Cell-Type Specific Metabolic Flux Analysis: A Challenge for Metabolic Phenotyping and a Potential Solution in Plants

Abstract
:1. Introduction
2. Results
2.1. Confounding Effect of Cellular Heterogeneity on Metabolic Flux Analysis
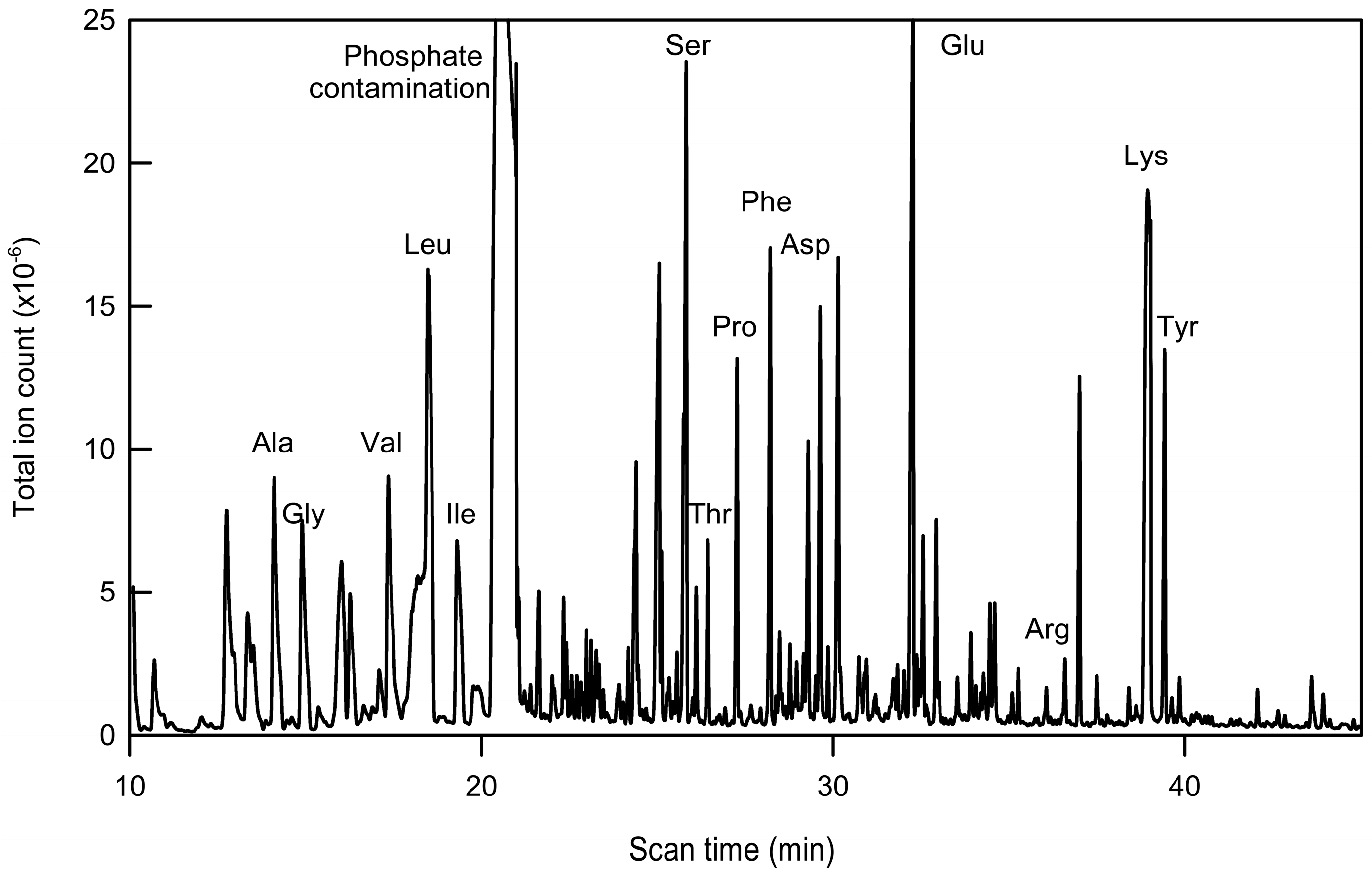
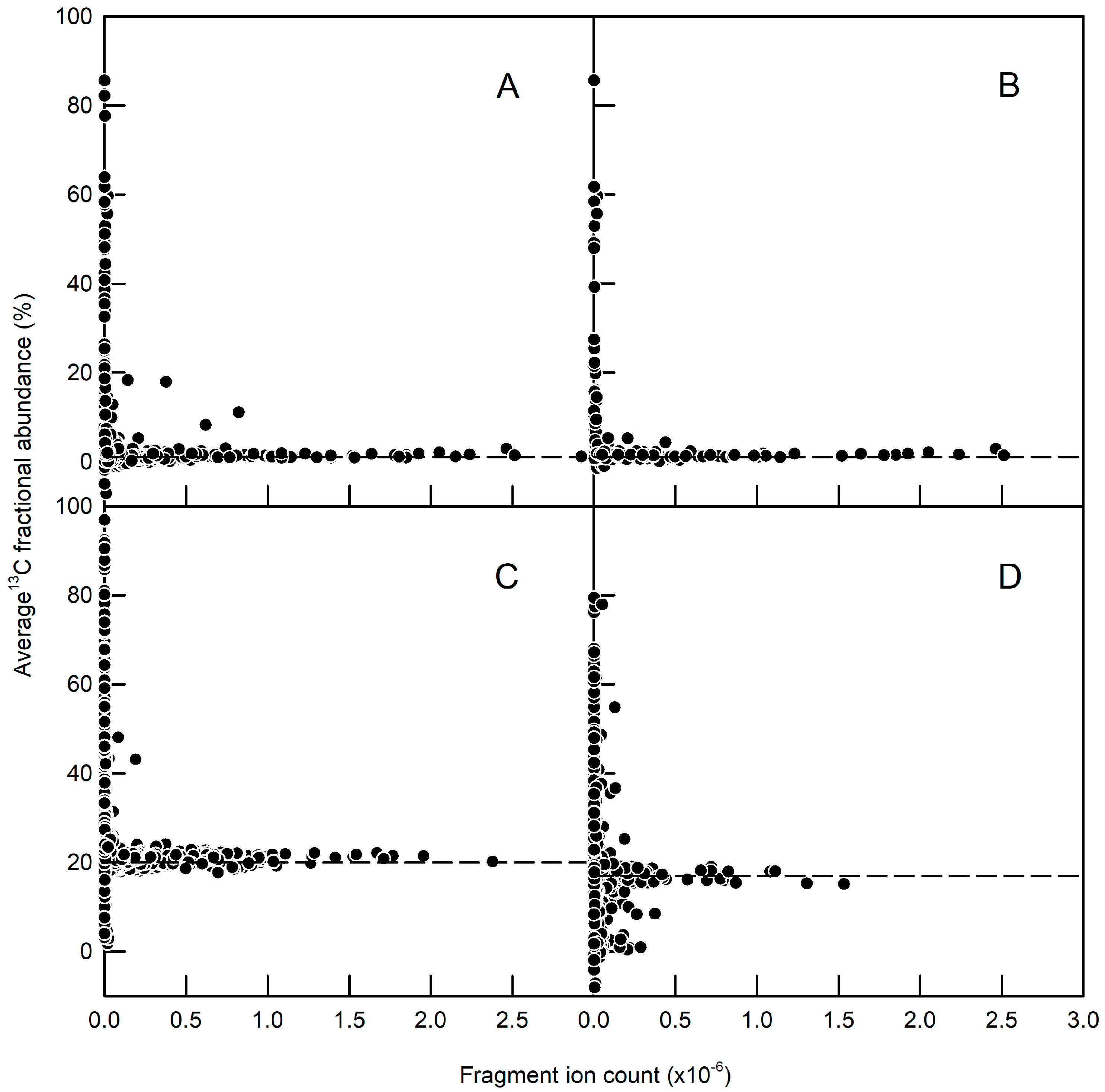
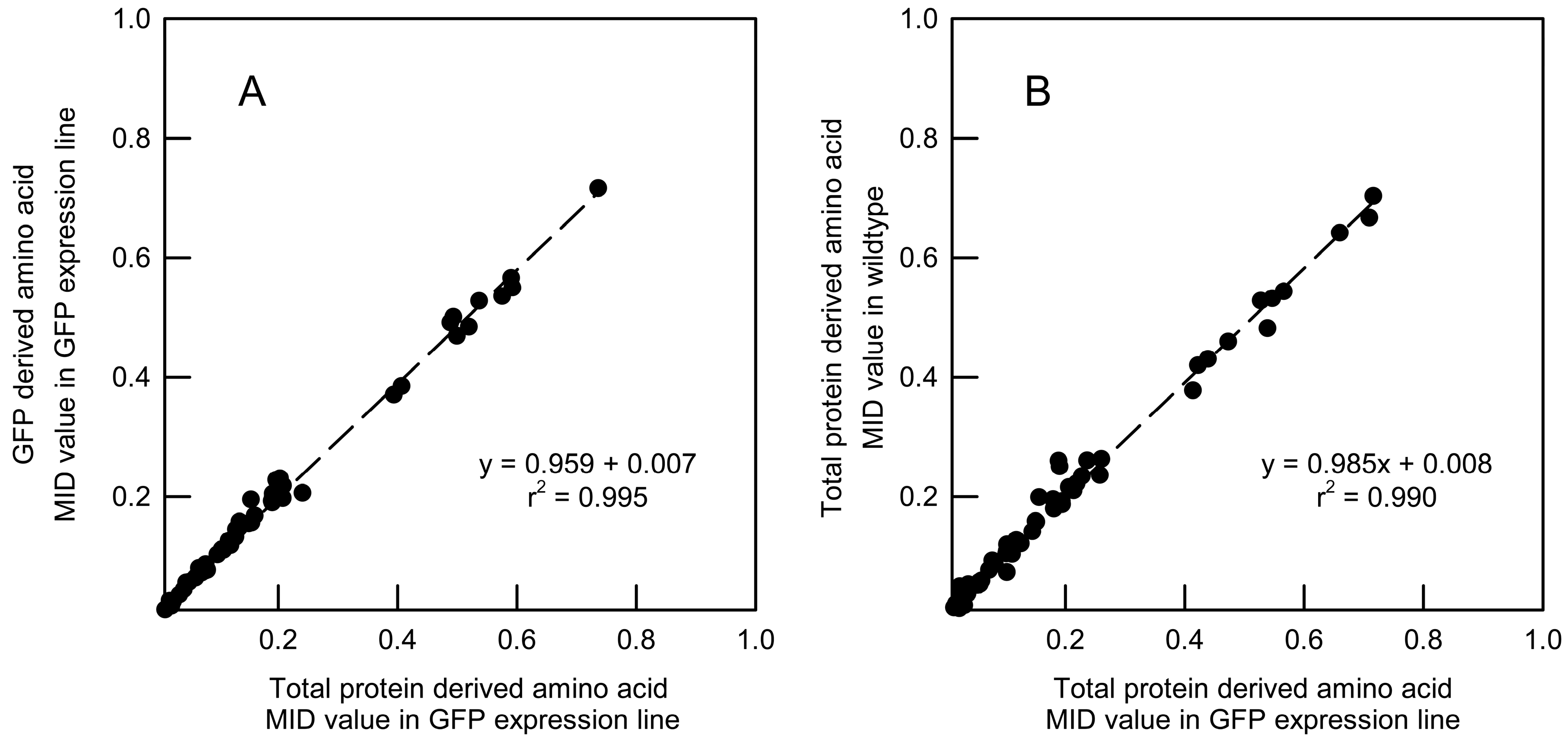
2.2. Immunopurification of GFP and MS Analysis of GFP Hydrolysates
2.3. Validation of GFP as a Reporter for the 13C Labelling of Total Root Protein in A. thaliana Seedlings with Constitutive GFP Expression
3. Discussion
4. Materials and Methods
4.1. Hydroponic Culture of Arabidopsis thaliana Seedlings
4.2. Extraction of Total Protein
4.3. Expression and Purification of the GFP Binding Protein in Escherichia coli
4.4. Immunopurification of GFP from Plant Protein Extracts
4.5. Preparation of GFP Samples for GC-MS
4.6. Gas Chromatography—Mass Spectrometry
4.7. Metabolic Modelling
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ratcliffe, R.G.; Shachar-Hill, Y. Revealing metabolic phenotypes in plants: Inputs from NMR analysis. Biol. Rev. 2005, 80, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Kruger, N.J.; Ratcliffe, R.G. Fluxes through plant metabolic networks: Measurements, predictions, insights and challenges. Biochem. J. 2015, 465, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Ratcliffe, R.G.; Shachar-Hill, Y. Measuring multiple fluxes through plant metabolic networks. Plant J. 2006, 45, 490–511. [Google Scholar] [CrossRef] [PubMed]
- Wiechert, W.; Möllney, M.; Petersen, S.; de Graaf, A.A. A universal framework for 13C metabolic flux analysis. Metab. Eng. 2001, 3, 265–283. [Google Scholar] [CrossRef] [PubMed]
- Masakapalli, S.K.; Le Lay, P.; Huddleston, J.E.; Pollock, N.L.; Kruger, N.J.; Ratcliffe, R.G. Subcellular flux analysis of central metabolism in a heterotrophic Arabidopsis cell suspension using steady-state stable isotope labeling. Plant Physiol. 2010, 152, 602–619. [Google Scholar] [CrossRef] [PubMed]
- Kruger, N.J.; Masakapalli, S.K.; Ratcliffe, R.G. Strategies for investigating the plant metabolic network with steady-state metabolic flux analysis: Lessons from an Arabidopsis cell culture and other systems. J. Exp. Bot. 2012, 63, 2309–2323. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.C.R.; Sweetlove, L.J.; Ratcliffe, R.G. Capturing metabolite channeling in metabolic flux phenotypes. Plant Physiol. 2011, 157, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Masakapalli, S.K.; Kruger, N.J.; Ratcliffe, R.G. The metabolic flux phenotype of heterotrophic Arabidopsis cells reveals a complex response to changes in nitrogen supply. Plant J. 2013, 74, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Masakapalli, S.K.; Bryant, F.M.; Kruger, N.J.; Ratcliffe, R.G. The metabolic flux phenotype of heterotrophic Arabidopsis cells reveals a flexible balance between the cytosolic and plastidic contributions to carbohydrate oxidation in response to phosphate limitation. Plant J. 2014, 78, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Liu, L.; Wu, C.; Yang, C.; Wu, Q. 13C-Tracer and gas chromatography-mass spectrometry analyses reveal metabolic flux distribution in the oleaginous microalga Chlorella protothecoides. Plant Physiol. 2010, 154, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Boyle, N.R.; Sengupta, N.; Morgan, J.A. Metabolic flux analysis of heterotrophic growth in Chlamydomonas rheinhardtii. PLoS ONE 2017, 12, e0177292. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.P.; Val, D.L.; Shachar-Hill, Y. Central metabolic fluxes in the endosperm of developing maize seeds and their implications for metabolic engineering. Metab. Eng. 2011, 13, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.K.; Young, J.D. Carbon and nitrogen provisions alter the metabolic flux in developing soybean embryos. Plant Physiol. 2013, 161, 1458–1475. [Google Scholar] [CrossRef] [PubMed]
- Pollard, M.; Delamarter, D.; Martin, T.M.; Shachar-Hill, Y. Lipid labelling from acetate or glycerol in cultured embryos of Camelina sativa seed: A tale of two substrates. Phytochemistry 2015, 118, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Schwender, J.; Hebbelmann, I.; Heinzel, N.; Hildebrandt, T.; Rogers, A.; Naik, D.; Klapperstück, M.; Braun, H.P.; Schreiber, F.; Denolf, P.; et al. Quantitative multilevel analysis of central metabolism in developing oilseeds of oilseed rape during in vitro culture. Plant Physiol. 2015, 168, 828–848. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.P.; Raymond, P.; Hernould, M.; Rondeau-Mouro, C.; de Graaf, A.; Chourey, P.; Lahaye, M.; Shachar-Hill, Y.; Rolin, D.; Dieuaide-Noubhani, M. A metabolic flux analysis to study the role of sucrose synthase in the regulation of carbon partitioning in central metabolism in maize root tips. Metab. Eng. 2007, 9, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Sriram, G.; Fulton, D.B.; Shanks, J.V. Flux quantification in central carbon metabolism of Catharanthus roseus hairy roots by 13C labeling and comprehensive bondomer balancing. Phytochemistry 2007, 68, 2243–2257. [Google Scholar] [CrossRef] [PubMed]
- Masakapalli, S.K.; Ritala, A.; Dong, L.; van der Krol, A.R.; Oksman-Caldentey, K.M.; Ratcliffe, R.G.; Sweetlove, L.J. Metabolic flux phenotype of tobacco hairy roots engineered for increased geraniol production. Phytochemistry 2014, 99, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Szecowka, M.; Heise, R.; Tohge, T.; Nunes-Nesi, A.; Vosloh, D.; Huege, J.; Feil, R.; Lunn, J.; Nikoloski, Z.; Stitt, M.; et al. Metabolic fluxes in an illuminated Arabidopsis rosette. Plant Cell 2013, 25, 694–714. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Jazmin, L.J.; Young, J.D.; Allen, D.K. Isotopically nonstationary 13C flux analysis of changes in Arabidopsis thaliana leaf metabolism due to high light acclimation. Proc. Natl. Acad. Sci. USA 2014, 111, 16967–16972. [Google Scholar] [CrossRef] [PubMed]
- Borisjuk, L.; Neuberger, T.; Schwender, J.; Heinzel, N.; Sunderhaus, S.; Fuchs, J.; Hay, J.O.; Tschiersch, H.; Braun, H.P.; Denolf, P.; et al. Seed architecture shapes embryo metabolism in oilseed rape. Plant Cell 2013, 25, 1625–1640. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.M.; Orlando, D.A.; Lee, J.Y.; Wang, J.Y.; Koch, J.; Dinneny, J.R.; Mace, D.; Ohler, U.; Benfey, P.N. A high-resolution root spatiotemporal map reveals dominant expression patterns. Science 2007, 318, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Petricka, J.J.; Schauer, M.A.; Megraw, M.; Breakfield, N.W.; Thompson, J.W.; Georgiev, S.; Soderblom, E.J.; Ohler, U.; Moseley, M.A.; Grossniklaus, U.; et al. The protein expression landscape of the Arabidopsis root. Proc. Natl. Acad. Sci. USA 2012, 109, 6811–6818. [Google Scholar] [CrossRef] [PubMed]
- Moussaieff, A.; Rogachev, I.; Brodsky, L.; Malitsky, S.; Toal, T.W.; Belcher, H.; Yativ, M.; Brady, S.M.; Benfey, P.N.; Aharoni, A. High-resolution metabolic mapping of cell types in plant roots. Proc. Natl. Acad. Sci. USA 2013, 110, E1232–E1241. [Google Scholar] [CrossRef] [PubMed]
- Vasdekis, A.E.; Stephanopoulos, G. Review of methods to probe single cell metabolism and bioenergetics. Metab. Eng. 2015, 27, 115–135. [Google Scholar] [CrossRef] [PubMed]
- De Vargas Roditi, L.; Claassen, M. Computational and experimental single cell biology techniques for the definition of cell type heterogeneity, interplay and intracellular dynamics. Curr. Opin. Biotechnol. 2015, 34, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Mills, E.S.; Deal, R.B. Technologies for systems-level analysis of specific cell types in plants. Plant Sci. 2012, 197, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Misra, B.B.; Assmann, S.M.; Chen, S. Plant single-cell and single-cell-type metabolomics. Trends Plant Sci. 2014, 19, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Petersson, S.V.; Lindén, P.; Moritz, T.; Ljung, K. Cell-type specific metabolic profiling of Arabidopsis thaliana protoplasts as a tool for plant systems biology. Metabolomics 2015, 11, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Etalo, D.W.; de Vos, R.C.H.; Joosten, M.H.A.J.; Hall, R.D. Spatially resolved plant metabolomics: Some potentials and limitations of laser-ablation electrospray ionization mass spectrometry metabolite imaging. Plant Physiol. 2015, 169, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Korte, A.R.; Yandeau-Nelson, M.D.; Nikolau, B.J.; Lee, Y.J. Subcellular-level resolution MALDI-MS imaging of maize leaf metabolites by MALDI-linear ion trap-Orbitrap mass spectrometer. Anal. Bioanal. Chem. 2015, 407, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Borisjuk, L.; Rolletschek, H.; Neuberger, T. Surveying the plant’s world by magnetic resonance imaging. Plant J. 2012, 70, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.K.; Laclair, R.W.; Ohlrogge, J.B.; Shachar-Hill, Y. Isotope labelling of Rubisco subunits provides in vivo information on subcellular biosynthesis and exchange of amino acids between compartments. Plant Cell Environ. 2012, 35, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Liu, H.; Blankenship, R.E.; Tang, Y.J. Using photosystem I as a reporter protein for 13C analysis in a coculture containing cyanobacterium and a heterotrophic bacterium. Anal. Biochem. 2015, 477, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, A.S.; Tang, Y.J.; Mukhopadhyay, A.; Keasling, J.D. Isotopomer distributions in amino acids from a highly expressed protein as a proxy for those from total protein. Anal. Chem. 2008, 80, 886–890. [Google Scholar] [CrossRef] [PubMed]
- Rühl, M.; Hardt, W.D.; Sauer, U. Sub-population-specific metabolic pathway usage in mixed cultures as revealed by reporter protein-based 13C analysis. Appl. Environ. Microbiol. 2011, 77, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Colinas, J.; Wang, J.Y.; Mace, D.; Ohler, U.; Benfey, P.N. Transcriptional and posttranscriptional regulation of transcription factor expression in Arabidopsis roots. Proc. Natl. Acad. Sci. USA 2006, 103, 6055–6060. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Xiao, Y.; Gebreselassie, N.; Zhang, F.; Antoniewicz, M.R.; Tang, Y.J.; Peng, L. Central metabolic responses to the overproduction of fatty acids in Escherichia coli based on 13C-metabolic flux analysis. Biotechnol. Bioeng. 2014, 111, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Kappelmann, J.; Wiechert, W.; Noack, S. Cutting the Gordian knot: Identifiability of anaplerotic reactions in Corynebacterium glutamicum by means of 13C-metabolic flux analysis. Biotechnol. Bioeng. 2016, 113, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Nilmeier, J.; Weaver, D.; Adams, P.D.; Keasling, J.D.; Mukhopadhyay, A.; Petzold, C.J.; Martín, H.G. A peptide-based method for 13C metabolic flux analysis in microbial communities. PLoS Comput. Biol. 2014, 10, e1003827. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, C. Fluxome analysis using GC-MS. Microb. Cell Fact. 2007, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Furbank, R.T. Walking the C4 pathway: Past, present and future. J. Exp. Bot. 2016, 67, 4057–4066. [Google Scholar] [CrossRef] [PubMed]
- Gebreselassie, N.A.; Antoniewicz, M.R. 13C-metabolic flux analysis of co-cultures: A novel approach. Metab. Eng. 2015, 31, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Schwarzländer, M.; Fricker, M.D.; Müller, C.; Marty, L.; Brach, T.; Novak, J.; Sweetlove, L.J.; Hell, R.; Meyer, A.J. Confocal imaging of glutathione redox potential in living plant cells. J. Microsc. 2008, 231, 299–316. [Google Scholar] [CrossRef] [PubMed]
- Mandy, D.E.; Goldford, J.E.; Yang, H.; Allen, D.K.; Libourel, I.G.L. Metabolic flux analysis using 13C peptide label measurements. Plant J. 2014, 77, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Kunz, S.; Pesquet, E.; Kleczkowski, L.A. Functional dissection of sugar signals affecting gene expression in Arabidopis thaliana. PLoS ONE 2014, 9, e100312. [Google Scholar] [CrossRef] [PubMed]
- Conrath, K.E.; Lauwereys, M.; Galleni, M.; Matagne, A.; Frère, J.M.; Kinne, J.; Wyns, L.; Muyldermans, S. β-lactamase inhibitors derived from single-domain antibody fragments elicited in the Camelidae. Antimicrob. Agents Chemother. 2001, 45, 2807–2812. [Google Scholar] [CrossRef] [PubMed]
- Rothbauer, U.; Zolghadr, K.; Tillib, S.; Nowak, D.; Schermelleh, L.; Gahl, A.; Backmann, N.; Conrath, K.; Muyldermans, S.; Cardoso, M.C.; et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods 2006, 3, 887–889. [Google Scholar] [CrossRef] [PubMed]
- Rothbauer, U.; Zolghadr, K.; Muyldermans, S.; Schepers, A.; Cardoso, M.C.; Leonhardt, H. A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol. Cell. Proteom. 2008, 7, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.C.R.; Poolman, M.G.; Howden, A.J.; Schwarzlander, M.; Fell, D.A.; Ratcliffe, R.G.; Sweetlove, L.J. A genome-scale metabolic model accurately predicts fluxes in central carbon metabolism under stress conditions. Plant Physiol. 2010, 154, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Lommen, A. MetAlign: Interface-driven, versatile metabolomics tool for hyphenated full-scan mass spectrometry data preprocessing. Anal. Chem. 2009, 81, 3079–3086. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.A.; Dauner, M.; Wiechert, W. New tools for mass isotopomer data evaluation in 13C flux analysis: Mass isotope correction, data consistency checking, and precursor relationships. Biotechnol. Bioeng. 2004, 85, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Young, J.D. INCA: A computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 2014, 30, 1333–1335. [Google Scholar] [CrossRef] [PubMed]
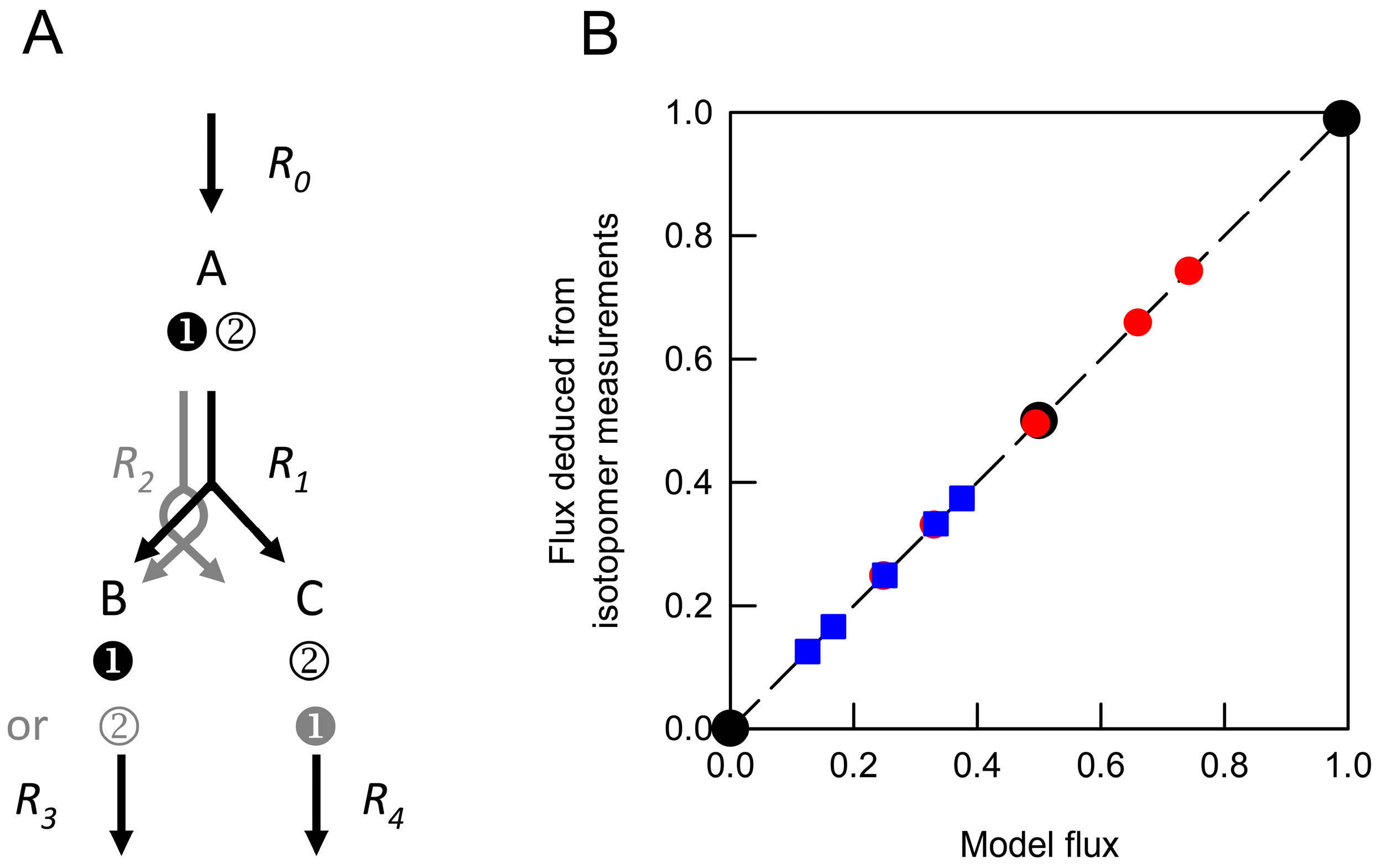
 ) and from weighted average datasets generated by combining values obtained from networks in which R1net was 0 and 1.0 (
) and from weighted average datasets generated by combining values obtained from networks in which R1net was 0 and 1.0 (  ) or 0 and 0.5 (
) or 0 and 0.5 (  ). These were compared with the anticipated model flux based on the relative contributions of different networks to the isotopomer dataset. Each deduced flux value is the mean of 100 Monte Carlo simulations for which the SD is smaller than the symbol. The dashed line indicates equivalence between the deduced flux estimate and model flux.
) and from weighted average datasets generated by combining values obtained from networks in which R1net was 0 and 1.0 ( ) or 0 and 0.5 ( ). These were compared with the anticipated model flux based on the relative contributions of different networks to the isotopomer dataset. Each deduced flux value is the mean of 100 Monte Carlo simulations for which the SD is smaller than the symbol. The dashed line indicates equivalence between the deduced flux estimate and model flux.
). These were compared with the anticipated model flux based on the relative contributions of different networks to the isotopomer dataset. Each deduced flux value is the mean of 100 Monte Carlo simulations for which the SD is smaller than the symbol. The dashed line indicates equivalence between the deduced flux estimate and model flux.
) and from weighted average datasets generated by combining values obtained from networks in which R1net was 0 and 1.0 ( ) or 0 and 0.5 ( ). These were compared with the anticipated model flux based on the relative contributions of different networks to the isotopomer dataset. Each deduced flux value is the mean of 100 Monte Carlo simulations for which the SD is smaller than the symbol. The dashed line indicates equivalence between the deduced flux estimate and model flux.
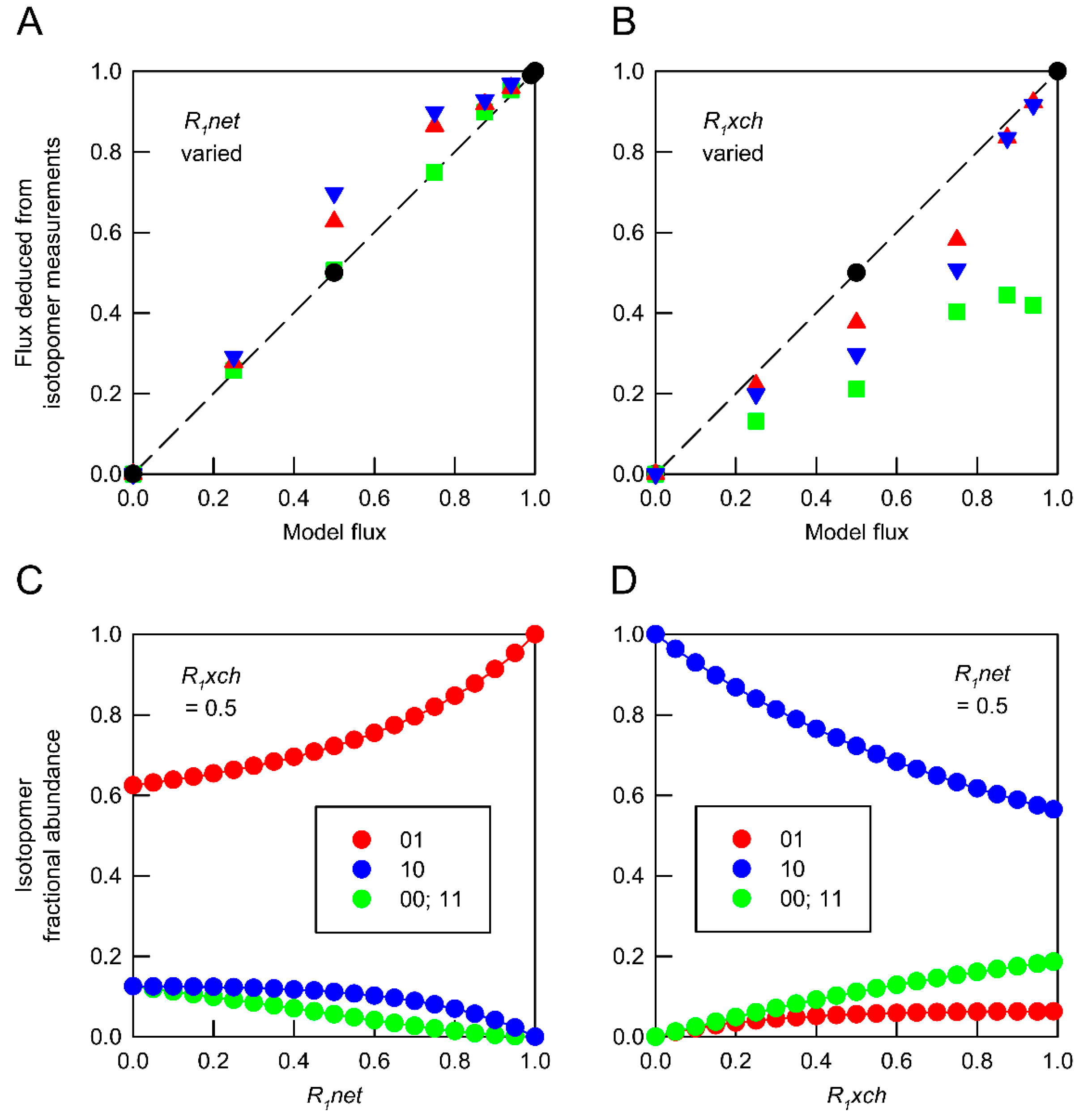
 ), 0.5 (
), 0.5 (  ) and 0.99 (
) and 0.99 (  ). (B) Deduced values of R1xch from combinations of networks in which R1net values were held at 0 ( ), 0.5 ( ) and 1.0 ( ). In both panels, isotopomer datasets from individual networks yielded the expected flux values under all conditions ( ). Each deduced flux value is the mean of 100 Monte Carlo simulations for which the SD is smaller than the symbol. The dashed line indicates equivalence between deduced flux estimate and model flux. The influence of flux distribution on the isotopomer composition of metabolic intermediates is exemplified by changes in the composition of metabolite A in response to (C) varying R1net while R1xch is held at 0.5, and (D) varying R1net while R1xch is held at 0.5. Isotopomers are identified using 0 to indicate 12C and 1 to indicate 13C in specified positions. In these simulations, the fractional abundances of 00 and 11 are identical.
), 0.5 ( ) and 0.99 ( ). (B) Deduced values of R1xch from combinations of networks in which R1net values were held at 0 ( ), 0.5 ( ) and 1.0 ( ). In both panels, isotopomer datasets from individual networks yielded the expected flux values under all conditions ( ). Each deduced flux value is the mean of 100 Monte Carlo simulations for which the SD is smaller than the symbol. The dashed line indicates equivalence between deduced flux estimate and model flux. The influence of flux distribution on the isotopomer composition of metabolic intermediates is exemplified by changes in the composition of metabolite A in response to (C) varying R1net while R1xch is held at 0.5, and (D) varying R1net while R1xch is held at 0.5. Isotopomers are identified using 0 to indicate 12C and 1 to indicate 13C in specified positions. In these simulations, the fractional abundances of 00 and 11 are identical.
). (B) Deduced values of R1xch from combinations of networks in which R1net values were held at 0 ( ), 0.5 ( ) and 1.0 ( ). In both panels, isotopomer datasets from individual networks yielded the expected flux values under all conditions ( ). Each deduced flux value is the mean of 100 Monte Carlo simulations for which the SD is smaller than the symbol. The dashed line indicates equivalence between deduced flux estimate and model flux. The influence of flux distribution on the isotopomer composition of metabolic intermediates is exemplified by changes in the composition of metabolite A in response to (C) varying R1net while R1xch is held at 0.5, and (D) varying R1net while R1xch is held at 0.5. Isotopomers are identified using 0 to indicate 12C and 1 to indicate 13C in specified positions. In these simulations, the fractional abundances of 00 and 11 are identical.
), 0.5 ( ) and 0.99 ( ). (B) Deduced values of R1xch from combinations of networks in which R1net values were held at 0 ( ), 0.5 ( ) and 1.0 ( ). In both panels, isotopomer datasets from individual networks yielded the expected flux values under all conditions ( ). Each deduced flux value is the mean of 100 Monte Carlo simulations for which the SD is smaller than the symbol. The dashed line indicates equivalence between deduced flux estimate and model flux. The influence of flux distribution on the isotopomer composition of metabolic intermediates is exemplified by changes in the composition of metabolite A in response to (C) varying R1net while R1xch is held at 0.5, and (D) varying R1net while R1xch is held at 0.5. Isotopomers are identified using 0 to indicate 12C and 1 to indicate 13C in specified positions. In these simulations, the fractional abundances of 00 and 11 are identical.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Step | Model Flux | Internal Fluxes Varied | Biomass Output Varied | ||
|---|---|---|---|---|---|
| Estimated Flux | % | Estimated Flux | % | ||
| [13C6]glucose input | 20 | 20.53 (20.48–20.57) | 102.6 | 19.98 (19.91–20.02) | 99.9 |
| Glucose 6-P dehydrogenase | 50 | 59.26 (59.04–59.47) | 118.5 | 56.48 (52.08–58.09) | 113.0 |
| Phosphoenolpyruvate carboxylase | 80 | 61.03 (60.11–62.11) | 76.3 | 136.19 (61.02–137.68) | 170.2 |
| Isocitrate lyase | 25 | 13.51 (11.78–15.24) | 54.1 | 25.19 (23.71–26.84) | 100.8 |
| Biomass output | 7.56 | 5.89 (5.73–6.06) | 78.0 | 8.79 (8.63–8.94) | 116.2 |
| Physiological output | - | - | - | - | - |
| CO2 production | 300 | 366.11 (359.54–372.78) | 122.0 | 251.29 (245.24–257.47) | 83.8 |
| O2 uptake | 283 | 352.55 (345.59–359.61) | 124.8 | 231.07 (224.67–237.61) | 81.8 |
| Respiratory quotient | 1.06 | 1.04 (1.04–1.04) | 97.8 | 1.09 (1.08–1.09) | 102.4 |
| Carbon conversion efficiency | 0.50 | 0.39 (0.38–0.42) | 78.0 | 0.58 (0.57–0.63) | 116.2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, M.T.; Kalde, M.; Srisakvarakul, C.; Kruger, N.J.; Ratcliffe, R.G. Cell-Type Specific Metabolic Flux Analysis: A Challenge for Metabolic Phenotyping and a Potential Solution in Plants. Metabolites 2017, 7, 59. https://doi.org/10.3390/metabo7040059
Rossi MT, Kalde M, Srisakvarakul C, Kruger NJ, Ratcliffe RG. Cell-Type Specific Metabolic Flux Analysis: A Challenge for Metabolic Phenotyping and a Potential Solution in Plants. Metabolites. 2017; 7(4):59. https://doi.org/10.3390/metabo7040059
Chicago/Turabian StyleRossi, Merja T., Monika Kalde, Chaiyakorn Srisakvarakul, Nicholas J. Kruger, and R. George Ratcliffe. 2017. "Cell-Type Specific Metabolic Flux Analysis: A Challenge for Metabolic Phenotyping and a Potential Solution in Plants" Metabolites 7, no. 4: 59. https://doi.org/10.3390/metabo7040059
APA StyleRossi, M. T., Kalde, M., Srisakvarakul, C., Kruger, N. J., & Ratcliffe, R. G. (2017). Cell-Type Specific Metabolic Flux Analysis: A Challenge for Metabolic Phenotyping and a Potential Solution in Plants. Metabolites, 7(4), 59. https://doi.org/10.3390/metabo7040059