
Level Structure and Properties of Open f-Shell Elements


Abstract
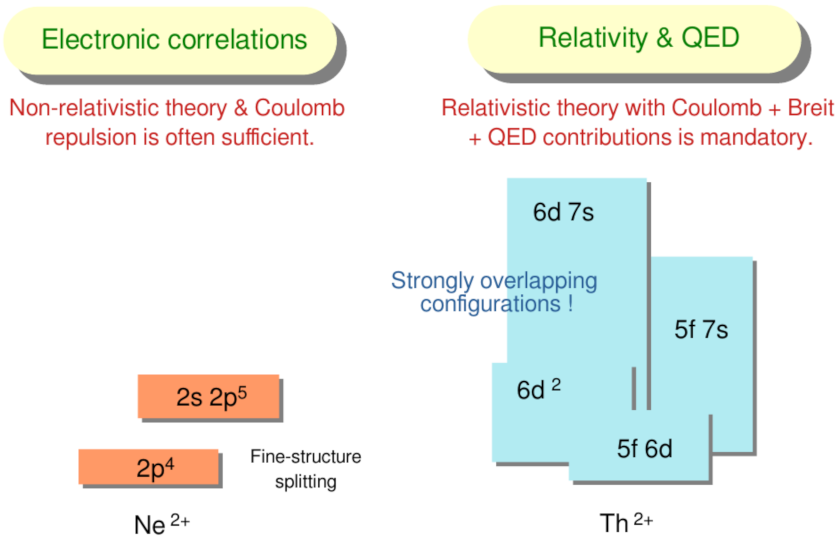
:1. Demands of Open -Shell Elements
2. Theory and Computations
2.1. Approximate Level Energies and Atomic State Functions
2.2. Configuration-Interaction Expansions for Open f-Shell Elements
2.3. The Jac Toolbox
2.3.1. Brief Overview of Jac
2.3.2. Needs of a Descriptive Language for Atomic Computations
2.3.3. Combining Syntax and Semantics: Jac’s Data Structures for Atomic Computations
2.4. Spectroscopic Notation for Open f-Shell Elements
2.5. Atomic Amplitudes and Properties
2.6. Atomic Excitation and Decay Processes of Open f-Shell Elements
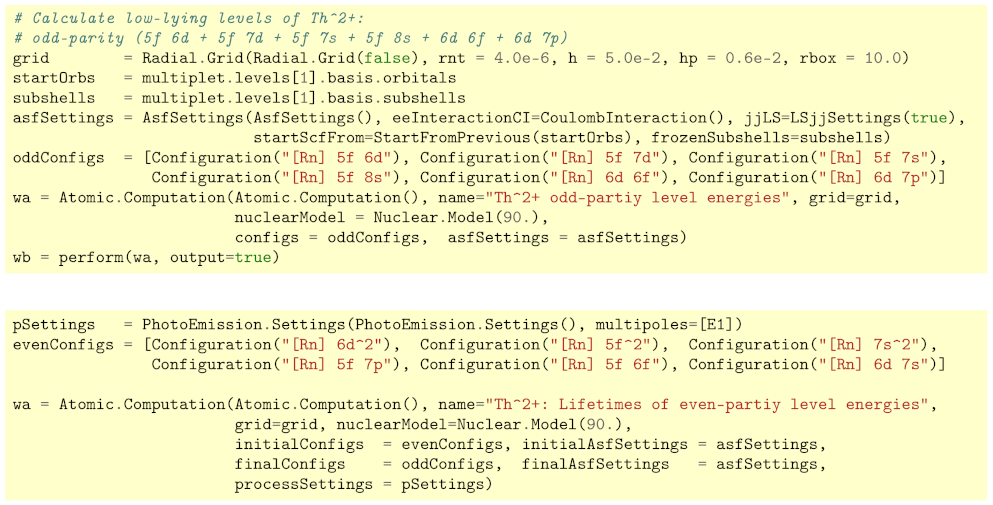
3. Low-Lying Level Structure of Th
3.1. Estimates on the Level Structure of Th
3.2. Transition Probabilities. Lifetimes and Branching Fractions
4. Summary and Conclusions
Funding
Conflicts of Interest
References
- Johnson, W.R. Atomic Structure Theory: Lectures on Atomic Physics; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Grant, I.P. Relativistic Quantum Theory of Atoms and Molecules: Theory and Computation; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Fritzsche, S. Large–scale accurate structure calculations for open–shell atoms and ions. Phys. Scr. 2002, T100, 37. [Google Scholar] [CrossRef]
- Raeder, S.; Ackermann, D.; Backe, H.; Block, M.; Cheal, B.; Chhetri, P.; Düllmann, C.E.; van Duppen, P.; Even, J.; Ferrer, R.; et al. Nuclear properties of nobelium isotopes from laser spectroscopy. Phys. Rev. Lett. 2018, 120, 232503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raeder, S.; Kneip, N.; Reich, T.; Studer, D.; Trautmann, N.; Wendt, K. Recent developments in resonance ionization mass spectrometry for ultra-trace analysis of actinide elements. Radiochim. Acta 2019, 107, 1515. [Google Scholar] [CrossRef]
- Rothe, S.; Andreyev, A.N.; Antalic, S.; Borschevsky, A.; Capponi, L.; Cocolios, T.E.; Witte, H.D.; Eliav, E.; Fedorov, D.V.; Fedosseev, V.N.; et al. Measurement of the first ionization potential of astatine by laser ionization. Nat. Commun. 2013, 4, 1835. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, K.T.; Lynch, K.M.; Billowes, J.; Bissell, M.L.; Budincevic, V.; Cocolios, T.E.; de Groote, R.P.; Schepper, S.D.; Fedosseev, V.N.; Franchoo, S.; et al. Collinear resonance ionization spectroscopy of neutron-deficient francium isotopes. Phys. Rev. Lett. 2013, 111, 212501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, R.; Barzakh, A.; Bastin, B.; Beerwerth, B.; Block, M.; Creemers, P.; Grawe, H.; de Groote, R.; Delahaye, P.; Fléchard, X.; et al. Towards high-resolution laser ionization spectroscopy of the heaviest elements in supersonic gas jet expansion. Nat. Commun. 2017, 8, 14520. [Google Scholar] [CrossRef] [Green Version]
- Granados, C.; Creemers, P.; Ferrer, R.; Gaffney, L.P.; Gins, W.; de Groote, R.; Huyse, M.; Kudryavtsev, Y.; Martnez, Y.; Raeder, S.; et al. In-gas laser ionization and spectroscopy of actinium isotopes near the N=126 closed shell. Phys. Rev. C 2017, 96, 054331. [Google Scholar] [CrossRef] [Green Version]
- Chhetri, P.; Ackermann, D.; Backe, H.; Block, M.; Cheal, B.; Droese, C.; Düllmann, C.E.; Even, J.; Ferrer, R.; Giacoppo, F.; et al. Precision measurement of the first ionization potential of nobelium. Phys. Rev. Lett. 2018, 120, 263003. [Google Scholar] [CrossRef] [PubMed]
- Laatiaoui, M. On the way to unveiling the atomic structure of superheavy elements. Eur. Phys. J. Conf. 2016, 131, 05002. [Google Scholar] [CrossRef] [Green Version]
- Laatiaoui, M.; Buchachenko, A.A.; Viehland, L.A. Exploiting transport properties for the detection of optical pumping in heavy ions. Phys. Rev. A 2020, 102, 013106. [Google Scholar] [CrossRef]
- Sato, T.K.; Asai, M.; Tsukada, K.; Kaneya, Y.; Toyoshima, A.; Mitsukai, A.; Nagame, Y.; Osa, A.; Toyoshima, A.; Tsukada, K.; et al. First ionization potentials of Fm, Md, No, and Lr: Verification of filling-up of 5f electrons and confirmation of the actinide series. J. Am. Chem. Soc. 2018, 140, 14609. [Google Scholar] [CrossRef] [Green Version]
- Sewtz, M.; Backe, H.; Dretzke, A.; Kube, G.; Lauth, W.; Schwamb, P.; Eberhardt, K.; Grüning, C.; Thörle, P.; Trautmann, N.; et al. First observation of atomic levels for the element fermium (Z = 100). Phys. Rev. Lett. 2003, 90, 163002. [Google Scholar] [CrossRef] [Green Version]
- Morrison, J.C.; Rajnak, K. Many-body calculations for the heavy atoms. Phys. Rev. 1971, A4, 536. [Google Scholar] [CrossRef]
- Borschevsky, A.; Pershina, V.; Eliav, E.; Kaldor, U. Ab initio predictions of atomic properties of element 120 and its lighter group-2 homologues. Phys. Rev. A 2013, 87, 022502. [Google Scholar] [CrossRef]
- Eliav, E.; Kaldor, U.; Ishikawa, Y. Transition energies of mercury and ekamercury (element 112) by the relativistic coupled-cluster method. Phys. Rev. 1995, 52, 2765. [Google Scholar] [CrossRef] [PubMed]
- Grant, I.P. Relativistic Effects in Atoms and Molecules. In Methods in Computational Chemistry; Wilson, S., Ed.; Plenum: New York, NY, USA, 1988; Volume 2, pp. 1–38. [Google Scholar]
- Sewtz, M.; Backe, H.; Dong, C.Z.; Dretzke, A.; Eberhardt, K.; Fritzsche, S.; Grüning, C.; Haire, R.G.; Kube, G.; Kunz, P.; et al. Resonance ionization spectroscopy of fermium (Z = 100). Spectrochim. Acta B 2003, 58, 1077. [Google Scholar] [CrossRef]
- Fritzsche, S. On the accuracy of valence–shell computations for heavy and super–heavy elements. Eur. Phys. J. D 2005, 33, 15. [Google Scholar] [CrossRef]
- Zou, Y.; Froese Fischer, C. Resonance transition energies and oscillator strengths in lutetium and lawrencium. Phys. Rev. Lett. 2002, 88, 183001. [Google Scholar] [CrossRef] [PubMed]
- Fritzsche, S.; Dong, C.Z.; Koike, F.; Uvarov, A. The low–ying level structure of atomic lawrencium (Z = 103): Energies and absorption rates. Eur. Phys. J. D 2007, 45, 107. [Google Scholar] [CrossRef]
- Yu, Y.J.; Li, J.G.; Dong, C.Z.; Ding, X.B.; Fritzsche, S.; Fricke, B. The excitation energies, ionization potentials and oscillator strengths of neutral and ionized species of Uub (Z = 112) and the homologue elements Zn, Cd and Hg. Eur. Phys. J. D 2007, 44, 51. [Google Scholar] [CrossRef]
- Fricke, B.; Greiner, W.; Waber, J.T. The continuation of the periodic table up to Z = 172. The chemistry of superheavy elements. Theor. Chim. Acta 1971, 21, 235. [Google Scholar] [CrossRef]
- Johnson, E.; Pershina, V.; Fricke, B. Ionization potentials of Seaborgium. J. Phys. Chem. 1999, 103, 8458. [Google Scholar] [CrossRef]
- Johnson, E.; Fricke, B.; Jacob, T.; Dong, C.Z.; Fritzsche, S.; Pershina, V. Ionization potentials and radii of neutral and ionized species of elements 107 (bohrium) and 108 (hassium) from extended multiconfiguration Dirac-Fock calculations. J. Phys. Chem. 2002, 116, 1862. [Google Scholar] [CrossRef]
- Eliav, E.; Fritzsche, S.; Kaldor, U. Electronic structure theory of the superheavy elements. Nucl. Phys. A 2015, 944, 518. [Google Scholar] [CrossRef]
- Kahl, E.V.; Berengut, J.C.; Laatiaoui, M.; Eliav, E.; Borschevsky, A. High-precision ab initio calculations of the spectrum of Lr+. Phys. Rev. 2019, A100, 062505. [Google Scholar] [CrossRef] [Green Version]
- Oleynichenko, A.V.; Zaitsevskii, A.; Skripnikov, L.V.; Eliav, E. Relativistic Fock space coupled cluster method for many-electron systems: Non-perturbative account for connected triple excitations. Symmetry 2020, 12, 1101. [Google Scholar] [CrossRef]
- Kumar, R.; Chattopadhyay, S.; Angom, D.; Mani, B.K. Relativistic coupled-cluster calculation of the electric dipole polarizability and correlation energy of Cn, Nh+, and Og: Correlation effects from lighter to superheavy elements. Phys. Rev. A 2021, 103, 062803. [Google Scholar] [CrossRef]
- Fritzsche, S.; Surzhykov, A.; Stöhlker, T. Dominance of the Breit interaction in the X-ray emission of highly charged ions following dielectronic recombination. Phys. Rev. Lett. 2009, 103, 113001. [Google Scholar] [CrossRef] [Green Version]
- Fritzsche, S. The Ratip program for relativistic calculations of atomic transition, ionization and recombination properties. Comp. Phys. Commun. 2012, 183, 1525. [Google Scholar] [CrossRef]
- Judd, B.R. Correlation crystal fields for lanthanide ions. Phys. Rev. Lett. 1977, 39, 242. [Google Scholar] [CrossRef]
- Dorenbos, P. Crystal field splitting of lanthanide 4fn-15d levels in inorganic compounds. J. Alloys Comp. 2002, 341, 156. [Google Scholar] [CrossRef]
- Suzuki, C.; Koike, F.; Murakami, I.; Tamura, N.; Sudo, S. Systematic observation of EUV spectra from highly charged lanthanide ions in the large helical device. Atoms 2018, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Fritzsche, S. A fresh computational approach to atomic structures, processes and cascades. Comp. Phys. Commun. 2019, 240, 1. [Google Scholar] [CrossRef]
- Seiferle, B.; von der Wense, L.; Thirolf, P.G. Lifetime measurement of the 229Th nuclear isomer. Phys. Rev. Lett. 2017, 118, 042501. [Google Scholar] [CrossRef] [Green Version]
- Gaigalas, G.; Fritzsche, S.; Rudzikas, Z. Reduced coefficients of fractional parentage and matrix elements of the tensor W(kqkj) in jj-coupling. At. D. Nucl. D. Tables 2002, 76, 235. [Google Scholar] [CrossRef]
- Shabaev, V.M.; Tupitsyn, I.I.; Yerokhin, V.A. Model operator approach to the Lamb shift calculations in relativistic many-electron atoms. Phys. Rev. A 2013, 88, 012513. [Google Scholar] [CrossRef] [Green Version]
- Indelicato, P.; Santos, J.P.; Boucard, S.; Desclaux, J.-P. QED and relativistic corrections in superheavy elements. Eur. Phys. J. D 2007, 45, 155. [Google Scholar] [CrossRef]
- Fritzsche, S. JAC: User Guide, Compendium & Theoretical Background. Unpublished. Available online: https://github.com/OpenJAC/JAC.jl/blob/master/UserGuide-Jac.pdf (accessed on 10 October 2021).
- Julia 1.7 Documentation. Available online: https://docs.julialang.org/ (accessed on 10 December 2021).
- Bezanson, J.; Chen, J.; Chung, B.; Karpinski, S.; Shah, V.B.; Vitek, J.; Zoubritzky, L. Julia: Dynamism and performance reconciled by design. Proc. ACM Program. Lang. 2018, 2, 120. [Google Scholar] [CrossRef] [Green Version]
- GitHub—OpenJAC / JAC.jl. Available online: https://github.com/OpenJAC/JAC.jl (accessed on 10 November 2021).
- Fritzsche, S.; Fricke, B.; Sepp, W.D. Reduced L1 level-width and Coster-Kronig yields by relaxation and continuum interactions in atomic zinc. Phys. Rev. A 1992, 45, 1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritzsche, S.; Palmeri, P.; Schippers, S. Atomic cascade computations. Symmetry 2021, 13, 520. [Google Scholar] [CrossRef]
- Schippers, S.; Martins, M.; Beerwerth, R.; Bari, S.; Holste, K.; Schubert, K.; Viefhaus, J.; Savin, D.W.; Fritzsche, S.; Müller, A. Near L-edge single and multiple photoionization of singly charged iron ions. Astrophys. J. 2017, 849, 5. [Google Scholar] [CrossRef] [Green Version]
- Fritzsche, S.; Froese Fischer, C.; Gaigalas, G. A program for relativistic configuration interaction calculations. Comput. Phys. Commun. 2002, 148, 103. [Google Scholar] [CrossRef]
- Fritzsche, S.; Surzhykov, A. Approximate atomic Green functions. Molecules 2021, 26, 2660. [Google Scholar] [CrossRef] [PubMed]
- Julia Comes with a Full-Featured Interactive and Command-Line REPL (Read-Eval-Print Loop) That Is Built into the Executable of the Language. Available online: https://docs.julialang.org/en/v1/stdlib/REPL/ (accessed on 10 December 2021).
- Gaigalas, G.; Zalandauskas, T.; Fritzsche, S. Spectroscopic LSJ notation for atomic levels as obtained from relativistic calculations. Comput. Phys. Commun. 2004, 157, 239. [Google Scholar] [CrossRef] [Green Version]
- Gaigalas, G.; Fritzsche, S. Angular coefficients for symmetry-adapted configuration states in jj-coupling. Comput. Phys. Commun. 2021, 267, 108086. [Google Scholar] [CrossRef]
- Gaigalas, G.; Fritzsche, S. Maple procedures for the coupling of angular momenta. VI. LS-jj transformations. Comput. Phys. Commun. 2002, 159, 39. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.; Canali, C.; Warring, U.; Kellerbauer, A.; Fritzsche, S. First optical hyperfine structure measurement in an atomic anion. Phys. Rev. Lett. 2010, 104, 073004. [Google Scholar] [CrossRef]
- Cocolios, T.E.; Dexters, W.; Seliverstov, M.D.; Andreyev, A.N.; Antalic, S.; Barzakh, A.E.; Bastin, B.; Büscher, J.; Darby, I.G.; Fedorov, D.V.; et al. Early onset of ground state deformation in neutron deficient polonium isotopes. Phys. Rev. Lett. 2011, 106, 052503. [Google Scholar] [CrossRef] [Green Version]
- Cheal, B.; Cocolios, T.E.; Fritzsche, S. Laser spectroscopy of radioactive isotopes: Role and limitations of accurate isotope-shift calculations. Phys. Rev. A 2012, 86, 042501. [Google Scholar] [CrossRef]
- Biemont, E.; Palmeri, P.; Quinet, P.; Zhang, Z.G.; Svanberg, S. Doubly ionized thorium: Laser lifetime measurements and transition probability determination of interest in cosmochronology. Astrophys. J. 2002, 567, 1276. [Google Scholar] [CrossRef] [Green Version]
- Kramida, A.; Ralchenko, Y.; Reader, J.; NIST ASD Team. NIST Atomic Spectra Database (ver. 5.8), [Online]. 2021. Available online: https://physics.nist.gov/asd (accessed on 25 October 2021).
- Wyart, J.-F.; Kaufman, V. Extended analysis of doubly ionized thorium (Th III). Phys. Scr. 1981, 24, 941. [Google Scholar] [CrossRef]
- Safronova, M.S.; Safronova, U.I.; Clark, C.W. Relativistic all-order calculations of Th, Th+, and Th2+ atomic properties. Phys. Rev. A 2014, 90, 032512. [Google Scholar] [CrossRef] [Green Version]
- Fritzsche, S. Symbolic evaluation of expressions from Racahs algebra. Symmetry 2021, 13, 1558. [Google Scholar] [CrossRef]
- Fritzsche, S. Dielectronic recombination strengths and plasma rate coefficients of multiply-charged ions. Astron. Astrophys. 2021, 656, A163. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Struct | Brief Explanation |
|---|---|
| AbstractEeInteraction | Abstract type to distinguish between different electron-electron interaction operators; it comprises the concrete (singleton) types BreitInteraction, CoulombInteraction, CoulombBreit. |
| AbstractExcitationScheme | Abstract type to support different excitation schemes, such as DeExciteSingleElectron, ExciteByCapture, and several others. |
| AbstractScField | Abstract type for dealing with different self-consistent-field (SCF) potentials. |
| AsfSettings | Settings to control the SCF and CI calculations for a given multiplet. |
| Atomic.Computation | An atomic computation of one or several multiplets, including the SCF and CI calculations, as well as of selected properties or processes. |
| Basis | (Relativistic) many-electron basis, including the specification of the configuration space and all radial orbitals. |
| Configuration | (Nonrelativistic) electron configuration as specified by the shell occupation. |
| EmMultipole | A multipole (component) of the electro-magnetic field as specified by its electric or magnetic character and the multipolarity. |
| Level | Atomic level in terms of its quantum numbers, symmetry, energy and its (possibly full) representation. |
| LevelSelection | List of levels that is specified by either the level numbers and/or level symmetries. |
| LevelSymmetry | specifies the (total) angular momentum and parity of a particular level. |
| LSjjSettings | Settings to control the transformation of the selected many-electron levels. |
| MeanFieldBasis | A simple representation of the electronic structure in terms of a mean-field orbital basis. |
| Multiplet | An ordered list of atomic levels, often associated with one or several configurations. |
| Nuclear.Model | A model of the nucleus to keep all nuclear parameters together. |
| Orbital | (Relativistic) radial orbital function that appears as building block in order to define the many-electron CSF; such an orbital comprises a large and small component and is typically given on a (radial) grid. |
| Radial.Grid | Radial grid to represent the (radial) orbital function and to perform all radial integration. |
| Radial.Potential | Radial potential function. |
| Representation | Representation of an atomic state in terms of either a mean-field basis, an approximate wave function, a many-electron Green function, or others. |
| RasExpansion | A restricted active-space representation of the levels from a given multiplet; cf. CiExpansion in Figure 3. |
| RasSettings | Settings to control the details of a RasExpansion. |
| RasStep | Single-step of a (systematically enlarged) restricted active-space computation. |
| Shell | Nonrelativistic shell, such as . |
| Subshell | Relativistic subshell, such as |
| Process & Brief Explanation |
|---|
| Photon emission : Transition probabilities; oscillator strengths; lifetimes; angular distributions. |
| Photoexcitation : Excitation cross sections, alignment parameters; statistical tensors. |
| Photoionization : Cross sections; angular parameters. |
| Photorecombination : Recombination cross sections; angular parameters. |
| Auger emission or autoionization : Auger rates; angular and polarization parameters. |
| Dielectronic recombination (DR) : Partial and total DR resonance strengths; DR plasma rate coefficients. |
| Photoexcitation with subsequent autoionization : Rates. |
| Photo-double ionization : Energy-differential and total cross sections. |
| Rayleigh & Compton scattering of light : Angle-differential cross sections. |
| Level | Energy [eV] | Lifetime [s] | ||||||
|---|---|---|---|---|---|---|---|---|
| This Work | Exp. [58] | Calc. [60] | This Work | Exp. [57] | Calc. [60] | |||
| 0 | 0 | 0 | ||||||
| 210 | 511 | 189 | ||||||
| 3686 | 2527 | 2436 | ||||||
| 4975 | 3182 | 2958 | ||||||
| 3026 | 3188 | 3207 | ||||||
| 5360 | 4827 | 4853 | ||||||
| 4863 | 4490 | 4802 | ||||||
| 6857 | 5060 | 5085 | ||||||
| 8466 | 6288 | 5797 | ||||||
| 6702 | 6311 | 6237 | ||||||
| 9279 | 7501 | 7609 | ||||||
| 8779 | 7921 | 8260 | ||||||
| 9192 | 8142 | 8197 | ||||||
| 8555 | 8437 | 8810 | ||||||
| 13,084 | 11,123 | 11,564 | ||||||
| 15,274 | 11,233 | 11,766 | ||||||
| 37,134 | 32,867 | 33,488 | [−8] | [−8] | [−8] | |||
| 43,256 | 38,581 | 38,980 | [−8] | [−9] | [−9] | |||
| 46,404 | 42,260 | - | [−9] | [−9] | [−9] | |||
| 47623 | 45,064 | - | [−9] | [−9] | [−9] | |||
| 55,884 | 53,052 | - | [−9] | [−9] | [−9] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fritzsche, S. Level Structure and Properties of Open f-Shell Elements. Atoms 2022, 10, 7. https://doi.org/10.3390/atoms10010007
Fritzsche S. Level Structure and Properties of Open f-Shell Elements. Atoms. 2022; 10(1):7. https://doi.org/10.3390/atoms10010007
Chicago/Turabian StyleFritzsche, Stephan. 2022. "Level Structure and Properties of Open f-Shell Elements" Atoms 10, no. 1: 7. https://doi.org/10.3390/atoms10010007
APA StyleFritzsche, S. (2022). Level Structure and Properties of Open f-Shell Elements. Atoms, 10(1), 7. https://doi.org/10.3390/atoms10010007