MolNetEnhancer: Enhanced Molecular Networks by Integrating Metabolome Mining and Annotation Tools

 , , , , , ,
, , , , , , 
Abstract
: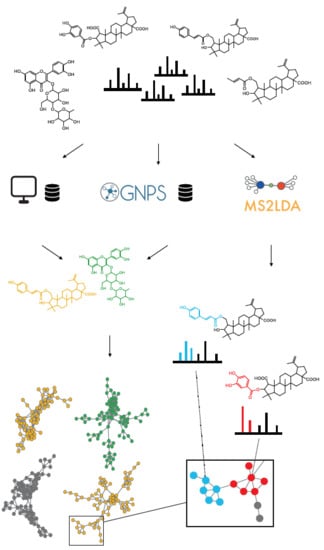
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
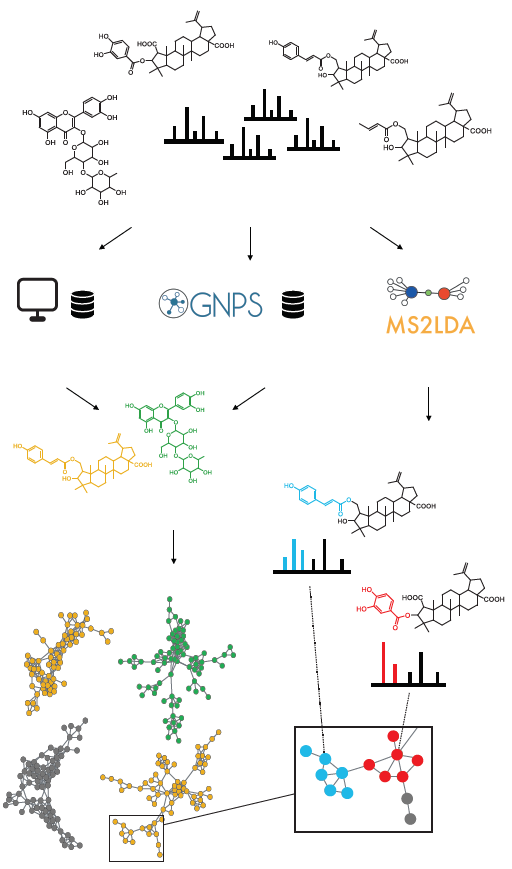
- Perform mass spectral molecular networking analysis through the Global Natural Products Social Molecular Networking platform (https://gnps.ucsd.edu).
- Perform unsupervised substructure discovery using MS2LDA (http://ms2lda.org).
- a
- Substructure information retrieved through MS2LDA is integrated with mass spectral molecular networks.
- b
- Most abundant chemical classes per molecular family are retrieved based on GNPS structural library hits and in silico chemical structural annotation and integrated within the mass spectral molecular networks.
3. Results
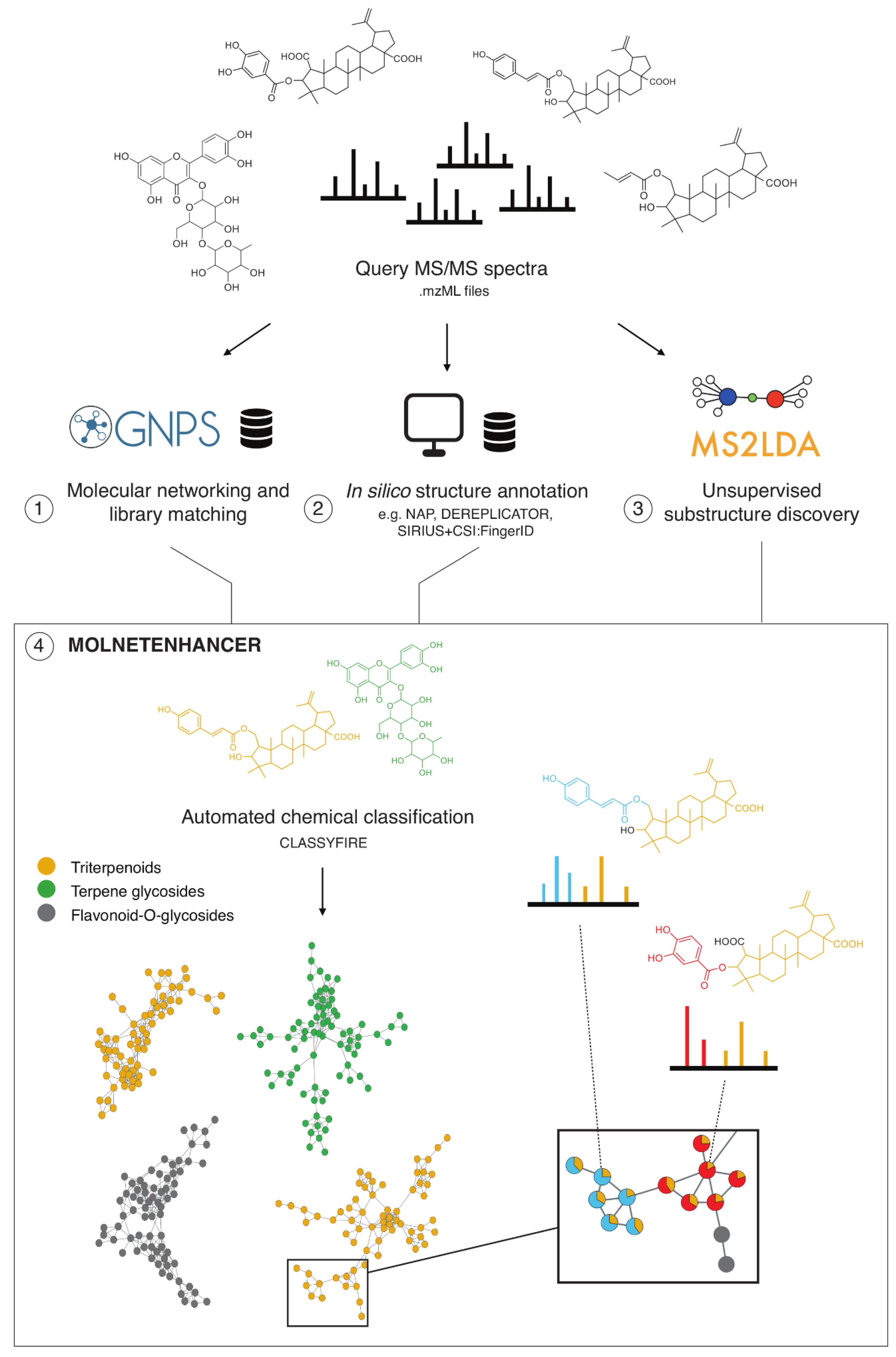
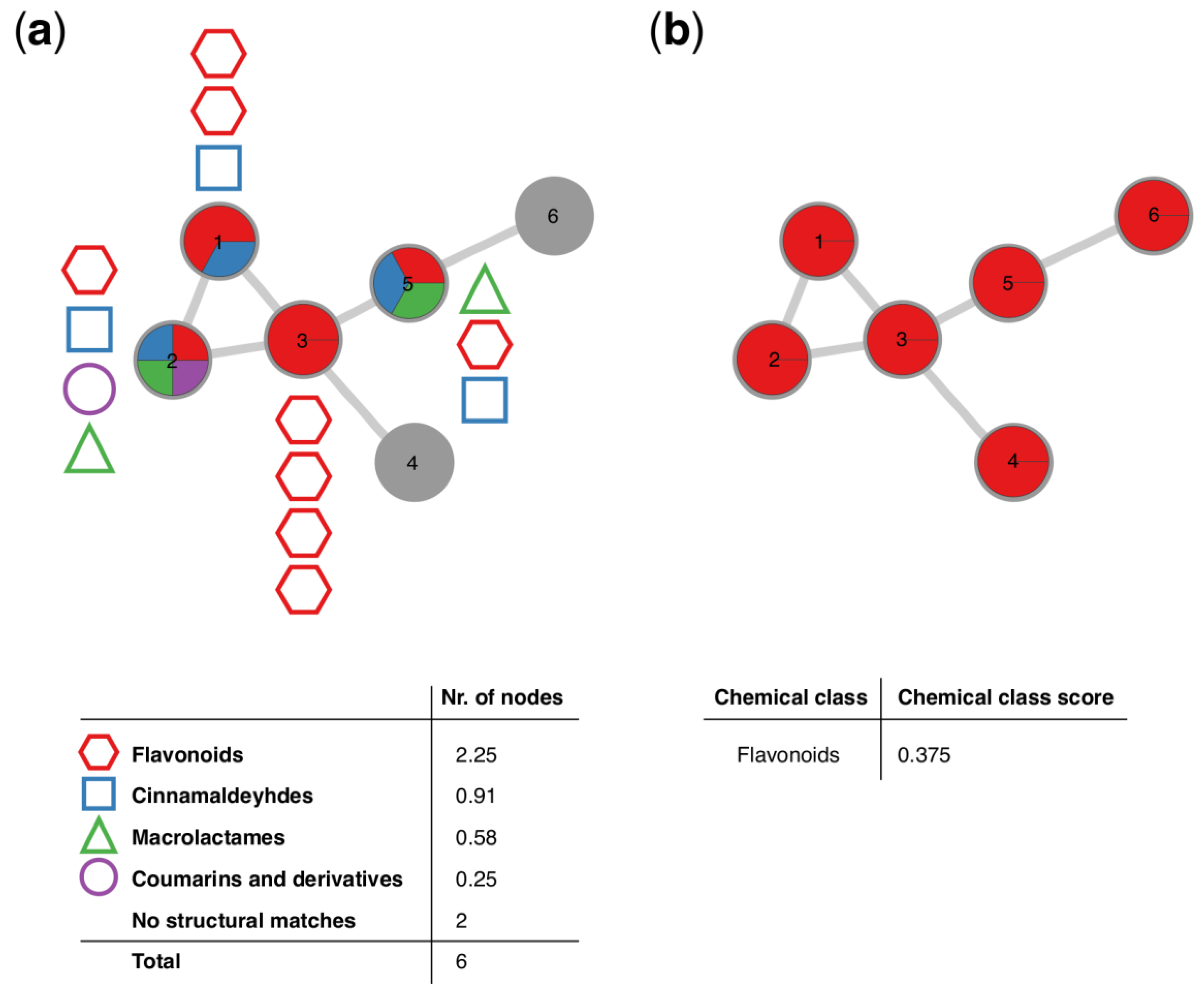
3.1. MolNetEnhancer Workflow
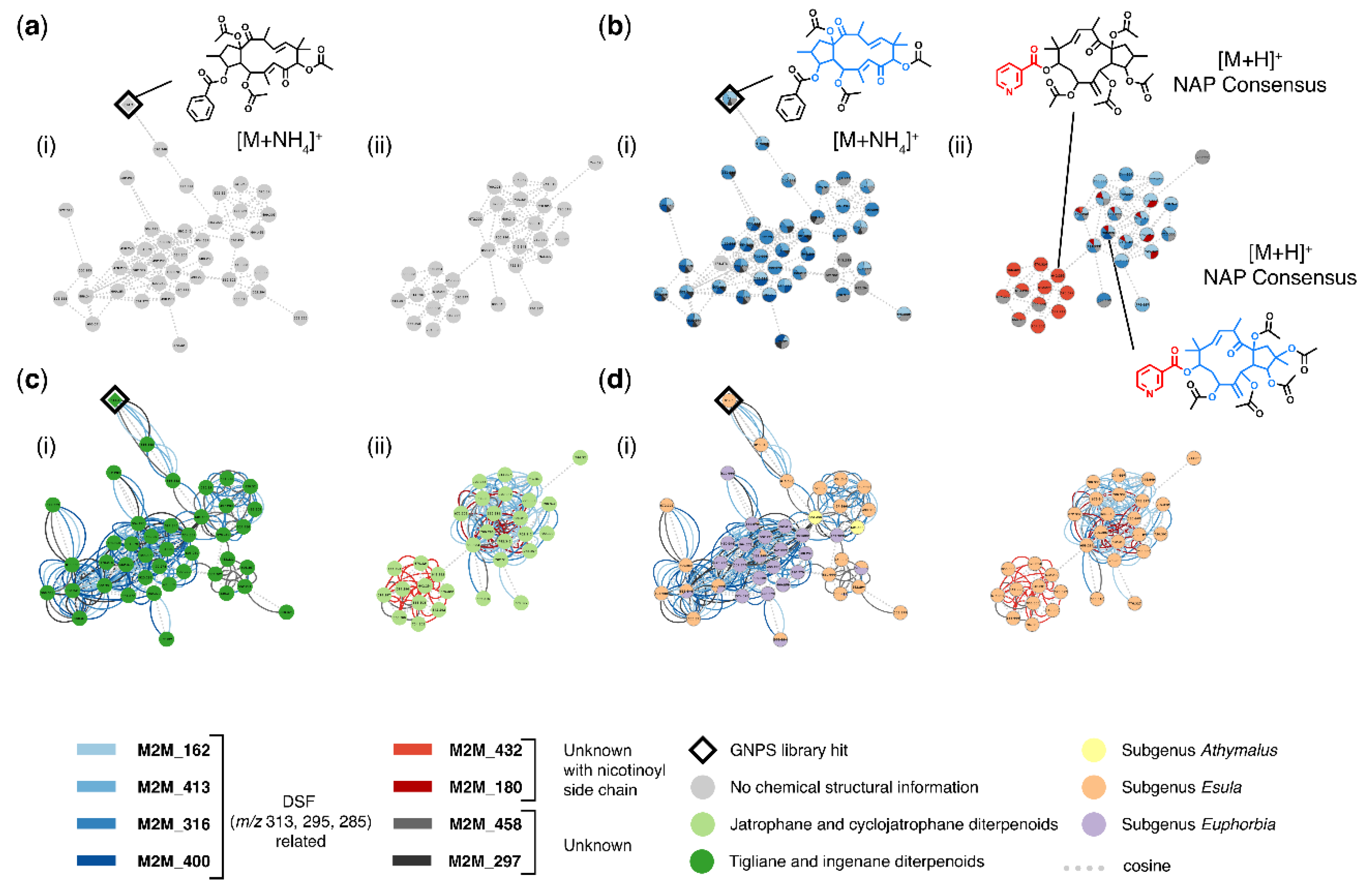
3.2. Case Study 1: Annotation of Euphorbia Specialized Metabolites Using MolNetEnhancer
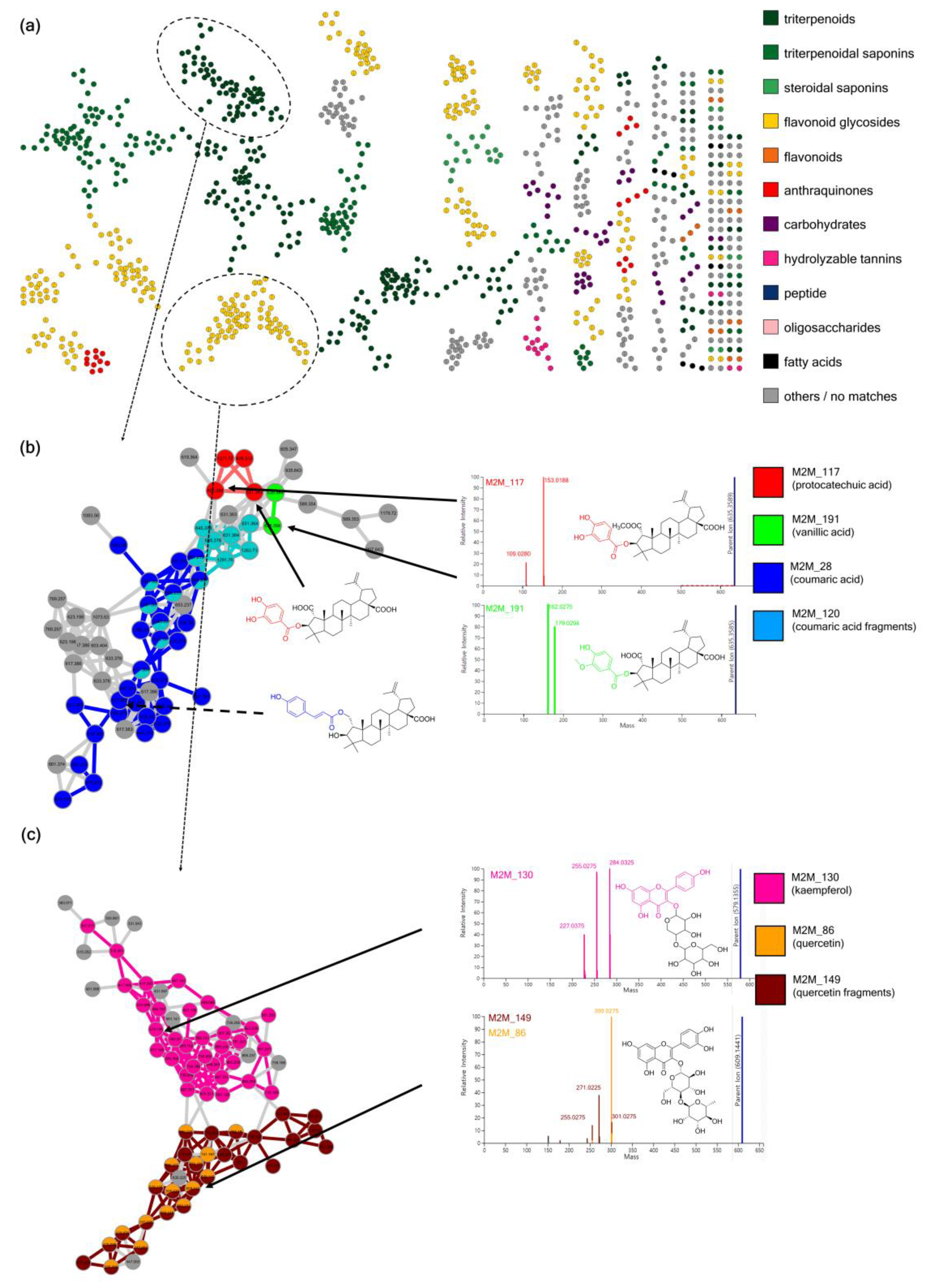
3.3. Case Study 2: Annotation of Rhamnaceae Specialized Metabolites
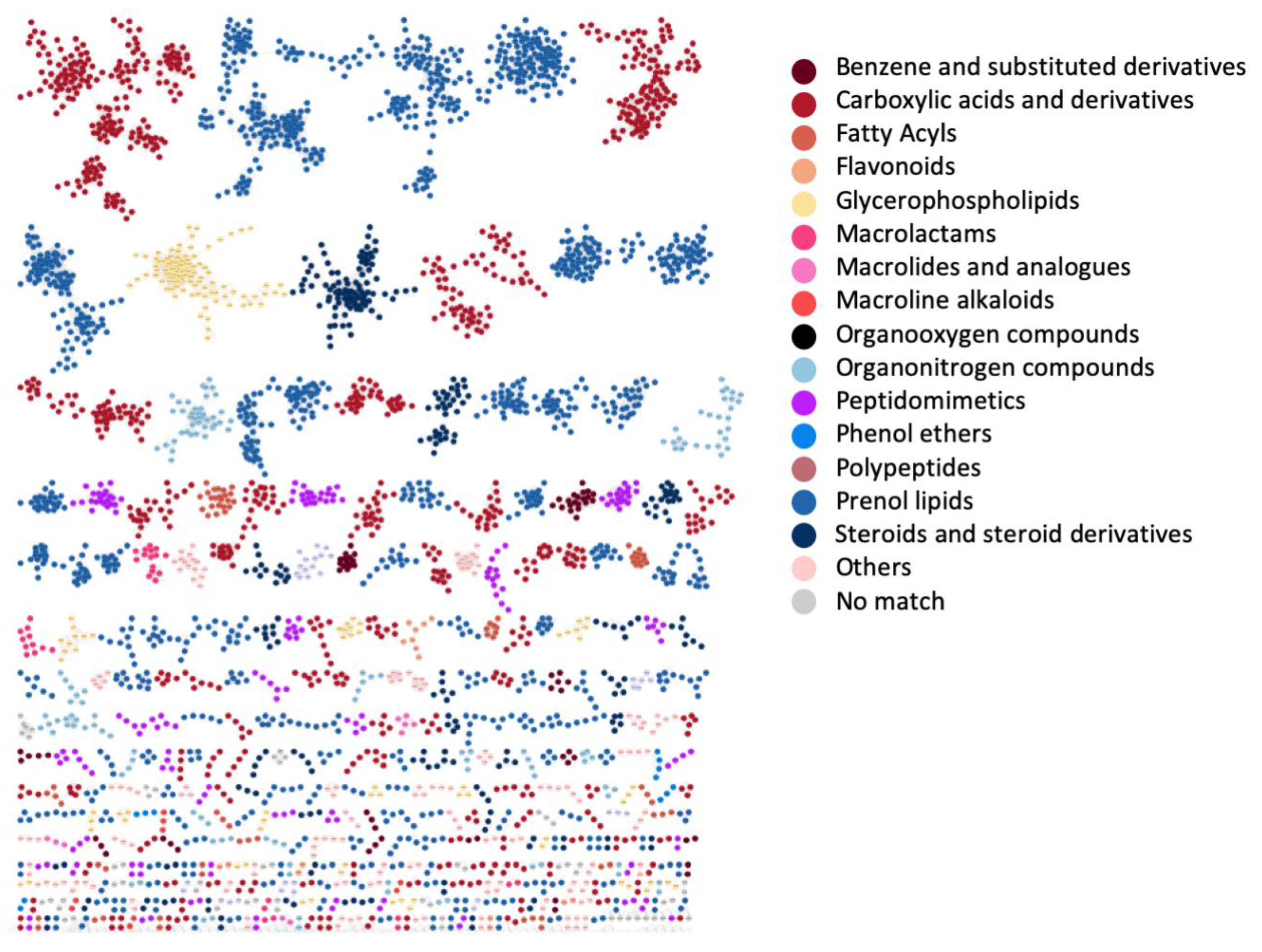
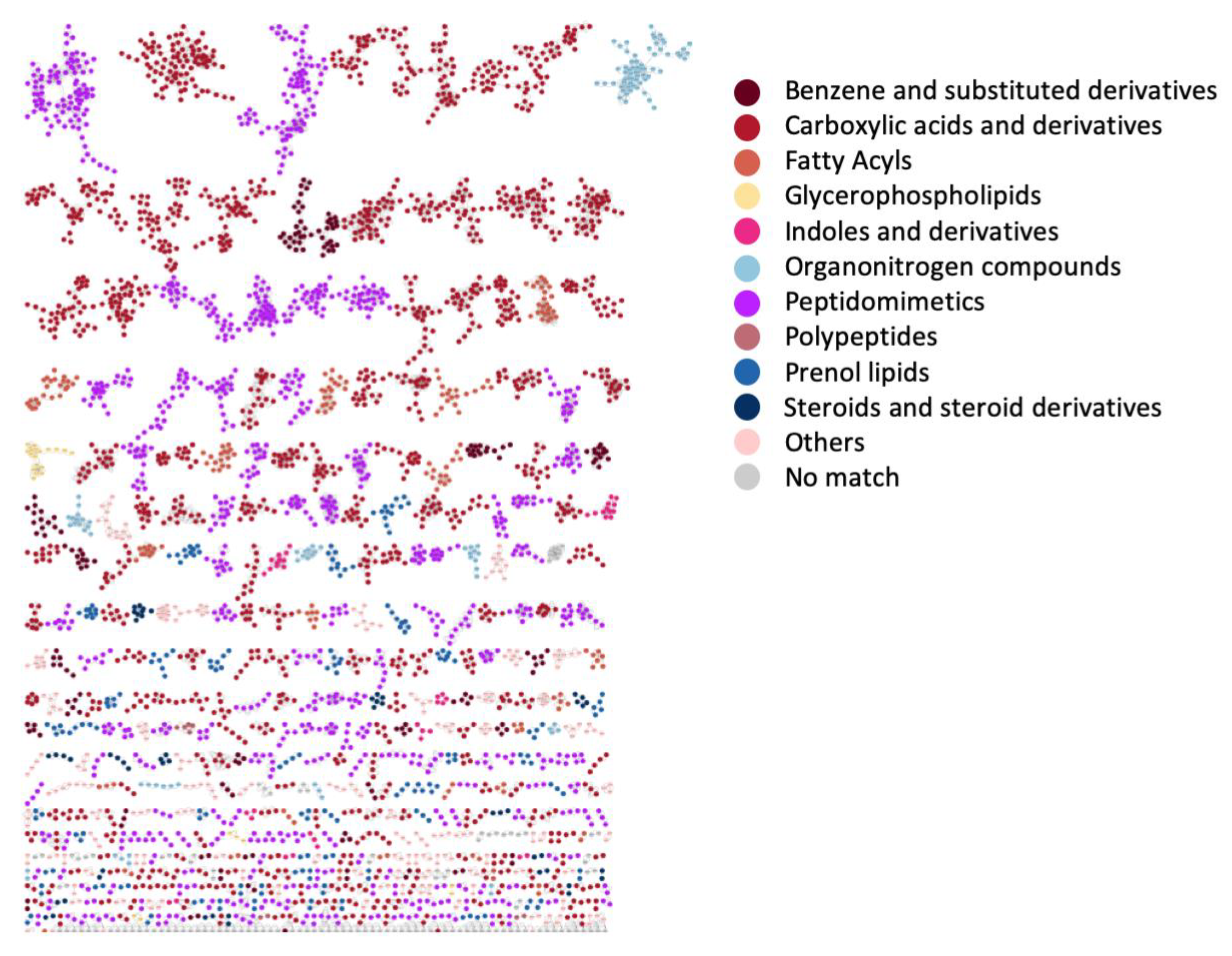
3.4. Case Study 3: Large Chemical Diversity Uncovered by Annotating Specialized Metabolites in Marine Sediment Streptomyces and Salinispora Bacterial Extracts
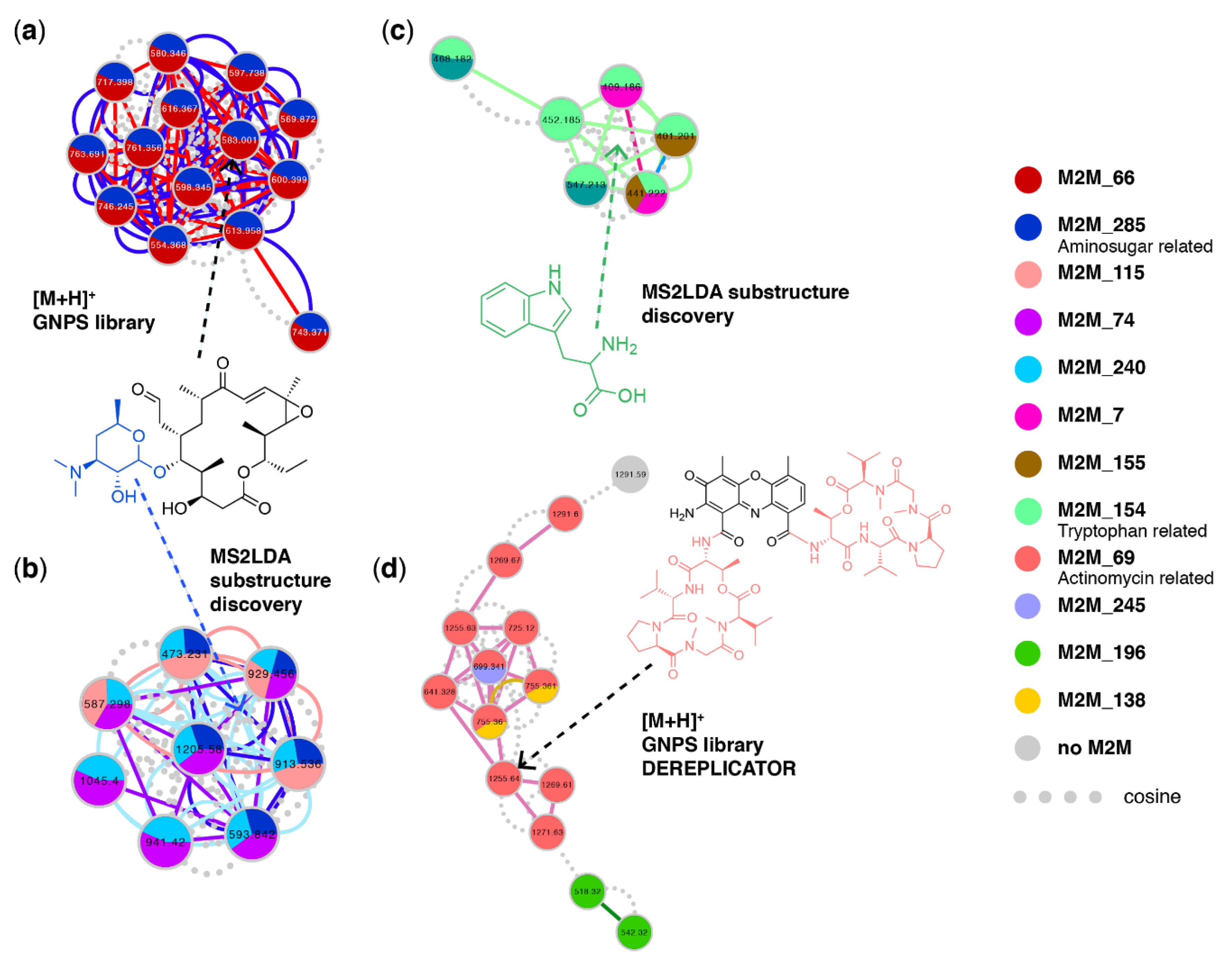
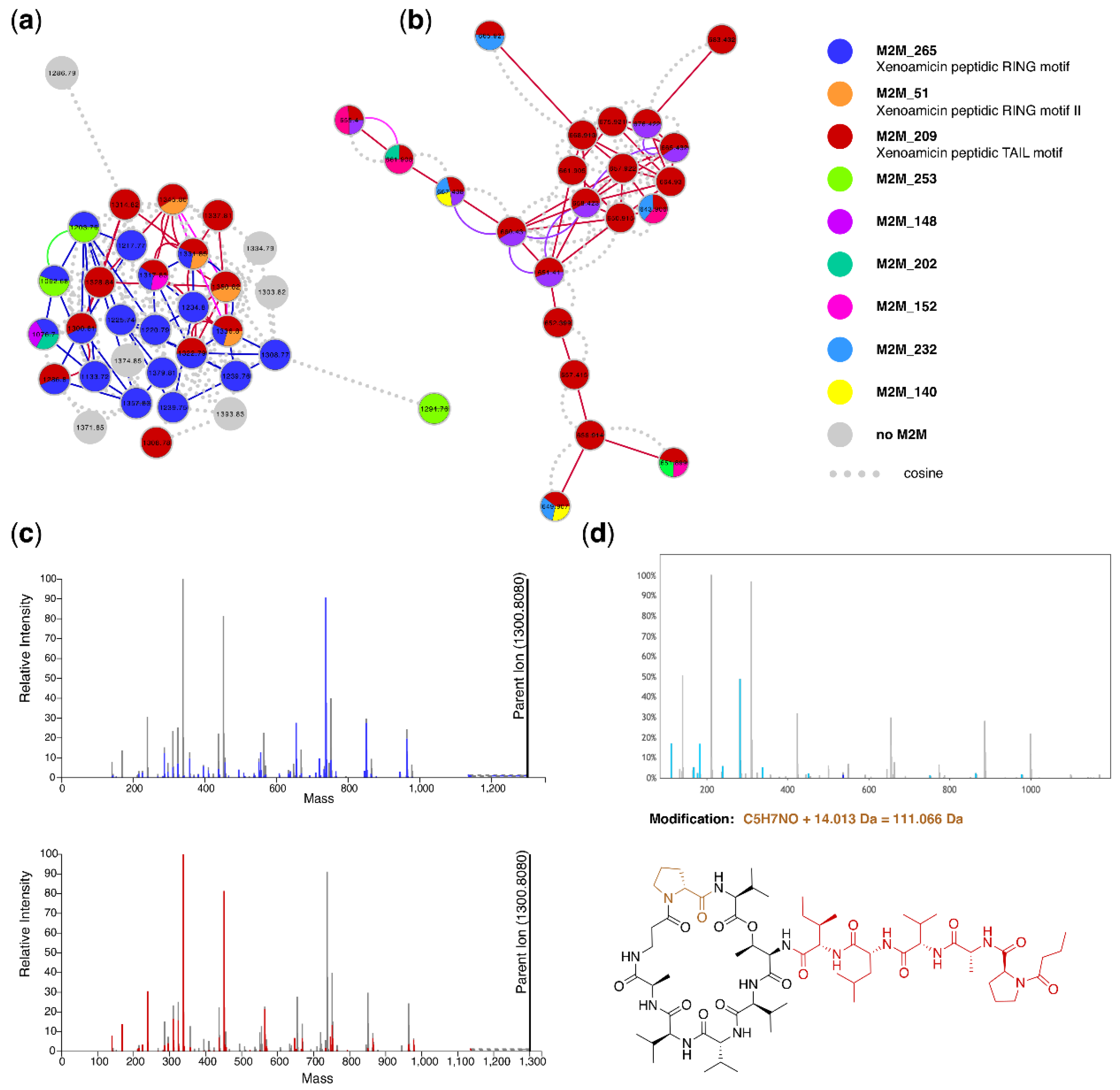
3.5. Case Study 4: Annotating Peptidic Motifs in Peptide-Rich Xenorhabdus/Photorhabdus Extracts
4. Discussion
5. Conclusions
6. Data Availability
- MASSIVE: MSV000081082 https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=c9f09d31a24c475e87a0a11f6e8889e7
- GNPS Molecular Networking job: https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=26326c233918419f8dc80e8af984cdae
- MS2LDA experiment: http://ms2lda.org/basicviz/summary/390
- MASSIVE: MSV000081805 https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=36f154d1c3844d31b9732fbaa72e9284
- GNPS Molecular Networking job: https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=e9e02c0ba3db473a9b1ddd36da72859b
- MS2LDA experiment: http://ms2lda.org/basicviz/summary/566
- MASSIVE: MSV000082678 https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=de2d18fd91804785bce8c225cc94a44
- GNPS Molecular Networking job: https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=b817262cb6114e7295fee4f73b22a3ad
- MS2LDA experiment: http://ms2lda.org/basicviz/summary/907
- GNPS Molecular Networking job: http://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=c36f90ba29fe44c18e96db802de0c6b9
- GNPS DEREPLICATOR 0.005 job: https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=0ad6535e34d449788f297e712f43068a
- GNPS DEREPLICATOR 0.05 job: https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=e494a63be6d34747a4b8cdfb838ef96e
- GNPS VARQUEST 0.005 job: https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=f1f00c1c20ba4f61ad471d340066df76
- GNPS VARQUEST 0.05 job: https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=f5ffcc8f63ab4e6f96a97caabc11048b
- MS2LDA annotation experiment: http://ms2lda.org/basicviz/summary/551
- MS2LDA MolNetEnhancer workflow experiment: http://ms2lda.org/basicviz/summary/912
- MASSIVE: MSV000081063 https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=dcc30b777c344d668a5626d01f26c9a0
- GNPS Molecular Networking job: https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=aaff4721951b4d92b54ecbd2fe4b9b4f
- GNPS DEREPLICATOR 0.005 job: http://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=338b422483d1432e82afd1bf848f1292
- GNPS DEREPLICATOR 0.05 job: http://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=83bca3c45665470891d41ead275dcae7
- GNPS VARQUEST 0.005 job: http://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=20cfb9af4a244feea102aa9c9da2651c
- GNPS VARQUEST 0.05 job: http://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=a4ffda169823476a9b1e81616aeccbda
- MS2LDA annotation experiment: http://ms2lda.org/basicviz/summary/570
- MS2LDA MolNetEnhancer workflow experiment: http://ms2lda.org/basicviz/summary/917
- GNPS Molecular Networking job: https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=b76dd5a123e54a7eb42765499f9163a5
- MS2LDA experiment: http://ms2lda.org/basicviz/summary/909
7. Code Availability
8. Tutorials
- GNPS molecular networking:
- DEREPLICATOR/VarQuest:
- Network annotation propagation:
- ClassyFire:
- MS2LDA:
- MolNetEnhancer workflow tutorials in both R and Python can be found here:
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mohimani, H.; Gurevich, A.; Mikheenko, A.; Garg, N.; Nothias, L.-F.; Ninomiya, A.; Takada, K.; Dorrestein, P.C.; Pevzner, P.A. Dereplication of peptidic natural products through database search of mass spectra. Nat. Chem. Biol. 2017, 13, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Mikheenko, A.; Shlemov, A.; Korobeynikov, A.; Mohimani, H.; Pevzner, P.A. Increased diversity of peptidic natural products revealed by modification-tolerant database search of mass spectra. Nat. Microbiol. 2018, 3, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Dührkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Böcker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, F.; Pon, A.; Wilson, M.; Greiner, R.; Wishart, D. CFM-ID: A web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014, 42, W94–W99. [Google Scholar] [CrossRef] [PubMed]
- Djoumbou-Feunang, Y.; Pon, A.; Karu, N.; Zheng, J.; Li, C.; Arndt, D.; Gautam, M.; Allen, F.; Wishart, D.S. CFM-ID 3.0: Significantly Improved ESI-MS/MS Prediction and Compound Identification. Metabolites 2019, 9, 72. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, R.R.; Wang, M.; Nothias, L.-F.; van der Hooft, J.J.J.; Caraballo-Rodríguez, A.M.; Fox, E.; Balunas, M.J.; Klassen, J.L.; Lopes, N.P.; Dorrestein, P.C. Propagating annotations of molecular networks using in silico fragmentation. PLoS Comput. Biol. 2018, 14, e1006089. [Google Scholar] [CrossRef]
- Ridder, L.; van der Hooft, J.J.J.; Verhoeven, S.; de Vos, R.C.H.; Vervoort, J.; Bino, R.J. In silico prediction and automatic LC-MS(n) annotation of green tea metabolites in urine. Anal. Chem. 2014, 86, 4767–4774. [Google Scholar] [CrossRef]
- Watrous, J.; Roach, P.; Alexandrov, T.; Heath, B.S.; Yang, J.Y.; Kersten, R.D.; van der Voort, M.; Pogliano, K.; Gross, H.; Raaijmakers, J.M.; et al. Mass spectral molecular networking of living microbial colonies. Proc. Natl. Acad. Sci. USA 2012, 109, E1743–E1752. [Google Scholar] [CrossRef] [Green Version]
- Gerlich, M.; Neumann, S. MetFusion: Integration of compound identification strategies. J. Mass Spectrom. 2013, 48, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Treutler, H.; Tsugawa, H.; Porzel, A.; Gorzolka, K.; Tissier, A.; Neumann, S.; Balcke, G.U. Discovering Regulated Metabolite Families in Untargeted Metabolomics Studies. Anal. Chem. 2016, 88, 8082–8090. [Google Scholar] [CrossRef] [PubMed]
- Van der Hooft, J.J.J.; Padmanabhan, S.; Burgess, K.E.V.; Barrett, M.P. Urinary antihypertensive drug metabolite screening using molecular networking coupled to high-resolution mass spectrometry fragmentation. Metabolomics 2016, 12, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Hooft, J.J.J.; Wandy, J.; Barrett, M.P.; Burgess, K.E.V.; Rogers, S. Topic modeling for untargeted substructure exploration in metabolomics. Proc. Natl. Acad. Sci. USA 2016, 113, 13738–13743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wandy, J.; Zhu, Y.; van der Hooft, J.J.J.; Daly, R.; Barrett, M.P.; Rogers, S. Ms2lda.org: web-based topic modelling for substructure discovery in mass spectrometry. Bioinformatics 2018, 34, 317–318. [Google Scholar] [CrossRef]
- Feunang, Y.D.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform. 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Olivon, F.; Elie, N.; Grelier, G.; Roussi, F.; Litaudon, M.; Touboul, D. MetGem Software for the Generation of Molecular Networks Based on the t-SNE Algorithm. Anal. Chem. 2018, 90, 13900–13908. [Google Scholar] [CrossRef]
- Ma, Y.; Kind, T.; Yang, D.; Leon, C.; Fiehn, O. MS2Analyzer: A software for small molecule substructure annotations from accurate tandem mass spectra. Anal. Chem. 2014, 86, 10724–10731. [Google Scholar] [CrossRef]
- Laponogov, I.; Sadawi, N.; Galea, D.; Mirnezami, R.; Veselkov, K.A. ChemDistiller: an engine for metabolite annotation in mass spectrometry. Bioinformatics 2018, 34, 2096–2102. [Google Scholar] [CrossRef]
- Edmands, W.M.B.; Petrick, L.; Barupal, D.K.; Scalbert, A.; Wilson, M.J.; Wickliffe, J.K.; Rappaport, S.M. compMS2Miner: An Automatable Metabolite Identification, Visualization, and Data-Sharing R Package for High-Resolution LC–MS Data Sets. Anal. Chem. 2017, 89, 3919–3928. [Google Scholar] [CrossRef]
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. MetFrag relaunched: incorporating strategies beyond in silico fragmentation. J. Cheminform. 2016, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Naake, T.; Gaquerel, E. MetCirc: Navigating mass spectral similarity in high-resolution MS/MS metabolomics data. Bioinformatics 2017, 33, 2419–2420. [Google Scholar] [CrossRef] [PubMed]
- Beauxis, Y.; Genta-Jouve, G. MetWork: A web server for natural products anticipation. Bioinformatics 2019, 35, 1795–1796. [Google Scholar] [CrossRef] [PubMed]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox Ramos, A.E.; Evanno, L.; Poupon, E.; Champy, P.; Beniddir, M.A. Natural products targeting strategies involving molecular networking: Different manners, one goal. Nat. Prod. Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, R.R.; Dorrestein, P.C.; Quinn, R.A. Illuminating the dark matter in metabolomics. Proc. Natl. Acad. Sci. USA 2015, 112, 12549–12550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, M.; Nothias, L.-F.; van der Hooft, J.J.J.; Silva, R.R.; Saslis-Lagoudakis, C.H.; Grace, O.M.; Martinez-Swatson, K.; Hassemer, G.; Funez, L.A.; Simonsen, H.T.; et al. Assessing Specialized Metabolite Diversity in the Cosmopolitan Plant Genus Euphorbia L. Front. Plant Sci. 2019, 10, 846. [Google Scholar] [CrossRef]
- Kang, K.B.; Ernst, M.; van der Hooft, J.J.J.; da Silva, R.R.; Park, J.; Medema, M.H.; Sung, S.H.; Dorrestein, P.C. Comprehensive mass spectrometry-guided phenotyping of plant specialized metabolites reveals metabolic diversity in the cosmopolitan plant family Rhamnaceae. Plant J. 2019. [Google Scholar] [CrossRef]
- Frank, A.M.; Bandeira, N.; Shen, Z.; Tanner, S.; Briggs, S.P.; Smith, R.D.; Pevzner, P.A. Clustering millions of tandem mass spectra. J. Proteome Res. 2008, 7, 113–122. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Van der Hooft, J.J.J.; Wandy, J.; Young, F.; Padmanabhan, S.; Gerasimidis, K.; Burgess, K.E.V.; Barrett, M.P.; Rogers, S. Unsupervised Discovery and Comparison of Structural Families Across Multiple Samples in Untargeted Metabolomics. Anal. Chem. 2017, 89, 7569. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Govaerts, R.; Fernández Casas, F.J.; Barker, C.; Carter, S.; Davies, S.; Esser, H.-J.; Gilbert, M.; Hoffmann, P.; Radcliffe-Smith, A.; Steinmann, V.; et al. World Checklist of Euphorbiaceae. Facilitated by the Royal Botanic Gardens, Kew. Available online: http://apps.kew.org/wcsp/ (accessed on 25 July 2014).
- Horn, J.W.; van Ee, B.W.; Morawetz, J.J.; Riina, R.; Steinmann, V.W.; Berry, P.E.; Wurdack, K.J. Phylogenetics and the evolution of major structural characters in the giant genus Euphorbia L. (Euphorbiaceae). Mol. Phylogenet. Evol. 2012, 63, 305–326. [Google Scholar] [CrossRef] [PubMed]
- Vasas, A.; Hohmann, J. Euphorbia Diterpenes: Isolation, Structure, Biological Activity, and Synthesis (2008–2012). Chem. Rev. 2014, 114, 8579–8612. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.-W.; Su, X.-H.; Kiyota, H. Chemical and pharmacological research of the plants in genus Euphorbia. Chem. Rev. 2008, 108, 4295–4327. [Google Scholar] [CrossRef] [PubMed]
- Berman, B. New developments in the treatment of actinic keratosis: Focus on ingenol mebutate gel. Clin. Cosmet. Investig. Dermatol. 2012, 5, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Callari, R.; Hamberger, B.; Wubshet, S.G.; Nielsen, M.T.; Andersen-Ranberg, J.; Hallström, B.M.; Cozzi, F.; Heider, H.; Møller, B.L.; et al. Oxidation and cyclization of casbene in the biosynthesis of Euphorbia factors from mature seeds of Euphorbia lathyris L. Proc. Natl. Acad. Sci. USA 2016, 113, E5082–E5089. [Google Scholar] [CrossRef] [PubMed]
- Appendino, G. Ingenane Diterpenoids. In Progress in the Chemistry of Organic Natural Products 102; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–90. [Google Scholar]
- Nothias-Scaglia, L.-F.; Schmitz-Afonso, I.; Renucci, F.; Roussi, F.; Touboul, D.; Costa, J.; Litaudon, M.; Paolini, J. Insights on profiling of phorbol, deoxyphorbol, ingenol and jatrophane diterpene esters by high performance liquid chromatography coupled to multiple stage mass spectrometry. J. Chromatogr. A 2015, 1422, 128–139. [Google Scholar] [CrossRef]
- Nothias, L.-F.; Boutet-Mercey, S.; Cachet, X.; De La Torre, E.; Laboureur, L.; Gallard, J.-F.; Retailleau, P.; Brunelle, A.; Dorrestein, P.C.; Costa, J.; et al. Environmentally Friendly Procedure Based on Supercritical Fluid Chromatography and Tandem Mass Spectrometry Molecular Networking for the Discovery of Potent Antiviral Compounds from Euphorbia semiperfoliata. J. Nat. Prod. 2017, 80, 2620–2629. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onstein, R.E.; Carter, R.J.; Xing, Y.; Richardson, J.E.; Linder, H.P. Do Mediterranean-type ecosystems have a common history?--insights from the Buckthorn family (Rhamnaceae). Evolution 2015, 69, 756–771. [Google Scholar] [CrossRef] [PubMed]
- March, R.E.; Lewars, E.G.; Stadey, C.J.; Miao, X.-S.; Zhao, X.; Metcalfe, C.D. A comparison of flavonoid glycosides by electrospray tandem mass spectrometry. Int. J. Mass Spectrom. 2006, 248, 61–85. [Google Scholar] [CrossRef]
- Van der Hooft, J.J.J.; Vervoort, J.; Bino, R.J.; de Vos, R.C.H. Spectral trees as a robust annotation tool in LC–MS based metabolomics. Metabolomics 2012, 8, 691–703. [Google Scholar] [CrossRef]
- Van der Hooft, J.J.J.; Vervoort, J.; Bino, R.J.; Beekwilder, J.; de Vos, R.C.H. Polyphenol identification based on systematic and robust high-resolution accurate mass spectrometry fragmentation. Anal. Chem. 2011, 83, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Crüsemann, M.; O’Neill, E.C.; Larson, C.B.; Melnik, A.V.; Floros, D.J.; da Silva, R.R.; Jensen, P.R.; Dorrestein, P.C.; Moore, B.S. Prioritizing Natural Product Diversity in a Collection of 146 Bacterial Strains Based on Growth and Extraction Protocols. J. Nat. Prod. 2017, 80, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Crnovčić, I.; Semsary, S.; Vater, J.; Keller, U. Biosynthetic rivalry of o-aminophenol-carboxylic acids initiates production of hemi-actinomycins in Streptomyces antibioticus. RSC Adv. 2014, 4, 5065. [Google Scholar] [CrossRef]
- Tobias, N.J.; Wolff, H.; Djahanschiri, B.; Grundmann, F.; Kronenwerth, M.; Shi, Y.-M.; Simonyi, S.; Grün, P.; Shapiro-Ilan, D.; Pidot, S.J.; et al. Natural product diversity associated with the nematode symbionts Photorhabdus and Xenorhabdus. Nat. Microbiol. 2017, 2, 1676. [Google Scholar] [CrossRef]
- Shi, Y.-M.; Bode, H.B. Chemical language and warfare of bacterial natural products in bacteria–nematode–insect interactions. Nat. Product Rep. 2018, 35, 309–335. [Google Scholar] [CrossRef]
- Tobias, N.; Parra-Rojas, C.; Shi, Y.-N.; Shi, Y.-M.; Simonyi, S.; Thanwisai, A.; Vitta, A.; Chantratita, N.; Hernandez-Vargas, E.A.; Bode, H.B. Focused natural product elucidation by prioritizing high-throughput metabolomic studies with machine learning. bioRxiv 2019, 535781. [Google Scholar]
- Zhao, L.; Kaiser, M.; Bode, H.B. Rhabdopeptide/Xenortide-like Peptides from Xenorhabdus innexi with Terminal Amines Showing Potent Antiprotozoal Activity. Org. Lett. 2018, 20, 5116–5120. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Dowling, A.; Heide, H.; Wöhnert, J.; Brandt, U.; Baum, J.; ffrench-Constant, R.; Bode, H.B. Xentrivalpeptides A–Q: Depsipeptide Diversification in Xenorhabdus. J. Nat. Prod. 2012, 75, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.W.; Proschak, A.; Jaskolla, T.W.; Karas, M.; Bode, H.B. Structure elucidation and biosynthesis of lysine-rich cyclic peptides in Xenorhabdus nematophila. Org. Biomol. Chem. 2011, 9, 3130–3132. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Grundmann, F.; Kaiser, M.; Schiell, M.; Gaudriault, S.; Batzer, A.; Kurz, M.; Bode, H.B. Structure and biosynthesis of xenoamicins from entomopathogenic Xenorhabdus. Chemistry 2013, 19, 16772–16779. [Google Scholar] [CrossRef] [PubMed]
- Metabolomics: Dark matter. Nature 2008, 455, 698. [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crupi, P.; Antonacci, D.; Savino, M.; Genghi, R.; Perniola, R.; Coletta, A. Girdling and gibberellic acid effects on yield and quality of a seedless red table grape for saving irrigation water supply. Eur. J. Agron. 2016, 80, 21–31. [Google Scholar] [CrossRef]
- Pan, A.; Chen, M.; Chowdhury, R.; Wu, J.H.Y.; Sun, Q.; Campos, H.; Mozaffarian, D.; Hu, F.B. α-Linolenic acid and risk of cardiovascular disease: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2012, 96, 1262–1273. [Google Scholar] [CrossRef]
- Oberacher, H.; Reinstadler, V.; Kreidl, M.; Stravs, M.A.; Hollender, J.; Schymanski, E.L. Annotating Nontargeted LC-HRMS/MS Data with Two Complementary Tandem Mass Spectral Libraries. Metabolites 2018, 9, 3. [Google Scholar] [CrossRef]
- Scheubert, K.; Hufsky, F.; Petras, D.; Wang, M.; Nothias, L.-F.; Dührkop, K.; Bandeira, N.; Dorrestein, P.C.; Böcker, S. Significance estimation for large scale metabolomics annotations by spectral matching. Nat. Commun. 2017, 8, 1494. [Google Scholar] [CrossRef]
- Olivon, F.; Grelier, G.; Roussi, F.; Litaudon, M.; Touboul, D. MZmine 2 Data-Preprocessing To Enhance Molecular Networking Reliability. Anal. Chem. 2017, 89, 7836–7840. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.; Ong, C.W.; Wandy, J.; Ernst, M.; Ridder, L.; van der Hooft, J.J.J. Deciphering complex metabolite mixtures by unsupervised and supervised substructure discovery and semi-automated annotation from MS/MS spectra. Faraday Discuss. 2019. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ernst, M.; Kang, K.B.; Caraballo-Rodríguez, A.M.; Nothias, L.-F.; Wandy, J.; Chen, C.; Wang, M.; Rogers, S.; Medema, M.H.; Dorrestein, P.C.; et al. MolNetEnhancer: Enhanced Molecular Networks by Integrating Metabolome Mining and Annotation Tools. Metabolites 2019, 9, 144. https://doi.org/10.3390/metabo9070144
Ernst M, Kang KB, Caraballo-Rodríguez AM, Nothias L-F, Wandy J, Chen C, Wang M, Rogers S, Medema MH, Dorrestein PC, et al. MolNetEnhancer: Enhanced Molecular Networks by Integrating Metabolome Mining and Annotation Tools. Metabolites. 2019; 9(7):144. https://doi.org/10.3390/metabo9070144
Chicago/Turabian StyleErnst, Madeleine, Kyo Bin Kang, Andrés Mauricio Caraballo-Rodríguez, Louis-Felix Nothias, Joe Wandy, Christopher Chen, Mingxun Wang, Simon Rogers, Marnix H. Medema, Pieter C. Dorrestein, and et al. 2019. "MolNetEnhancer: Enhanced Molecular Networks by Integrating Metabolome Mining and Annotation Tools" Metabolites 9, no. 7: 144. https://doi.org/10.3390/metabo9070144
APA StyleErnst, M., Kang, K. B., Caraballo-Rodríguez, A. M., Nothias, L. -F., Wandy, J., Chen, C., Wang, M., Rogers, S., Medema, M. H., Dorrestein, P. C., & van der Hooft, J. J. J. (2019). MolNetEnhancer: Enhanced Molecular Networks by Integrating Metabolome Mining and Annotation Tools. Metabolites, 9(7), 144. https://doi.org/10.3390/metabo9070144