
Mechanisms of Electron-Induced Chemistry in Molecular Ices

 , , , and
, , , and
Abstract
:1. Introduction
- (i)
- provides a summary of our experimental approach which relies on a sufficient resolution in electron energy to identify resonant enhancements and energetic thresholds in the energy dependence of product yields and thus the electron-molecule interactions that initiate product formation,
- (ii)
- shows how information on the mechanisms of electron-driven chemistry in ices can be obtained by investigating the energy dependence of several products in the same sample or by comparing different ice systems, and
- (iii)
- discusses that the chemistry following the initial electron-molecule encounter can be understood based on established reaction mechanisms known from organic chemistry combined with quantum chemical calculations.
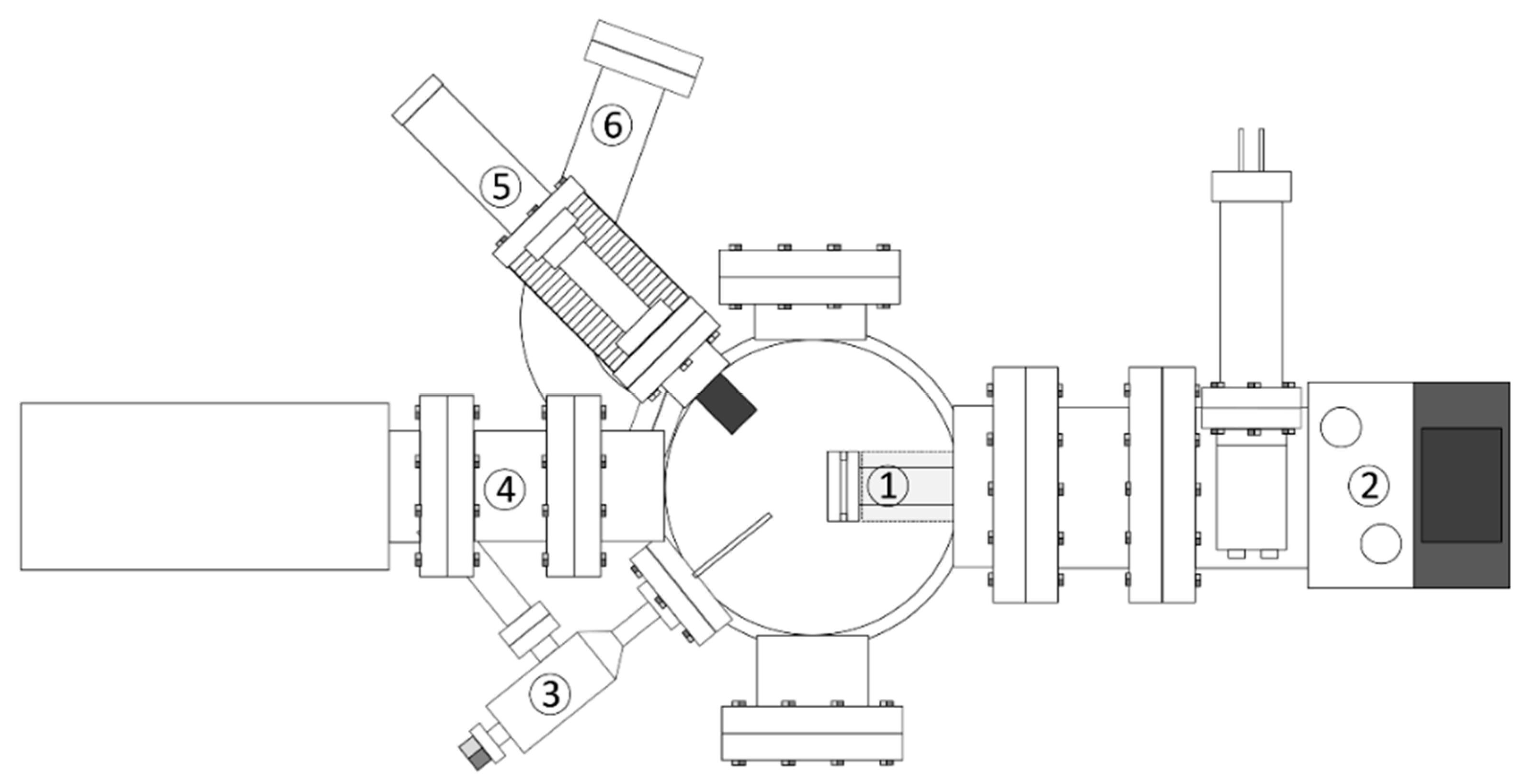
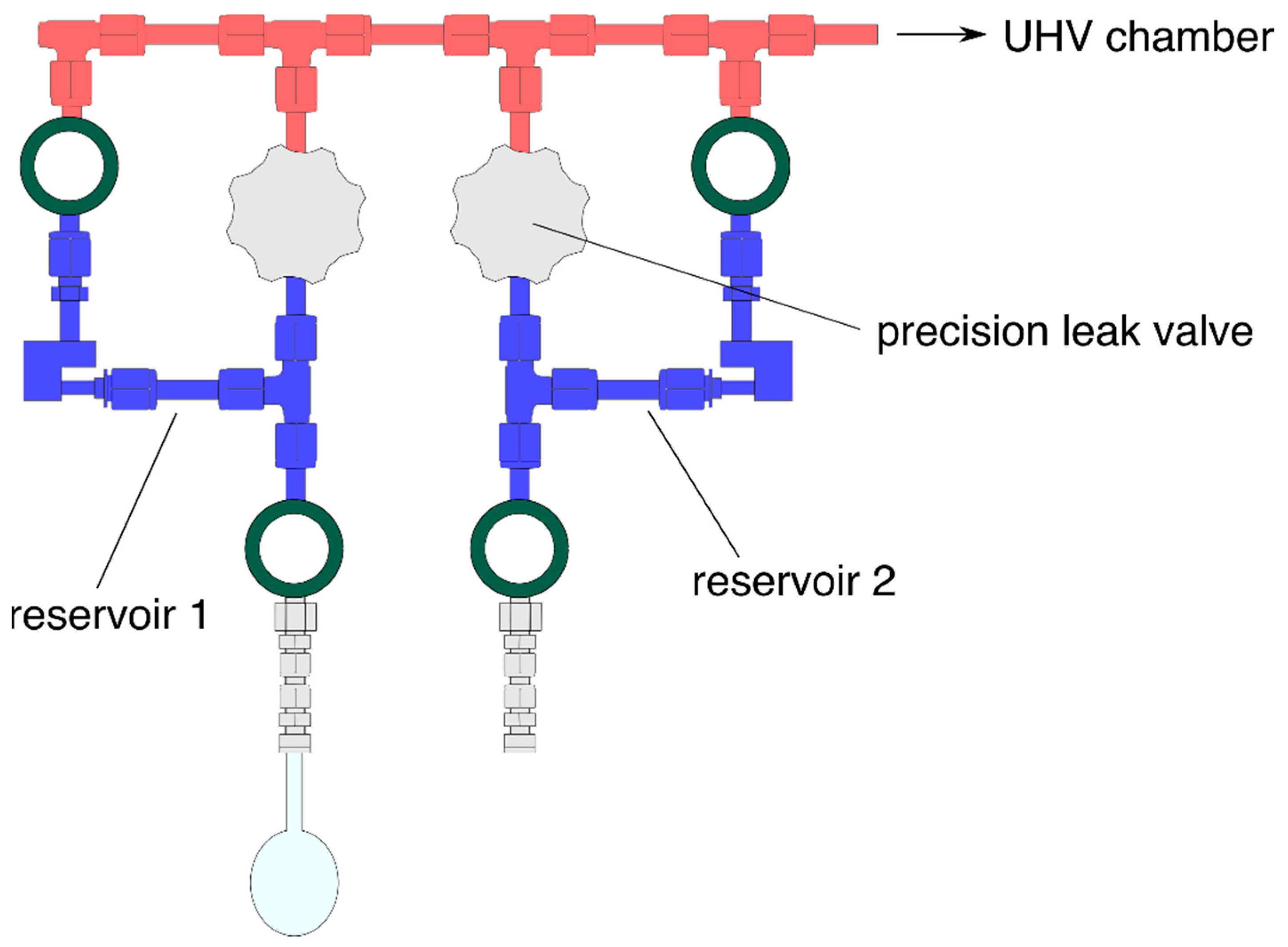
2. Experimental Approach
- (i)
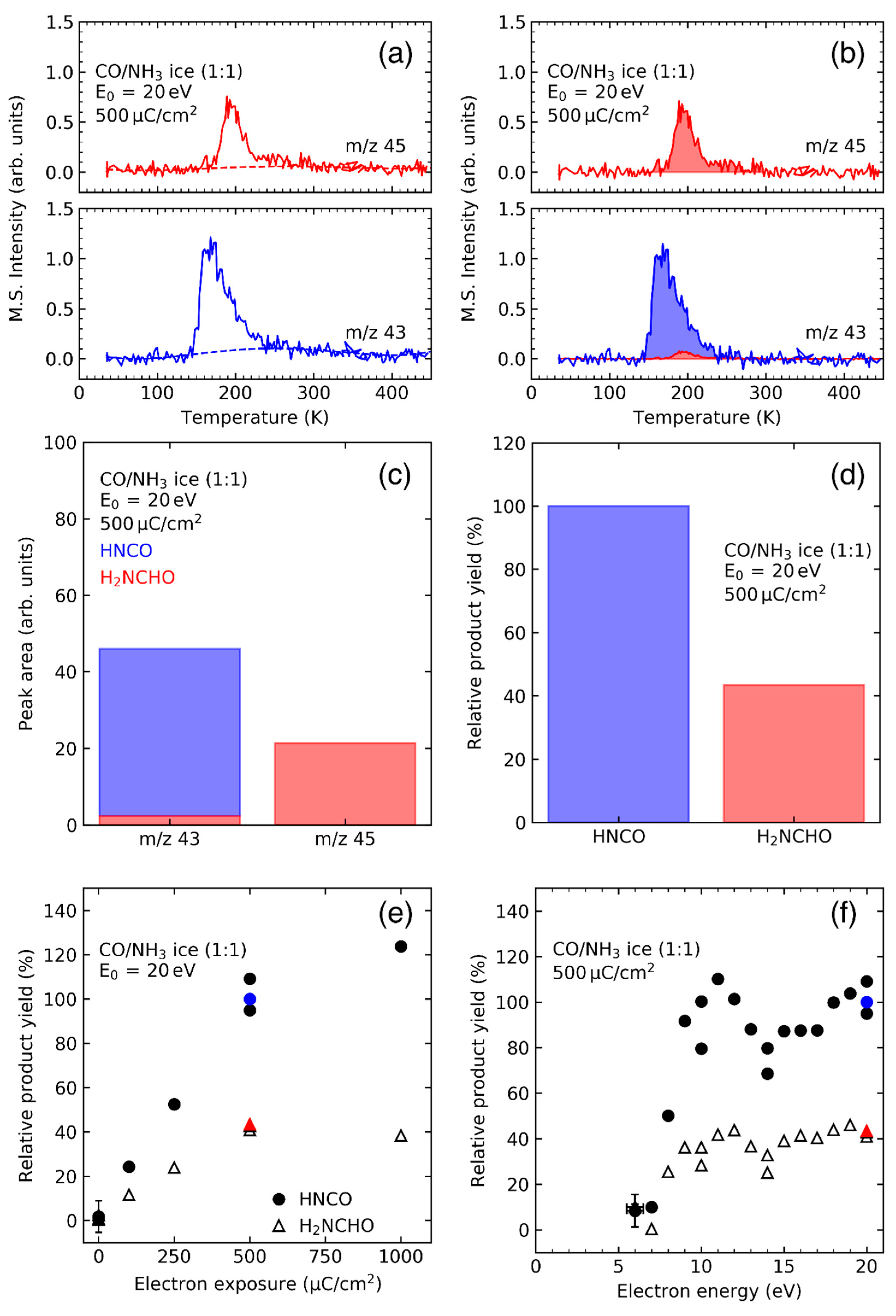
- Products that are formed after electron irradiation are identified from TDS data for characteristic m/z values. In the ideal case, the desorption of a particular species can be seen in TDS curves for several m/z values and the intensity of the desorption peaks for these different m/z reflects the intensities in the mass spectra (MS) of this compound recorded on the same instrument when leaking the pure vapor of the product into the UHV chamber.
- (ii)
- Product formation is quantified from integrated desorption signals for a characteristic m/z ratio. The absolute product yields can be derived by comparing these integrated desorption signals to those obtained from ices that have been prepared with defined composition and surface coverage, as can be deduced from TDS, so that the amount of the respective product is known [31,35,36]. Relative amounts of different products can also be obtained without such a standard sample by using partial ionization cross sections for the characteristic m/z ratios used to monitor the products. The partial ionization cross sections for a given m/z ratio with intensity of a specific product is defined by , where are the intensities of all m/z ratios of that product and is its total ionization cross section. These cross sections, that refer to the electron energy applied for ionization in the mass spectrometer, define the intensities with which specific m/z ratios appear in the mass spectrum. If known, they can be used as correction factor that is applied to compare the intensities for specific m/z ratios and thus also the desorption peak areas of two different products and to convert them to a relative yield [24,29,31].
- (iii)
- The thus obtained product yields are determined as function of electron exposure, which is defined as the transmitted charge per surface area as measured on the metal foil, to establish over which range they increase linearly with exposure. In this linear regime, the decay of the initial reactants is still negligible and the product is not yet consumed by electron irradiation [24,26,29,30,31,32,35,36]. Under this condition, the rate with which a product is formed is directly proportional to the applied electron exposure. This is the basis on which the rates of formation can be compared between different products.
- (iv)
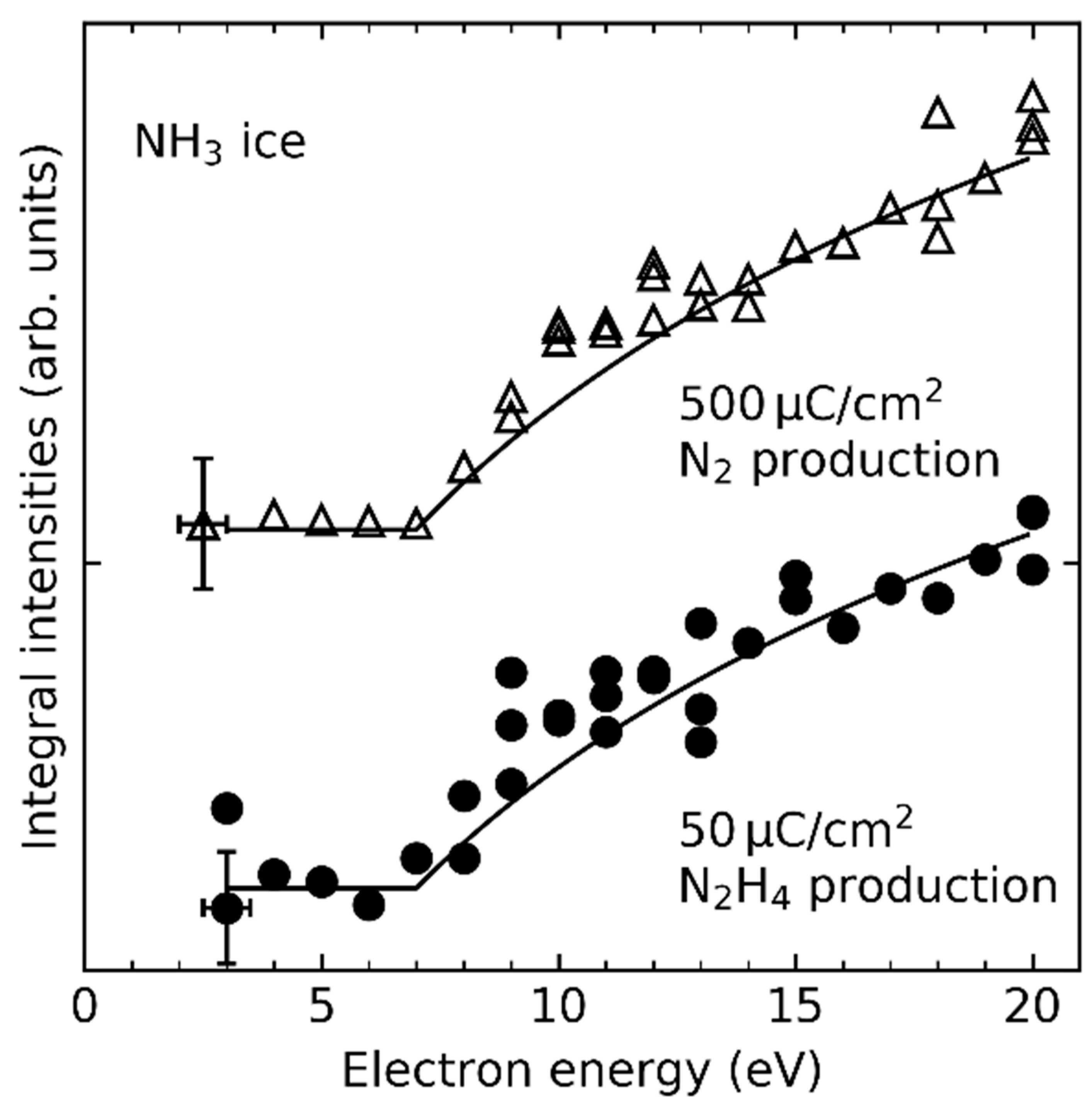
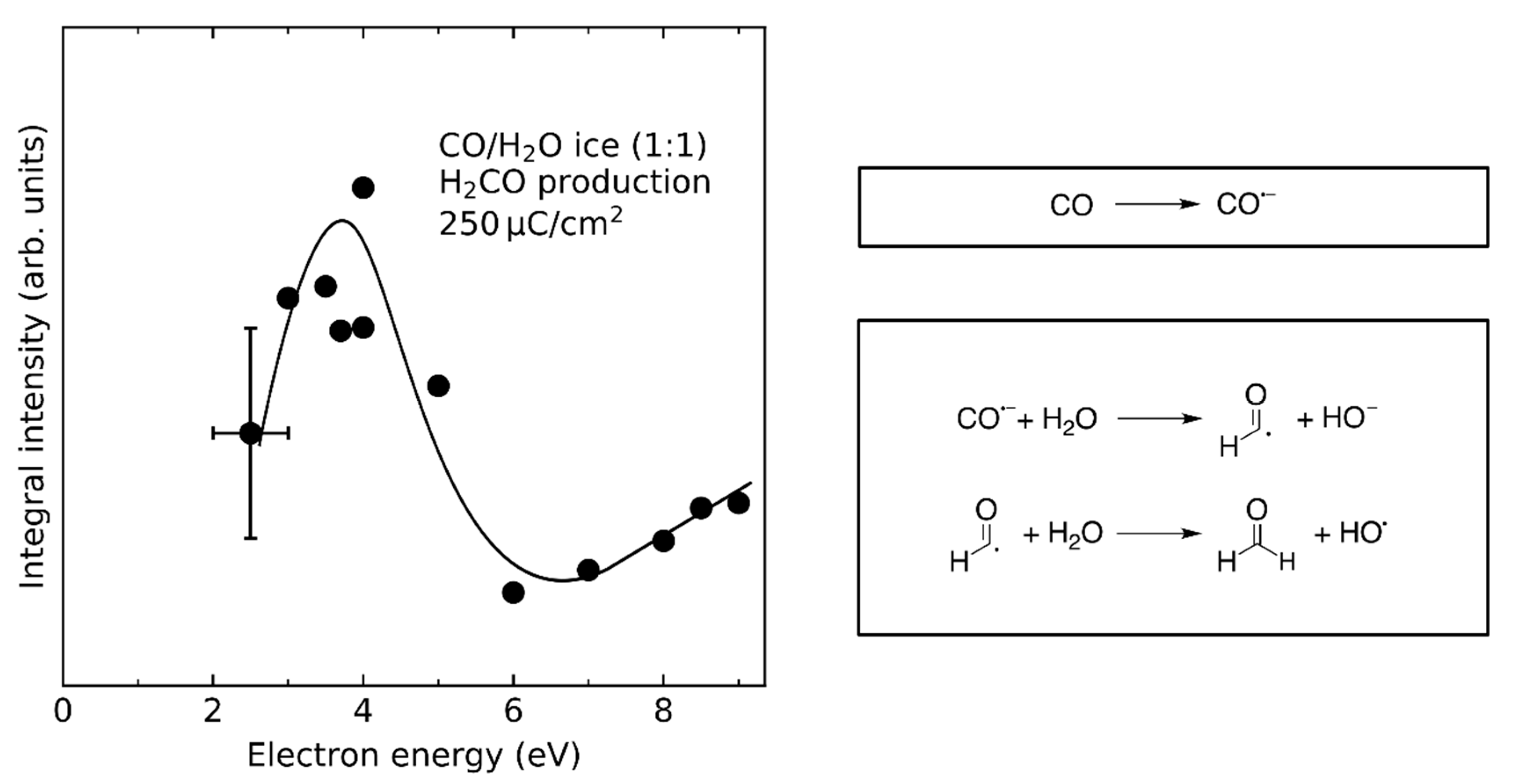
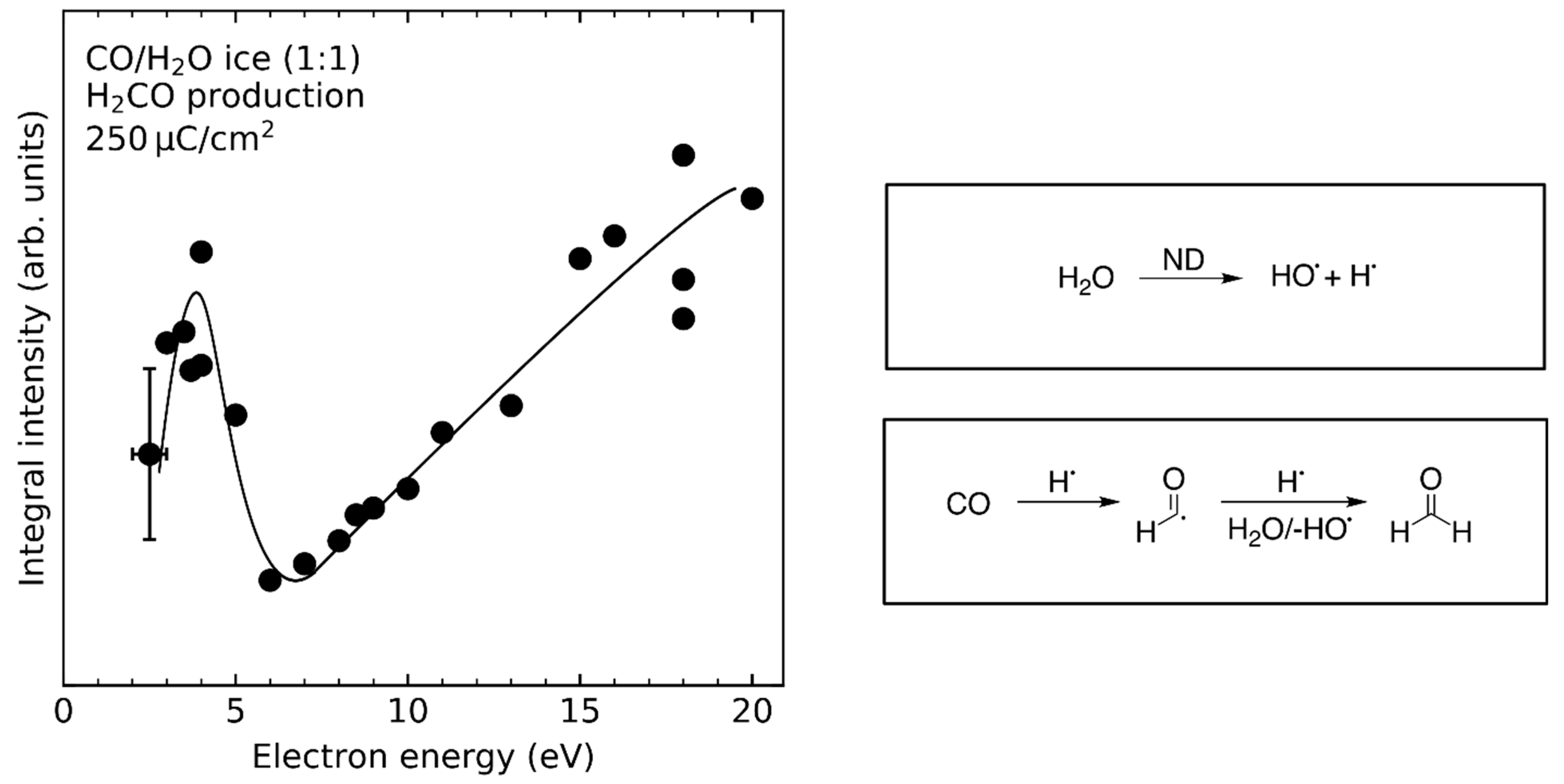
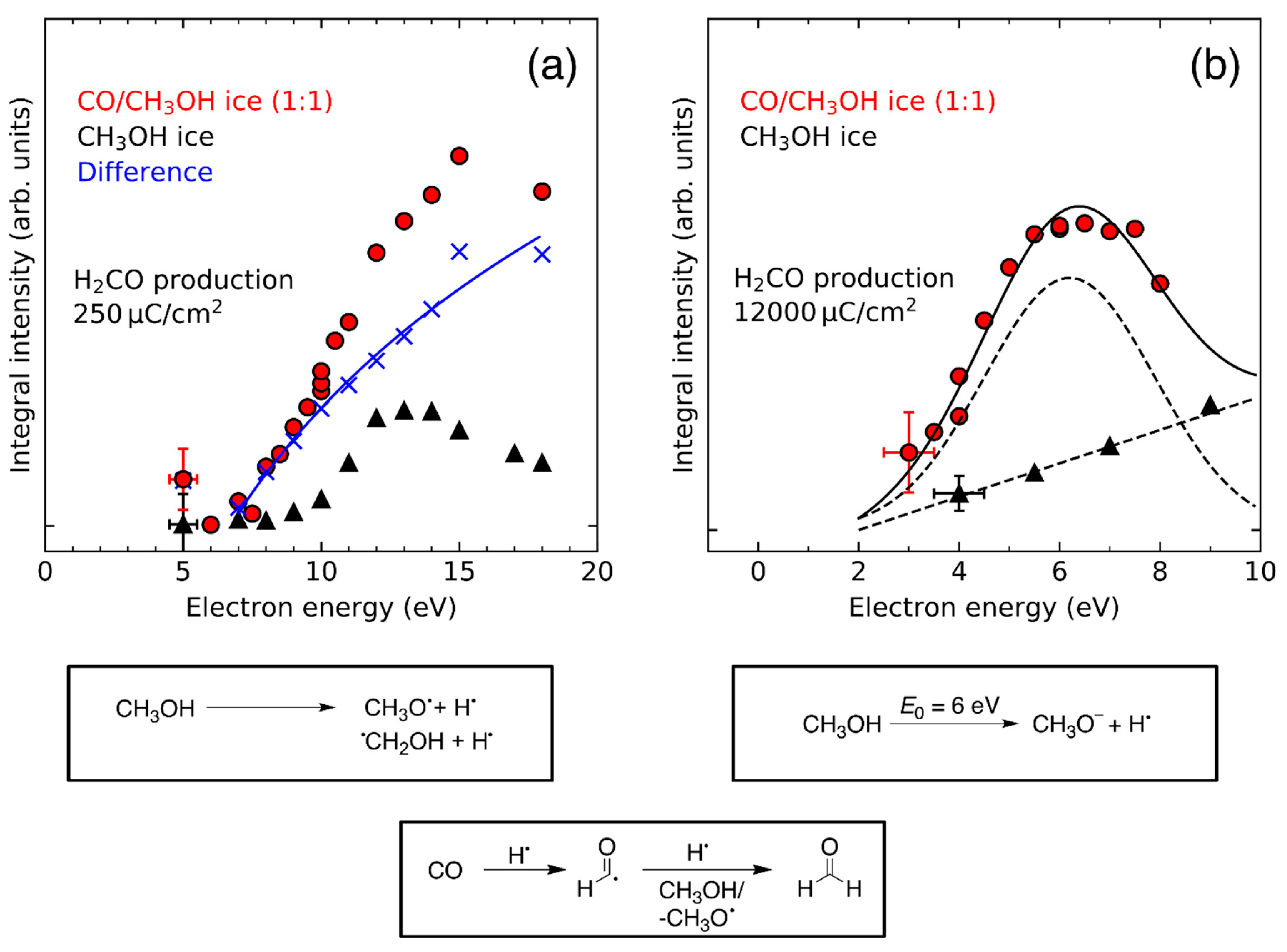
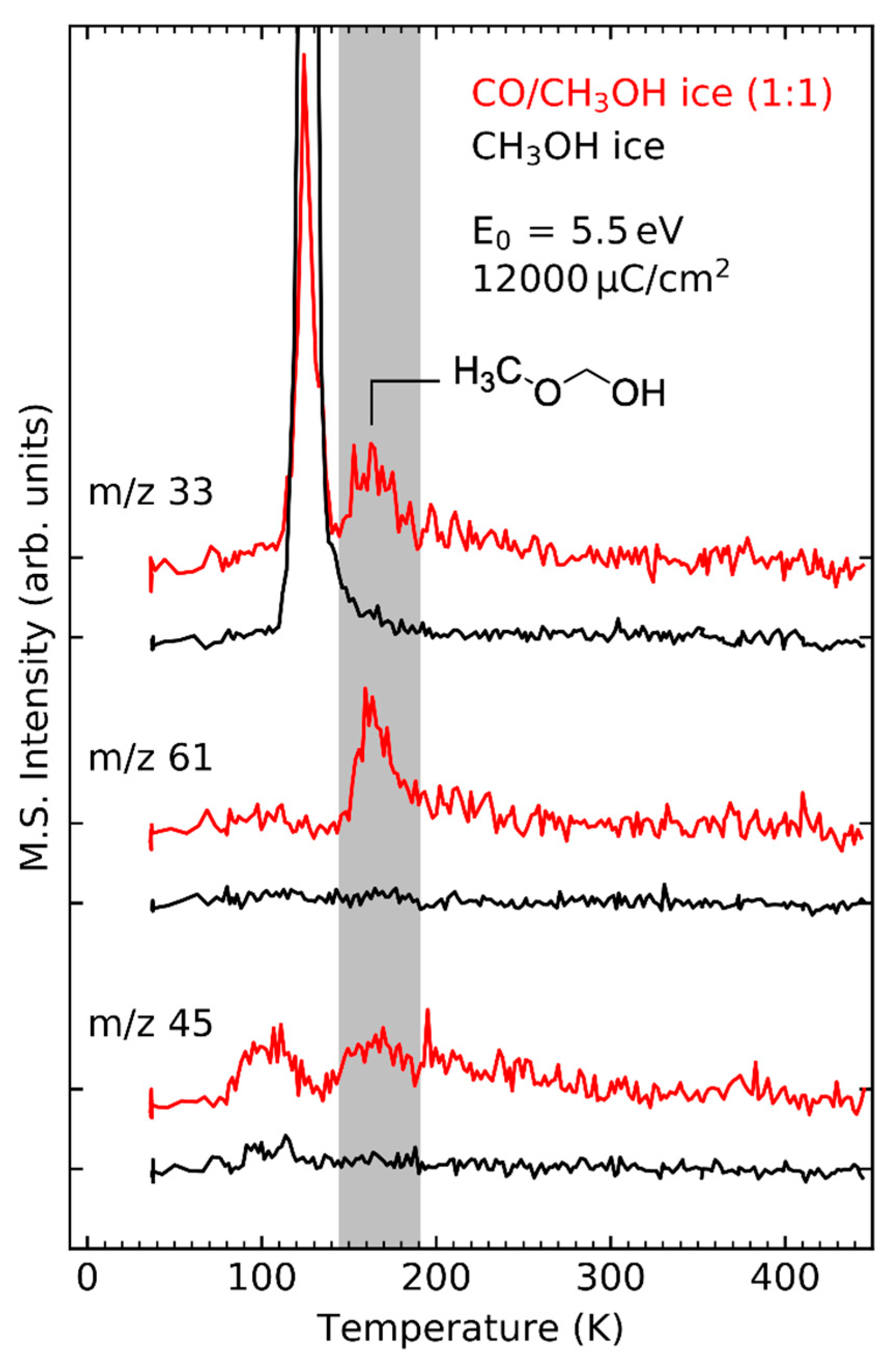
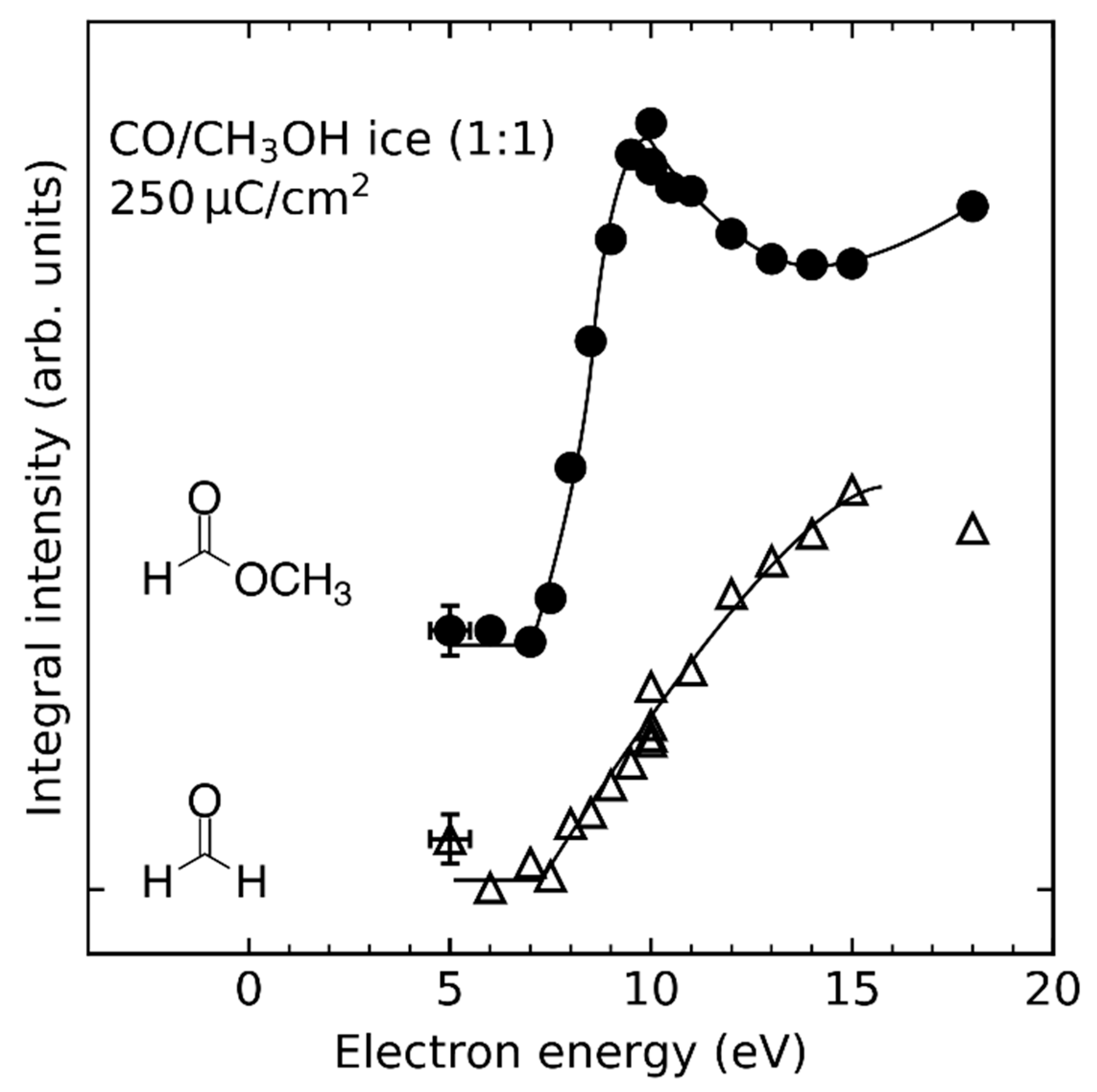
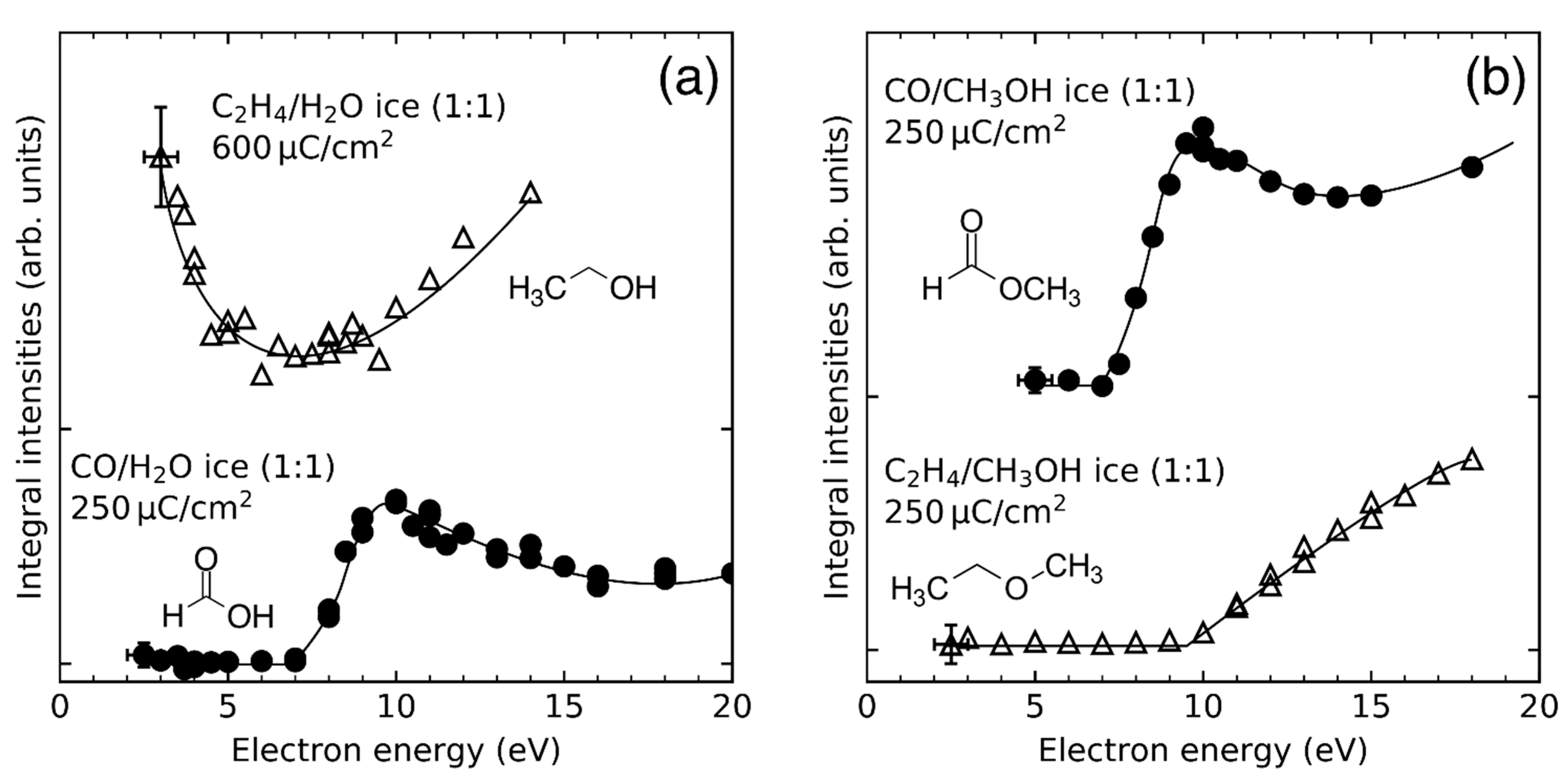
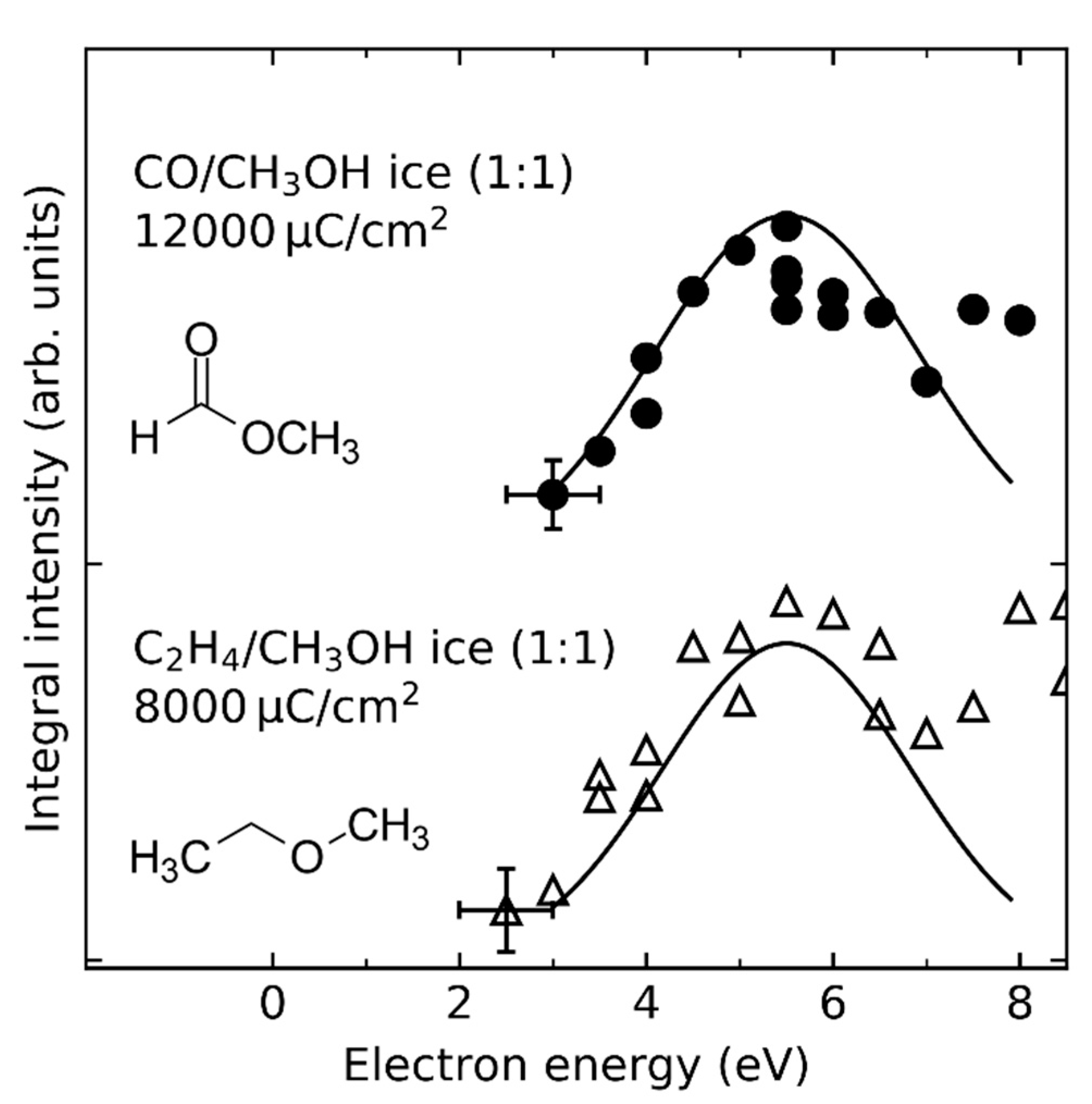
- Product yields are then measured as function of electron energy (E0) for an exposure that is within the linear regime for all investigated electron energies. The energy dependence of the product yields gives information on the electron-molecule interactions that initiate product formation. This includes thresholds for electronic excitation or ionization as well as maxima within specific energy ranges that point to a contribution of electron attachment. These processes are summarized in Section 3. When monitoring product formation in mixed ices, it is not obvious from the energy dependence alone which constituent undergoes the initiating electron-molecule interaction if both show similar DI or ND thresholds or energy ranges for DEA. In these cases, more detailed mechanistic information can be derived by comparing the energy dependent yields of several products [24,26] as will be exemplified in Section 4. In particular cases, a unique assignment can be obtained by exchanging one component of the ice for another and checking whether product formation still occurs [37].
3. How Do Reactive Species Form under Electron Irradiation?
3.1. Basics of Electron Molecule Interactions
3.2. Reactive Intermediates Formed by EI/DI
3.3. Reactive Intermediates from ND
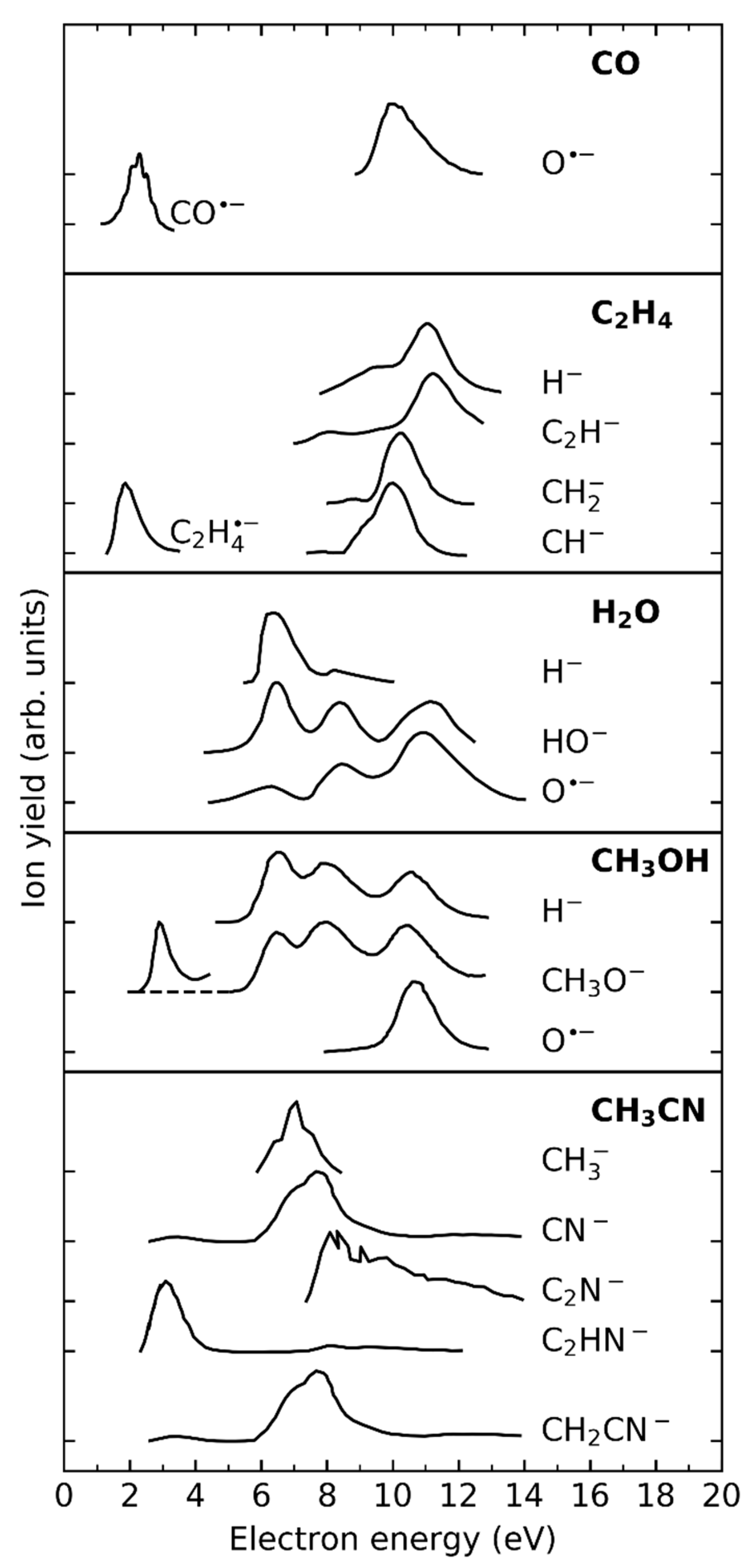
3.4. Reactive Intermediates from (D)EA
3.5. Effect of Condensed Phase
4. How Do Reactive Species Undergo Chemical Transformation in Condensed Phase?
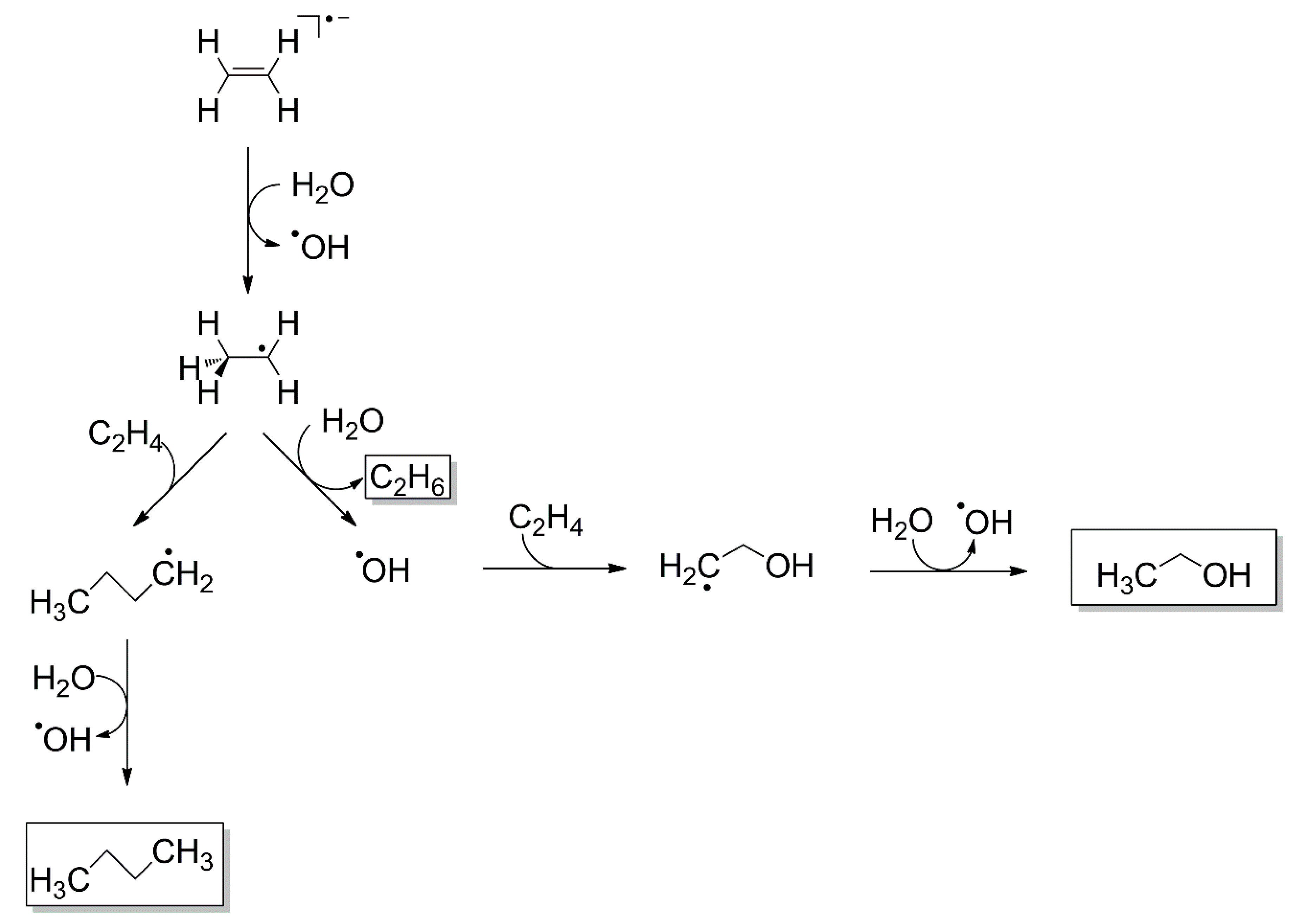
4.1. Radical Recombination
4.2. Transfer Hydrogenation
4.3. Reduction by Hydrogen Radicals
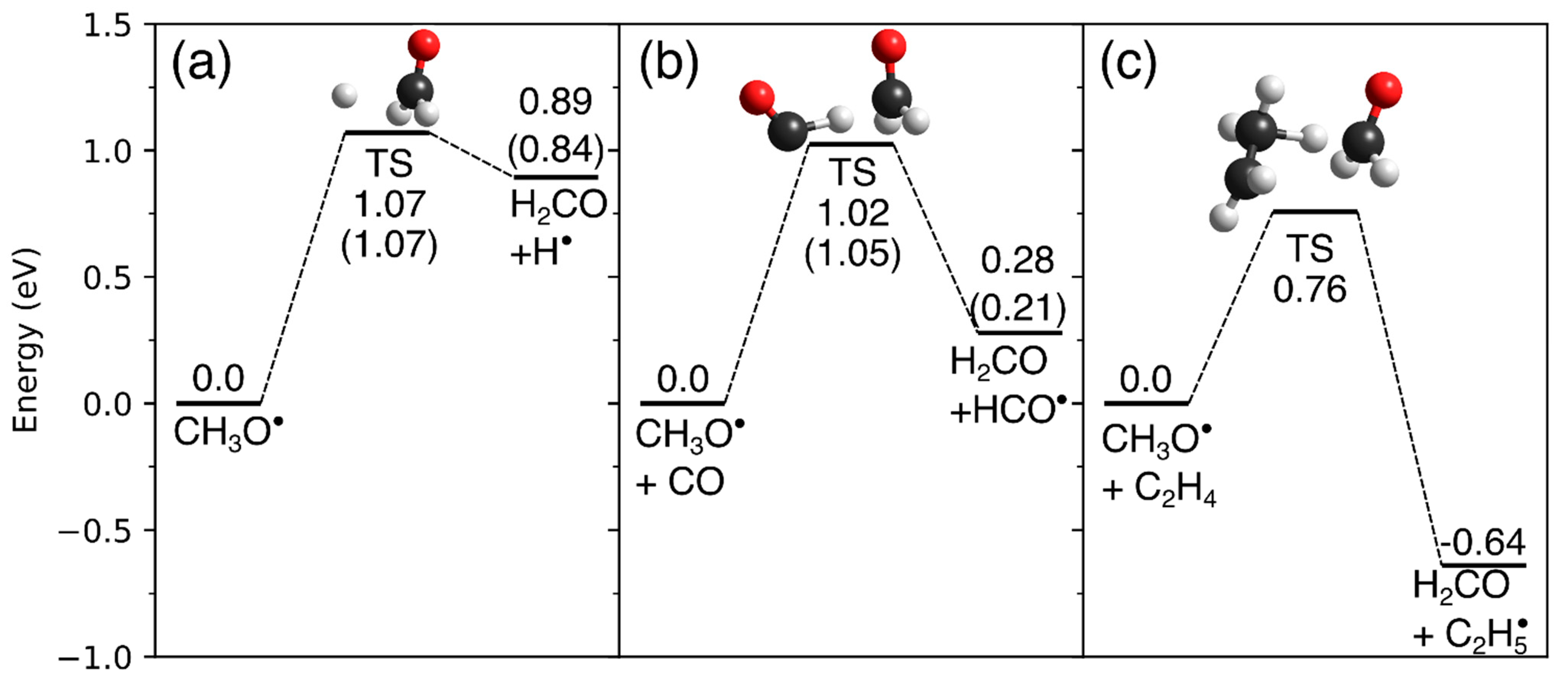
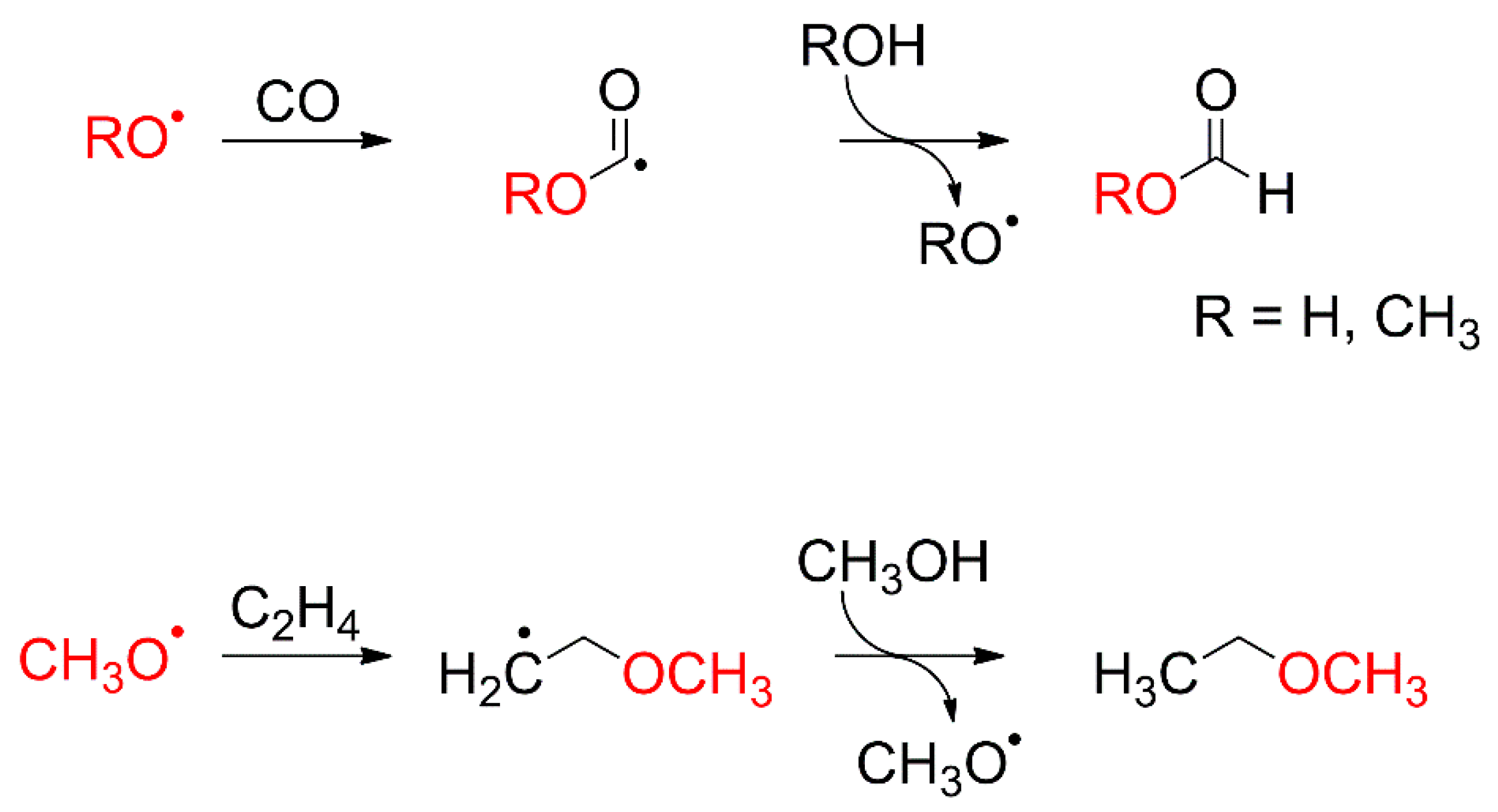
4.4. Hydrogenation by Methoxy Radicals
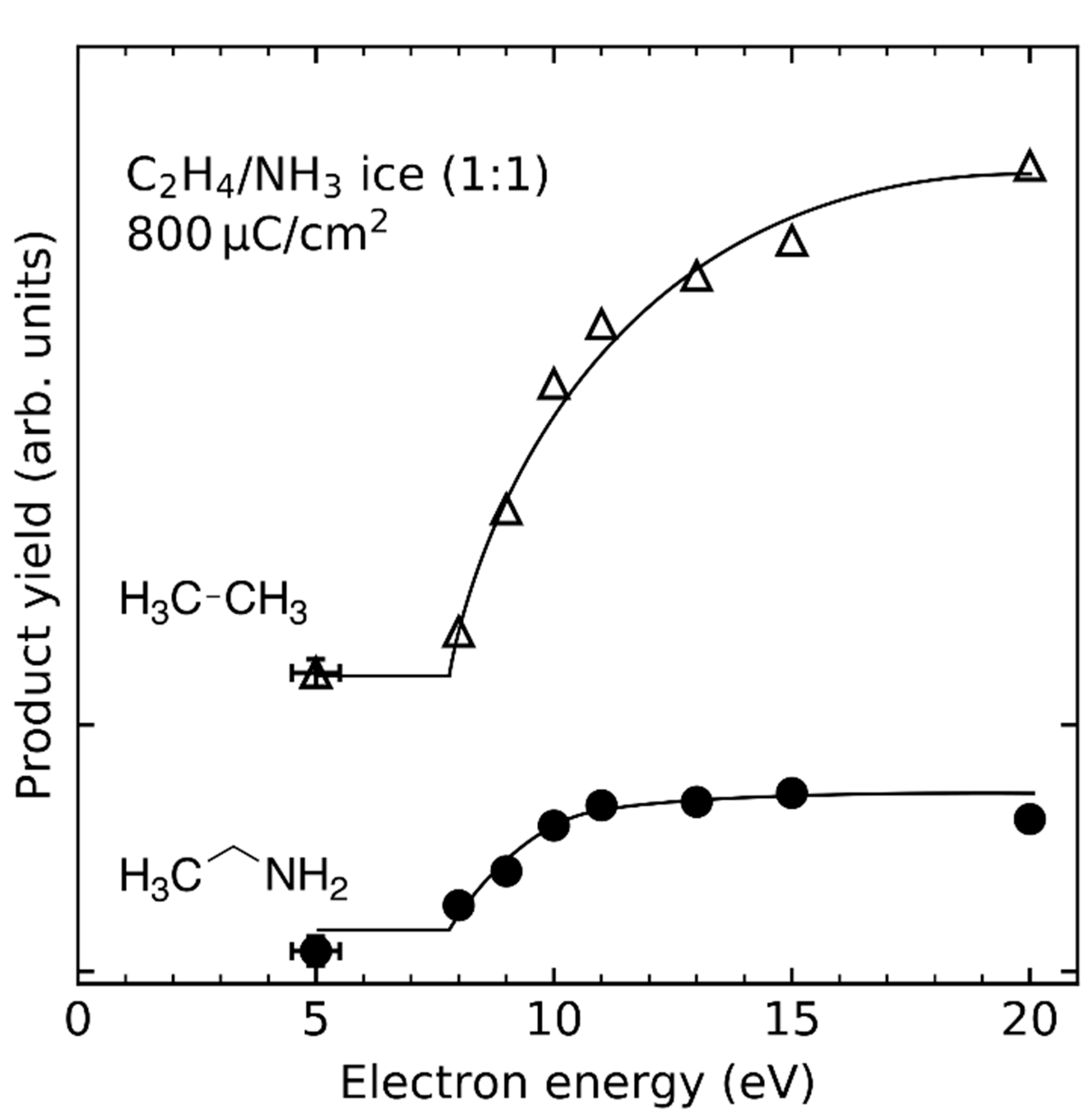
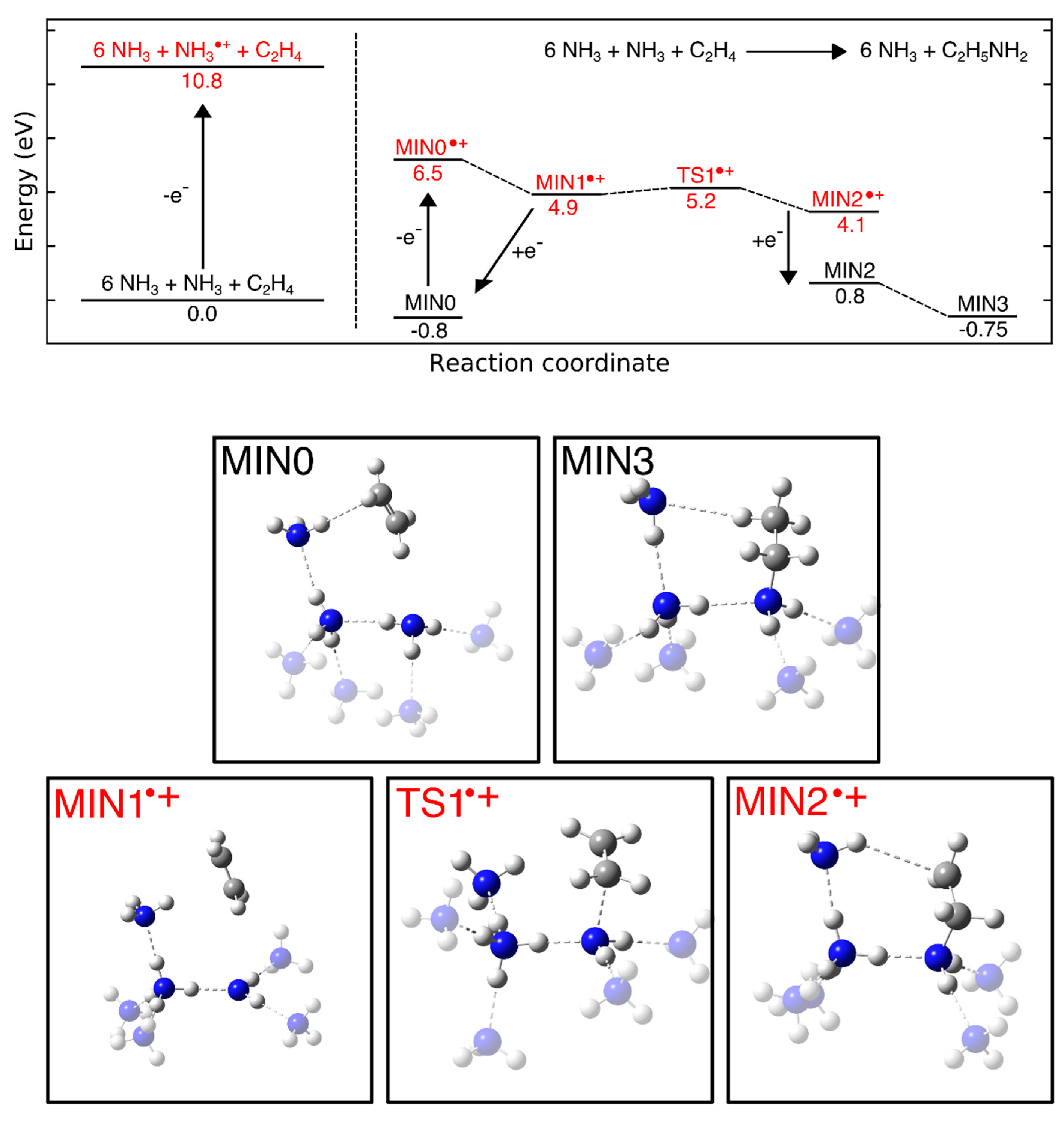
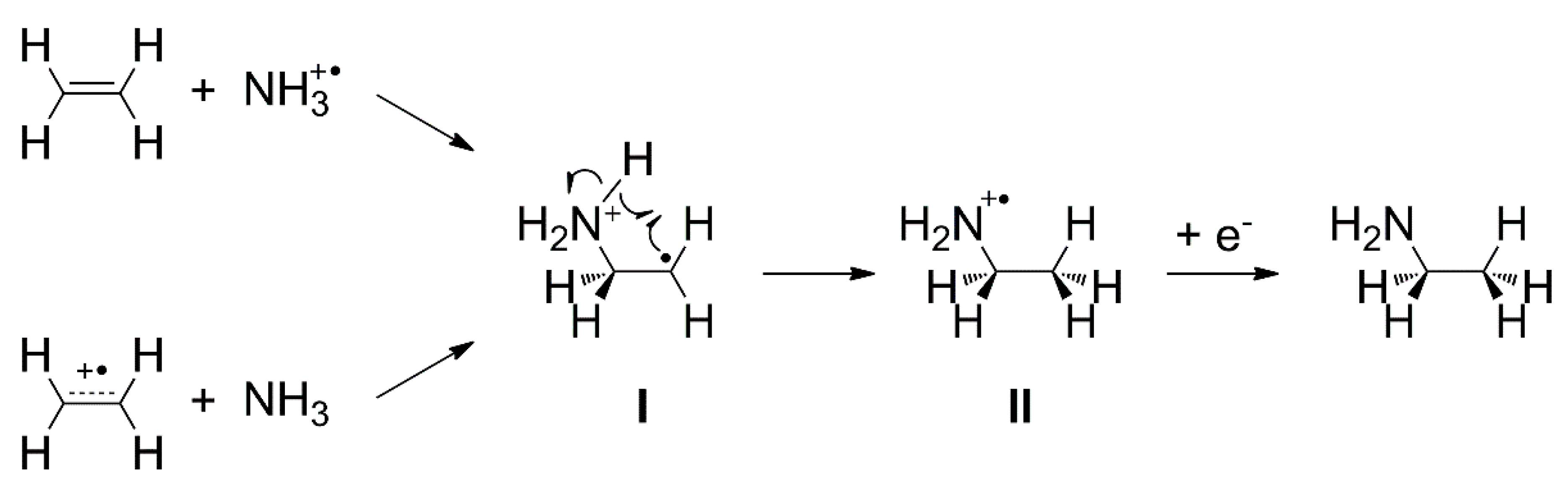
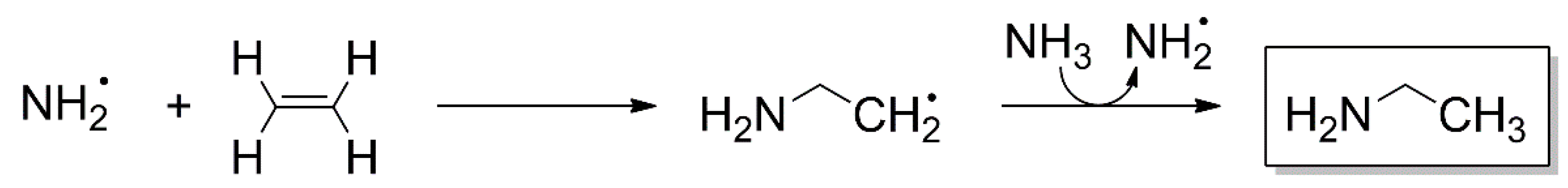
4.5. Addition Reactions Involving Radical Cations or Neutral Radicals
4.6. Oxidation of CO to CO2
5. Open Questions and Perspectives for Future Studies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ingólfsson, O. Low-Energy Electrons: Fundamentals and Applications; Pan Stanford Publishing: Singapore, 2019. [Google Scholar]
- Kumar, A.; Becker, D.; Adhikary, A.; Sevilla, M.D. Reaction of Electrons with DNA: Radiation Damage to Radiosensitization. Int. J. Mol. Sci. 2019, 20, 3998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.K.; Chaki, T.K.; Bhowmick, A.K. Electron Beam Processing of Polymers. In Advanced Polymer Processing Operations; Cheremisinoff, N.P., Ed.; William Andrew Publishing: Norwich, NY, USA, 1998; pp. 157–186. [Google Scholar]
- Dispenza, C.; Giacomazza, D.; Jonsson, M. Micro-to Nanoscale Bio-Hybrid Hydrogels Engineered by Ionizing Radiation. Biomolecules 2021, 11, 47. [Google Scholar] [CrossRef] [PubMed]
- Utke, I.; Hoffmann, P.; Melngailis, J. Gas-assisted focused electron beam and ion beam processing and fabrication. J. Vac. Sci. Technol. B 2008, 26, 1197–1276. [Google Scholar] [CrossRef] [Green Version]
- Huth, M.; Porrati, F.; Dobrovolskiy, O.V. Focused electron beam induced deposition meets materials science. Microelectron. Eng. 2018, 185–186, 9–28. [Google Scholar] [CrossRef] [Green Version]
- Wagner, C.; Harned, N. Lithography gets extreme. Nat. Photonics 2010, 4, 24–26. [Google Scholar] [CrossRef]
- Arumainayagam, C.R.; Garrod, R.T.; Boyer, M.C.; Hay, A.K.; Bao, S.T.; Campbell, J.S.; Wang, J.; Nowak, C.M.; Arumainayagam, M.R.; Hodge, P.J. Extraterrestrial prebiotic molecules: Photochemistry vs. radiation chemistry of interstellar ices. Chem. Soc. Rev. 2019, 48, 2293–2314. [Google Scholar] [CrossRef] [Green Version]
- Mason, N.J.; Nair, B.; Jheeta, S.; Szymańska, E. Electron induced chemistry: A new frontier in astrochemistry. Faraday Discuss. 2014, 168, 235–247. [Google Scholar] [CrossRef] [Green Version]
- Arumainayagam, C.R.; Lee, H.-L.; Nelson, R.B.; Haines, D.R.; Gunawardane, R.P. Low-energy electron-induced reactions in condensed matter. Surf. Sci. Rep. 2010, 65, 1–44. [Google Scholar] [CrossRef]
- Boyer, M.C.; Rivas, N.; Tran, A.A.; Verish, C.A.; Arumainayagam, C.R. The role of low-energy (≤20 eV) electrons in astrochemistry. Surf. Sci. 2016, 652, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.H.; Naulleau, P.; Ahmed, M.; Kostko, O. Determination of effective attenuation length of slow electrons in polymer films. J. Appl. Phys. 2020, 127, 245301. [Google Scholar] [CrossRef]
- Thorman, R.M.; Kumar, T.P.R.; Fairbrother, D.H.; Ingólfsson, O. The role of low-energy electrons in focused electron beam induced deposition: Four case studies of representative precursors. Beilstein J. Nanotechnol. 2015, 6, 1904–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, J.A.; Rosenberg, S.G.; Barclay, M.; Wu, Y.-C.; McElwee-White, L.; Fairbrother, D.H. Understanding the electron-stimulated surface reactions of organometallic complexes to enable design of precursors for electron beam-induced deposition. Appl. Phys. A 2014, 117, 1631–1644. [Google Scholar] [CrossRef]
- Rohdenburg, M.; Thakur, N.; Cartaya, R.; Castellanos, S.; Swiderek, P. Role of low-energy electrons in the solubility switch of Zn-based oxocluster photoresist for extreme ultraviolet lithography. Phys. Chem. Chem. Phys. 2021, 23, 16646–16657. [Google Scholar] [CrossRef] [PubMed]
- Böhler, E.; Warneke, J.; Swiderek, P. Control of chemical reactions and synthesis by low-energy electrons. Chem. Soc. Rev. 2013, 42, 9219–9231. [Google Scholar] [CrossRef]
- Moore, J.H.; Swiderek, P.; Matejcik, S.; Allan, M. Fundamentals of interactions of electrons with molecules. In Nanofabrication Using Focused Ion and Electron Beams: Principles and Applications; Russel, P., Moshkalev, S., Utke, I., Eds.; Oxford University Press: New York, NY, USA, 2012; pp. 184–225. [Google Scholar]
- Goesmann, F.; Raulin, F.; Bredehöft, J.H.; Cabane, M.; Ehrenfreund, P.; MacDermott, A.J.; McKenna-Lawlor, S.; Meierhenrich, U.J.; Muñoz Caro, G.M.; Szopa, C.; et al. COSAC prepares for sampling and in situ analysis of cometary matter from comet 67P/Churyumov–Gerasimenko. Planet. Space Sci. 2014, 103, 318–330. [Google Scholar] [CrossRef]
- Goesmann, F.; McKenna-Lawlor, S.; Roll, R.; Bredehöft, J.H.; Meierhenrich, U.; Raulin, F.; Thiemann, W.; Muñoz Caro, G.M.; Szopa, C. Interpretation of COSAC mass spectrometer data acquired during Rosetta’s Lutetia fly-by 10 July 2010. Planet. Space Sci. 2012, 66, 187–191. [Google Scholar] [CrossRef]
- Gardener, J.A.; Golovchenko, J.A. Ice-assisted electron beam lithography of graphene. Nanotechnology 2012, 23, 185302. [Google Scholar] [CrossRef]
- de Rooij, A. Corrosion in Space. In Encyclopedia of Aerospace Engineering; Blockley, R., Shyy, W., Eds.; John Wiley & Sons: Chichester, UK, 2010. [Google Scholar] [CrossRef]
- Lafosse, A.; Bertin, M.; Azria, R. Electron driven processes in ices: Surface functionalization and synthesis reactions. Prog. Surf. Sci. 2009, 84, 177–198. [Google Scholar] [CrossRef]
- Ipolyi, I.; Michaelis, W.; Swiderek, P. Electron-induced reactions in condensed films of acetonitrile and ethane. Phys. Chem. Chem. Phys. 2007, 9, 180–191. [Google Scholar] [CrossRef]
- Schmidt, F.; Swiderek, P.; Bredehöft, J.H. Formation of Formic Acid, Formaldehyde, and Carbon Dioxide by Electron-Induced Chemistry in Ices of Water and Carbon Monoxide. ACS Earth Space Chem. 2019, 3, 1974–1986. [Google Scholar] [CrossRef]
- Bass, A.D.; Bredehöft, J.H.; Böhler, E.; Sanche, L.; Swiderek, P. Reactions and anion desorption induced by low-energy electron exposure of condensed acetonitrile. Eur. Phys. J. D 2012, 66, 53. [Google Scholar] [CrossRef]
- Schmidt, F.; Swiderek, P.; Scheele, T.; Bredehöft, J.H. Mechanisms of methyl formate production during electron-induced processing of methanol–carbon monoxide ices. Phys. Chem. Chem. Phys. 2021, 23, 11649–11662. [Google Scholar] [CrossRef] [PubMed]
- Hamann, T.; Böhler, E.; Swiderek, P. Low-Energy-Electron-Induced Hydroamination of an Alkene. Angew. Chem. Int. Ed. 2009, 48, 4643–4645. [Google Scholar] [CrossRef] [PubMed]
- Warneke, J.; Wang, Z.; Swiderek, P.; Bredehöft, J.H. Electron-Induced Hydration of an Alkene: Alternative Reaction Pathways. Angew. Chem. Int. Ed. 2015, 54, 4397–4400. [Google Scholar] [CrossRef]
- Böhler, E.; Bredehöft, J.H.; Swiderek, P. Low-Energy Electron-Induced Hydroamination Reactions between Different Amines and Olefins. J. Phys. Chem. C 2014, 118, 6922–6933. [Google Scholar] [CrossRef]
- Bredehöft, J.H.; Böhler, E.; Schmidt, F.; Borrmann, T.; Swiderek, P. Electron-Induced Synthesis of Formamide in Condensed Mixtures of Carbon Monoxide and Ammonia. ACS Earth Space Chem. 2017, 1, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, F.; Swiderek, P.; Bredehöft, J.H. Electron-Induced Processing of Methanol Ice. ACS Earth Space Chem. 2021, 5, 391–408. [Google Scholar] [CrossRef]
- Schmidt, F.; Swiderek, P.; Bredehöft, J.H. Electron-Induced Formation of Ethyl Methyl Ether in Condensed Mixtures of Methanol and Ethylene. J. Phys. Chem. A 2019, 123, 37–47. [Google Scholar] [CrossRef]
- Öberg, K.I. Photochemistry and Astrochemistry: Photochemical Pathways to Interstellar Complex Organic Molecules. Chem. Rev. 2016, 116, 9631–9663. [Google Scholar] [CrossRef] [Green Version]
- Boamah, M.D.; Sullivan, K.K.; Shulenberger, K.E.; Soe, C.M.; Jacob, L.M.; Yhee, F.C.; Atkinson, K.E.; Boyer, M.C.; Haines, D.R.; Arumainayagam, C.R. Low-energy electron-induced chemistry of condensed methanol: Implications for the interstellar synthesis of prebiotic molecules. Faraday Discuss. 2014, 168, 249–266. [Google Scholar] [CrossRef]
- Burean, E.; Swiderek, P. Thermal desorption measurement of cross section for reactions in condensed acetaldehyde induced by low-energy electrons. Surf. Sci. 2008, 602, 3194–3198. [Google Scholar] [CrossRef]
- Burean, E.; Swiderek, P. Electron-Induced Reactions in Condensed Acetaldehyde: Identification of Products and Energy-Dependent Cross Sections. J. Phys. Chem. C 2008, 112, 19456–19464. [Google Scholar] [CrossRef]
- Schmidt, F.; Mues, M.P.; Bredehöft, J.H.; Swiderek, P. Molecular synthesis in ices triggered by dissociative electron attachment to carbon monoxide. Eur. Phys. J. D 2021, 75, 302. [Google Scholar] [CrossRef]
- Python (Version 3.8.1). Python Software Foundation. 2019. Available online: https://www.python.org/downloads/release/python-381/ (accessed on 19 December 2021).
- van der Walt, S.; Colbert, S.C.; Varoquaux, G. The NumPy Array: A Structure for Efficient Numerical Computation. Comput. Sci. Eng. 2011, 13, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Press, W.H.; Teukolsky, S.A. Savitzky-Golay Smoothing Filters. Comput. Phys. 1990, 4, 669–672. [Google Scholar] [CrossRef]
- Bald, I.; Langer, J.; Tegeder, P.; Ingólfsson, O. From isolated molecules through clusters and condensates to the building blocks of life. Int. J. Mass Spectrom. 2008, 277, 4–25. [Google Scholar] [CrossRef]
- McConkey, J.W.; Malone, C.P.; Johnson, P.V.; Winstead, C.; McKoy, V.; Kanik, I. Electron impact dissociation of oxygen-containing molecules-A critical review. Phys. Rep. 2008, 466, 1–103. [Google Scholar] [CrossRef]
- Christophorou, L.G.; Olthoff, J.K. Fundamental Electron Interactions with Plasma Processing Gases; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2004. [Google Scholar]
- Szymańska, E.; Čadež, I.; Krishnakumar, E.; Mason, N.J. Electron impact induced anion production in acetylene. Phys. Chem. Chem. Phys. 2014, 16, 3425–3432. [Google Scholar] [CrossRef] [PubMed]
- Lepage, M.; Michaud, M.; Sanche, L. Low Energy Electron Total Scattering Cross Section for the Production of CO Within Condensed Methanol. J. Chem. Phys. 1997, 107, 3478–3484. [Google Scholar] [CrossRef]
- Davis, D.; Vysotskiy, V.P.; Sajeev, Y.; Cederbaum, L.S. Electron Impact Catalytic Dissociation: Two-Bond Breaking by a Low-Energy Catalytic Electron. Angew. Chem. Int. Ed. 2011, 50, 4119–4122. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.; Vysotskiy, V.P.; Sajeev, Y.; Cederbaum, L.S. A One-Step Four-Bond-Breaking Reaction Catalyzed by an Electron. Angew. Chem. Int. Ed. 2012, 51, 8003–8007. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.; Kundu, S.; Prabhudesai, V.S.; Sajeev, Y.; Krishnakumar, E. Formation of CO2 from formic acid through catalytic electron channel. J. Chem. Phys. 2018, 149, 064308. [Google Scholar] [CrossRef] [PubMed]
- Pimblott, S.M.; LaVerne, J.A. Production of low-energy electrons by ionizing radiation. Radiat. Phys. Chem. 2007, 76, 1244–1247. [Google Scholar] [CrossRef]
- Völkel, B.; Gölzhäuser, A.; Müller, H.U.; David, C.; Grunze, M. Influence of secondary electrons in proximal probe lithography. J. Vac. Sci. Technol. B 1997, 15, 2877–2881. [Google Scholar] [CrossRef]
- Lias, S.G.; Rosenstock, H.M.; Draxl, K.; Steiner, B.W.; Herron, J.T.; Holmes, J.L.; Levin, R.D.; Liebman, J.F.; Kafafi, S.A. Ionization Energetics Data. In NIST Chemistry WebBook; Linstrom, P.J., Mallard, W.G., Eds.; NIST Standard Reference Database No. 69; National Institute for Standards and Technology: Gaithersburg, MD, USA.
- Rempt, R.D. Electron-Impact Excitation of Carbon Monoxide Near Threshold in the 1.5- to 3-eV Incident Energy Range. Phys. Rev. Lett. 1969, 22, 1034–1036. [Google Scholar] [CrossRef]
- Gope, K.; Tadsare, V.; Prabhudesai, V.S.; Mason, N.J.; Krishnakumar, E. Negative ion resonances in carbon monoxide. Eur. Phys. J. D 2016, 70, 134. [Google Scholar] [CrossRef]
- Walker, I.C.; Stamatovic, A.; Wong, S.F. Vibrational excitation of ethylene by electron impact: 1–11 eV. J. Chem. Phys. 1978, 69, 5532–5537. [Google Scholar] [CrossRef]
- Thynne, J.C.J.; MacNeil, K.A.G. Negative Ion Formation by Ethylene and 1,1-Difluoroethylene. J. Phys. Chem. 1971, 75, 2584–2591. [Google Scholar] [CrossRef]
- Trepka, L.V.; Neuert, H. Über die Entstehung von negativen Ionen aus einigen Kohlenwasserstoffen und Alkoholen durch Elektronenstoß. Z. Naturforsch. A Phys. Sci. 1963, 18, 1295–1303. [Google Scholar] [CrossRef]
- Itikawa, Y.; Mason, N. Cross Sections for Electron Collisions with Water Molecules. J. Phys. Chem. Ref. Data 2005, 34, 1–22. [Google Scholar] [CrossRef]
- Ibănescu, B.C.; May, O.; Monney, A.; Allan, M. Electron-induced chemistry of alcohols. Phys. Chem. Chem. Phys. 2007, 9, 3163–3173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heni, M.; Illenberger, E. Electron attachment by saturated nitriles, acrylonitrile (C2H3CN), and benzonitrile (C6H5CN). Int. J. Mass Spectrom. Ion Processes 1986, 73, 127–144. [Google Scholar] [CrossRef]
- Bredehöft, J.H. Electron-Induced Chemistry in the Condensed Phase. Atoms 2019, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.Y.; McMenamin, J.C.; Spicer, W.E. UPS measurements of molecular energy level of condensed gases. Surf. Sci. 1975, 50, 149–156. [Google Scholar] [CrossRef]
- Ellis-Gibbings, L.; Bass, A.D.; Cloutier, P.; García, G.; Sanche, L. Electron stimulated desorption from condensed pyrimidine and pyridazine. Phys. Chem. Chem. Phys. 2017, 19, 13038–13048. [Google Scholar] [CrossRef]
- Shukla, A.K.; Stace, A.J. Intermolecular Ion-Molecule Reactions in Clusters: The Reactions of Aliphatic Alcohols. J. Phys. Chem. 1988, 92, 2579–2583. [Google Scholar] [CrossRef]
- El-Shall, M.S.; Marks, C.; Sieck, L.W.; Meot-Ner, M. Reactions and Thermochemistry of Protonated Methanol Clusters Produced by Electron Impact Ionization. J. Phys. Chem. 1992, 96, 2045–2051. [Google Scholar] [CrossRef]
- Lengyel, J.; Papp, P.; Matejčík, Š.; Kočišek, J.; Fárník, M.; Fedor, J. Suppression of low-energy dissociative electron attachment in Fe(CO)5 upon clustering. Beilstein J. Nanotechnol. 2017, 8, 2200–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imhoff, M.; Parenteau, L.; Sanche, L.; Huels, M.A. Low energy electron and O− reactions in films of O2 coadsorbed with benzene or toluene. Phys. Chem. Chem. Phys. 2005, 7, 3359. [Google Scholar] [CrossRef] [PubMed]
- Schmittel, M.; Burghart, A. Understanding Reactivity Patterns of Radical Cations. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550–2589. [Google Scholar] [CrossRef]
- Matyushov, D.V.; Newton, M.D. Electron-Induced Proton Transfer. J. Phys. Chem. B 2021, 125, 12264–12273. [Google Scholar] [CrossRef] [PubMed]
- Borrmann, T.; Swiderek, P. Formation of 2-propanol in condensed molecular films of acetaldehyde following electron impact ionisation-induced proton transfer. Eur. Phys. J. D 2016, 70, 133. [Google Scholar] [CrossRef]
- Martinez, O.; Yang, Z.; Demarais, N.J.; Snow, T.P.; Bierbaum, V.M. Gas-phase reactions of hydride anion, H-. Astrophys. J. 2010, 720, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Garrett, B.C.; Dixon, D.A.; Camaioni, D.M.; Chipman, D.M.; Johnson, M.A.; Jonah, C.D.; Kimmel, G.A.; Miller, J.H.; Rescigno, T.N.; Rossky, P.J.; et al. Role of Water in Electron-Initiated Processes and Radical Chemistry: Issues and Scientific Advances. Chem. Rev. 2005, 105, 355–389. [Google Scholar] [CrossRef]
- Balog, R.; Langer, J.; Gohlke, S.; Stano, M.; Abdoul-Carime, H.; Illenberger, E. Low energy electron driven reactions in free and bound molecules: From unimolecular processes in the gas phase to complex reactions in a condensed environment. Int. J. Mass Spectrom. 2004, 233, 267–291. [Google Scholar] [CrossRef]
- Swiderek, P.; Deschamps, M.C.; Michaud, M.; Sanche, L. Bond Formation in Reactions of Solid Cyclopropane Induced by Low-Energy Electrons. J. Phys. Chem. B 2003, 107, 563–567. [Google Scholar] [CrossRef]
- Weeks, L.D.; Zhu, L.L.; Pellon, M.; Haines, D.R.; Arumainayagam, C.R. Low-Energy Electron-Induced Oligomerization of Condensed Carbon Tetrachloride. J. Phys. Chem. C 2007, 111, 4815–4822. [Google Scholar] [CrossRef]
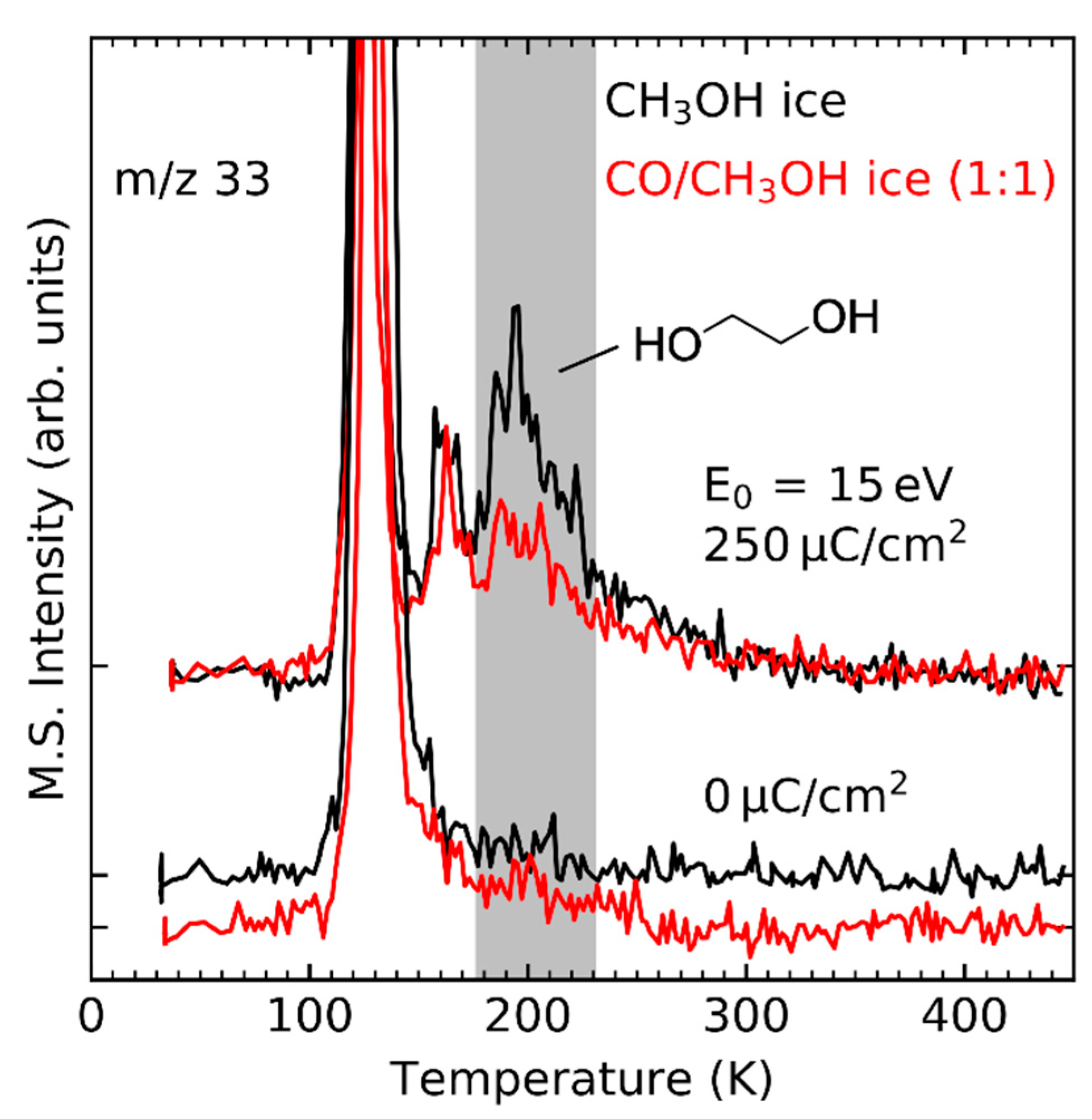
- Maity, S.; Kaiser, R.I.; Jones, B.M. Formation of complex organic molecules in methanol and methanol-carbon monoxide ices exposed to ionizing radiation—A combined FTIR and reflectron time-of-flight mass spectrometry study. Phys. Chem. Chem. Phys. 2015, 17, 3081–3114. [Google Scholar] [CrossRef]
- Öberg, K.I.; Garrod, R.T.; van Dishoeck, E.F.; Linnartz, H. Formation rates of complex organics in UV irradiated CH3OH-rich ices. Astron. Astrophys. 2009, 504, 891–913. [Google Scholar] [CrossRef] [Green Version]
- Maity, S.; Kaiser, R.I.; Jones, B.M. Infrared and reflectron time-of-flight mass spectroscopic study on the synthesis of glycolaldehyde in methanol (CH3OH) and methanol-carbon monoxide (CH3OH-CO) ices exposed to ionization radiation. Faraday Discuss. 2014, 168, 485–516. [Google Scholar] [CrossRef] [PubMed]
- Rajappan, M.; Zhu, L.L.; Bass, A.D.; Sanche, L.; Arumainayagam, C.R. Chemical Synthesis Induced by Dissociative Electron Attachment. J. Chem. Phys. C 2008, 112, 17319–17323. [Google Scholar] [CrossRef]
- Butscher, T.; Duvernay, F.; Rimola, A.; Segado-Centellas, M.; Chiavassa, T. Radical recombination in interstellar ices, a not so simple mechanism. Phys. Chem. Chem. Phys. 2017, 19, 2857–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enrique-Romero, J.; Rimola, A.; Ceccarelli, C.; Ugliengo, P.; Balucani, N.; Skouteris, D. Reactivity of HCO with CH3 and NH2 on Water Ice Surfaces. A Comprehensive Accurate Quantum Chemistry Study. ACS Earth Space Chem. 2019, 3, 2158–2170. [Google Scholar] [CrossRef] [Green Version]
- Enrique-Romero, J.; Rimola, A.; Ceccarelli, C.; Balucani, N. The (impossible?) formation of acetaldehyde on the grain surfaces: Insights from quantum chemical calculations. Mon. Not. R. Astron. Soc. Lett. 2016, 459, L6–L10. [Google Scholar] [CrossRef] [Green Version]
- Rimola, A.; Skouteris, D.; Balucani, N.; Ceccarelli, C.; Enrique-Romero, J.; Taquet, V.; Ugliengo, P. Can Formamide Be Formed on Interstellar Ice? An Atomistic Perspective. ACS Earth Space Chem. 2018, 2, 720–734. [Google Scholar] [CrossRef]
- Shulenberger, K.E.; Zhu, J.L.; Tran, K.; Abdullahi, S.; Belvin, C.; Lukens, J.; Peeler, Z.; Mullikin, E.; Cumberbatch, H.M.; Huang, J.; et al. Electron-Induced Radiolysis of Astrochemically Relevant Ammonia Ices. ACS Earth Space Chem. 2019, 3, 800–810. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.M.; Sloan, D.; Ihm, H.; White, J.M. Electron-induced surface chemistry: Production and characterization of NH2 and NH species on Pt(111). J. Vac. Sci. Technol. A 1996, 14, 1516–1521. [Google Scholar] [CrossRef]
- Zhu, C.; Frigge, R.; Bergantini, A.; Fortenberry, R.C.; Kaiser, R.I. Untangling the Formation of Methoxymethanol (CH3OCH2OH) and Dimethyl Peroxide (CH3OOCH3) in Star-forming Regions. Astrophys. J. 2019, 881, 156. [Google Scholar] [CrossRef]
- Esmaili, S.; Bass, A.D.; Cloutier, P.; Sanche, L.; Huels, M.A. Synthesis of complex organic molecules in simulated methane rich astrophysical ices. J. Chem. Phys. 2017, 147, 224704. [Google Scholar] [CrossRef] [PubMed]
- Sailer, W.; Pelc, A.; Limão-Vieira, P.; Mason, N.J.; Limtrakul, J.; Scheier, P.; Probst, M.; Märk, T.D. Low energy electron attachment to CH3CN. Chem. Phys. Lett. 2003, 381, 216–222. [Google Scholar] [CrossRef]
- Garrod, R.T.; Weaver, S.L.W.; Herbst, E. Complex Chemistry in Star-Forming Regions: An Expanded Gas-Grain Warum-up Chemical Model. Astrophys. J. 2008, 682, 283–302. [Google Scholar] [CrossRef] [Green Version]
- Afeefy, H.Y.; Liebman, J.F.; Stein, S.E.; Burgess, D.R., Jr. Neutral Thermochemical Data. In NIST Chemistry WebBook; Linstrom, P.J., Mallard, W.G., Eds.; NIST Standard Reference Database Number 69; National Institute of Standards and Technology: Gaithersburg, MD, USA.
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef] [PubMed]
- Birch, A.J. Reduction by Dissolving Metals. Part I. J. Chem. Soc. 1944, 430–436. [Google Scholar] [CrossRef]
- Birch, A.J. The Birch reduction in organic synthesis. Pure Appl. Chem. 1996, 68, 553–556. [Google Scholar] [CrossRef]
- Birch, A.J.; Subba Rao, G.S. Reduction by dissolving metals. XV. Reactions of some cyclohexadienes with metal-ammonia solutions. Aust. J. Chem. 1970, 23, 1641–1649. [Google Scholar] [CrossRef]
- Peters, B.K.; Rodriguez, K.X.; Reisberg, S.H.; Beil, S.B.; Hickey, D.P.; Kawamata, Y.; Collins, M.; Starr, J.; Chen, L.; Udyavara, S.; et al. Scalable and safe synthetic organic electroreduction inspired by Li-ion battery chemistry. Science 2019, 363, 838–845. [Google Scholar] [CrossRef]
- Glaser, F.; Kerzig, C.; Wenger, O.S. Multi-Photon Excitation in Photoredox Catalysis: Concepts, Applications, Methods. Angew. Chem. Int. Ed. 2020, 59, 10266–10284. [Google Scholar] [CrossRef]
- Brauman, J.I.; Biairlb, L.K. Gas-Phase Acidities of Alcohols. J. Am. Chem. Soc. 1970, 92, 5986–5992. [Google Scholar] [CrossRef]
- Mackay, G.I.; Bohme, D.K. Bridging the Gap between the Gas Phase and Solution: Transition in the Relative Acidity of Water and Methanol at 296 ± 2 K. J. Am. Chem. Soc. 1978, 100, 327–329. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Ibrahim, M. Ab Initio Studies of RO−…HOR’ Complexes. Solvent Effects on the Relative Acidities of Water and Methanol. J. Comput. Chem. 1981, 2, 7–11. [Google Scholar] [CrossRef]
- Saenko, E.V.; Laikov, D.N.; Baranova, I.A.; Feldman, V.I. Communication: Stabilization of radical anions with weakly bound electron in condensed media: A case study of diacetonyl radical anion. J. Chem. Phys. 2011, 135, 101103. [Google Scholar] [CrossRef]
- Melton, C.E. Cross Sections and Interpretation of Dissociative Attachment Reactions Producing OH−, O−, and H− in H2O. J. Chem. Phys. 1972, 57, 4218–4225. [Google Scholar] [CrossRef]
- Kühn, A.; Fenzlaff, H.P.; Illenberger, E. Formation and dissociation of negative ion resonances in methanol and allylalcohol. J. Chem. Phys. 1988, 88, 7453–7458. [Google Scholar] [CrossRef]
- Boyer, M.C.; Boamah, M.D.; Sullivan, K.K.; Arumainayagam, C.R.; Bazin, M.; Bass, A.D.; Sanche, L. Dynamics of Dissociative Electron-Molecule Interactions in Condensed Methanol. J. Phys. Chem. C 2014, 118, 22592–22600. [Google Scholar] [CrossRef]
- Kimmel, G.A.; Orlando, T.M. Low-Energy (5–120 eV) Electron-Stimulated Dissociation of Amorphous D2O Ice: D(2S), O(3P2,1,0), and O(1D2) Yields and Velocity Distributions. Phys. Rev. Lett. 1995, 75, 2606–2609. [Google Scholar] [CrossRef]
- Petrik, N.G.; Monckton, R.J.; Koehler, S.P.K.; Kimmel, G.A. Distance-Dependent Radiation Chemistry: Oxidation versus Hydrogenation of CO in Electron-Irradiated H2O/CO/H2O Ices. J. Phys. Chem. C 2014, 118, 27483–27492. [Google Scholar] [CrossRef]
- Yamamoto, S.; Beniya, A.; Mukai, K.; Yamashita, Y.; Yoshinobu, J. Low-energy electron-stimulated chemical reactions of CO in water ice. Chem. Phys. Lett. 2004, 388, 384–388. [Google Scholar] [CrossRef]
- Petrik, N.G.; Monckton, R.J.; Koehler, S.P.K.; Kimmel, G.A. Electron-stimulated reactions in layered CO/H2O films: Hydrogen atom diffusion and the sequential hydrogenation of CO to methanol. J. Chem. Phys. 2014, 140, 204710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varela, K.; Hargreaves, L.R.; Ralphs, K.; Khakoo, M.A.; Winstead, C.; McKoy, V.; Rescigno, T.N.; Orel, A.E. Excitation of the 4 lowest electronic transitions in methanol by low-energy electrons. J. Phys. B At. Mol. Opt. Phys. 2015, 48, 115208. [Google Scholar] [CrossRef]
- Michaud, M.; Fraser, M.-J.; Sanche, L. Low-energy electron-energy-loss spectroscopy of solid methanol: Vibrational and electronic excitations. J. Chim. Phys. 1994, 91, 1223–1227. [Google Scholar] [CrossRef]
- Curtis, M.G.; Walker, I.C. Dissociative Electron Attachment in Water and Methanol (5–14 eV). J. Chem. Soc. Faraday Trans. 1992, 88, 2805–2810. [Google Scholar] [CrossRef]
- Rohdenburg, M.; Martinović, P.; Ahlenhoff, K.; Koch, S.; Emmrich, D.; Gölzhäuser, A.; Swiderek, P. Cisplatin as a Potential Platinum Focused Electron Beam Induced Deposition Precursor: NH3 Ligands Enhance the Electron-Induced Removal of Chlorine. J. Phys. Chem. C 2019, 123, 21774–21787. [Google Scholar] [CrossRef] [Green Version]
- Rohdenburg, M.; Boeckers, H.; Brewer, C.R.; McElwee-White, L.; Swiderek, P. Efficient NH3-based process to remove chlorine from electron beam deposited ruthenium produced from (η3-C3H5)Ru(CO)3Cl. Sci. Rep. 2020, 10, 10901. [Google Scholar] [CrossRef]
- Lee, J.; Grabowski, J.J. Reactions of the Atomic Oxygen Radical Anion and the Synthesis of Organic Reactive Intermediates. Chem. Rev. 1992, 92, 1611–1647. [Google Scholar] [CrossRef]
- Kamarchik, E.; Rodrigo, C.; Bowman, J.M.; Reisler, H.; Krylov, A.I. Overtone-induced dissociation and isomerization dynamics of the hydroxymethyl radical (CH2OH and CD2OH). I. A theoretical study. J. Chem. Phys. 2012, 136, 084304. [Google Scholar] [CrossRef]
- Wang, B.; Hou, H.; Gu, Y. Ab Initio/Density Functional Theory and Multichannel RRKM Calculations for the CH3O + CO Reaction. J. Phys. Chem. A 1999, 103, 8021–8029. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA Quantum Chemistry Program Package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Sitha, S.; Jewell, L.L. Non-catalytic hydroamination of alkenes: A computational study. Tetrahedron 2010, 66, 3030–3036. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzweski, V.G.; Montgomery, J.A.; Stratman, R.E.; Burant, J.C.; et al. Gaussian 98, Revision A.7; Gaussian, Inc.: Pittsburgh, PA, USA, 1998. [Google Scholar]
- Mullikin, E.; van Mulbregt, P.; Perea, J.; Kasule, M.; Huang, J.; Buffo, C.; Campbell, J.; Gates, L.; Cumberbatch, H.M.; Peeler, Z.; et al. Condensed-Phase Photochemistry in the Absence of Radiation Chemistry. ACS Earth Space Chem. 2018, 2, 863–868. [Google Scholar] [CrossRef] [Green Version]
- Azria, R.; Parenteau, L.; Sanche, L. Mechanisms for O− electron stimulated desorption via dissociative attachment in condensed CO. J. Chem. Phys. 1988, 88, 5166–5170. [Google Scholar] [CrossRef]
- Kimmel, G.A.; Orlando, T.M. Observation of Negative Ion Resonances in Amorphous Ice via Low-Energy (5–40 eV) Electron-Stimulated Production of Molecular Hydrogen. Phys. Rev. Lett. 1996, 77, 3983–3986. [Google Scholar] [CrossRef]
- Klyachko, D.; Rowntree, P.; Sanche, L. Oxidation of hydrogen-passivated silicon surfaces induced by dissociative electron attachment to physisorbed H2O. Surf. Sci. 1996, 346, L49–L54. [Google Scholar] [CrossRef]
- Simpson, W.C.; Sieger, M.T.; Orlando, T.M.; Parenteau, L.; Nagesha, K.; Sanche, L. Dissociative electron attachment in nanoscale ice films: Temperature and morphology effects. J. Chem. Phys. 1997, 107, 8668–8677. [Google Scholar] [CrossRef] [Green Version]
- Lane, C.D.; Orlando, T.M. Inelastic electron scattering and energy-selective negative ion reactions in molecular films on silicon surfaces. Appl. Surf. Sci. 2007, 253, 6646–6656. [Google Scholar] [CrossRef]
- Prabhudesai, V.S.; Nandi, D.; Kelkar, A.H.; Krishnakumar, E. Functional group dependent dissociative electron attachment to simple organic molecules. J. Chem. Phys. 2008, 128, 154309. [Google Scholar] [CrossRef]
- Szymańska, E.; Mason, N.J.; Krishnakumar, E.; Matias, C.; Mauracher, A.; Scheier, P.; Denifl, S. Dissociative electron attachment and dipolar dissociation in ethylene. Int. J. Mass Spectrom. 2014, 365–366, 356–364. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.J.; Chen, S.-H.; Sun, B.; Chang, A.H.H.; Kaiser, R.I. Mechanistical Studies on the Irradiation of Methanol in Extraterrestrial Ices. Astrophys. J. 2007, 660, 1588–1608. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.J.; Hama, T.; Kim, Y.S.; Kawasaki, M.; Kaiser, R.I. Laboratory studies on the formation of formic acid (HCOOH) in interstellar and cometary ices. Astrophys. J. 2011, 727, 27. [Google Scholar] [CrossRef] [Green Version]
- McCunn, L.R.; Lau, K.C.; Krisch, M.J.; Butler, L.J.; Tsung, J.W.; Lin, J.J. Unimolecular Dissociation of the CH3OCO Radical: An Intermediate in the CH3O + CO Reaction. J. Phys. Chem. A 2006, 110, 1625–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, C.J.; Kaiser, R.I. On the Formation of Glycolaldehyde (HCOCH2OH) and Methyl Formate (HCOOCH3) in Interstellar Ice Analogs. Astrophys. J. 2007, 661, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Arasa, C.; van Hemert, M.C.; van Dishoeck, E.F.; Kroes, G.J. Molecular Dynamics Simulations of CO2 Formation in Interstellar Ices. J. Phys. Chem. A 2013, 117, 7064–7074. [Google Scholar] [CrossRef] [Green Version]
- Tronc, M.; Azria, R.; Arfa, M.B. Differential cross section for H- and NH2- ions in NH3. J. Phys. B At. Mol. Opt. Phys. 1988, 21, 2497–2506. [Google Scholar] [CrossRef]
- Ingólfsson, O.; Weik, F.; Illenberger, E. The reactivity of slow electrons with molecules at different degrees of aggregation: Gas phase, clusters and condensed phase. Int. J. Mass Spectrom. Ion Processes 1996, 155, 1–68. [Google Scholar] [CrossRef]
- Nag, P.; Nandi, D. Fragmentation dynamics in dissociative electron attachment to CO probed by velocity slice imaging. Phys. Chem. Chem. Phys. 2015, 17, 7130–7137. [Google Scholar] [CrossRef] [Green Version]
- López-Sepulcre, A.; Jaber, A.A.; Mendoza, E.; Lefloch, B.; Ceccarelli, C.; Vastel, C.; Bachiller, R.; Cernicharo, J.; Codella, C.; Kahane, C.; et al. Shedding light on the formation of the pre-biotic molecule formamide with ASAI. Mon. Not. R. Astron. Soc. 2015, 449, 2438–2458. [Google Scholar] [CrossRef] [Green Version]
- Gadoum, A.; Benyoucef, D. Set of the Electron Collision Cross Sections for Methane Molecule. IEEE Trans. Plasma Sci. 2019, 47, 1505–1513. [Google Scholar] [CrossRef]
- Bouwman, D.; Martinez, A.; Braams, B.J.; Ebert, U. Neutral dissociation of methane by electron impact and a complete and consistent cross section set. Plasma Sources Sci. Technol. 2021, 30, 075012. [Google Scholar] [CrossRef]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NH3 | H2O | CH3OH | |
| C2H4 |  Ethylamine [29] |  Ethanol [28] |  Ethyl methyl ether [32] |
| CO |  Formamide [30] |  Formic acid [24] |  Methyl formate [26] |
| Source Molecule | Reactive Species | Ionization Threshold (Gas Phase) 1 | Threshold for Product Formation (Ice) | References (Ice) |
|---|---|---|---|---|
| C2H4 | C2H4•+ | 10.5 eV | 8–10 eV | [28,29,32] |
| CO | CO•+ | 14.014 eV | 13–14 eV | [30] |
| NH3 | NH3•+ | 10.070 eV | ≤12 eV/10 eV | [27,30] |
| H2O | H2O•+ | 12.621 eV | 11 eV | [28] |
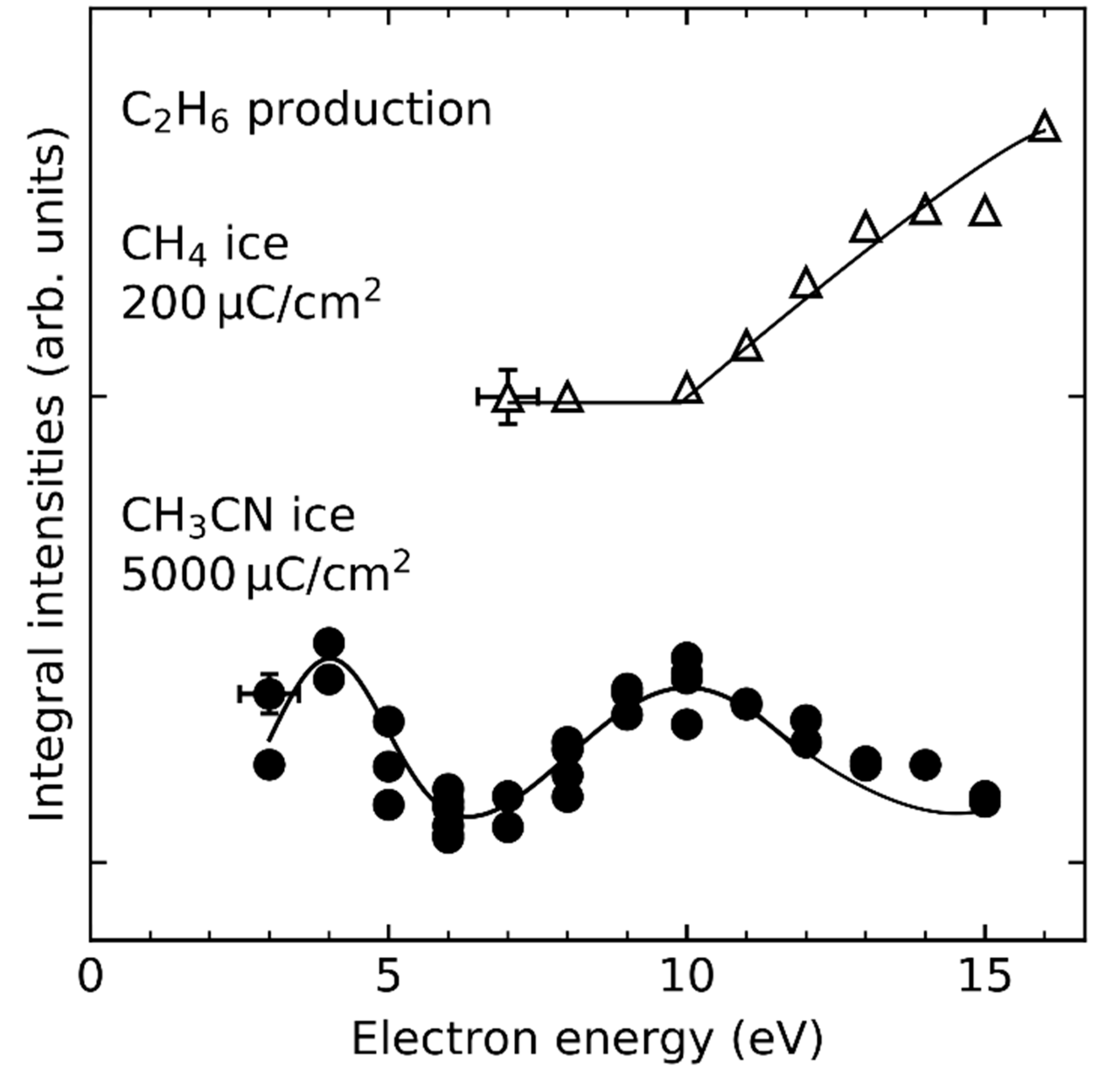
| CH4 | CH4•+ | 12.61 eV | 12 eV | this work |
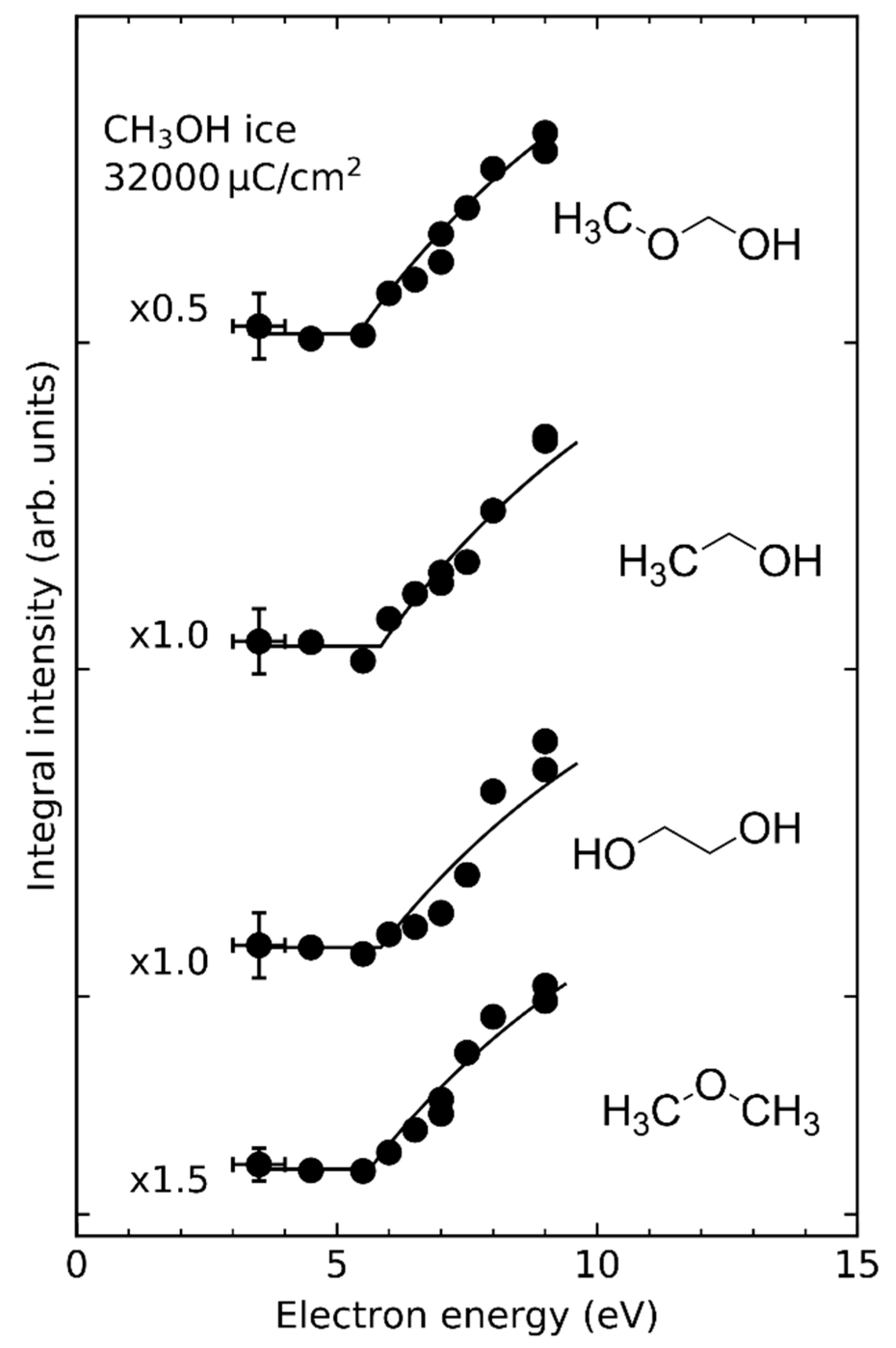
| CH3OH | CH3OH•+ CH3O•+ | 10.84 eV 11.67 eV | 9.8 eV ≤11 eV | [31,32] [32] |
| Source Molecule | Reactive Species | Threshold for Product Formation (Ice) | References (Ice) |
|---|---|---|---|
| NH3 | H• + NH2• | ~7 eV | this work |
| H2O | H2 + O H• + HO• | 6–7 eV 6–7 eV | [24] [24,28] |
| CH4 | H• + CH3• | ~10 eV | this work |
| CH3OH | CH3O• + H• H• + •CH2OH CH3• + HO• | ~6 eV ~6 eV ~6 eV | [31] [31] [31] |
| Source Molecule | Reactive Species | Resonance in Product Formation (Ice) | References (Ice) |
|---|---|---|---|
| C2H4 | C2H4•− | <5 eV | [28] |
| CO | CO•− C + O•− | ~4 eV 11 eV/16 eV | [24] [37] |
| H2O | H• + HO− O•− + H2 | 6.5 eV 10 eV | [24] [24] |
| CH3OH | CH3O• + H− CH3O− + H• | 5.5 eV 5.5 eV | [32] [26,31] |
| CH3CN | CH3• + CN− | 4 eV/10 eV | [25] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, F.; Borrmann, T.; Mues, M.P.; Benter, S.; Swiderek, P.; Bredehöft, J.H. Mechanisms of Electron-Induced Chemistry in Molecular Ices. Atoms 2022, 10, 25. https://doi.org/10.3390/atoms10010025
Schmidt F, Borrmann T, Mues MP, Benter S, Swiderek P, Bredehöft JH. Mechanisms of Electron-Induced Chemistry in Molecular Ices. Atoms. 2022; 10(1):25. https://doi.org/10.3390/atoms10010025
Chicago/Turabian StyleSchmidt, Fabian, Tobias Borrmann, Martin Philipp Mues, Sanna Benter, Petra Swiderek, and Jan Hendrik Bredehöft. 2022. "Mechanisms of Electron-Induced Chemistry in Molecular Ices" Atoms 10, no. 1: 25. https://doi.org/10.3390/atoms10010025
APA StyleSchmidt, F., Borrmann, T., Mues, M. P., Benter, S., Swiderek, P., & Bredehöft, J. H. (2022). Mechanisms of Electron-Induced Chemistry in Molecular Ices. Atoms, 10(1), 25. https://doi.org/10.3390/atoms10010025