

Anaplerotic Therapy Using Triheptanoin in Two Brothers Suffering from Aconitase 2 Deficiency

 , , , , , ,
, , , , , ,
Abstract
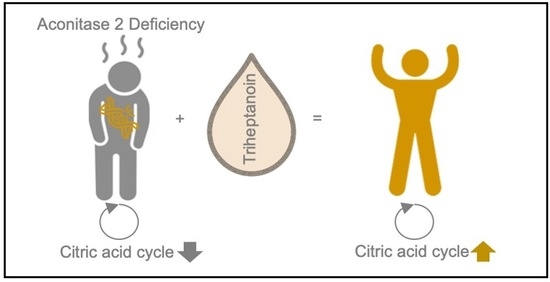
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Therapy with Triheptanoin
2.2. Therapy Monitoring
2.3. Isolation of Mitochondria from Cell Cultures
2.4. Western Blotting
2.5. Aconitase Activity
2.6. Monitoring of Plasma C7 Fatty Acids
3. Results
3.1. Clinical Report
3.2. Genetic Analysis
3.3. Fibroblasts Analysis
3.4. Therapy with Triheptanoin
3.5. Motor Ability Analysis
3.6. Organic Metabolites
3.7. C7 Fatty Acids
3.8. Follow-Up
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sadat, R.; Barca, E.; Masand, R.; Donti, T.R.; Naini, A.; De Vivo, D.C.; DiMauro, S.; Hanchard, N.A.; Graham, B.H. Functional Cellular Analyses Reveal Energy Metabolism Defect and Mitochondrial DNA Depletion in a Case of Mitochondrial Aconitase Deficiency. Mol. Genet. Metab. 2016, 118, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cai, G.H.; Xia, B.; Wang, X.; Zhang, C.C.; Xie, B.C.; Shi, X.C.; Liu, H.; Lu, J.F.; Zhang, R.X.; et al. Mitochondrial Aconitase Controls Adipogenesis through Mediation of Cellular ATP Production. FASEB J. 2020, 34, 6688–6702. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.A.C.; Grossmann, D.; Schimpf-Linzenbold, S.; Dayan, D.; Stingl, K.; Ben-Menachem, R.; Pines, O.; Massart, F.; Delcambre, S.; Ghelfi, J.; et al. Haploinsufficiency Due to a Novel ACO2 Deletion Causes Mitochondrial Dysfunction in Fibroblasts from a Patient with Dominant Optic Nerve Atrophy. Sci. Rep. 2020, 10, 16736. [Google Scholar] [CrossRef] [PubMed]
- Ciccarone, F.; Vegliante, R.; Di Leo, L.; Ciriolo, M.R. The TCA Cycle as a Bridge between Oncometabolism and DNA Transactions in Cancer. Semin. Cancer Biol. 2017, 47, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Xin, J.C.; Wang, X.; Kaufman, B.A.; Butow, R.A. Aconitase Couples Metabolic Regulation to Mitochondrial DNA Maintenance. Science 2005, 307, 714–717. [Google Scholar] [CrossRef]
- Guehlouz, K.; Foulonneau, T.; Amati-Bonneau, P.; Charif, M.; Colin, E.; Bris, C.; Desquiret-Dumas, V.; Milea, D.; Gohier, P.; Procaccio, V.; et al. ACO2 Clinicobiological Dataset with Extensive Phenotype Ontology Annotation. Sci. Data 2021, 8, 205. [Google Scholar] [CrossRef] [PubMed]
- Charif, M.; Gueguen, N.; Ferré, M.; Elkarhat, Z.; Khiati, S.; LeMao, M.; Chevrollier, A.; Desquiret-Dumas, V.; Goudenège, D.; Bris, C.; et al. Dominant ACO2 Mutations Are a Frequent Cause of Isolated Optic Atrophy. Brain Commun. 2021, 3, 205. [Google Scholar] [CrossRef] [PubMed]
- Sharkia, R.; Wierenga, K.J.; Kessel, A.; Azem, A.; Bertini, E.; Carrozzo, R.; Torraco, A.; Goffrini, P.; Berti, C.C.; McCormick, M.E.; et al. Clinical, Radiological, and Genetic Characteristics of 16 Patients with ACO2 Gene Defects: Delineation of an Emerging Neurometabolic Syndrome. J. Inherit. Metab. Dis. 2019, 42, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, R.; Pines, O.; Ta-Shma, A.; Burak, E.; Shaag, A.; Halvardson, J.; Edvardson, S.; Mahajna, M.; Zenvirt, S.; Saada, A.; et al. Infantile Cerebellar-Retinal Degeneration Associated with a Mutation in Mitochondrial Aconitase, ACO2. Am. J. Hum. Genet. 2012, 90, 518–523. [Google Scholar] [CrossRef]
- Reilmann, R.; Schubert, R. Motor Outcome Measures in Huntington Disease Clinical Trials; Elsevier B.V.: Amsterdam, The Netherladns, 2017; Volume 144, ISBN 978-0-12-801893-4. [Google Scholar]
- Main, M.; Kairon, H.; Mercuri, E.; Muntoni, F. The Hammersmith Functional Motor Scale for Children with Spinal Muscular Atrophy: A Scale to Test Ability and Monitor Progress in Children with Limited Ambulation. Eur. J. Paediatr. Neurol. 2003, 7, 155–159. [Google Scholar] [CrossRef]
- Glanzman, A.M.; Mazzone, E.; Main, M.; Pelliccioni, M.; Wood, J.; Swoboda, K.J.; Scott, C.; Pane, M.; Messina, S.; Bertini, E.; et al. The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): Test Development and Reliability The CHOP INTEND Is a Reliable Measure of Motor Skills in Patients with SMA-I and Neuromuscular Disorders Presenting in Infancy. Neuromuscul. Disord. 2010, 20, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. Mutationtaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Predicting Deleterious Amino Acid Substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Kisiel, M.A.; Klar, A.S. Isolation and Culture of Human Dermal Fibroblasts. In Skin Tissue Engineering; Böttcher-Haberzeth, S., Biedermann, T., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1993, pp. 71–78. ISBN 978-1-4939-9472-4. [Google Scholar]
- Bartolucci, G.; Pallecchi, M.; Menicatti, M.; Moracci, L.; Pucciarelli, S.; Agostini, M.; Crotti, S. A Method for Assessing Plasma Free Fatty Acids from C2 to C18 and Its Application for the Early Detection of Colorectal Cancer. J. Pharm. Biomed. Anal. 2022, 215, 114762. [Google Scholar] [CrossRef]
- Metodiev, M.D.; Gerber, S.; Hubert, L.; Delahodde, A.; Chretien, D.; Gérard, X.; Amati-Bonneau, P.; Giacomotto, M.C.; Boddaert, N.; Kaminska, A.; et al. Mutations in the Tricarboxylic Acid Cycle Enzyme, Aconitase 2, Cause Either Isolated or Syndromic Optic Neuropathy with Encephalopathy and Cerebellar Atrophy. J. Med. Genet. 2014, 51, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Heretsch, P.; Thomas, F.; Aurich, A.; Krautscheid, H.; Sicker, D.; Giannis, A. Synthesen Mit Einem Chiralen Baustein Aus Dem Citratzyklus: (2R,3S)-Isocitronensäure Aus Einer Fermentation Mit Sonnenblumenöl. Angew. Chem. 2008, 120, 1984–1986. [Google Scholar] [CrossRef]
- Longo, N.; Price, L.B.; Gappmaier, E.; Cantor, N.L.; Ernst, S.L.; Bailey, C.; Pasquali, M. Anaplerotic Therapy in Propionic Acidemia. Mol. Genet. Metab. 2017, 122, 51–59. [Google Scholar] [CrossRef]
- Wehbe, Z.; Tucci, S. Therapeutic Potential of Triheptanoin in Metabolic and Neurodegenerative Diseases. J. Inherit. Metab. Dis. 2019, 43, 385–391. [Google Scholar] [CrossRef]
- Roe, C.R.; Sweetman, L.; Roe, D.S.; David, F.; Brunengraber, H. Treatment of Cardiomyopathy and Rhabdomyolysis in Long-Chain Fat Oxidation Disorders Using an Anaplerotic Odd-Chain Triglyceride. J. Clin. Investig. 2002, 110, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Mochel, F.; DeLonlay, P.; Touati, G.; Brunengraber, H.; Kinman, R.P.; Rabier, D.; Roe, C.R.; Saudubray, J.M. Pyruvate Carboxylase Deficiency: Clinical and Biochemical Response to Anaplerotic Diet Therapy. Mol. Genet. Metab. 2005, 84, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Schwarzkopf, T.M.; Koch, K.; Klein, J. Reduced Severity of Ischemic Stroke and Improvement of Mitochondrial Function after Dietary Treatment with the Anaplerotic Substance Triheptanoin. Neuroscience 2015, 300, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Lemarié, F.; Beauchamp, E.; Legrand, P.; Rioux, V. Revisiting the Metabolism and Physiological Functions of Caprylic Acid (C8:0) with Special Focus on Ghrelin Octanoylation. Biochimie 2016, 120, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Kinman, R.P.; Kasumov, T.; Jobbins, K.A.; Thomas, K.R.; Adams, J.E.; Brunengraber, L.N.; Kutz, G.; Brewer, W.U.; Roe, C.R.; Brunengraber, H. Parenteral and Enteral Metabolism of Anaplerotic Triheptanoin in Normal Rats. Am. J. Physiol. Endocrinol. Metab. 2006, 291, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhang, G.F.; Kombu, R.S.; Allen, F.; Kutz, G.; Brewer, W.U.; Roe, C.R.; Brunengraber, H. Parenteral and Enteral Metabolism of Anaplerotic Triheptanoin in Normal Rats. II. Effects on Lipolysis, Glucose Production, and Liver Acyl-CoA Profile. Am. J. Physiol. Endocrinol. Metab. 2010, 298, 362–371. [Google Scholar] [CrossRef]
- Aoyama, T.; Nosaka, N.; Kasai, M. Research on the Nutritional Characteristics of Medium-Chain Fatty Acids. J. Med. Investig. 2007, 54, 385–388. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penkl, M.; Mayr, J.A.; Feichtinger, R.G.; Reilmann, R.; Debus, O.; Fobker, M.; Penkl, A.; Reunert, J.; Rust, S.; Marquardt, T. Anaplerotic Therapy Using Triheptanoin in Two Brothers Suffering from Aconitase 2 Deficiency. Metabolites 2024, 14, 238. https://doi.org/10.3390/metabo14040238
Penkl M, Mayr JA, Feichtinger RG, Reilmann R, Debus O, Fobker M, Penkl A, Reunert J, Rust S, Marquardt T. Anaplerotic Therapy Using Triheptanoin in Two Brothers Suffering from Aconitase 2 Deficiency. Metabolites. 2024; 14(4):238. https://doi.org/10.3390/metabo14040238
Chicago/Turabian StylePenkl, Maximilian, Johannes A. Mayr, René G. Feichtinger, Ralf Reilmann, Otfried Debus, Manfred Fobker, Anja Penkl, Janine Reunert, Stephan Rust, and Thorsten Marquardt. 2024. "Anaplerotic Therapy Using Triheptanoin in Two Brothers Suffering from Aconitase 2 Deficiency" Metabolites 14, no. 4: 238. https://doi.org/10.3390/metabo14040238
APA StylePenkl, M., Mayr, J. A., Feichtinger, R. G., Reilmann, R., Debus, O., Fobker, M., Penkl, A., Reunert, J., Rust, S., & Marquardt, T. (2024). Anaplerotic Therapy Using Triheptanoin in Two Brothers Suffering from Aconitase 2 Deficiency. Metabolites, 14(4), 238. https://doi.org/10.3390/metabo14040238