indicator for Pancreatic Cancer Outcomes. J. Clin. Med. 2018, 7, 502. [Google Scholar] [CrossRef]
- Fogelman, D.R.; Morris, J.; Xiao, L.; Hassan, M.; Vadhan, S.; Overman, M.; Javle, S.; Shroff, R.; Varadhachary, G.; Wolff, R.; et al. A predictive model of inflammatory markers and patient-reported symptoms for cachexia in newly diagnosed pancreatic cancer patients. Support. Care Cancer 2017, 25, 1809–1817. [Google Scholar] [CrossRef]
- Xiong, X.; Liao, X.; Qiu, S.; Xu, H.; Zhang, S.; Wang, S.; Ai, J.; Yang, L. CXCL8 in Tumor Biology and Its Implications for Clinical Translation. Front. Mol. Biosci. 2022, 9, 723846. [Google Scholar] [CrossRef] [PubMed]
- Assadi, A.; Zahabi, A.; Hart, R.A. GDF15, an update of the physiological and pathological roles it plays: A review. Pflug. Arch. 2020, 472, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Martucci, M.; Mosconi, G.; Chiariello, A.; Cappuccilli, M.; Totti, V.; Santoro, A.; Franceschi, C.; Salvioli, S. GDF15 plasma level is inversely associated with level of physical activity and correlates with markers of inflammation and muscle weakness. Front. Immunol. 2020, 11, 915. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Ostan, R.; Fabbri, C.; Santoro, A.; Guidarelli, G.; Vitale, G.; Mari, D.; Sevini, F.; Capri, M.; Sandri, M.; et al. Human Aging and Longevity Are Characterized by High Levels of Mitokines. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Giuliani, C.; Chiariello, A.; Iannuzzi, V.; Franceschi, C.; Salvioli, S. GDF15, an emerging key player in human aging. Ageing Res. Rev. 2022, 75, 101569. [Google Scholar] [CrossRef] [PubMed]
- Tsai, V.W.W.; Husaini, Y.; Sainsbury, A.; Brown, D.A.; Breit, S.N. The MIC-1/GDF15-GFRAL Pathway in Energy Homeostasis: Implications for Obesity, Cachexia, and Other Associated Diseases. Cell Metab. 2018, 28, 353–368. [Google Scholar] [CrossRef]
- Zimmers, T.A.; Jin, X.; Hsiao, E.C.; McGrath, S.A.; Esquela, A.F.; Koniaris, L.G. Growth differentiation factor-15/macrophage inhibitory cytokine-1 induction after kidney and lung injury. Shock 2005, 23, 543–548. [Google Scholar] [PubMed]
- Suzuki, H.; Mitsunaga, S.; Ikeda, M.; Aoyama, T.; Yoshizawa, K.; Yoshimatsu, H.; Kawai, N.; Masuda, M.; Miura, T.; Ochiai, A. Clinical and Tumor Characteristics of Patients with High Serum Levels of Growth Differentiation Factor 15 in Advanced Pancreatic Cancer. Cancers 2021, 13, 4842. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, W.; Gu, X.; Miao, C.; Feng, L.; Shen, Q.; Liu, X.; Zhang, X. GDF-15 in tumor-derived exosomes promotes muscle atrophy via Bcl-2/caspase-3 pathway. Cell Death Discov. 2022, 8, 162. [Google Scholar] [CrossRef]
- Argilés, J.M.; López-Soriano, F.J.; Busquets, S. Mediators of cachexia in cancer patients. Nutrition 2019, 66, 11–15. [Google Scholar] [CrossRef]
- Biswas, A.K.; Acharyya, S. Understanding cachexia in the context of metastatic progression. Nat. Rev. Cancer 2020, 20, 274–284. [Google Scholar] [CrossRef]
- Bai, S.; Wang, Z.; Wang, M.; Li, J.; Wei, Y.; Xu, R.; Du, J. Tumor-Derived Exosomes Modulate Primary Site Tumor Metastasis. Front. Cell Dev. Biol. 2022, 10, 752818. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, J.; Kong, J.; Tang, J.; Wu, Y.; Xu, E.; Zhang, H.; Lai, M. GDF15 promotes EMT and metastasis in colorectal cancer. Oncotarget 2016, 7, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Lindmark, F.; Stattin, P.; Bälter, K.; Adami, H.O.; Zheng, S.L.; Xu, J.; Isaacs, W.B.; Grönberg, H.; Breit, S.N.; et al. Macrophage inhibitory cytokine 1: A new prognostic marker in prostate cancer. Clin. Cancer Res. 2009, 15, 6658–6664. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Bennett, D.; Brosnan, J.; Calle, R.; Collins, S.; Esquejo, R.; Joaquim, S.; Joyce, A.; Kim, H.; LaCarubba, B. 2O growth differentiation factor 15 (GDF-15) neutralization reverses cancer cachexia, restores physical performance and mitigates emesis associated with platinum-based chemotherapy. Ann. Oncol. 2020, 31, S245. [Google Scholar] [CrossRef]
- Tisdale, M.J. Mechanisms of cancer cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef]
- Kimball, S.R.; Jefferson, L.S. Control of translation initiation through integration of signals generated by hormones, nutrients, and exercise. J. Biol. Chem. 2010, 285, 29027–29032. [Google Scholar] [CrossRef]
- Attard-Montalto, S.; Camacho-Hübner, C.; Cotterill, A.; D’Souza-Li, L.; Daley, S.; Bartlett, K.; Halliday, D.; Eden, O. Changes in protein turnover, IGF-I and IGF binding proteins in children with cancer. Acta Paediatr. 1998, 87, 54–60. [Google Scholar] [CrossRef]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef]
- Sandri, M.; Barberi, L.; Bijlsma, A.Y.; Blaauw, B.; Dyar, K.A.; Milan, G.; Mammucari, C.; Meskers, C.G.; Pallafacchina, G.; Paoli, A.; et al. Signalling pathways regulating muscle mass in ageing skeletal muscle: The role of the IGF1-Akt-mTOR-FoxO pathway. Biogerontology 2013, 14, 303–323. [Google Scholar] [CrossRef]
- Khal, J.; Wyke, S.M.; Russell, S.T.; Hine, A.V.; Tisdale, M.J. Expression of the ubiquitin-proteasome pathway and muscle loss in experimental cancer cachexia. Br. J. Cancer 2005, 93, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Cho, J.; Song, E.J. Ubiquitin–proteasome system (UPS) as a target for anticancer treatment. Arch. Pharmacal Res. 2020, 43, 1144–1161. [Google Scholar] [CrossRef]
- Baracos, V.E.; DeVivo, C.; Hoyle, D.; Goldberg, A.L. Activation of the ATP-ubiquitin-proteasome pathway in skeletal muscle of cachectic rats bearing a hepatoma. Am. J. Physiol.-Endocrinol. Metab. 1995, 268, E996–E1006. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 2002, 9, 459–470. [Google Scholar] [CrossRef]
- Belizário, J.E.; Lorite, M.J.; Tisdale, M.J. Cleavage of caspases-1, -3, -6, -8 and -9 substrates by proteases in skeletal muscles from mice undergoing cancer cachexia. Br. J. Cancer 2001, 84, 1135–1140. [Google Scholar] [CrossRef]
- de Castro, G.S.; Simoes, E.; Lima, J.D.C.C.; Ortiz-Silva, M.; Festuccia, W.T.; Tokeshi, F.; Alcântara, P.S.; Otoch, J.P.; Coletti, D.; Seelaender, M. Human Cachexia Induces Changes in Mitochondria, Autophagy and Apoptosis in the Skeletal Muscle. Cancers 2019, 11, 1264. [Google Scholar] [CrossRef]
- Silva, K.A.; Dong, J.; Dong, Y.; Schor, N.; Tweardy, D.J.; Zhang, L.; Mitch, W.E. Inhibition of Stat3 activation suppresses caspase-3 and the ubiquitin-proteasome system, leading to preservation of muscle mass in cancer cachexia. J. Biol. Chem. 2015, 290, 11177–11187. [Google Scholar] [CrossRef]
- Allen, D.L.; Roy, R.R.; Edgerton, V.R. Myonuclear domains in muscle adaptation and disease. Muscle Nerve 1999, 22, 1350–1360. [Google Scholar] [CrossRef]
- D’Emilio, A.; Biagiotti, L.; Burattini, S.; Battistelli, M.; Canonico, B.; Evangelisti, C.; Ferri, P.; Papa, S.; Martelli, A.M.; Falcieri, E. Morphological and biochemical patterns in skeletal muscle apoptosis. Histol. Histopathol. 2010, 25, 21–32. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes. Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Gunadi, J.W.; Welliangan, A.S.; Soetadji, R.S.; Jasaputra, D.K.; Lesmana, R. The Role of Autophagy Modulated by Exercise in Cancer Cachexia. Life 2021, 11, 781. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Ballarò, R.; Beltrá, M.; De Lucia, S.; Costelli, P. Modulating Metabolism to Improve Cancer-Induced Muscle Wasting. Oxid. Med. Cell Longev. 2018, 2018, 7153610. [Google Scholar] [CrossRef]
- Penna, F.; Costamagna, D.; Pin, F.; Camperi, A.; Fanzani, A.; Chiarpotto, E.M.; Cavallini, G.; Bonelli, G.; Baccino, F.M.; Costelli, P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am. J. Pathol. 2013, 182, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Vara-Perez, M.; Felipe-Abrio, B.; Agostinis, P. Mitophagy in cancer: A tale of adaptation. Cells 2019, 8, 493. [Google Scholar] [CrossRef] [PubMed]
- Marzetti, E.; Lorenzi, M.; Landi, F.; Picca, A.; Rosa, F.; Tanganelli, F.; Galli, M.; Doglietto, G.B.; Pacelli, F.; Cesari, M. Altered mitochondrial quality control signaling in muscle of old gastric cancer patients with cachexia. Exp. Gerontol. 2017, 87, 92–99. [Google Scholar] [CrossRef]
- White, J.P.; Puppa, M.J.; Sato, S.; Gao, S.; Price, R.L.; Baynes, J.W.; Kostek, M.C.; Matesic, L.E.; Carson, J.A. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the Apc Min/+ mouse. Skelet. Muscle 2012, 2, 14. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial diseases in man and mouse. Science 1999, 283, 1482–1488. [Google Scholar] [CrossRef]
- Blackwell, T.A.; Cervenka, I.; Khatri, B.; Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Perry, R.A.; Brown, L.A.; Haynie, W.S.; Wiggs, M.P.; et al. Transcriptomic analysis of the development of skeletal muscle atrophy in cancer-cachexia in tumor-bearing mice. Physiol. Genom. 2018, 50, 1071–1082. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Mohan G, M.; G, S.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef]
- Nohl, H.; Kozlov, A.V.; Gille, L.; Staniek, K. Cell respiration and formation of reactive oxygen species: Facts and artefacts. Biochem. Soc. Trans. 2003, 31, 1308–1311. [Google Scholar] [CrossRef] [PubMed]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Oxidatively modified proteins in aging and disease. Free Radic. Biol. Med. 2002, 32, 797–803. [Google Scholar] [CrossRef]
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative stress in cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Miccheli, A.; Tomassini, A.; Puccetti, C.; Valerio, M.; Peluso, G.; Tuccillo, F.; Calvani, M.; Manetti, C.; Conti, F. Metabolic profiling by 13C-NMR spectroscopy: [1,2-13C2]glucose reveals a heterogeneous metabolism in human leukemia T cells. Biochimie 2006, 88, 437–448. [Google Scholar] [CrossRef]
- Lee, M.; Yoon, J.-H. Metabolic interplay between glycolysis and mitochondrial oxidation: The reverse Warburg effect and its therapeutic implication. World J. Biol. Chem. 2015, 6, 148. [Google Scholar] [CrossRef]
- Zu, X.L.; Guppy, M. Cancer metabolism: Facts, fantasy, and fiction. Biochem. Biophys. Res. Commun. 2004, 313, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Herst, P.M.; Berridge, M.V. Cell surface oxygen consumption: A major contributor to cellular oxygen consumption in glycolytic cancer cell lines. Biochim. Biophys. Acta-Bioenerg. 2007, 1767, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Liemburg-Apers, D.C.; Willems, P.H.; Koopman, W.J.; Grefte, S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 2015, 89, 1209–1226. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, S.; Nakanomyo, I.; Kudo-Sakamoto, Y.; Akazawa, H.; Komuro, I.; Shimizu, S. Identification of a novel compound that inhibits both mitochondria-mediated necrosis and apoptosis. Biochem. Biophys. Res. Commun. 2015, 467, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Christofferson, D.E.; Yuan, J. Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol. 2010, 22, 263–268. [Google Scholar] [CrossRef]
- Cazzaniga, M.; Bonanni, B. Relationship between Metabolic Reprogramming and Mitochondrial Activity in Cancer Cells. Understanding the Anticancer Effect of Metformin and Its Clinical Implications. Anticancer. Res. 2015, 35, 5789–5796. [Google Scholar]
- Gaude, E.; Frezza, C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014, 2, 10. [Google Scholar] [CrossRef]
- Hu, H.; Juvekar, A.; Lyssiotis, C.A.; Lien, E.C.; Albeck, J.G.; Oh, D.; Varma, G.; Hung, Y.P.; Ullas, S.; Lauring, J.; et al. Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton. Cell 2016, 164, 433–446. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Deng, H.; Chen, Y.; Li, P.; Hang, Q.; Zhang, P.; Jin, Y.; Chen, M. PI3K/AKT/mTOR pathway, hypoxia, and glucose metabolism: Potential targets to overcome radioresistance in small cell lung cancer. Cancer Pathog. Ther. 2023, 1, 56–66. [Google Scholar] [CrossRef]
- Kierans, S.; Taylor, C. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Pezzuto, A.; Carico, E. Role of HIF-1 in cancer progression: Novel insights. A review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Grasso, D.; Zampieri, L.X.; Capelôa, T.; Van de Velde, J.A.; Sonveaux, P. Mitochondria in cancer. Cell Stress. 2020, 4, 114–146. [Google Scholar] [CrossRef] [PubMed]
- Bost, F.; Kaminski, L. The metabolic modulator PGC-1α in cancer. Am. J. Cancer Res. 2019, 9, 198. [Google Scholar] [PubMed]
- Adams, G.R. Invited Review: Autocrine/paracrine IGF-I and skeletal muscle adaptation. J. Appl. Physiol. 2002, 93, 1159–1167. [Google Scholar] [CrossRef]
- Lee, S.J. Regulation of muscle mass by myostatin. Annu. Rev. Cell Dev. Biol. 2004, 20, 61–86. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef]
- Ruas, J.L.; White, J.P.; Rao, R.R.; Kleiner, S.; Brannan, K.T.; Harrison, B.C.; Greene, N.P.; Wu, J.; Estall, J.L.; Irving, B.A.; et al. A PGC-1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 2012, 151, 1319–1331. [Google Scholar] [CrossRef]
- Terada, S.; Tabata, I. Effects of acute bouts of running and swimming exercise on PGC-1alpha protein expression in rat epitrochlearis and soleus muscle. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E208–E216. [Google Scholar] [CrossRef]
- Pilegaard, H.; Saltin, B.; Neufer, P.D. Exercise induces transient transcriptional activation of the PGC-1alpha gene in human skeletal muscle. J. Physiol. 2003, 546, 851–858. [Google Scholar] [CrossRef]
- Adhihetty, P.J.; Uguccioni, G.; Leick, L.; Hidalgo, J.; Pilegaard, H.; Hood, D.A. The role of PGC-1α on mitochondrial function and apoptotic susceptibility in muscle. Am. J. Physiol.-Cell Physiol. 2009, 297, C217–C225. [Google Scholar] [CrossRef]
- Pin, F.; Busquets, S.; Toledo, M.; Camperi, A.; Lopez-Soriano, F.J.; Costelli, P.; Argilés, J.M.; Penna, F. Combination of exercise training and erythropoietin prevents cancer-induced muscle alterations. Oncotarget 2015, 6, 43202–43215. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lawler, A.M.; Lee, S.-J. Regulation of skeletal muscle mass in mice by a new TGF-p superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Langley, B.; Berry, C.; Sharma, M.; Kirk, S.; Bass, J.; Kambadur, R. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J. Biol. Chem. 2000, 275, 40235–40243. [Google Scholar] [CrossRef]
- Sartori, R.; Milan, G.; Patron, M.; Mammucari, C.; Blaauw, B.; Abraham, R.; Sandri, M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am. J. Physiol. Cell Physiol. 2009, 296, C1248–C1257. [Google Scholar] [CrossRef] [PubMed]
- Trendelenburg, A.U.; Meyer, A.; Rohner, D.; Boyle, J.; Hatakeyama, S.; Glass, D.J. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am. J. Physiol. Cell Physiol. 2009, 296, C1258–C1270. [Google Scholar] [CrossRef] [PubMed]
- Lokireddy, S.; McFarlane, C.; Ge, X.; Zhang, H.; Sze, S.K.; Sharma, M.; Kambadur, R. Myostatin induces degradation of sarcomeric proteins through a Smad3 signaling mechanism during skeletal muscle wasting. Mol. Endocrinol. 2011, 25, 1936–1949. [Google Scholar] [CrossRef]
- Guerci, A.; Lahoute, C.; Hébrard, S.; Collard, L.; Graindorge, D.; Favier, M.; Cagnard, N.; Batonnet-Pichon, S.; Précigout, G.; Garcia, L.; et al. Srf-dependent paracrine signals produced by myofibers control satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 2012, 15, 25–37. [Google Scholar] [CrossRef]
- Wagner, K.R.; Liu, X.; Chang, X.; Allen, R.E. Muscle regeneration in the prolonged absence of myostatin. Proc. Natl. Acad. Sci. USA 2005, 102, 2519–2524. [Google Scholar] [CrossRef]
- Suh, J.; Lee, Y.S. Myostatin Inhibitors: Panacea or Predicament for Musculoskeletal Disorders? J. Bone Metab. 2020, 27, 151–165. [Google Scholar] [CrossRef]
- Haidet, A.M.; Rizo, L.; Handy, C.; Umapathi, P.; Eagle, A.; Shilling, C.; Boue, D.; Martin, P.T.; Sahenk, Z.; Mendell, J.R.; et al. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 4318–4322. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; McPherron, A.C. Myostatin inhibition induces muscle fibre hypertrophy prior to satellite cell activation. J. Physiol. 2012, 590, 2151–2165. [Google Scholar] [CrossRef] [PubMed]
- LeBrasseur, N.K.; Schelhorn, T.M.; Bernardo, B.L.; Cosgrove, P.G.; Loria, P.M.; Brown, T.A. Myostatin inhibition enhances the effects of exercise on performance and metabolic outcomes in aged mice. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Benny Klimek, M.E.; Aydogdu, T.; Link, M.J.; Pons, M.; Koniaris, L.G.; Zimmers, T.A. Acute inhibition of myostatin-family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem. Biophys. Res. Commun. 2010, 391, 1548–1554. [Google Scholar] [CrossRef]
- Costelli, P.; Muscaritoli, M.; Bonetto, A.; Penna, F.; Reffo, P.; Bossola, M.; Bonelli, G.; Doglietto, G.B.; Baccino, F.M.; Rossi Fanelli, F. Muscle myostatin signalling is enhanced in experimental cancer cachexia. Eur. J. Clin. Investig. 2008, 38, 531–538. [Google Scholar] [CrossRef]
- Hanada, K.; Fukasawa, K.; Hinata, H.; Imai, S.; Takayama, K.; Hirai, H.; Ohfusa, R.; Hayashi, Y.; Itoh, F. Combination therapy with anamorelin and a myostatin inhibitor is advantageous for cancer cachexia in a mouse model. Cancer Sci. 2022, 113, 3547–3557. [Google Scholar] [CrossRef]
- Nissinen, T.A.; Hentilä, J.; Penna, F.; Lampinen, A.; Lautaoja, J.H.; Fachada, V.; Holopainen, T.; Ritvos, O.; Kivelä, R.; Hulmi, J.J. Treating cachexia using soluble ACVR2B improves survival, alters mTOR localization, and attenuates liver and spleen responses. J. Cachexia Sarcopenia Muscle 2018, 9, 514–529. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, C.; Hui, G.Z.; Amanda, W.Z.; Lau, H.Y.; Lokireddy, S.; Xiaojia, G.; Mouly, V.; Butler-Browne, G.; Gluckman, P.D.; Sharma, M.; et al. Human myostatin negatively regulates human myoblast growth and differentiation. Am. J. Physiol. Cell Physiol. 2011, 301, C195–C203. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef]
- Winbanks, C.E.; Murphy, K.T.; Bernardo, B.C.; Qian, H.; Liu, Y.; Sepulveda, P.V.; Beyer, C.; Hagg, A.; Thomson, R.E.; Chen, J.L.; et al. Smad7 gene delivery prevents muscle wasting associated with cancer cachexia in mice. Sci. Transl. Med. 2016, 8, 348ra398. [Google Scholar] [CrossRef]
- Saitoh, M.; Ishida, J.; Ebner, N.; Anker, S.D.; Springer, J.; von Haehling, S. Myostatin inhibitors as pharmacological treatment for muscle wasting and muscular dystrophy. JCSM Clin. Rep. 2017, 2, 1–10. [Google Scholar] [CrossRef]
- Lee, J.H.; Jun, H.S. Role of Myokines in Regulating Skeletal Muscle Mass and Function. Front. Physiol. 2019, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Bina, H.A.; Ouchi, N.; Akasaki, Y.; Kharitonenkov, A.; Walsh, K. FGF21 is an Akt-regulated myokine. FEBS Lett. 2008, 582, 3805–3810. [Google Scholar] [CrossRef] [PubMed]
- Akbulut, M.C.; Erbaş, O. Possible therapeutics: Myokines. Demiroglu Sci. Univ. Florence Nightingale J. Transplant. 2022, 7, 32–39. [Google Scholar]
- Refsgaard Holm, M.; Christensen, H.; Rasmussen, J.; Johansen, M.L.; Schou, M.; Faber, J.; Kistorp, C. Fibroblast growth factor 21 in patients with cardiac cachexia: A possible role of chronic inflammation. ESC Heart Fail. 2019, 6, 983–991. [Google Scholar] [CrossRef]
- Franz, K.; Ost, M.; Otten, L.; Herpich, C.; Coleman, V.; Endres, A.S.; Klaus, S.; Müller-Werdan, U.; Norman, K. Higher serum levels of fibroblast growth factor 21 in old patients with cachexia. Nutrition 2019, 63–64, 81–86. [Google Scholar] [CrossRef]
- Oost, L.J.; Sandri, M.; Romanello, V. The authors reply: Letter on: “Fibroblast growth factor 21 controls mitophagy and muscle mass” by Oost et al. J. Cachexia Sarcopenia Muscle 2020, 11, 338–340. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Akerstrom, T.C.; Nielsen, A.R.; Fischer, C.P. Role of myokines in exercise and metabolism. J. Appl. Physiol. 2007, 103, 1093–1098. [Google Scholar] [CrossRef]
- Pedersen, B.K. Muscles and their myokines. J. Exp. Biol. 2011, 214, 337–346. [Google Scholar] [CrossRef]
- Tamura, Y.; Watanabe, K.; Kantani, T.; Hayashi, J.; Ishida, N.; Kaneki, M. Upregulation of circulating IL-15 by treadmill running in healthy individuals: Is IL-15 an endocrine mediator of the beneficial effects of endurance exercise? Endocr. J. 2011, 58, 211–215. [Google Scholar] [CrossRef]
- Waldmann, T.A. Interleukin-15 in the treatment of cancer. Expert. Rev. Clin. Immunol. 2014, 10, 1689–1701. [Google Scholar] [CrossRef] [PubMed]
- Beyer, M.; Schultze, J.L. Regulatory T cells in cancer. Blood 2006, 108, 804–811. [Google Scholar] [CrossRef]
- Sarvaria, A.; Madrigal, J.A.; Saudemont, A. B cell regulation in cancer and anti-tumor immunity. Cell. Mol. Immunol. 2017, 14, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Brittenden, J.; Heys, S.D.; Ross, J.; Eremin, O. Natural killer cells and cancer. Cancer 1996, 77, 1226–1243. [Google Scholar] [CrossRef]
- Martínez-Hernández, P.L.; Hernanz-Macías, Á.; Gómez-Candela, C.; Grande-Aragón, C.; Feliu-Batlle, J.; Castro-Carpeño, J.; Martínez-Muñoz, I.; Zurita-Rosa, L.; Villarino-Sanz, M.; Prados-Sánchez, C.; et al. Serum interleukin-15 levels in cancer patients with cachexia. Oncol. Rep. 2012, 28, 1443–1452. [Google Scholar] [CrossRef]
- Carbó, N.; López-Soriano, J.; Costelli, P.; Busquets, S.; Alvarez, B.; Baccino, F.M.; Quinn, L.S.; López-Soriano, F.J.; Argilés, J.M. Interleukin-15 antagonizes muscle protein waste in tumour-bearing rats. Br. J. Cancer 2000, 83, 526–531. [Google Scholar] [CrossRef]
- Shamsi, M.M.; Chekachak, S.; Soudi, S.; Quinn, L.; Ranjbar, K.; Chenari, J.; Yazdi, M.; Mahdavi, M. Combined effect of aerobic interval training and selenium nanoparticles on expression of IL-15 and IL-10/TNF-α ratio in skeletal muscle of 4T1 breast cancer mice with cachexia. Cytokine 2017, 90, 100–108. [Google Scholar] [CrossRef]
- Murphy, R.; Wilke, M.; Perrine, M.; Pawlowicz, M.; Mourtzakis, M.; Lieffers, J.; Maneshgar, M.; Bruera, E.; Clandinin, M.; Baracos, V. Loss of adipose tissue and plasma phospholipids: Relationship to survival in advanced cancer patients. Clin. Nutr. 2010, 29, 482–487. [Google Scholar] [CrossRef]
- Fouladiun, M.; Körner, U.; Bosaeus, I.; Daneryd, P.; Hyltander, A.; Lundholm, K.G. Body composition and time course changes in regional distribution of fat and lean tissue in unselected cancer patients on palliative care—Correlations with food intake, metabolism, exercise capacity, and hormones. Cancer: Interdiscip. Int. J. Am. Cancer Soc. 2005, 103, 2189–2198. [Google Scholar] [CrossRef]
- Daas, S.I.; Rizeq, B.R.; Nasrallah, G.K. Adipose tissue dysfunction in cancer cachexia. J. Cell Physiol. 2018, 234, 13–22. [Google Scholar] [CrossRef]
- Petruzzelli, M.; Schweiger, M.; Schreiber, R.; Campos-Olivas, R.; Tsoli, M.; Allen, J.; Swarbrick, M.; Rose-John, S.; Rincon, M.; Robertson, G. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014, 20, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Kir, S.; White, J.P.; Kleiner, S.; Kazak, L.; Cohen, P.; Baracos, V.E.; Spiegelman, B.M. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 2014, 513, 100–104. [Google Scholar] [CrossRef]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Kim, W.K.; Bae, K.H. Distinction of white, beige and brown adipocytes derived from mesenchymal stem cells. World J. Stem Cells 2014, 6, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Arends, J.; Strasser, F.; Gonella, S.; Solheim, T.S.; Madeddu, C.; Ravasco, P.; Buonaccorso, L.; de van der Schueren, M.A.E.; Baldwin, C.; Chasen, M.; et al. Cancer cachexia in adult patients: ESMO Clinical Practice Guidelines. ESMO Open 2021, 6, 100092. [Google Scholar] [CrossRef]
- Bland, K.A.; Harrison, M.; Zopf, E.M.; Sousa, M.S.; Currow, D.C.; Ely, M.; Agar, M.; Butcher, B.E.; Vaughan, V.; Dowd, A.; et al. Quality of Life and Symptom Burden Improve in Patients Attending a Multidisciplinary Clinical Service for Cancer Cachexia: A Retrospective Observational Review. J. Pain. Symptom Manag. 2021, 62, e164–e176. [Google Scholar] [CrossRef]
- Nishikawa, H.; Goto, M.; Fukunishi, S.; Asai, A.; Nishiguchi, S.; Higuchi, K. Cancer Cachexia: Its Mechanism and Clinical Significance. Int. J. Mol. Sci. 2021, 22, 8491. [Google Scholar] [CrossRef] [PubMed]
- Flock, M.R.; Harris, W.S.; Kris-Etherton, P.M. Long-chain omega-3 fatty acids: Time to establish a dietary reference intake. Nutr. Rev. 2013, 71, 692–707. [Google Scholar] [CrossRef]
- Zhang, Z.; Fulgoni, V.L.; Kris-Etherton, P.M.; Mitmesser, S.H. Dietary intakes of EPA and DHA omega-3 fatty acids among US childbearing-age and pregnant women: An analysis of NHANES 2001–2014. Nutrients 2018, 10, 416. [Google Scholar] [CrossRef]
- Lalia, A.Z.; Dasari, S.; Robinson, M.M.; Abid, H.; Morse, D.M.; Klaus, K.A.; Lanza, I.R. Influence of omega-3 fatty acids on skeletal muscle protein metabolism and mitochondrial bioenergetics in older adults. Aging 2017, 9, 1096–1129. [Google Scholar] [CrossRef]
- Barber, M.D. Cancer cachexia and its treatment with fish-oil-enriched nutritional supplementation. Nutrition 2001, 17, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Wigmore, S.J.; Barber, M.D.; Ross, J.A.; Tisdale, M.J.; Fearon, K.C. Effect of oral eicosapentaenoic acid on weight loss in patients with pancreatic cancer. Nutr. Cancer 2000, 36, 177–184. [Google Scholar] [CrossRef]
- Gogos, C.A.; Ginopoulos, P.; Salsa, B.; Apostolidou, E.; Zoumbos, N.C.; Kalfarentzos, F. Dietary omega-3 polyunsaturated fatty acids plus vitamin E restore immunodeficiency and prolong survival for severely ill patients with generalized malignancy: A randomized control trial. Cancer 1998, 82, 395–402. [Google Scholar] [CrossRef]
- Tarnopolsky, M.A.; Mahoney, D.J.; Vajsar, J.; Rodriguez, C.; Doherty, T.J.; Roy, B.D.; Biggar, D. Creatine monohydrate enhances strength and body composition in Duchenne muscular dystrophy. Neurology 2004, 62, 1771–1777. [Google Scholar] [CrossRef] [PubMed]
- Candow, D.G.; Forbes, S.C.; Chilibeck, P.D.; Cornish, S.M.; Antonio, J.; Kreider, R.B. Effectiveness of Creatine Supplementation on Aging Muscle and Bone: Focus on Falls Prevention and Inflammation. J. Clin. Med. 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.N.; Agharkar, A.S.; Gonzales, E.B. A review of creatine supplementation in age-related diseases: More than a supplement for athletes. F1000Res 2014, 3, 222. [Google Scholar] [CrossRef]
- Kreider, R.B.; Kalman, D.S.; Antonio, J.; Ziegenfuss, T.N.; Wildman, R.; Collins, R.; Candow, D.G.; Kleiner, S.M.; Almada, A.L.; Lopez, H.L. International Society of Sports Nutrition position stand: Safety and efficacy of creatine supplementation in exercise, sport, and medicine. J. Int. Soc. Sports Nutr. 2017, 14, 18. [Google Scholar] [CrossRef]
- Fairman, C.M.; Kendall, K.L.; Hart, N.H.; Taaffe, D.R.; Galvão, D.A.; Newton, R.U. The potential therapeutic effects of creatine supplementation on body composition and muscle function in cancer. Crit. Rev. Oncol. Hematol. 2019, 133, 46–57. [Google Scholar] [CrossRef]
- Wei, L.; Wang, R.; Lin, K.; Jin, X.; Li, L.; Wazir, J.; Pu, W.; Lian, P.; Lu, R.; Song, S.; et al. Creatine modulates cellular energy metabolism and protects against cancer cachexia-associated muscle wasting. Front. Pharmacol. 2022, 13, 1086662. [Google Scholar] [CrossRef]
- Ter Borg, S.; Luiking, Y.C.; van Helvoort, A.; Boirie, Y.; Schols, J.M.G.A.; de Groot, C.P.G.M. Low Levels of Branched Chain Amino Acids, Eicosapentaenoic Acid and Micronutrients Are Associated with Low Muscle Mass, Strength and Function in Community-Dwelling Older Adults. J. Nutr. Health Aging 2019, 23, 27–34. [Google Scholar] [CrossRef]
- Kimball, S.R.; Jefferson, L.S. Regulation of protein synthesis by branched-chain amino acids. Curr. Opin. Clin. Nutr. Metab. Care 2001, 4, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Rennie, M.J.; Bohé, J.; Smith, K.; Wackerhage, H.; Greenhaff, P. Branched-chain amino acids as fuels and anabolic signals in human muscle. J. Nutr. 2006, 136, 264S–268S. [Google Scholar] [CrossRef] [PubMed]
- D’Antona, G.; Nisoli, E. mTOR signaling as a target of amino acid treatment of the age-related sarcopenia. Interdiscip. Top. Gerontol. 2010, 37, 115–141. [Google Scholar] [CrossRef]
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Tayek, J.A.; Bistrian, B.R.; Hehir, D.J.; Martin, R.; Moldawer, L.L.; Blackburn, G.L. Improved protein kinetics and albumin synthesis by branched chain amino acid-enriched total parenteral nutrition in cancer cachexia. A prospective randomized crossover trial. Cancer 1986, 58, 147–157. [Google Scholar] [CrossRef]
- Hunter, D.C.; Weintraub, M.; Blackburn, G.L.; Bistrian, B.R. Branched chain amino acids as the protein component of parenteral nutrition in cancer cachexia. Br. J. Surg. 1989, 76, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Ventrucci, G.; Mello, M.A.; Gomes-Marcondes, M.C. Leucine-rich diet alters the eukaryotic translation initiation factors expression in skeletal muscle of tumour-bearing rats. BMC Cancer 2007, 7, 42. [Google Scholar] [CrossRef]
- Eley, H.L.; Russell, S.T.; Tisdale, M.J. Effect of branched-chain amino acids on muscle atrophy in cancer cachexia. Biochem. J. 2007, 407, 113–120. [Google Scholar] [CrossRef]
- Nissen, S.L.; Abumrad, N.N. Nutritional role of the leucine metabolite β-hydroxy β-methylbutyrate (HMB). J. Nutr. Biochem. 1997, 8, 300–311. [Google Scholar] [CrossRef]
- Holecek, M.; Muthny, T.; Kovarik, M.; Sispera, L. Effect of beta-hydroxy-beta-methylbutyrate (HMB) on protein metabolism in whole body and in selected tissues. Food Chem. Toxicol. 2009, 47, 255–259. [Google Scholar] [CrossRef]
- Wilkinson, D.J.; Hossain, T.; Limb, M.C.; Phillips, B.E.; Lund, J.; Williams, J.P.; Brook, M.S.; Cegielski, J.; Philp, A.; Ashcroft, S. Impact of the calcium form of β-hydroxy-β-methylbutyrate upon human skeletal muscle protein metabolism. Clin. Nutr. 2018, 37, 2068–2075. [Google Scholar] [CrossRef] [PubMed]
- Courel-Ibáñez, J.; Vetrovsky, T.; Dadova, K.; Pallarés, J.G.; Steffl, M. Health benefits of β-Hydroxy-β-Methylbutyrate (HMB) supplementation in addition to physical exercise in older adults: A systematic review with meta-analysis. Nutrients 2019, 11, 2082. [Google Scholar] [CrossRef] [PubMed]
- Maykish, A.; Sikalidis, A.K. Utilization of hydroxyl-methyl butyrate, leucine, glutamine and arginine supplementation in nutritional management of sarcopenia—Implications and clinical considerations for Type 2 diabetes mellitus risk modulation. J. Pers. Med. 2020, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Kougias, D.G.; Das, T.; Perez, A.B.; Pereira, S.L. A role for nutritional intervention in addressing the aging neuromuscular junction. Nutr. Res. 2018, 53, 1–14. [Google Scholar] [CrossRef]
- May, P.E.; Barber, A.; D’Olimpio, J.T.; Hourihane, A.; Abumrad, N.N. Reversal of cancer-related wasting using oral supplementation with a combination of beta-hydroxy-beta-methylbutyrate, arginine, and glutamine. Am. J. Surg. 2002, 183, 471–479. [Google Scholar] [CrossRef]
- Ritch, C.R.; Cookson, M.S.; Clark, P.E.; Chang, S.S.; Fakhoury, K.; Ralls, V.; Thu, M.H.; Penson, D.F.; Smith, J.A.; Silver, H.J. Perioperative Oral Nutrition Supplementation Reduces Prevalence of Sarcopenia following Radical Cystectomy: Results of a Prospective Randomized Controlled Trial. J. Urol. 2019, 201, 470–477. [Google Scholar] [CrossRef]
- Parlak, E.; Atalay, B.G. The effects of protein support with various content on nutrition status and clinical outcomes in elderly malnourished cancer patients. Prog. Nutr. 2020, 22, e2020061. [Google Scholar]
- Shukla, S.K.; Gebregiworgis, T.; Purohit, V.; Chaika, N.V.; Gunda, V.; Radhakrishnan, P.; Mehla, K.; Pipinos, I.I.; Powers, R.; Yu, F.; et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. 2014, 2, 18. [Google Scholar] [CrossRef]
- Maurer, G.D.; Brucker, D.P.; Bähr, O.; Harter, P.N.; Hattingen, E.; Walenta, S.; Mueller-Klieser, W.; Steinbach, J.P.; Rieger, J. Differential utilization of ketone bodies by neurons and glioma cell lines: A rationale for ketogenic diet as experimental glioma therapy. BMC Cancer 2011, 11, 315. [Google Scholar] [CrossRef]
- Poff, A.M.; Ari, C.; Arnold, P.; Seyfried, T.N.; D’Agostino, D.P. Ketone supplementation decreases tumor cell viability and prolongs survival of mice with metastatic cancer. Int. J. Cancer 2014, 135, 1711–1720. [Google Scholar] [CrossRef]
- Rouiller, C. Physiological and pathological changes in mitochondrial morphology. In International Review of Cytology; Elsevier: Amsterdam, The Netherlands, 1960; Volume 9, pp. 227–292. [Google Scholar]
- Nakamura, K.; Tonouchi, H.; Sasayama, A.; Ashida, K. A Ketogenic Formula Prevents Tumor Progression and Cancer Cachexia by Attenuating Systemic Inflammation in Colon 26 Tumor-Bearing Mice. Nutrients 2018, 10, 206. [Google Scholar] [CrossRef]
- Schmidt, M.; Pfetzer, N.; Schwab, M.; Strauss, I.; Kämmerer, U. Effects of a ketogenic diet on the quality of life in 16 patients with advanced cancer: A pilot trial. Nutr. Metab. 2011, 8, 54. [Google Scholar] [CrossRef]
- Poff, A.M.; Ari, C.; Seyfried, T.N.; D’Agostino, D.P. The ketogenic diet and hyperbaric oxygen therapy prolong survival in mice with systemic metastatic cancer. PLoS ONE 2013, 8, e65522. [Google Scholar] [CrossRef]
- Poff, A.M.; Ward, N.; Seyfried, T.N.; Arnold, P.; D’Agostino, D.P. Non-Toxic Metabolic Management of Metastatic Cancer in VM Mice: Novel Combination of Ketogenic Diet, Ketone Supplementation, and Hyperbaric Oxygen Therapy. PLoS ONE 2015, 10, e0127407. [Google Scholar] [CrossRef]
- Fine, E.J.; Segal-Isaacson, C.J.; Feinman, R.D.; Herszkopf, S.; Romano, M.C.; Tomuta, N.; Bontempo, A.F.; Negassa, A.; Sparano, J.A. Targeting insulin inhibition as a metabolic therapy in advanced cancer: A pilot safety and feasibility dietary trial in 10 patients. Nutrition 2012, 28, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Arcidiacono, B.; Iiritano, S.; Nocera, A.; Possidente, K.; Nevolo, M.T.; Ventura, V.; Foti, D.; Chiefari, E.; Brunetti, A. Insulin resistance and cancer risk: An overview of the pathogenetic mechanisms. Exp. Diabetes Res. 2012, 2012, 789174. [Google Scholar] [CrossRef] [PubMed]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef]
- Bozzetti, F.; Santarpia, L.; Pironi, L.; Thul, P.; Klek, S.; Gavazzi, C.; Tinivella, M.; Joly, F.; Jonkers, C.; Baxter, J. The prognosis of incurable cachectic cancer patients on home parenteral nutrition: A multi-centre observational study with prospective follow-up of 414 patients. Ann. Oncol. 2014, 25, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Park, Y.K. Rationale, Feasibility and Acceptability of Ketogenic Diet for Cancer Treatment. J. Cancer Prev. 2017, 22, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Bojková, B.; Winklewski, P.J.; Wszedybyl-Winklewska, M. Dietary fat and cancer—Which is good, which is bad, and the body of evidence. Int. J. Mol. Sci. 2020, 21, 4114. [Google Scholar] [CrossRef]
- Duan, Y.; Zeng, L.; Zheng, C.; Song, B.; Li, F.; Kong, X.; Xu, K. Inflammatory links between high fat diets and diseases. Front. Immunol. 2018, 9, 2649. [Google Scholar] [CrossRef] [PubMed]
- Clements, V.K.; Long, T.; Long, R.; Figley, C.; Smith, D.M.C.; Ostrand-Rosenberg, S. Frontline Science: High fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. J. Leukoc. Biol. 2018, 103, 395–407. [Google Scholar] [CrossRef]
- Das, S.K.; Hoefler, G. The role of triglyceride lipases in cancer associated cachexia. Trends Mol. Med. 2013, 19, 292–301. [Google Scholar] [CrossRef]
- Ho, V.W.; Leung, K.; Hsu, A.; Luk, B.; Lai, J.; Shen, S.Y.; Minchinton, A.I.; Waterhouse, D.; Bally, M.B.; Lin, W.; et al. A low carbohydrate, high protein diet slows tumor growth and prevents cancer initiation. Cancer Res. 2011, 71, 4484–4493. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J.; Brennan, R.A.; Fearon, K.C. Reduction of weight loss and tumour size in a cachexia model by a high fat diet. Br. J. Cancer 1987, 56, 39–43. [Google Scholar] [CrossRef]
- Martuscello, R.T.; Vedam-Mai, V.; McCarthy, D.J.; Schmoll, M.E.; Jundi, M.A.; Louviere, C.D.; Griffith, B.G.; Skinner, C.L.; Suslov, O.; Deleyrolle, L.P.; et al. A Supplemented High-Fat Low-Carbohydrate Diet for the Treatment of Glioblastoma. Clin. Cancer Res. 2016, 22, 2482–2495. [Google Scholar] [CrossRef]
- Li, R.J.; Liu, Y.; Liu, H.Q.; Li, J. Ketogenic diets and protective mechanisms in epilepsy, metabolic disorders, cancer, neuronal loss, and muscle and nerve degeneration. J. Food Biochem. 2020, 44, e13140. [Google Scholar] [CrossRef]
- Paoli, A.; Cancellara, P.; Pompei, P.; Moro, T. Ketogenic Diet and Skeletal Muscle Hypertrophy: A Frenemy Relationship? J. Hum. Kinet. 2019, 68, 233–247. [Google Scholar] [CrossRef]
- Muscaritoli, M.; Arends, J.; Bachmann, P.; Baracos, V.; Barthelemy, N.; Bertz, H.; Bozzetti, F.; Hütterer, E.; Isenring, E.; Kaasa, S.; et al. ESPEN practical guideline: Clinical Nutrition in cancer. Clin. Nutr. 2021, 40, 2898–2913. [Google Scholar] [CrossRef]
- Nishie, K.; Sato, S.; Hanaoka, M. Anamorelin for cancer cachexia. Drugs Today 2022, 58, 97–104. [Google Scholar] [CrossRef]
- Hain, B.A.; Narasimhan, A.; Ballinger, T.J.; Guise, T.A.; Waning, D.L. Cancer-associated muscle dysfunction. In Encyclopedia of Bone Biology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 379–389. [Google Scholar]
- Katakami, N.; Uchino, J.; Yokoyama, T.; Naito, T.; Kondo, M.; Yamada, K.; Kitajima, H.; Yoshimori, K.; Sato, K.; Saito, H.; et al. Anamorelin (ONO-7643) for the treatment of patients with non-small cell lung cancer and cachexia: Results from a randomized, double-blind, placebo-controlled, multicenter study of Japanese patients (ONO-7643-04). Cancer 2018, 124, 606–616. [Google Scholar] [CrossRef]
- Hamauchi, S.; Furuse, J.; Takano, T.; Munemoto, Y.; Furuya, K.; Baba, H.; Takeuchi, M.; Choda, Y.; Higashiguchi, T.; Naito, T.; et al. A multicenter, open-label, single-arm study of anamorelin (ONO-7643) in advanced gastrointestinal cancer patients with cancer cachexia. Cancer 2019, 125, 4294–4302. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.M.; Polvino, W.J. Pharmacodynamic hormonal effects of anamorelin, a novel oral ghrelin mimetic and growth hormone secretagogue in healthy volunteers. Growth Horm. IGF Res. 2009, 19, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Currow, D.C.; Skipworth, R.J. The emerging role of anamorelin hydrochloride in the management of patients with cancer anorexia-cachexia. Future Oncol. 2017, 13, 1767–1783. [Google Scholar] [CrossRef] [PubMed]
- Pietra, C.; Takeda, Y.; Tazawa-Ogata, N.; Minami, M.; Yuanfeng, X.; Duus, E.M.; Northrup, R. Anamorelin HCl (ONO-7643), a novel ghrelin receptor agonist, for the treatment of cancer anorexia-cachexia syndrome: Preclinical profile. J. Cachexia Sarcopenia Muscle 2014, 5, 329–337. [Google Scholar] [CrossRef]
- Temel, J.S.; Abernethy, A.P.; Currow, D.C.; Friend, J.; Duus, E.M.; Yan, Y.; Fearon, K.C. Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): Results from two randomised, double-blind, phase 3 trials. Lancet Oncol. 2016, 17, 519–531. [Google Scholar] [CrossRef]
- Naito, T.; Uchino, J.; Kojima, T.; Matano, Y.; Minato, K.; Tanaka, K.; Mizukami, T.; Atagi, S.; Higashiguchi, T.; Muro, K.; et al. A multicenter, open-label, single-arm study of anamorelin (ONO-7643) in patients with cancer cachexia and low body mass index. Cancer 2022, 128, 2025–2035. [Google Scholar] [CrossRef]
- Ji, H.; Englmaier, F.; Morigny, P.; Giroud, M.; Gräsle, P.; Brings, S.; Szendrödi, J.; Berriel Diaz, M.; Plettenburg, O.; Herzig, S.; et al. Development of a peptide drug restoring AMPK and adipose tissue functionality in cancer cachexia. Mol. Ther. 2023, 31, 2408–2421. [Google Scholar] [CrossRef]
- Bilgic, S.N.; Domaniku, A.; Toledo, B.; Agca, S.; Weber, B.Z.; Arabaci, D.H.; Ozornek, Z.; Lause, P.; Thissen, J.-P.; Loumaye, A. EDA2R–NIK signalling promotes muscle atrophy linked to cancer cachexia. Nature 2023, 617, 827–834. [Google Scholar] [CrossRef]
- Wen, H.S.; Li, X.; Cao, Y.Z.; Zhang, C.C.; Yang, F.; Shi, Y.M.; Peng, L.M. Clinical studies on the treatment of cancer cachexia with megestrol acetate plus thalidomide. Chemotherapy 2012, 58, 461–467. [Google Scholar] [CrossRef]
- Roeland, E.J.; Bohlke, K.; Baracos, V.E.; Bruera, E.; Del Fabbro, E.; Dixon, S.; Fallon, M.; Herrstedt, J.; Lau, H.; Platek, M.; et al. Management of Cancer Cachexia: ASCO Guideline. J. Clin. Oncol. 2020, 38, 2438–2453. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.N.; Trebble, T.M.; Ellis, R.D.; Duncan, H.D.; Johns, T.; Goggin, P.M. Thalidomide in the treatment of cancer cachexia: A randomised placebo controlled trial. Gut 2005, 54, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.L.; Teoh, S.E.; Yaow, C.Y.L.; Lin, D.J.; Masuda, Y.; Han, M.X.; Yeo, W.S.; Ng, Q.X. A Systematic Review and Meta-Analysis of the Clinical Use of Megestrol Acetate for Cancer-Related Anorexia/Cachexia. J. Clin. Med. 2022, 11, 3756. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, T.J.; Smith, J.T.; Schuster, M.; Dragnev, K.H.; Rigas, J.R. A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer. Expert. Opin. Biol. Ther. 2011, 11, 1663–1668. [Google Scholar] [CrossRef]
- Mantovani, G.; Macciò, A.; Madeddu, C.; Serpe, R.; Antoni, G.; Massa, E.; Dessì, M.; Panzone, F. Phase II nonrandomized study of the efficacy and safety of COX-2 inhibitor celecoxib on patients with cancer cachexia. J. Mol. Med. 2010, 88, 85–92. [Google Scholar] [CrossRef]
- Deuster, P.A.; Morrison, S.D.; Ahrens, R.A. Endurance exercise modifies cachexia of tumor growth in rats. Med. Sci. Sports Exerc. 1985, 17, 385–392. [Google Scholar] [CrossRef]
- Ballarò, R.; Beltrà, M.; De Lucia, S.; Pin, F.; Ranjbar, K.; Hulmi, J.J.; Costelli, P.; Penna, F. Moderate exercise in mice improves cancer plus chemotherapy-induced muscle wasting and mitochondrial alterations. FASEB J. 2019, 33, 5482–5494. [Google Scholar] [CrossRef]
- Baracos, V.E. Exercise inhibits progressive growth of the Morris hepatoma 7777 in male and female rats. Can. J. Physiol. Pharmacol. 1989, 67, 864–870. [Google Scholar] [CrossRef]
- Daneryd, P.L.-E.; Hafström, L.R.; Karlberg, I.H. Effects of spontaneous physical exercise on experimental cancer anorexia and cachexia. Eur. J. Cancer Clin. Oncol. 1990, 26, 1083–1088. [Google Scholar] [CrossRef]
- Ranjbar, K.; Ballarò, R.; Bover, Q.; Pin, F.; Beltrà, M.; Penna, F.; Costelli, P. Combined Exercise Training Positively Affects Muscle Wasting in Tumor-Bearing Mice. Med. Sci. Sports Exerc. 2019, 51, 1387–1395. [Google Scholar] [CrossRef]
- Puppa, M.J.; Murphy, E.A.; Fayad, R.; Hand, G.A.; Carson, J.A. Cachectic skeletal muscle response to a novel bout of low-frequency stimulation. J. Appl. Physiol. 2014, 116, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Moreira, V.M.; da Silva Franco, C.C.; Prates, K.V.; Gomes, R.M.; De Moraes, A.M.P.; Ribeiro, T.A.; Martins, I.P.; Previate, C.; Pavanello, A.; Matiusso, C.C.I. Aerobic exercise training attenuates tumor growth and reduces insulin secretion in Walker 256 tumor-bearing rats. Front. Physiol. 2018, 9, 465. [Google Scholar] [CrossRef] [PubMed]
- Schcolnik-Cabrera, A.; Chávez-Blanco, A.; Domínguez-Gómez, G.; Dueñas-González, A. Understanding tumor anabolism and patient catabolism in cancer-associated cachexia. Am. J. Cancer Res. 2017, 7, 1107–1135. [Google Scholar] [PubMed]
- PANEL, E. American college of sports medicine roundtable on exercise guidelines for cancer survivors. J. ACSM 2010, 42, 1409–1426. [Google Scholar]
- Grande, A.J.; Silva, V.; Maddocks, M. Exercise for cancer cachexia in adults: Executive summary of a Cochrane Collaboration systematic review. J. Cachexia Sarcopenia Muscle 2015, 6, 208–211. [Google Scholar] [CrossRef]
- Lira, F.S.; Koyama, C.H.; Yamashita, A.S.; Rosa, J.C.; Zanchi, N.E.; Batista, M.L.; Seelaender, M.C. Chronic exercise decreases cytokine production in healthy rat skeletal muscle. Cell Biochem. Funct. 2009, 27, 458–461. [Google Scholar] [CrossRef]
- Jee, H.; Chang, J.E.; Yang, E.J. Positive Prehabilitative Effect of Intense Treadmill Exercise for Ameliorating Cancer Cachexia Symptoms in a Mouse Model. J. Cancer 2016, 7, 2378–2387. [Google Scholar] [CrossRef]
- Mader, T.; Chaillou, T.; Alves, E.S.; Jude, B.; Cheng, A.J.; Kenne, E.; Mijwel, S.; Kurzejamska, E.; Vincent, C.T.; Rundqvist, H. Exercise reduces intramuscular stress and counteracts muscle weakness in mice with breast cancer. J. Cachexia Sarcopenia Muscle 2022, 13, 1151–1163. [Google Scholar] [CrossRef]
- Re Cecconi, A.D.; Forti, M.; Chiappa, M.; Zhu, Z.; Zingman, L.V.; Cervo, L.; Beltrame, L.; Marchini, S.; Piccirillo, R.; Musclin, A. Myokine Induced by Aerobic Exercise, Retards Muscle Atrophy During Cancer Cachexia in Mice. Cancers 2019, 11, 1541. [Google Scholar] [CrossRef]
- Khamoui, A.V.; Park, B.S.; Kim, D.H.; Yeh, M.C.; Oh, S.L.; Elam, M.L.; Jo, E.; Arjmandi, B.H.; Salazar, G.; Grant, S.C.; et al. Aerobic and resistance training dependent skeletal muscle plasticity in the colon-26 murine model of cancer cachexia. Metabolism 2016, 65, 685–698. [Google Scholar] [CrossRef]
- Pigna, E.; Berardi, E.; Aulino, P.; Rizzuto, E.; Zampieri, S.; Carraro, U.; Kern, H.; Merigliano, S.; Gruppo, M.; Mericskay, M.; et al. Aerobic Exercise and Pharmacological Treatments Counteract Cachexia by Modulating Autophagy in Colon Cancer. Sci. Rep. 2016, 6, 26991. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, M.; Sako, N.; Isobe, M.; Lee-Hotta, S.; Sugiura, H.; Kametaka, S. Aerobic Exercise Ameliorates Cancer Cachexia-Induced Muscle Wasting through Adiponectin Signaling. Int. J. Mol. Sci. 2021, 22, 3110. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Gao, S.; Puppa, M.J.; Kostek, M.C.; Wilson, L.B.; Carson, J.A. High-Frequency Stimulation on Skeletal Muscle Maintenance in Female Cachectic Mice. Med. Sci. Sports Exerc. 2019, 51, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Hardee, J.P.; Mangum, J.E.; Gao, S.; Sato, S.; Hetzler, K.L.; Puppa, M.J.; Fix, D.K.; Carson, J.A. Eccentric contraction-induced myofiber growth in tumor-bearing mice. J. Appl. Physiol. 2016, 120, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Testa, M.T.J.; Cella, P.S.; Marinello, P.C.; Frajacomo, F.T.T.; Padilha, C.S.; Perandini, P.C.; Moura, F.A.; Duarte, J.A.; Cecchini, R.; Guarnier, F.A.; et al. Resistance Training Attenuates Activation of STAT3 and Muscle Atrophy in Tumor-Bearing Mice. Front. Oncol. 2022, 12, 880787. [Google Scholar] [CrossRef] [PubMed]
- Al-Majid, S.; McCarthy, D.O. Resistance exercise training attenuates wasting of the extensor digitorum longus muscle in mice bearing the colon-26 adenocarcinoma. Biol. Res. Nurs. 2001, 2, 155–166. [Google Scholar] [CrossRef]
- Mavropalias, G.; Sim, M.; Taaffe, D.R.; Galvão, D.A.; Spry, N.; Kraemer, W.J.; Häkkinen, K.; Newton, R.U. Exercise medicine for cancer cachexia: Targeted exercise to counteract mechanisms and treatment side effects. J. Cancer Res. Clin. Oncol. 2022, 148, 1389–1406. [Google Scholar] [CrossRef]
- Kamel, F.H.; Basha, M.A.; Alsharidah, A.S.; Salama, A.B. Resistance Training Impact on Mobility, Muscle Strength and Lean Mass in Pancreatic Cancer Cachexia: A Randomized Controlled Trial. Clin. Rehabil. 2020, 34, 1391–1399. [Google Scholar] [CrossRef]
- Geremia, A.; Sartori, R.; Baraldo, M.; Nogara, L.; Balmaceda, V.; Dumitras, G.A.; Ciciliot, S.; Scalabrin, M.; Nolte, H.; Blaauw, B. Activation of Akt–mTORC1 signalling reverts cancer-dependent muscle wasting. J. Cachexia Sarcopenia Muscle 2022, 13, 648–661. [Google Scholar] [CrossRef]
- Tanaka, M.; Sugimoto, K.; Fujimoto, T.; Xie, K.; Takahashi, T.; Akasaka, H.; Kurinami, H.; Yasunobe, Y.; Matsumoto, T.; Fujino, H. Preventive effects of low-intensity exercise on cancer cachexia–induced muscle atrophy. FASEB J. 2019, 33, 7852–7862. [Google Scholar] [CrossRef]
- Segal, R.; Zwaal, C.; Green, E.; Tomasone, J.R.; Loblaw, A.; Petrella, T.; The Exercise for People with Cancer Guideline Development Group. Exercise for people with cancer: A systematic review. Curr. Oncol. 2017, 24, e290–e315. [Google Scholar] [CrossRef] [PubMed]
- Solheim, T.S.; Laird, B.J.A.; Balstad, T.R.; Stene, G.B.; Bye, A.; Johns, N.; Pettersen, C.H.; Fallon, M.; Fayers, P.; Fearon, K.; et al. A randomized phase II feasibility trial of a multimodal intervention for the management of cachexia in lung and pancreatic cancer. J. Cachexia Sarcopenia Muscle 2017, 8, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Oldervoll, L.M.; Loge, J.H.; Lydersen, S.; Paltiel, H.; Asp, M.B.; Nygaard, U.V.; Oredalen, E.; Frantzen, T.L.; Lesteberg, I.; Amundsen, L.; et al. Physical exercise for cancer patients with advanced disease: A randomized controlled trial. Oncologist 2011, 16, 1649–1657. [Google Scholar] [CrossRef]
- Wood, N.R.; Garritson, J.; Mathias, A.; Haughian, J.M.; Hayward, R. Moderate Intensity Endurance and Resistance Exercise Attenuates Cachexia in Tumor-bearing Mice. Anticancer. Res. 2022, 42, 397–405. [Google Scholar] [CrossRef]
- Møller, A.B.; Lønbro, S.; Farup, J.; Voss, T.S.; Rittig, N.; Wang, J.; Højris, I.; Mikkelsen, U.R.; Jessen, N. Molecular and cellular adaptations to exercise training in skeletal muscle from cancer patients treated with chemotherapy. J. Cancer Res. Clin. Oncol. 2019, 145, 1449–1460. [Google Scholar] [CrossRef]
- Herrero, F.; San Juan, A.F.; Fleck, S.J.; Balmer, J.; Pérez, M.; Cañete, S.; Earnest, C.P.; Foster, C.; Lucía, A. Combined aerobic and resistance training in breast cancer survivors: A randomized, controlled pilot trial. Int. J. Sports Med. 2006, 27, 573–580. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Directo, D.; Lee, S.-R. Cancer Cachexia: Underlying Mechanisms and Potential Therapeutic Interventions. Metabolites 2023, 13, 1024. https://doi.org/10.3390/metabo13091024
Directo D, Lee S-R. Cancer Cachexia: Underlying Mechanisms and Potential Therapeutic Interventions. Metabolites. 2023; 13(9):1024. https://doi.org/10.3390/metabo13091024
Chicago/Turabian StyleDirecto, Dean, and Sang-Rok Lee. 2023. "Cancer Cachexia: Underlying Mechanisms and Potential Therapeutic Interventions" Metabolites 13, no. 9: 1024. https://doi.org/10.3390/metabo13091024
APA StyleDirecto, D., & Lee, S. -R. (2023). Cancer Cachexia: Underlying Mechanisms and Potential Therapeutic Interventions. Metabolites, 13(9), 1024. https://doi.org/10.3390/metabo13091024