Metabolic Profiling of Human Plasma and Urine, Targeting Tryptophan, Tyrosine and Branched Chain Amino Acid Pathways

 , , , , and
, , , , and 
Abstract
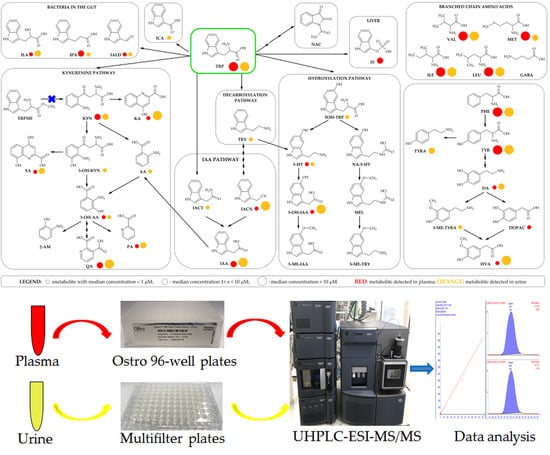
:
1. Introduction
2. Results
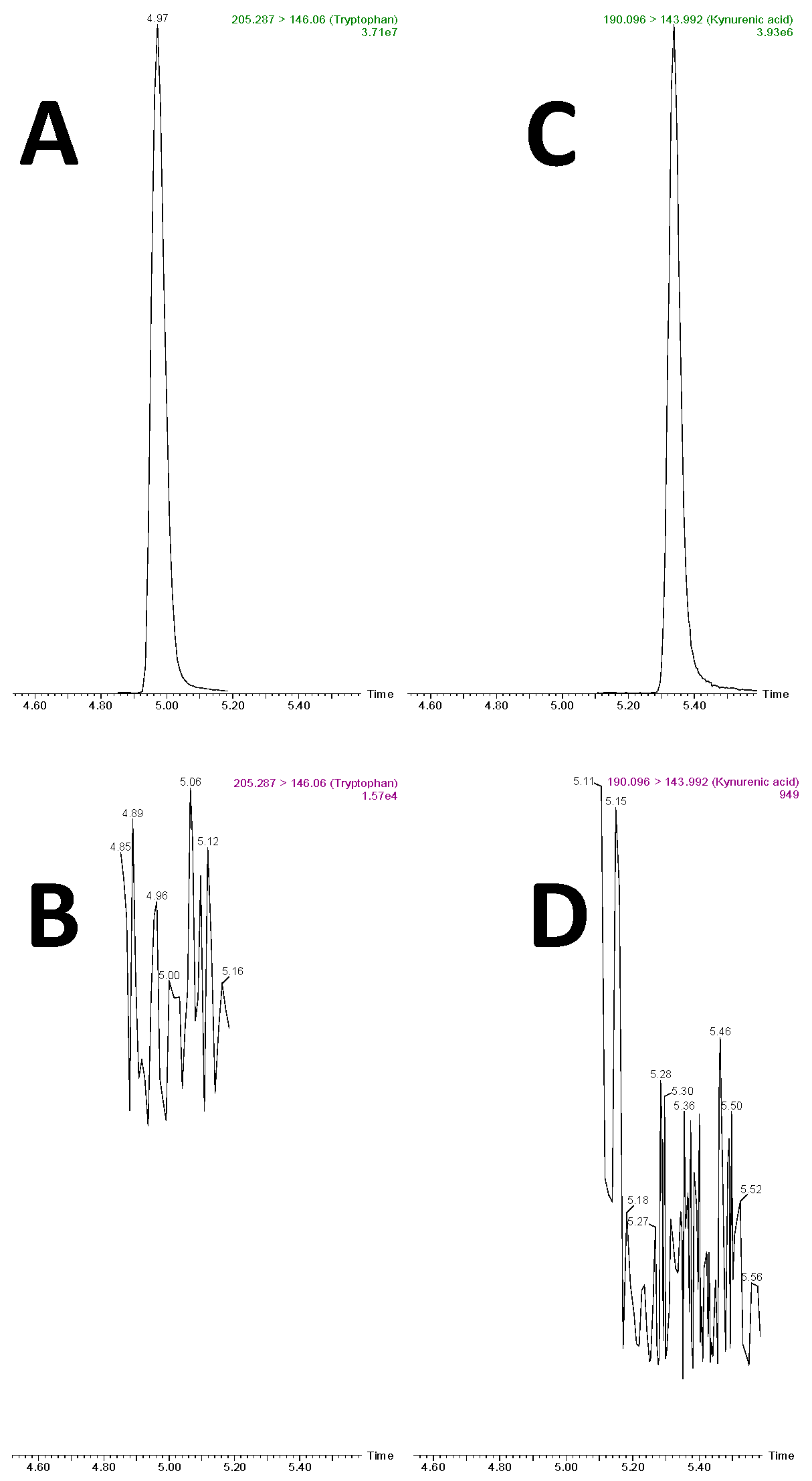
2.1. Liquid Chromatography and Mass Spectrometry
2.2. Linearity and Limit of Quantification (LOQ)
2.3. Retention Time Stability
2.4. Matrix Effects
2.5. Recovery, Intra- and Inter-Day Accuracy and Precision
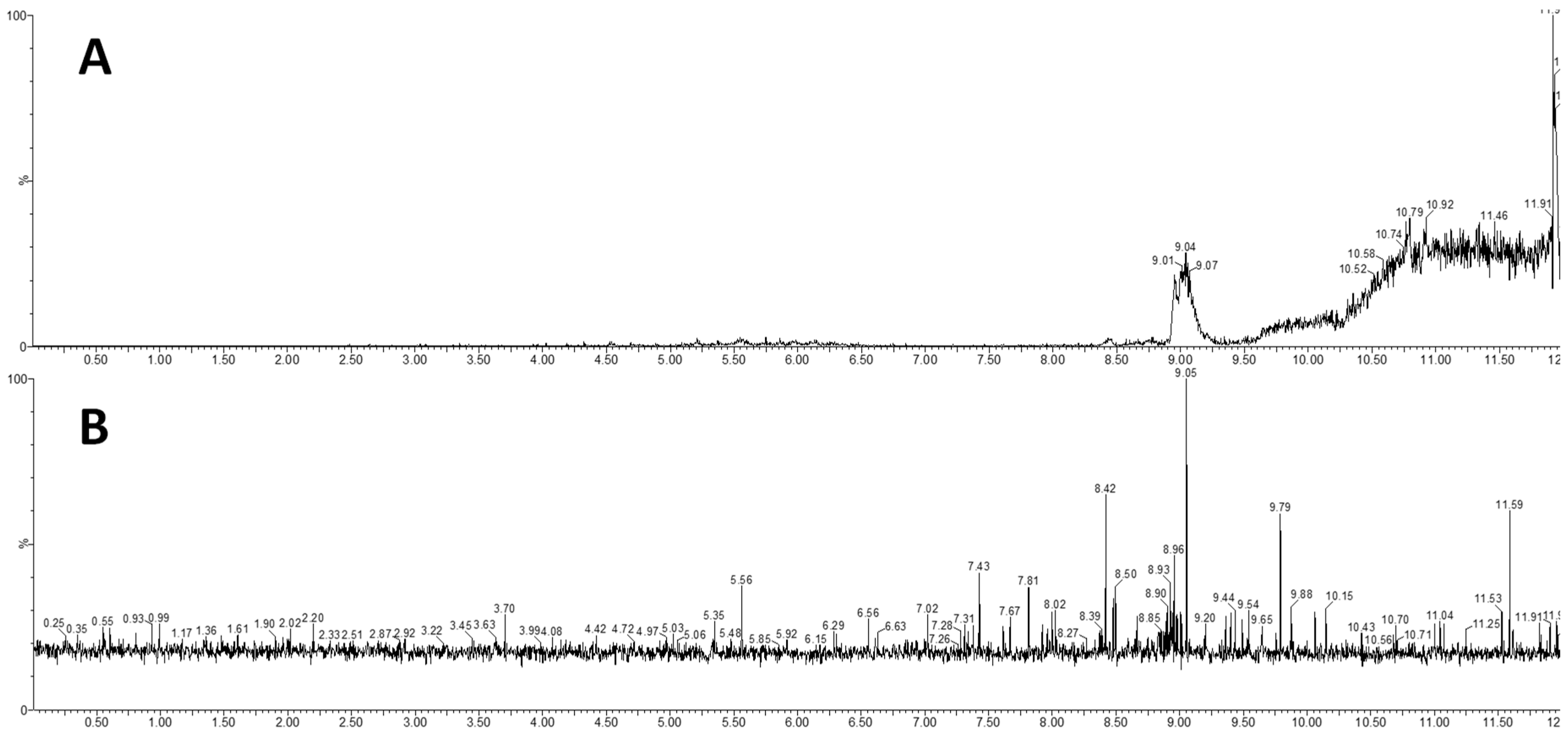
2.6. Carryover Effect and Phospholipid Removal
2.7. Method Application to Biological Samples
3. Discussion
3.1. Optimisation of MS Parameters and Analytical Specificity
3.2. Choice of Chromatographic Technique
3.3. Optimisation of Chromatography on C18 Stationary Phase
3.4. Comparison of Proposed Extraction Procedures and Analytical Performance: Efficiency and Efficacy
3.5. Method Validation
3.6. Method Application
3.7. Study Strengths and Limitations
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Preparation of Stock Solution and Calibration Curves
4.3. Method Validation
4.3.1. Linearity and LOQs
4.3.2. Recovery, Intra- and Inter-Day Accuracy and Precision, and RT Stability
4.3.3. RT Stability
4.3.4. Analysis of Blank Samples and ME
4.3.5. Carryover Effect and Phospholipid Removal
4.4. Extraction Procedures
4.4.1. Urine
Plasma
LLE of Urine and Plasma
4.5. Ultra High Performance Liquid Chromatography-Electrospray Ionization-Triple Quadrupole-Mass Spectrometry (UHPLC-ESI-QqQ-MS)
4.5.1. RP C18 Chromatography
4.5.2. HILIC Chromatography
4.6. Method Application to Biological Samples
4.6.1. The DONALD Study
4.6.2. The Rhineland Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carey, M.; Small, H.; Yoong, S.L.; Boyes, A.; Bisquera, A.; Sanson-Fisher, R. Prevalence of comorbid depression and obesity in general practice: A cross-sectional survey. Br. J. Gen. Pract. 2014, 64, e122–e127. [Google Scholar] [CrossRef]
- Jantaratnotai, N.; Mosikanon, K.; Lee, Y.; McIntyre, R.S. The interface of depression and obesity. Obes. Res. Clin. Pract. 2017, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, S.M.; Marchesi, J.R.; Scully, P.; Codling, C.; Ceolho, A.-M.; Quigley, E.M.M.; Cryan, J.F.; Dinan, T.G. Early Life Stress Alters Behavior, Immunity, and Microbiota in Rats: Implications for Irritable Bowel Syndrome and Psychiatric Illnesses. Biol. Psychiatry 2009, 65, 263–267. [Google Scholar] [CrossRef]
- Lee, S.P.; Sung, I.-K.; Kim, J.H.; Lee, S.-Y.; Park, H.S.; Shim, C.S. The effect of emotional stress and depression on the prevalence of digestive diseases. J. Neurogastroenterol. Motil. 2015, 21, 273–282. [Google Scholar] [CrossRef]
- Walter, F.M.; Emery, J.D.; Mendonca, S.; Hall, N.; Morris, H.C.; Mills, K.; Dobson, C.; Bankhead, C.; Johnson, M.; Abel, G.A.; et al. Symptoms and patient factors associated with longer time to diagnosis for colorectal cancer: Results from a prospective cohort study. Br. J. Cancer 2016, 115, 533–541. [Google Scholar] [CrossRef]
- Mosher, C.E.; Winger, J.G.; Given, B.A.; Helft, P.R.; O’Neil, B.H. Mental health outcomes during colorectal cancer survivorship: A review of the literature. Psychooncology 2016, 25, 1261–1270. [Google Scholar] [CrossRef]
- Walker, J.; Hansen, C.H.; Martin, P.; Symeonides, S.; Ramessur, R.; Murray, G.; Sharpe, M. Prevalence, associations, and adequacy of treatment of major depression in patients with cancer: A cross-sectional analysis of routinely collected clinical data. Lancet Psychiatry 2014, 1, 343–350. [Google Scholar] [CrossRef]
- Gigic, B.; Boeing, H.; Toth, R.; Böhm, J.; Habermann, N.; Scherer, D.; Schrotz-King, P.; Abbenhardt-Martin, C.; Skender, S.; Brenner, H.; et al. Associations between dietary patterns and longitudinal quality of life changes in colorectal cancer patients: The colocare study. Nutr. Cancer 2018, 70, 51–60. [Google Scholar] [CrossRef]
- Song, M.; Garrett, W.S.; Chan, A.T. Nutrients, Foods, and Colorectal Cancer Prevention. Gastroenterology 2015, 148, 1244–1260.e16. [Google Scholar] [CrossRef] [Green Version]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef]
- Bellono, N.W.; Bayrer, J.R.; Leitch, D.B.; Castro, J.; Zhang, C.; O’Donnell, T.A.; Brierley, S.M.; Ingraham, H.A.; Julius, D. Enterochromaffin Cells Are Gut Chemosensors that Couple to Sensory Neural Pathways. Cell 2017, 170, 185–198.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.A.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.; Bohórquez, D.V. A gut-brain neural circuit for nutrient sensory transduction. Science 2018, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golubeva, A.V.; Joyce, S.A.; Moloney, G.; Burokas, A.; Sherwin, E.; Arboleya, S.; Flynn, I.; Khochanskiy, D.; Moya-Pérez, A.; Peterson, V.; et al. Microbiota-related Changes in Bile Acid & Tryptophan Metabolism are Associated with Gastrointestinal Dysfunction in a Mouse Model of Autism. EBioMedicine 2017, 24, 166–178. [Google Scholar] [CrossRef] [Green Version]
- Dinan, T.G.; Cryan, J.F. Gut-brain axis in 2016: Brain-gut-microbiota axis-mood, metabolism and behaviour. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 69–70. [Google Scholar] [CrossRef]
- Dinan, T.G.; Cryan, J.F. The Microbiome-Gut-Brain Axis in Health and Disease. Gastroenterol. Clin. N. Am. 2017, 46, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.R.; Borre, Y.; O’ Brien, C.; Patterson, E.; El Aidy, S.; Deane, J.; Kennedy, P.J.; Beers, S.; Scott, K.; Moloney, G.; et al. Transferring the blues: Depression-associated gut microbiota induces neurobehavioural changes in the rat. J. Psychiatr. Res. 2016, 82, 109–118. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Review Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Valles-Colomer, M.; Falony, G.; Darzi, Y.; Tigchelaar, E.F.; Wang, J.; Tito, R.Y.; Schiweck, C.; Kurilshikov, A.; Joossens, M.; Wijmenga, C.; et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol. 2019, 4, 623–632. [Google Scholar] [CrossRef]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357. [Google Scholar] [CrossRef]
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; Rehman, A.; Tran, F.; Aden, K.; Häsler, R.; et al. Increased Tryptophan Metabolism Is Associated with Activity of Inflammatory Bowel Diseases. Gastroenterology 2017, 153, 1504–1516.e2. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology 2017, 112, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.B. Kynurenine pathway of tryptophan metabolism: Regulatory and functional aspects. Int. J. Tryptophan Res. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Crotti, S.; D’Angelo, E.; Bedin, C.; Fassan, M.; Pucciarelli, S.; Nitti, D.; Bertazzo, A.; Agostini, M. Tryptophan metabolism along the kynurenine and serotonin pathways reveals substantial differences in colon and rectal cancer. Metabolomics 2017, 13. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef]
- Zhang, L.S.; Davies, S.S. Microbial metabolism of dietary components to bioactive metabolites: Opportunities for new therapeutic interventions. Genome Med. 2016, 8, 1–18. [Google Scholar] [CrossRef]
- Bansal, T.; Alaniz, R.C.; Wood, T.K.; Jayaraman, A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 228–233. [Google Scholar] [CrossRef]
- Shimada, Y.; Kinoshita, M.; Harada, K.; Mizutani, M.; Masahata, K.; Kayama, H.; Takeda, K. Commensal bacteria-dependent indole production enhances epithelial barrier function in the colon. PLoS ONE 2013, 8, e80604. [Google Scholar] [CrossRef]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Lin, C.J.; Chen, H.H.; Pan, C.F.; Chuang, C.K.; Wang, T.J.; Sun, F.J.; Wu, C.J. P-Cresylsulfate and Indoxyl Sulfate Level At Different Stages of Chronic Kidney Disease. J. Clin. Lab. Anal. 2011, 25, 191–197. [Google Scholar] [CrossRef]
- Chyan, Y.-J.; Poeggeler, B.; Omar, R.A.; Chain, D.G.; Frangione, B.; Ghiso, J.; Pappolla, M.A. Potent Neuroprotective Properties against the Alzheimer β-Amyloid by an Endogenous Melatonin-related Indole Structure, Indole-3-propionic Acid. J. Biol. Chem. 1999, 274, 21937–21942. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic Bacterial Metabolites Regulate Gastrointestinal Barrier Function via the Xenobiotic Sensor PXR and Toll-like Receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Aragozzini, F.; Ferrari, A.; Pacini, N.; Gualandris, R. Indole-3-lactic acid as a tryptophan metabolite produced by Bifidobacterium spp. Appl. Environ. Microbiol. 1979, 38, 544–546. [Google Scholar]
- Manna, S.K.; Patterson, A.D.; Yang, Q.; Krausz, K.W.; Idle, J.R.; Fornace, A.J.; Gonzalez, F.J. UPLC-MS-based urine metabolomics reveals indole-3-lactic acid and phenyllactic acid as conserved biomarkers for alcohol-induced liver disease in the Ppara-null mouse model. J. Proteome Res. 2011, 10, 4120–4133. [Google Scholar] [CrossRef]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; DeLuca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef]
- Nie, C.; He, T.; Zhang, W.; Zhang, G.; Ma, X. Branched chain amino acids: Beyond nutrition metabolism. Int. J. Mol. Sci. 2018, 19, 954. [Google Scholar] [CrossRef]
- Shoaie, S.; Ghaffari, P.; Kovatcheva-Datchary, P.; Mardinoglu, A.; Sen, P.; Pujos-Guillot, E.; De Wouters, T.; Juste, C.; Rizkalla, S.; Chilloux, J.; et al. Quantifying Diet-Induced Metabolic Changes of the Human Gut Microbiome. Cell Metab. 2015, 22, 320–331. [Google Scholar] [CrossRef] [Green Version]
- Baranyi, A.; Amouzadeh-Ghadikolai, O.; von Lewinski, D.; Rothenhäusler, H.-B.; Theokas, S.; Robier, C.; Mangge, H.; Reicht, G.; Hlade, P.; Meinitzer, A. Branched-Chain Amino Acids as New Biomarkers of Major Depression—A Novel Neurobiology of Mood Disorder. PLoS ONE 2016, 11, e0160542. [Google Scholar] [CrossRef]
- Delphan, M.; Lin, T.; Liesenfeld, D.B.; Nattenmüller, J.; Böhm, J.T.; Gigic, B.; Habermann, N.; Zielske, L.; Schrotz-King, P.; Schneider, M.; et al. Associations of branched-chain amino acids with parameters of energy balance and survival in colorectal cancer patients: results from the ColoCare study. Metabolomics 2018, 14. [Google Scholar] [CrossRef]
- Morita, I.; Kawamoto, M.; Hattori, M.; Eguchi, K.; Sekiba, K.; Yoshida, H. Determination of tryptophan and its metabolites in human plasma and serum by high-performance liquid chromatography with automated sample clean-up system. J. Chromatogr. B Biomed. Sci. Appl. 1990, 526, 367–374. [Google Scholar] [CrossRef]
- Huang, Y.; Louie, A.; Yang, Q.; Massenkoff, N.; Xu, C.; Hunt, P.W.; Gee, W. A simple LC-MS/MS method for determination of kynurenine and tryptophan concentrations in human plasma from HIV-infected patients. Bioanalysis 2013, 5, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hou, W.; Han, B.; Liu, G.; Gong, J.; Li, Y.; Zhong, D.; Liao, Q.; Xie, Z. Target-based metabolomics for the quantitative measurement of 37 pathway metabolites in rat brain and serum using hydrophilic interaction ultra-high-performance liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2016, 408, 2527–2542. [Google Scholar] [CrossRef] [PubMed]
- Fuertig, R.; Ceci, A.; Camus, S.M.; Bezard, E.; Luippold, A.H.; Hengerer, B. LC–MS/MS-based quantification of kynurenine metabolites, tryptophan, monoamines and neopterin in plasma, cerebrospinal fluid and brain. Bioanalysis 2016, 8, 1903–1917. [Google Scholar] [CrossRef]
- Hu, L.J.; Li, X.F.; Hu, J.Q.; Ni, X.J.; Lu, H.Y.; Wang, J.J.; Huang, X.N.; Lin, C.X.; Shang, D.W.; Wen, Y.G. A Simple HPLC-MS/MS method for determination of tryptophan, kynurenine and kynurenic acid in human serum and its potential for monitoring antidepressant therapy. J. Anal. Toxicol. 2017, 41, 37–44. [Google Scholar] [CrossRef]
- Torii, Y.; Kawano, Y.; Sato, H.; Fujimori, T.; Sasaki, K.; Kawada, J.I.; Takikawa, O.; Lim, C.K.; Guillemin, G.J.; Ohashi, Y.; et al. Metabolome analysis reveals the association between the kynurenine pathway and human herpesvirus 6 encephalopathy in immunocompetent children. Metabolomics 2017, 13, 1–10. [Google Scholar] [CrossRef]
- Miller, D.; Tan, L.; Dorshorst, D.; Morrissey, K.; Mahrus, S.; Milanowski, D.; McKnight, J.; Cape, S.; Dean, B.; Liang, X. A validated surrogate analyte LC-MS/MS assay for quantitation of endogenous kynurenine and tryptophan in human plasma. Bioanalysis 2018, 10, 1307–1317. [Google Scholar] [CrossRef]
- Wang, W.; Zhuang, X.; Liu, W.; Dong, L.; Sun, H.; Du, G.; Ye, L. Determination of kynurnine and tryptophan, biomarkers of indoleamine 2,3-dioxygenase by LC-MS/MS in plasma and tumor. Bioanalysis 2018, 10, 1335–1344. [Google Scholar] [CrossRef]
- Zhu, W.; Stevens, A.P.; Dettmer, K.; Gottfried, E.; Hoves, S.; Kreutz, M.; Holler, E.; Canelas, A.B.; Kema, I.; Oefner, P.J. Quantitative profiling of tryptophan metabolites in serum, urine, and cell culture supernatants by liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2011, 401, 3249–3261. [Google Scholar] [CrossRef]
- Marcos, J.; Renau, N.; Valverde, O.; Aznar-Laín, G.; Gracia-Rubio, I.; Gonzalez-Sepulveda, M.; Pérez-Jurado, L.A.; Ventura, R.; Segura, J.; Pozo, O.J. Targeting tryptophan and tyrosine metabolism by liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2016, 1434, 91–101. [Google Scholar] [CrossRef]
- Whiley, L.; Nye, L.C.; Grant, I.; Andreas, N.; Chappell, K.E.; Sarafian, M.H.; Misra, R.; Plumb, R.S.; Lewis, M.R.; Nicholson, J.K.; et al. Ultrahigh-Performance Liquid Chromatography Tandem Mass Spectrometry with Electrospray Ionization Quantification of Tryptophan Metabolites and Markers of Gut Health in Serum and Plasma—Application to Clinical and Epidemiology Cohorts. Anal. Chem. 2019, 91, 5207–5216. [Google Scholar] [CrossRef] [PubMed]
- Gika, H.G.; Theodoridis, G.A.; Vrhovsek, U.; Mattivi, F. Quantitative profiling of polar primary metabolites using hydrophilic interaction ultrahigh performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2012, 1259, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Guder, W.G. Preanalytical factors and their influence on analytical quality specifications. Scand. J. Clin. Lab. Investig. 1999, 59, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Sun, H.; Wang, P.; Han, Y.; Wang, X. Recent and potential developments of biofluid analyses in metabolomics. J. Proteomics 2012, 75, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Peter, A.; Franken, H.; Zhao, X.; Neukamm, S.S.; Rosenbaum, L.; Lucio, M.; Zell, A.; Häring, H.U.; Xu, G.; et al. Preanalytical aspects and sample quality assessment in metabolomics studies of human blood. Clin. Chem. 2013, 59, 833–845. [Google Scholar] [CrossRef]
- Lima-Oliveira, G.; Volanski, W.; Lippi, G.; Picheth, G.; Guidi, G.C. Pre-analytical phase management: A review of the procedures from patient preparation to laboratory analysis. Scand. J. Clin. Lab. Investig. 2017, 77, 153–163. [Google Scholar] [CrossRef]
- Ulaszewska, M.M.; Weinert, C.H.; Trimigno, A.; Portmann, R.; Andres Lacueva, C.; Badertscher, R.; Brennan, L.; Brunius, C.; Bub, A.; Capozzi, F.; et al. Nutrimetabolomics: An Integrative Action for Metabolomic Analyses in Human Nutritional Studies. Mol. Nutr. Food Res. 2019, 63, 1–38. [Google Scholar] [CrossRef]
- Kema, I.P.; De Vries, E.G.E.; Schellings, A.M.J.; Postmus, P.E.; Muskiet, F.A.J. Improved diagnosis of carcinoid tumors by measurement of platelet serotonin. Clin. Chem. 1992, 38, 534–540. [Google Scholar]
- Xiao, R.; Beck, O.; Hjemdahl, P. On the accurate measurement of serotonin in whole blood. Scand. J. Clin. Lab. Investig. 1998, 58, 505–510. [Google Scholar] [CrossRef]
- Carling, R.S.; Degg, T.J.; Allen, K.R.; Bax, N.D.S.; Barth, J.H. Evaluation of whole blood serotonin and plasma and urine 5-hydroxyindole acetic acid in diagnosis of carcinoid disease. Ann. Clin. Biochem. 2002, 39, 577–582. [Google Scholar] [CrossRef]
- Bowen, R.A.R.; Remaley, A.T. Interferences from blood collection tube components on clinical chemistry assays. Biochem. Med. 2014, 24, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Cook, J.D.; Strauss, K.A.; Caplan, Y.H.; LoDico, C.P.; Bush, D.M. Urine pH: The effects of time and temperature after collection. J. Anal. Toxicol. 2007, 31, 486–496. [Google Scholar] [CrossRef]
- Dinan, T.G.; Stanton, C.; Cryan, J.F. Psychobiotics: A novel class of psychotropic. Biol. Psychiatry 2013, 74, 720–726. [Google Scholar] [CrossRef]
- Sarkar, A.; Lehto, S.M.; Harty, S.; Dinan, T.G.; Cryan, J.F.; Burnet, P.W.J. Psychobiotics and the Manipulation of Bacteria–Gut–Brain Signals. Trends Neurosci. 2016, 39, 763–781. [Google Scholar] [CrossRef]
- Cheng, L.H.; Liu, Y.W.; Wu, C.C.; Wang, S.; Tsai, Y.C. Psychobiotics in mental health, neurodegenerative and neurodevelopmental disorders. J. Food Drug Anal. 2019, 27, 632–648. [Google Scholar] [CrossRef] [Green Version]
- Buyken, A.E.; Alexy, U.; Kersting, M.; Remer, T. Die DONALD Kohorte: Ein aktueller Überblick zu 25 Jahren Forschung im Rahmen der Dortmund Nutritional and Anthropometric Longitudinally Designed Study. Bundesgesundheitsblatt Gesundheitsforsch. Gesundheitsschutz 2012, 55, 875–884. [Google Scholar] [CrossRef]
- Remer, T.; Neubert, A.; Maser-Gluth, C. Anthropometry-based reference values for 24-h urinary creatinine excretion during growth and their use in endocrine and nutritional research. Am. J. Clin. Nutr. 2002, 75, 561–569. [Google Scholar] [CrossRef] [Green Version]
 : metabolite with median concentration of < 1 μM;
: metabolite with median concentration of < 1 μM;  : metabolite with median concentration of 1 < x < 10 μM;
: metabolite with median concentration of 1 < x < 10 μM;  : metabolite with median concentration of > 10 μM.
: metabolite with median concentration of < 1 μM; : metabolite with median concentration of 1 < x < 10 μM; : metabolite with median concentration of > 10 μM.
: metabolite with median concentration of > 10 μM.
: metabolite with median concentration of < 1 μM; : metabolite with median concentration of 1 < x < 10 μM; : metabolite with median concentration of > 10 μM.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Internal Standard | RT (min) | Parent m/z | ESI | Q m/z | q m/z | CV (V) | CE (eV) |
|---|---|---|---|---|---|---|---|---|
| γ-aminobutyric acid | MET-d4 | 1.16 | 104.03 | + | 68.95 | 86.14 | 12 | 14 |
| l-valine | MET-d4 | 1.47 | 118.03 | + | 55.01 | 72.02 | 12 | 18 |
| picolinic acid | MET-d4 | 1.53 | 124.00 | + | 77.96 | 105.87 | 26 | 10 |
| dopamine-d4 | 1.66 | 158.16 | + | 94.85 | 122.4 | 12 | 22 | |
| dopamine | DA-d4 | 1.67 | 154.22 | + | 91.02 | 119.01 | 12 | 20 |
| methionine-d4 | 1.68 | 154.09 | + | 59.17 | 62.95 | 12 | 16 | |
| methionine | MET-d4 | 1.68 | 150.22 | + | 104.02 | 56.04 | 12 | 10 |
| 2-aminophenol | TRP-d5 | 1.70 | 110.16 | + | 92.00 | 65.01 | 20 | 14 |
| quinolinic acid | MET-d4 | 1.80 | 168.22 | + | 77.98 | 106.03 | 14 | 16 |
| 3-hydroxykynurenine | TRP-d5 | 2.01 | 225.176 | + | 110.02 | 162.01 | 14 | 18 |
| tyrosine-d4 | 2.04 | 186.16 | + | 140.11 | 93.95 | 12 | 14 | |
| tyrosine | TYR-d4 | 2.07 | 182.17 | + | 136.07 | 90.96 | 18 | 16 |
| l-isoleucine | MET-d4 | 2.25 | 132.09 | + | 86.00 | 69.00 | 10 | 12 |
| tyramine | TYR-d4 | 2.25 | 138.12 | + | 76.68 | 103.97 | 10 | 24 |
| l-leucine | MET-d4 | 2.38 | 132.09 | + | 86.00 | 43.00 | 10 | 12 |
| serotonin-d4 | 2.93 | 181.16 | + | 118.14 | 146.05 | 12 | 26 | |
| serotonin | 5-HT-d4 | 3.02 | 177.22 | + | 115.09 | 132.18 | 10 | 26 |
| 5-hydroxy-tryptophan | TRP-d5 | 3.00 | 221.29 | + | 162.01 | 134.02 | 12 | 18 |
| 3-methoxy-p-tyramine | TYR-d4 | 3.02 | 168.22 | + | 91.00 | 119.05 | 8 | 20 |
| kynurenine | TRP-d5 | 3.53 | 209.12 | + | 94.01 | 146.08 | 14 | 16 |
| dl-phenylalanine | TYR-d4 | 3.61 | 166.22 | + | 120.10 | 103.01 | 14 | 20 |
| 3-hydroxyanthranilic acid | TRP-d5 | 4.75 | 154.22 | + | 80.01 | 108.01 | 10 | 22 |
| tryptophan-d5 | 4.90 | 210.16 | + | 150.09 | 122.11 | 12 | 18 | |
| tryptophan | TRP-d5 | 4.94 | 205.29 | + | 146.06 | 118.01 | 12 | 16 |
| 1-acetylisatin | TRP-d5 | 4.94 | 190.01 | + | 148.01 | 162.01 | 18 | 10 |
| DOPAC-d5 | 4.99 | 172.11 | - | 128.04 | 99.99 | 14 | 8 | |
| 3,4-dihydroxyphenyl acetic acid | DOPAC-d5 | 5.04 | 167.07 | - | 123.05 | 94.99 | 14 | 8 |
| xanthurenic acid | TRP-d5 | 5.03 | 206.09 | + | 160.00 | 132.02 | 20 | 18 |
| kynurenic acid-d5 | 5.41 | 195.09 | + | 149.06 | 121.08 | 24 | 18 | |
| kynurenic acid | KA-d5 | 5.44 | 190.09 | + | 143.99 | 116.00 | 20 | 20 |
| tryptamine | TRP-d5 | 5.45 | 161.13 | + | 127.20 | 117.40 | 12 | 24 |
| 5-methoxytryptamine | TRP-d5 | 5.60 | 191.20 | + | 159.09 | 143.08 | 12 | 22 |
| 5-hydroxyindole acetic acid-d5 | 5.71 | 197.16 | + | 150.16 | 122.17 | 16 | 14 | |
| 5-hydroxyindole acetic acid | 5-OH-IAA- d5 | 5.74 | 192.23 | + | 146.27 | 91.00 | 18 | 14 |
| N-acetyl-5-hydroxytryptamine | TRP-d5 | 5.86 | 219.20 | + | 160.07 | 115.09 | 16 | 16 |
| tryptophan methyl ester | TRP-d5 | 6.07 | 219.14 | + | 160.00 | 132.02 | 12 | 18 |
| homovanillic acid | DOPAC-d5 | 6.20 | 181.09 | - | 137.08 | 121.99 | 8 | 10 |
| indoxyl sulfate | TRP-d5 | 6.24 | 212.04 | - | 80.08 | 132.02 | 24 | 20 |
| indole-3-acetamide | TRP-d5 | 6.53 | 175.05 | + | 102.99 | 76.95 | 14 | 30 |
| anthranilic acid | TRP-d5 | 6.78 | 138.22 | + | 91.99 | 65.04 | 10 | 22 |
| indole-3-lactic acid | TRP-d5 | 6.96 | 206.11 | + | 160.09 | 130.02 | 18 | 10 |
| indole-3-carboxylic acid | TRP-d5 | 7.15 | 162.08 | + | 116.03 | 88.95 | 16 | 20 |
| melatonin | TRP-d5 | 7.31 | 233.22 | + | 174.08 | 159.05 | 16 | 14 |
| 5-methoxyindole acetic acid | TRP-d5 | 7.35 | 206.17 | + | 160.17 | 145.05 | 16 | 16 |
| indole-3-carboxaldehyde | TRP-d5 | 7.36 | 146.09 | + | 118.05 | 90.97 | 22 | 24 |
| indole-3-acetonitrile | TRP-d5 | 7.52 | 130.22 | + | 76.95 | 102.99 | 30 | 22 |
| indole-3-acetic acid | TRP-d5 | 7.53 | 176.09 | + | 130.00 | 102.99 | 18 | 12 |
| indole-3-propionic acid | TRP-d5 | 8.06 | 190.11 | + | 130.02 | 54.96 | 12 | 16 |
| Plasma (μM) | Urine (μM) | |||||
|---|---|---|---|---|---|---|
| Metabolite | Min | Median | Max | Min | Median | Max |
| l-valine | 12.09 | 62.12 | 130 | 1.678 | 29.15 | 94.71 |
| picolinic acid | 0.00179 | 0.0198 | 0.057 | 0.649 | 1.402 | 2.488 |
| dopamine | 0 | 0.0128 | 0.0718 | 0.29 | 2.089 | 8.029 |
| methionine | 3.09 | 11.45 | 25.10 | 0 | 2.158 | 36.23 |
| 2-aminophenol | n.d. | n.d. | ||||
| quinolinic acid | 0.414 | 1.404 | 9.694 | 10.58 | 40.16 | 146 |
| 3-hydroxykynurenine | n.d. | 0 | 0.357 | 3.870 | ||
| tyrosine | 6.721 | 27.86 | 71.30 | 6.013 | 136 | 849 |
| l-isoleucine | 4.924 | 26.46 | 77.95 | 0.106 | 12.69 | 55.82 |
| tyramine | n.d. | 0.197 | 4.518 | 139 | ||
| l-leucine | 10.71 | 57.07 | 120.0 | 1.763 | 33.05 | 158 |
| serotonin | 0 | 0.167 | 1.047 | 0.04 | 0.492 | 1.905 |
| 5-hydroxy-tryptophan | n.d. | 0.0394 | 0.151 | 0.723 | ||
| 3-methoxy-p-tyramine | n.d. | 0.0846 | 0.346 | 1.722 | ||
| kynurenine | 0.450 | 1.270 | 3.479 | 0.215 | 3.703 | 43.88 |
| dl-phenylalanine | 8.096 | 27.86 | 71.30 | 2.342 | 18.08 | 107 |
| 3-hydroxyanthranilic acid | 0.177 | 0.203 | 0.322 | 0.0808 | 0.412 | 4.494 |
| tryptophan | 8.499 | 29.82 | 81.49 | 7.051 | 64.02 | 366 |
| 1-acetylisatin | n.d. | n.d. | ||||
| 3,4-dihydroxyphenyl acetic acid | 0 | 0.0477 | 73.7 | n.d. | ||
| xanthurenic acid | 0.02 | 0.0661 | 0.183 | 0.561 | 5.424 | 36.10 |
| kynurenic acid | 0.00553 | 0.0185 | 0.167 | 3.334 | 20.38 | 91.75 |
| tryptamine | n.d. | 0.0272 | 0.449 | 2.467 | ||
| 5-methoxytryptamine | n.d. | n.d. | ||||
| 5-hydroxyindole acetic acid | 0.0164 | 0.0447 | 0.456 | 0.0268 | 19.68 | 89.54 |
| N-acetyl-5-hydroxytryptamine | n.d. | n.d. | ||||
| tryptophan methyl ester | n.d. | n.d. | ||||
| homovanillic acid | 0.0118 | 0.0782 | 1.0 | 9.313 | 35.58 | 136 |
| indoxyl sulfate | 0.0491 | 2.744 | 12.99 | n.a. | ||
| indole-3-acetamide | n.d. | 0.0170 | 0.272 | 10.07 | ||
| anthranilic acid | n.d. | 0.0950 | 0.401 | 2.058 | ||
| indole-3-lactic acid | 0.0759 | 0.697 | 4.009 | 0.198 | 1.165 | 18.90 |
| indole-3-carboxylic acid | n.d. | 0.0305 | 0.0994 | 7.279 | ||
| melatonin | n.d. | n.d. | ||||
| 5-methoxyindole acetic acid | n.d. | n.d. | ||||
| indole-3-carboxaldehyde | 0.0103 | 0.0494 | 0.186 | 0.00245 | 0.123 | 3.992 |
| indole-3-acetonitrile | 0.326 | 2.003 | 31.72 | 3.116 | 15.60 | 96.82 |
| indole-3-acetic acid | 0.292 | 1.51 | 23.01 | 6.114 | 30.09 | 205 |
| indole-3-propionic acid | 0 | 1.156 | 12.75 | 0.0187 | 0.0557 | 2.197 |
| Recovery (Average = 5) | Extraction Methods | |
|---|---|---|
| 21 pre-selected metabolites | LLE | Ostro 96-Well plate |
| <50 | 1 | 1 |
| 50–60 | 1 | 1 |
| 60–70 | 5 | 0 |
| 70–80 | 3 | 1 |
| 80–90 | 4 | 1 |
| 90–100 | 3 | 15 |
| >100 | 4 | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anesi, A.; Rubert, J.; Oluwagbemigun, K.; Orozco-Ruiz, X.; Nöthlings, U.; Breteler, M.M.B.; Mattivi, F. Metabolic Profiling of Human Plasma and Urine, Targeting Tryptophan, Tyrosine and Branched Chain Amino Acid Pathways. Metabolites 2019, 9, 261. https://doi.org/10.3390/metabo9110261
Anesi A, Rubert J, Oluwagbemigun K, Orozco-Ruiz X, Nöthlings U, Breteler MMB, Mattivi F. Metabolic Profiling of Human Plasma and Urine, Targeting Tryptophan, Tyrosine and Branched Chain Amino Acid Pathways. Metabolites. 2019; 9(11):261. https://doi.org/10.3390/metabo9110261
Chicago/Turabian StyleAnesi, Andrea, Josep Rubert, Kolade Oluwagbemigun, Ximena Orozco-Ruiz, Ute Nöthlings, Monique M.B. Breteler, and Fulvio Mattivi. 2019. "Metabolic Profiling of Human Plasma and Urine, Targeting Tryptophan, Tyrosine and Branched Chain Amino Acid Pathways" Metabolites 9, no. 11: 261. https://doi.org/10.3390/metabo9110261
APA StyleAnesi, A., Rubert, J., Oluwagbemigun, K., Orozco-Ruiz, X., Nöthlings, U., Breteler, M. M. B., & Mattivi, F. (2019). Metabolic Profiling of Human Plasma and Urine, Targeting Tryptophan, Tyrosine and Branched Chain Amino Acid Pathways. Metabolites, 9(11), 261. https://doi.org/10.3390/metabo9110261