Metabolic Modeling of Human Gut Microbiota on a Genome Scale: An Overview



Abstract
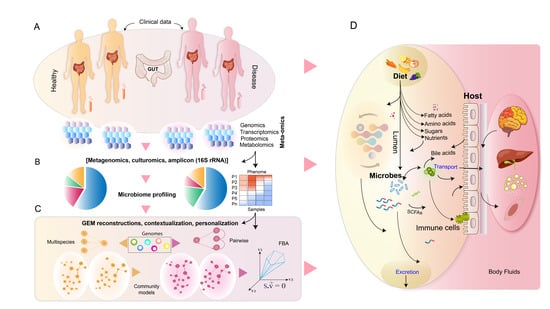
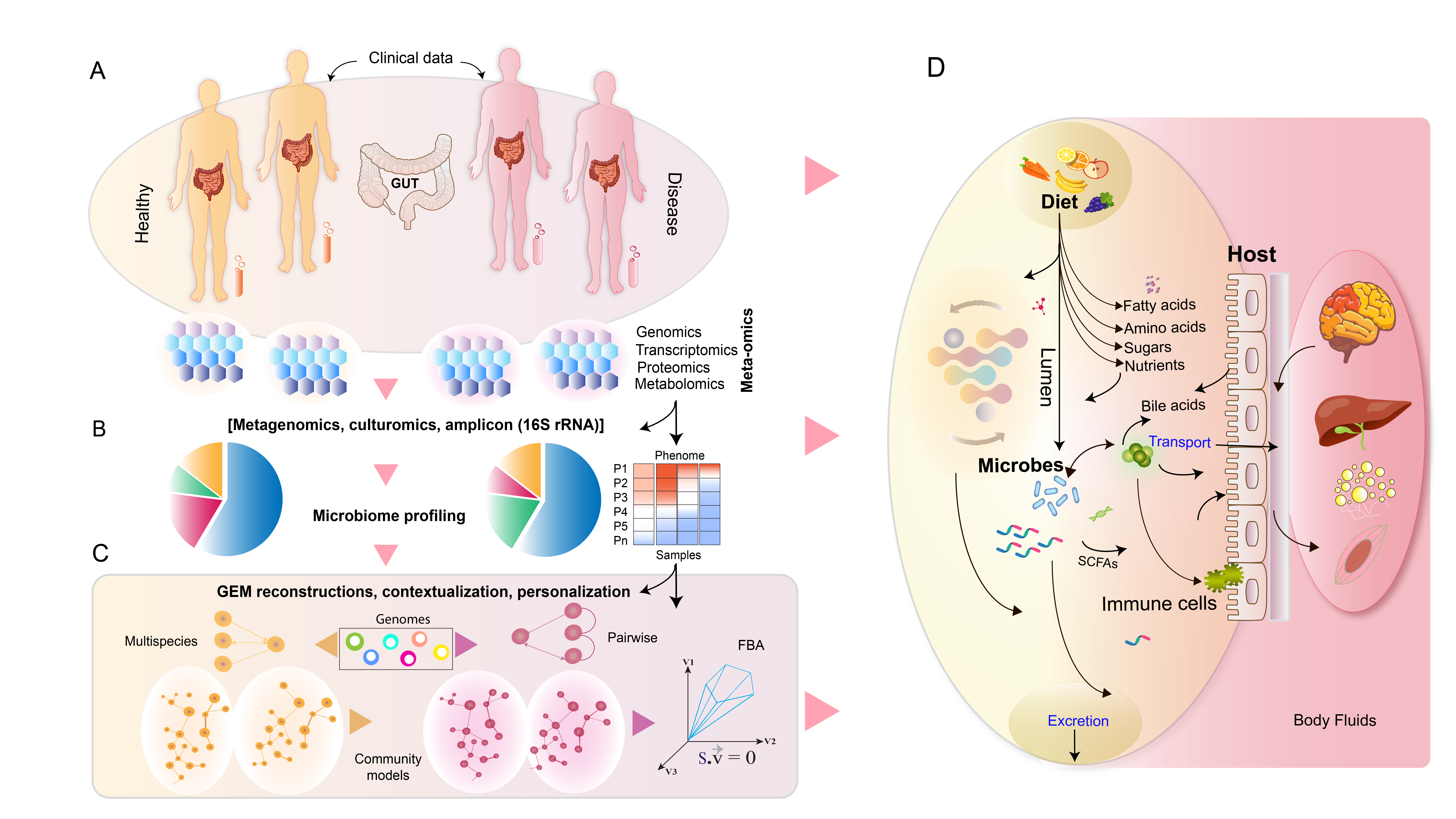
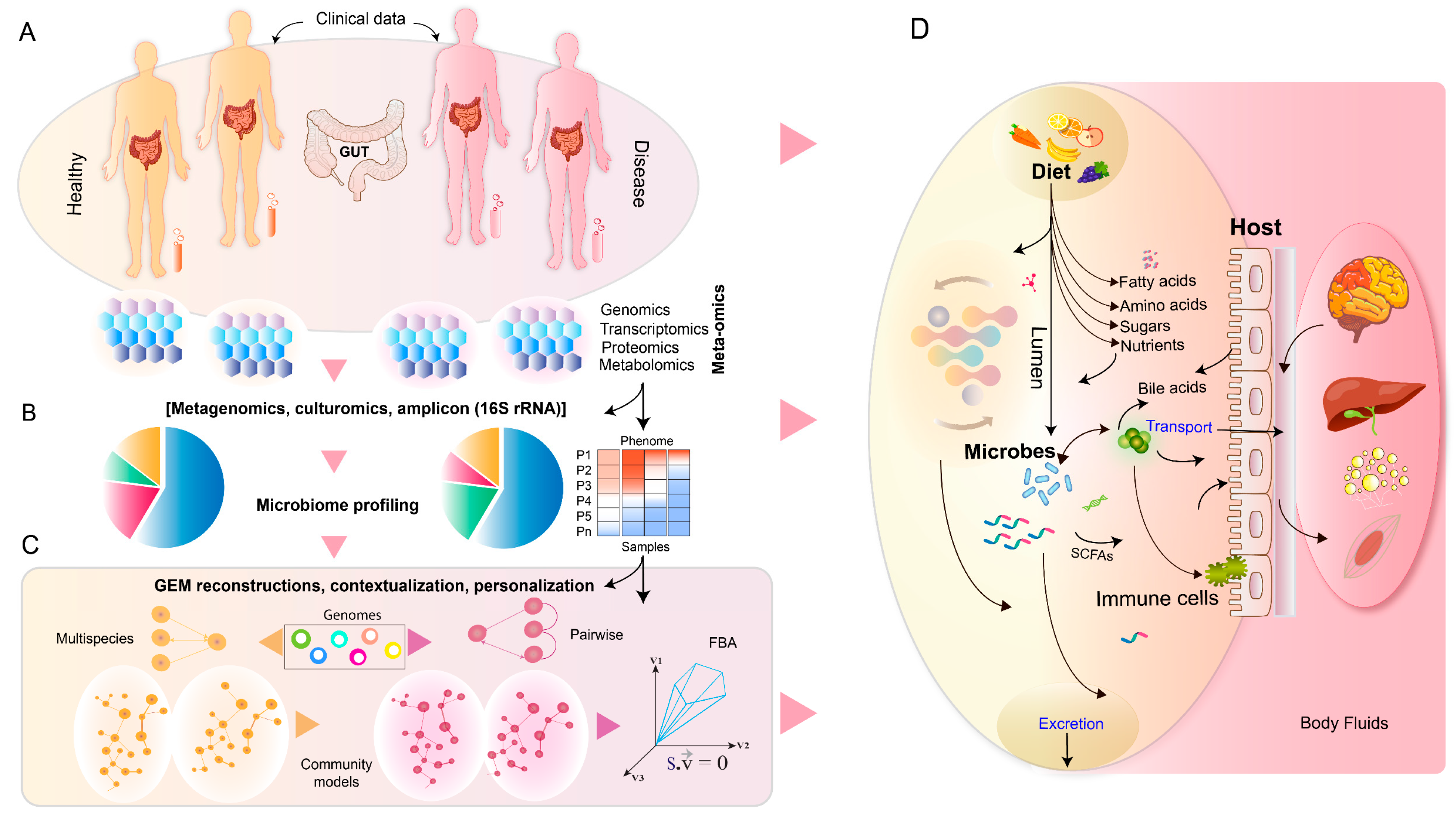
:
1. Introduction
2. Colonization and Shaping of the Gut Ecosystem
3. Gut Microbiome Profiling and Functional Annotation
4. A Constraint-Based Strategy and Tools for Genome-Scale Metabolic Modeling of Gut Microbiota
5. Reconstruction of Condition-Specific Personalized Gut Microbiota Models
6. Modeling the Effect of Diet on Gut Microbiome
7. Multispecies Modeling and Interactions in the Gut Community
8. Metabolic Modeling of Host–Microbiome Interactions
9. Model Predictions and Experimental Validation
10. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NGS | Next-Generation Sequencing |
| GSMM | Genome-Scale Metabolic Modeling |
| HMP | Human Microbiome Project |
| SCFAs | Short-Chain Fatty Acids |
| BCFAs | Branched Chain Fatty Acids |
| BCAAs | Branched Chain Amino Acids |
| BAs | Bile Acids |
| TLR | Toll-Like Receptor |
| APCs | Antigen Presenting Cells |
| WGS | Whole Genomes Shotgun metagenomics sequencing |
| GEMs | Genome-Scale Models |
| T1D | Type 1 Diabetes |
| RA | Rheumatoid Arthritis |
| T2D | Type 2 Diabetes |
| NAFLD | Non-Alcoholic Fatty Liver Disease |
| IDB | Bowel Disease |
| CDS | Protein Coding Sequences |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| HUMAnN2 | HMP Unified Metabolic Analysis Network |
| MEGAN | MEtaGenome ANalyzer |
| MG-RAST | Metagenomics Rast |
| RAST | Rapid Annotation using Subsystem Technology |
| IMG/M | Integrated Microbial Genomes and Microbiomes |
| FBA | Flux Balance Analysis |
| CBA | Constraint-Based Approach |
| AGORA | Assembly of Gut Organisms through Reconstruction and Analysis |
| VMH | Virtual Metabolic Human |
| COBRA | COnstraint-Based Reconstruction and Analysis |
| RAVEN | Reconstruction, Analysis, and Visualization of Metabolic Networks |
| FVA | Flux Variability Analysis |
| SP | Shadow Price |
| UDCA | Ursodeoxycholate |
| RUTFs | Ready-to-Use Therapeutic Foods |
| EHMN | Edinburgh Human Metabolic Network |
| HMR | Human Metabolic Reaction |
| sIECs | small Intestinal Epithelial Cells |
| WBM | Whole-Body Metabolism |
| GF | Germ-Free |
| CONV-R | Conventionally Raised |
References
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.S.B.; Raes, J.; Bork, P. The Human Gut Microbiome: From Association to Modulation. Cell 2018, 172, 1198–1215. [Google Scholar] [CrossRef]
- Hugon, P.; Dufour, J.C.; Colson, P.; Fournier, P.E.; Sallah, K.; Raoult, D. A comprehensive repertoire of prokaryotic species identified in human beings. Lancet Infect. Dis. 2015, 15, 1211–1219. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016, 535, 376–381. [Google Scholar] [CrossRef]
- Gevers, D.; Knight, R.; Petrosino, J.F.; Huang, K.; McGuire, A.L.; Birren, B.W.; Nelson, K.E.; White, O.; Methe, B.A.; Huttenhower, C. The Human Microbiome Project: A community resource for the healthy human microbiome. PLoS Biol. 2012, 10, e1001377. [Google Scholar] [CrossRef]
- Integrative, H. The Integrative Human Microbiome Project: Dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe 2014, 16, 276. [Google Scholar]
- McDonald, D.; Hyde, E.; Debelius, J.W.; Morton, J.T.; Gonzalez, A.; Ackermann, G.; Aksenov, A.A.; Behsaz, B.; Brennan, C.; Chen, Y.; et al. American Gut: An Open Platform for Citizen Science Microbiome Research. mSystems 2018, 3. [Google Scholar] [CrossRef]
- Rodriguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C.; et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis. 2015, 26, 26050. [Google Scholar] [CrossRef]
- Faith, J.J.; Guruge, J.L.; Charbonneau, M.; Subramanian, S.; Seedorf, H.; Goodman, A.L.; Clemente, J.C.; Knight, R.; Heath, A.C.; Leibel, R.L.; et al. The long-term stability of the human gut microbiota. Science 2013, 341, 1237439. [Google Scholar] [CrossRef]
- Costea, P.I.; Coelho, L.P.; Sunagawa, S.; Munch, R.; Huerta-Cepas, J.; Forslund, K.; Hildebrand, F.; Kushugulova, A.; Zeller, G.; Bork, P. Subspecies in the global human gut microbiome. Mol. Syst. Biol. 2017, 13, 960. [Google Scholar] [CrossRef]
- Hisada, T.; Endoh, K.; Kuriki, K. Inter- and intra-individual variations in seasonal and daily stabilities of the human gut microbiota in Japanese. Arch. Microbiol. 2015, 197, 919–934. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.; Duffy, A. Factors influencing the gut microbiota, inflammation, and type 2 diabetes. J. Nutr. 2017, 147, 1468S–1475S. [Google Scholar] [CrossRef]
- Ji, B.; Nielsen, J. New insight into the gut microbiome through metagenomics. Adv. Genom. Genet. 2015, 5, 77–91. [Google Scholar] [Green Version]
- Shoaie, S.; Karlsson, F.; Mardinoglu, A.; Nookaew, I.; Bordel, S.; Nielsen, J. Understanding the interactions between bacteria in the human gut through metabolic modeling. Sci. Rep. 2013, 3, 2532. [Google Scholar] [CrossRef]
- Lamichhane, S.; Sen, P.; Dickens, A.M.; Oresic, M.; Bertram, H.C. Gut metabolome meets microbiome: A methodological perspective to understand the relationship between host and microbe. Methods 2018. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Molloy, M.J.; Bouladoux, N.; Belkaid, Y. Intestinal microbiota: Shaping local and systemic immune responses. Semin. Immunol. 2012, 24, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Valentini, M.; Piermattei, A.; Di Sante, G.; Migliara, G.; Delogu, G.; Ria, F. Immunomodulation by gut microbiota: Role of Toll-like receptor expressed by T cells. J. Immunol. Res. 2014, 2014, 586939. [Google Scholar] [CrossRef]
- Hamady, M.; Knight, R. Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome Res. 2009, 19, 1141–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoaie, S.; Nielsen, J. Elucidating the interactions between the human gut microbiota and its host through metabolic modeling. Front. Genet. 2014, 5, 86. [Google Scholar] [CrossRef] [PubMed]
- Bauer, E.; Thiele, I. From Network Analysis to Functional Metabolic Modeling of the Human Gut Microbiota. mSystems 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Ji, B.; Babaei, P.; Das, P.; Lappa, D.; Ramakrishnan, G.; Fox, T.E.; Haque, R.; Petri, W.A., Jr.; Bäckhed, F. Gut microbiota dysbiosis is associated with malnutrition and reduced plasma amino acid levels: Lessons from genome-scale metabolic modeling. Metab. Eng. 2018, 49, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Shoaie, S.; Ghaffari, P.; Kovatcheva-Datchary, P.; Mardinoglu, A.; Sen, P.; Pujos-Guillot, E.; de Wouters, T.; Juste, C.; Rizkalla, S.; Chilloux, J. Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 2015, 22, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Magnúsdóttir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T. A human gut microbial gene catalog established by metagenomic sequencing. Nature 2010, 464, 59. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, N.D.; Reed, J.L.; Palsson, B.Ø. Genome-scale models of microbial cells: Evaluating the consequences of constraints. Nat. Rev. Microbiol. 2004, 2, 886–897. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.J.; Monk, J.M.; Palsson, B.O. Using genome-scale models to predict biological capabilities. Cell 2015, 161, 971–987. [Google Scholar] [CrossRef] [PubMed]
- Bordbar, A.; Monk, J.M.; King, Z.A.; Palsson, B.O. Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 2014, 15, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L.; et al. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol. Mol. Biol. Rev. 2017, 81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Geng, J.; Tang, X.; Fan, H.; Xu, J.; Wen, X.; Ma, Z.S.; Shi, P. Spatial heterogeneity and co-occurrence patterns of human mucosal-associated intestinal microbiota. ISME J. 2014, 8, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Boerner, B.P.; Sarvetnick, N.E. Type 1 diabetes: Role of intestinal microbiome in humans and mice. Ann. N. Y. Acad. Sci. 2011, 1243, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi-Roodsaz, S.; Abramson, S.B.; Scher, J.U. The metabolic role of the gut microbiota in health and rheumatic disease: Mechanisms and interventions. Nat. Rev. Rheumatol. 2016, 12, 446–455. [Google Scholar] [CrossRef]
- Sears, C.L.; Garrett, W.S. Microbes, microbiota, and colon cancer. Cell Host Microbe 2014, 15, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, A.L.; Backhed, F. Role of gut microbiota in atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 79–87. [Google Scholar] [CrossRef]
- Spencer, M.D.; Hamp, T.J.; Reid, R.W.; Fischer, L.M.; Zeisel, S.H.; Fodor, A.A. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 2011, 140, 976–986. [Google Scholar] [CrossRef]
- He, X.; Ji, G.; Jia, W.; Li, H. Gut Microbiota and Nonalcoholic Fatty Liver Disease: Insights on Mechanism and Application of Metabolomics. Int. J. Mol. Sci. 2016, 17, 300. [Google Scholar] [CrossRef]
- Wlodarska, M.; Kostic, A.D.; Xavier, R.J. An integrative view of microbiome-host interactions in inflammatory bowel diseases. Cell Host Microbe 2015, 17, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Daniel, R. Metagenomic analyses: Past and future trends. Appl. Environ. Microbiol. 2011, 77, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Carlos, N.; Tang, Y.W.; Pei, Z. Pearls and pitfalls of genomics-based microbiome analysis. Emerg. Microbes Infect. 2012, 1, e45. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Kumar, N.; Prakash, T.; Taylor, T.D. MetaBioME: A database to explore commercially useful enzymes in metagenomic datasets. Nucleic Acids Res. 2010, 38, D468–D472. [Google Scholar] [CrossRef] [PubMed]
- Kultima, J.R.; Coelho, L.P.; Forslund, K.; Huerta-Cepas, J.; Li, S.S.; Driessen, M.; Voigt, A.Y.; Zeller, G.; Sunagawa, S.; Bork, P. MOCAT2: A metagenomic assembly, annotation and profiling framework. Bioinformatics 2016, 32, 2520–2523. [Google Scholar] [CrossRef] [PubMed]
- Abubucker, S.; Segata, N.; Goll, J.; Schubert, A.M.; Izard, J.; Cantarel, B.L.; Rodriguez-Mueller, B.; Zucker, J.; Thiagarajan, M.; Henrissat, B.; et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol. 2012, 8, e1002358. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2015, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016, 44, D471–D480. [Google Scholar] [CrossRef]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N.; et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Glass, E.M.; Wilkening, J.; Wilke, A.; Antonopoulos, D.; Meyer, F. Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. Cold Spring Harbor Protocols 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.A.; Markowitz, V.M.; Chu, K.; Palaniappan, K.; Szeto, E.; Pillay, M.; Ratner, A.; Huang, J.; Andersen, E.; Huntemann, M.; et al. IMG/M: Integrated genome and metagenome comparative data analysis system. Nucleic Acids Res. 2017, 45, D507–D516. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.; Taylor, T.D. Functional assignment of metagenomic data: Challenges and applications. Brief. Bioinform. 2012, 13, 711–727. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Field, D.; Swift, P.; Thomas, S.; Cummings, D.; Temperton, B.; Weynberg, K.; Huse, S.; Hughes, M.; Joint, I. The taxonomic and functional diversity of microbes at a temperate coastal site: A ‘multi-omic’study of seasonal and diel temporal variation. PLoS ONE 2010, 5, e15545. [Google Scholar] [CrossRef]
- Thiele, I.; Palsson, B.Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protocols 2010, 5, 93. [Google Scholar] [CrossRef]
- Thiele, I.; Swainston, N.; Fleming, R.M.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D.; et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Sen, P.; Kemppainen, E.; Orešič, M. Perspectives on Systems Modeling of Human Peripheral Blood Mononuclear Cells. Front. Mol. Biosci. 2018, 4, 96. [Google Scholar] [CrossRef] [Green Version]
- Sen, P.; Mardinogulu, A.; Nielsen, J. Selection of complementary foods based on optimal nutritional values. Sci. Rep. 2017, 7, 5413. [Google Scholar] [CrossRef]
- Becker, S.A.; Feist, A.M.; Mo, M.L.; Hannum, G.; Palsson, B.O.; Herrgard, M.J. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox. Nat. Protoc. 2007, 2, 727–738. [Google Scholar] [CrossRef]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdottir, H.S.; Keating, S.M.; Vlasov, V.; Wachowiak, J. Creation and analysis of biochemical constraint-based models: The COBRA Toolbox v3.0. arXiv, 2017; arXiv:1710.04038. [Google Scholar]
- Schellenberger, J.; Que, R.; Fleming, R.M.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2.0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef] [PubMed]
- Agren, R.; Liu, L.; Shoaie, S.; Vongsangnak, W.; Nookaew, I.; Nielsen, J. The RAVEN toolbox and its use for generating a genome-scale metabolic model for Penicillium chrysogenum. PLoS Comput. Biol. 2013, 9, e1002980. [Google Scholar] [CrossRef] [PubMed]
- Arkin, A.P.; Stevens, R.L.; Cottingham, R.W.; Maslov, S.; Henry, C.S.; Dehal, P.; Ware, D.; Perez, F.; Harris, N.L.; Canon, S. The DOE Systems Biology Knowledgebase (KBase). bioRxiv 2016. [Google Scholar] [CrossRef]
- Bauer, E.; Zimmermann, J.; Baldini, F.; Thiele, I.; Kaleta, C. BacArena: Individual-based metabolic modeling of heterogeneous microbes in complex communities. PLoS Comput. Biol. 2017, 13, e1005544. [Google Scholar] [CrossRef] [PubMed]
- Harcombe, W.R.; Riehl, W.J.; Dukovski, I.; Granger, B.R.; Betts, A.; Lang, A.H.; Bonilla, G.; Kar, A.; Leiby, N.; Mehta, P.; et al. Metabolic resource allocation in individual microbes determines ecosystem interactions and spatial dynamics. Cell Rep. 2014, 7, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Louca, S.; Doebeli, M. Calibration and analysis of genome-based models for microbial ecology. eLife 2015, 4, e08208. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, K.; Izallalen, M.; Mouser, P.; Richter, H.; Risso, C.; Mahadevan, R.; Lovley, D.R. Genome-scale dynamic modeling of the competition between Rhodoferax and Geobacter in anoxic subsurface environments. ISME J. 2011, 5, 305. [Google Scholar] [CrossRef] [PubMed]
- Zomorrodi, A.R.; Islam, M.M.; Maranas, C.D. d-OptCom: Dynamic multi-level and multi-objective metabolic modeling of microbial communities. ACS Synth. Biol. 2014, 3, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.J.; Simons, M.N.; Maranas, C.D. SteadyCom: Predicting microbial abundances while ensuring community stability. PLoS Comput. Biol. 2017, 13, e1005539. [Google Scholar] [CrossRef] [PubMed]
- Cottret, L.; Wildridge, D.; Vinson, F.; Barrett, M.P.; Charles, H.; Sagot, M.F.; Jourdan, F. MetExplore: A web server to link metabolomic experiments and genome-scale metabolic networks. Nucleic Acids Res. 2010, 38, W132–W137. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Soares, H.; Mundy, M.; Soares, L.M.; Chia, N. MMinte: An application for predicting metabolic interactions among the microbial species in a community. BMC Bioinform. 2016, 17, 343. [Google Scholar] [CrossRef] [PubMed]
- Birkel, G.W.; Ghosh, A.; Kumar, V.S.; Weaver, D.; Ando, D.; Backman, T.W.H.; Arkin, A.P.; Keasling, J.D.; Martin, H.G. The JBEI quantitative metabolic modeling library (jQMM): A python library for modeling microbial metabolism. BMC Bioinform. 2017, 18, 205. [Google Scholar] [CrossRef]
- King, Z.A.; Lu, J.; Dräger, A.; Miller, P.; Federowicz, S.; Lerman, J.A.; Ebrahim, A.; Palsson, B.O.; Lewis, N.E. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2015, 44, D515–D522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noronha, A.; Modamio, J.; Jarosz, Y.; Sompairac, N.; Gonzalez, G.P.; Danielsdottir, A.D.; Krecke, M.; Merten, D.; Haraldsdottir, H.S.; Heinken, A. The Virtual Metabolic Human database: Integrating human and gut microbiome metabolism with nutrition and disease. bioRxiv 2018. [Google Scholar] [CrossRef]
- Henry, C.S.; DeJongh, M.; Best, A.A.; Frybarger, P.M.; Linsay, B.; Stevens, R.L. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol 2010, 28, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Pornputtapong, N.; Nookaew, I.; Nielsen, J. Human metabolic atlas: An online resource for human metabolism. Database 2015, 2015, bav068. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2013, 42, D199–D205. [Google Scholar] [CrossRef]
- Schomburg, I.; Jeske, L.; Ulbrich, M.; Placzek, S.; Chang, A.; Schomburg, D. The BRENDA enzyme information system-From a database to an expert system. J. Biotechnol. 2017, 261, 194–206. [Google Scholar] [CrossRef]
- D’Eustachio, P. Reactome knowledgebase of human biological pathways and processes. Methods Mol. Biol. 2011, 694, 49–61. [Google Scholar] [CrossRef]
- UniProt Consortium, T. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef] [PubMed]
- Baldini, F.; Heinken, A.; Heirendt, L.; Magnusdottir, S.; Fleming, R.M.; Thiele, I. The Microbiome Modeling Toolbox: From microbial interactions to personalized microbial communities. bioRxiv 2018. [Google Scholar] [CrossRef] [PubMed]
- Heinken, A.; Ravcheev, D.A.; Baldini, F.; Heirendt, L.; Fleming, R.M.; Thiele, I. Personalized modeling of the human gut microbiome reveals distinct bile acid deconjugation and biotransformation potential in healthy and IBD individuals. bioRxiv 2017. [Google Scholar] [CrossRef]
- Heinken, A.; Sahoo, S.; Fleming, R.M.; Thiele, I. Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes 2013, 4, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, S.; Thiele, I. Computationally efficient flux variability analysis. BMC Bioinform. 2010, 11, 489. [Google Scholar] [CrossRef] [PubMed]
- Gorvitovskaia, A.; Holmes, S.P.; Huse, S.M. Interpreting Prevotella and Bacteroides as biomarkers of diet and lifestyle. Microbiome 2016, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Biggs, M.B.; Papin, J.A. Novel multiscale modeling tool applied to Pseudomonas aeruginosa biofilm formation. PLoS ONE 2013, 8, e78011. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Van Treuren, W.; Lozupone, C.; Faust, K.; Friedman, J.; Deng, Y.; Xia, L.C.; Xu, Z.Z.; Ursell, L.; Alm, E.J.; et al. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 2016, 10, 1669–1681. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, Z.D.; Muller, C.L.; Miraldi, E.R.; Littman, D.R.; Blaser, M.J.; Bonneau, R.A. Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput. Biol. 2015, 11, e1004226. [Google Scholar] [CrossRef]
- Das, P.; Ji, B.; Kovatcheva-Datchary, P.; Bäckhed, F.; Nielsen, J. In vitro co-cultures of human gut bacterial species as predicted from co-occurrence network analysis. PLoS ONE 2018, 13, e0195161. [Google Scholar] [CrossRef]
- Duarte, N.C.; Becker, S.A.; Jamshidi, N.; Thiele, I.; Mo, M.L.; Vo, T.D.; Srivas, R.; Palsson, B.O. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl. Acad. Sci. USA 2007, 104, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Sorokin, A.; Mazein, A.; Selkov, A.; Selkov, E.; Demin, O.; Goryanin, I. The Edinburgh human metabolic network reconstruction and its functional analysis. Mol. Syst. Biol. 2007, 3, 135. [Google Scholar] [CrossRef] [PubMed]
- Swainston, N.; Smallbone, K.; Hefzi, H.; Dobson, P.D.; Brewer, J.; Hanscho, M.; Zielinski, D.C.; Ang, K.S.; Gardiner, N.J.; Gutierrez, J.M. Recon 2.2: From reconstruction to model of human metabolism. Metabolomics 2016, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Dräger, A.; Mih, N.; Gatto, F.; Nilsson, A.; Gonzalez, G.A.P.; Aurich, M.K.; et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Nookaew, I.; Jacobson, P.; Walley, A.J.; Froguel, P.; Carlsson, L.M.; Uhlen, M.; et al. Integration of clinical data with a genome-scale metabolic model of the human adipocyte. Mol. Syst. Biol. 2013, 9, 649. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahoo, S.; Thiele, I. Predicting the impact of diet and enzymopathies on human small intestinal epithelial cells. Hum. Mol. Genet. 2013, 22, 2705–2722. [Google Scholar] [CrossRef]
- Sahoo, S.; Aurich, M.K.; Jonsson, J.J.; Thiele, I. Membrane transporters in a human genome-scale metabolic knowledgebase and their implications for disease. Front. Physiol. 2014, 5, 91. [Google Scholar] [CrossRef]
- Lewis, N.E.; Schramm, G.; Bordbar, A.; Schellenberger, J.; Andersen, M.P.; Cheng, J.K.; Patel, N.; Yee, A.; Lewis, R.A.; Eils, R. Large-scale in silico modeling of metabolic interactions between cell types in the human brain. Nat. Biotechnol. 2010, 28, 1279. [Google Scholar] [CrossRef]
- Väremo, L.; Scheele, C.; Broholm, C.; Mardinoglu, A.; Kampf, C.; Asplund, A.; Nookaew, I.; Uhlén, M.; Pedersen, B.K.; Nielsen, J. Proteome-and Transcriptome-Driven Reconstruction of the Human Myocyte Metabolic Network and Its Use for Identification of Markers for Diabetes. Cell Rep. 2016, 14, 1567. [Google Scholar] [CrossRef]
- Agren, R.; Bordel, S.; Mardinoglu, A.; Pornputtapong, N.; Nookaew, I.; Nielsen, J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput. Biol. 2012, 8, e1002518. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Sahoo, S.; Heinken, A.; Heirendt, L.; Aurich, M.K.; Noronha, A.; Fleming, R.M. When metabolism meets physiology: Harvey and Harvetta. bioRxiv 2018. [Google Scholar] [CrossRef]
- Roesch, L.F.; Lorca, G.L.; Casella, G.; Giongo, A.; Naranjo, A.; Pionzio, A.M.; Li, N.; Mai, V.; Wasserfall, C.H.; Schatz, D.; et al. Culture-independent identification of gut bacteria correlated with the onset of diabetes in a rat model. ISME J. 2009, 3, 536–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, L.; Ley, R.E.; Volchkov, P.Y.; Stranges, P.B.; Avanesyan, L.; Stonebraker, A.C.; Hu, C.; Wong, F.S.; Szot, G.L.; Bluestone, J.A.; et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008, 455, 1109–1113. [Google Scholar] [CrossRef] [Green Version]
- Brugman, S.; Klatter, F.; Visser, J.; Wildeboer-Veloo, A.; Harmsen, H.; Rozing, J.; Bos, N. Antibiotic treatment partially protects against type 1 diabetes in the Bio-Breeding diabetes-prone rat. Is the gut flora involved in the development of type 1 diabetes? Diabetologia 2006, 49, 2105–2108. [Google Scholar] [CrossRef] [Green Version]
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyötyläinen, T.; Hämäläinen, A.-M.; Peet, A.; Tillmann, V.; Pöhö, P.; Mattila, I. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 2015, 17, 260–273. [Google Scholar] [CrossRef]
- Dimeloe, S.; Burgener, A.V.; Grahlert, J.; Hess, C. T-cell metabolism governing activation, proliferation and differentiation; a modular view. Immunology 2017, 150, 35–44. [Google Scholar] [CrossRef]
- Bordbar, A.; Mo, M.L.; Nakayasu, E.S.; Schrimpe-Rutledge, A.C.; Kim, Y.M.; Metz, T.O.; Jones, M.B.; Frank, B.C.; Smith, R.D.; Peterson, S.N.; et al. Model-driven multi-omic data analysis elucidates metabolic immunomodulators of macrophage activation. Mol. Syst. Biol. 2012, 8, 558. [Google Scholar] [CrossRef]
- Segata, N.; Boernigen, D.; Tickle, T.L.; Morgan, X.C.; Garrett, W.S.; Huttenhower, C. Computational meta’omics for microbial community studies. Mol. Syst. Biol. 2013, 9, 666. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Shoaie, S.; Bergentall, M.; Ghaffari, P.; Zhang, C.; Larsson, E.; Bäckhed, F.; Nielsen, J. The gut microbiota modulates host amino acid and glutathione metabolism in mice. Mol. Syst. Biol. 2015, 11, 834. [Google Scholar] [CrossRef]
- Lagier, J.C.; Khelaifia, S.; Alou, M.T.; Ndongo, S.; Dione, N.; Hugon, P.; Caputo, A.; Cadoret, F.; Traore, S.I.; Seck, E.H.; et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nat. Microbiol. 2016, 1, 16203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagier, J.-C.; Hugon, P.; Khelaifia, S.; Fournier, P.-E.; La Scola, B.; Raoult, D. The rebirth of culture in microbiology through the example of culturomics to study human gut microbiota. Clin. Microbiol. Rev. 2015, 28, 237–264. [Google Scholar] [CrossRef] [PubMed]
- David, L.A. Toward Personalized Control of Human Gut Bacterial Communities. mSystems 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Clancy, C.M.; Heinken, A.; Fleming, R.M. Quantitative systems pharmacology and the personalized drug–microbiota–diet axis. Curr. Opin. Syst. Biol. 2017, 4, 43–52. [Google Scholar] [CrossRef]
- Sen, P.; Vial, H.J.; Radulescu, O. Mathematical modeling and omic data integration to understand dynamic adaptation of Apicomplexan parasites and identify pharmaceutical targets. Compr. Anal. Parasite Biol. Metab. Drug Discov. 2016, 7, 457. [Google Scholar]
- Sen, P.; Vial, H.J.; Radulescu, O. Kinetic modelling of phospholipid synthesis in Plasmodium knowlesi unravels crucial steps and relative importance of multiple pathways. BMC Syst. Biol. 2013, 7, 123. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Toolboxes | Short Description | Source or Reference |
|---|---|---|
| Modeling Tools | ||
| COBRA (Microbiome Modeling Toolbox) | A MATLAB suite for constraint-based modeling (CBM), includes tools and methods for pairwise and community modeling of microbiota. COBRA can be used for GEM reconstruction and analysis. | [59,60,61] |
| RAVEN (CASINO) | A MATLAB suite for CBM, includes tools for modeling diet-microbiota interactions. It can be used for GEM reconstruction and analysis. | [62] |
| Kbase | A web-based tool for systems biology and metabolic modeling. It can be used for automatic GEM reconstruction and analysis. | [63] |
| BacArena | An R-package for individual-based and CBM of microbes in a gut community. | [64] |
| COMETS | A software platform for stoichiometric modeling of individual microbial species using dynamic flux balance analysis (FBA). | [65] |
| MCM | A tool for CBM of microbial community model, based on conventional FBA. | [66] |
| DyMMM | A tool for CBM that integrates multiple microbial species into a dynamic community model. | [67] |
| OptCom | A modeling framework to perform FBA of microbial communities. | [68] |
| SteadyCom | A toolbox that can be used to predict the changes in microbial species abundance in response to the dietary changes. | [69] |
| MetExplore | An open access web-server for integrative analysis of metabolomic datasets and genome-scale metabolic networks. | [70] |
| MMinte | An integrated pipeline for modeling the pairwise interactions within a microbial network. | [71] |
| jQMM library | An open-source, Python-based framework for modeling internal metabolic fluxes. The toolbox can be used for FBA and 13C Metabolic Flux Analysis (MFA). | [72] |
| Model repositories and databases | ||
| BiGG database | An open access database for gold standard GEMs. | [73] |
| Virtual Metabolic Human (VMH) | An open access database for human and gut microbial metabolism (GEMs). | [74] |
| ModelSEED | A web-based resource for metabolic modeling. | [75] |
| Human Metabolic Atlas (HMA) | An open access web-based resource for human metabolism. | [76] |
| Metabolic Pathways and Enzyme databases | ||
| MetaCyc/HumanCyc | A curated database of experimentally validated metabolic pathways. HumanCyc is a database of curated human metabolic pathways. | [48] |
| KEGG | A resource comprised of databases including large-scale molecular datasets and detailed pathway information. | [77,78] |
| BRENDA | An information retrieval system focusing on enzymes and their ligands. | [79] |
| REACTOME | An open access database of biological pathways. | [80] |
| UniProt. | An open access database of curated protein information. | [81] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sen, P.; Orešič, M. Metabolic Modeling of Human Gut Microbiota on a Genome Scale: An Overview. Metabolites 2019, 9, 22. https://doi.org/10.3390/metabo9020022
Sen P, Orešič M. Metabolic Modeling of Human Gut Microbiota on a Genome Scale: An Overview. Metabolites. 2019; 9(2):22. https://doi.org/10.3390/metabo9020022
Chicago/Turabian StyleSen, Partho, and Matej Orešič. 2019. "Metabolic Modeling of Human Gut Microbiota on a Genome Scale: An Overview" Metabolites 9, no. 2: 22. https://doi.org/10.3390/metabo9020022
APA StyleSen, P., & Orešič, M. (2019). Metabolic Modeling of Human Gut Microbiota on a Genome Scale: An Overview. Metabolites, 9(2), 22. https://doi.org/10.3390/metabo9020022