
Independently Optimized Orbital Sets in GRASP—The Case of Hyperfine Structure in Li I

 ,
,  , ,
, ,  ,
,  , , and
, , and
Abstract
:1. Introduction
2. Variational Calculations
2.1. The MCDHF Method
2.2. Localization of the Radial Orbitals and Their Dependence on the Energy Functional
3. Computed Properties and Their Dependence on Correlation Effects
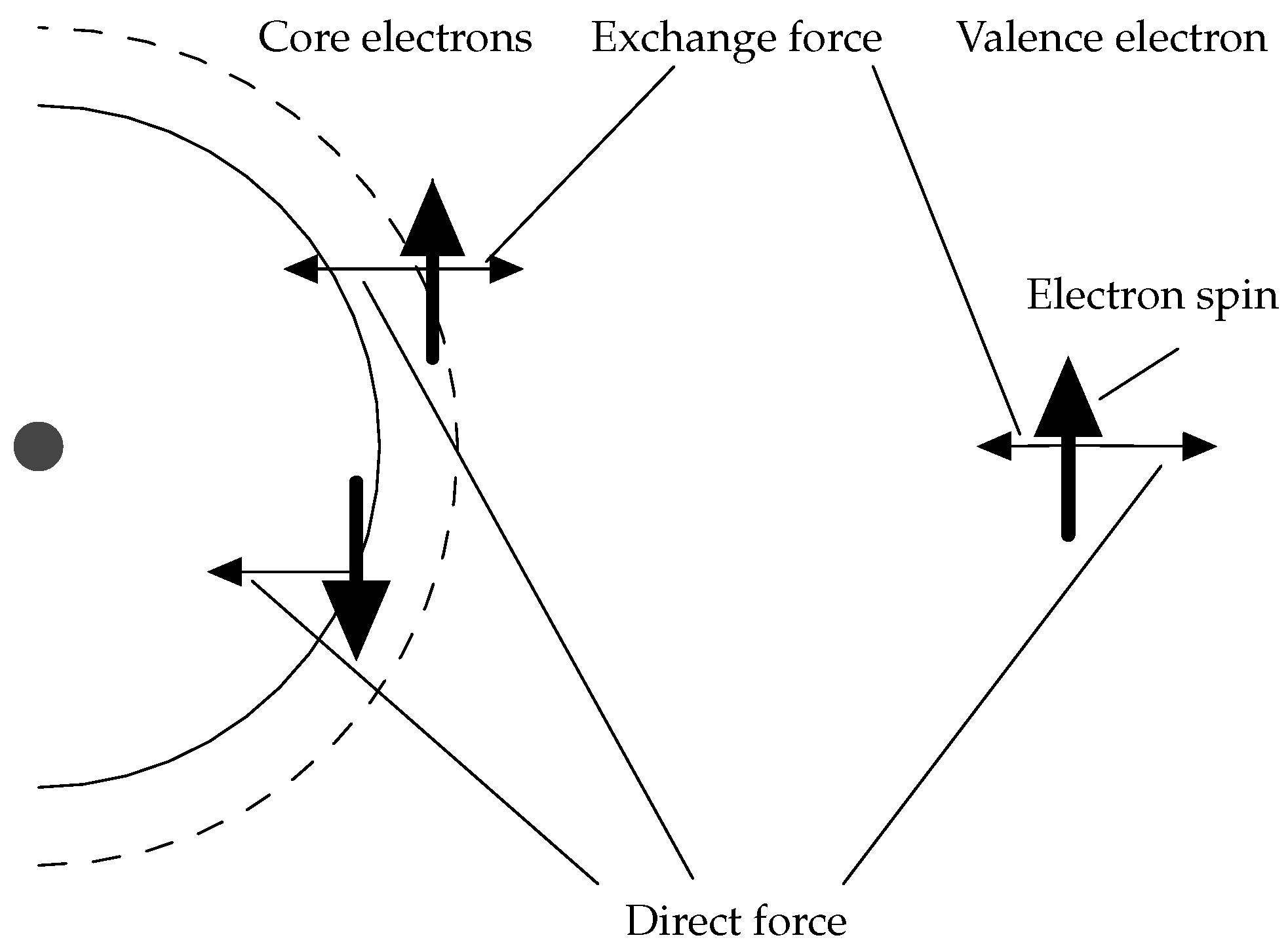
3.1. Hyperfine Structure
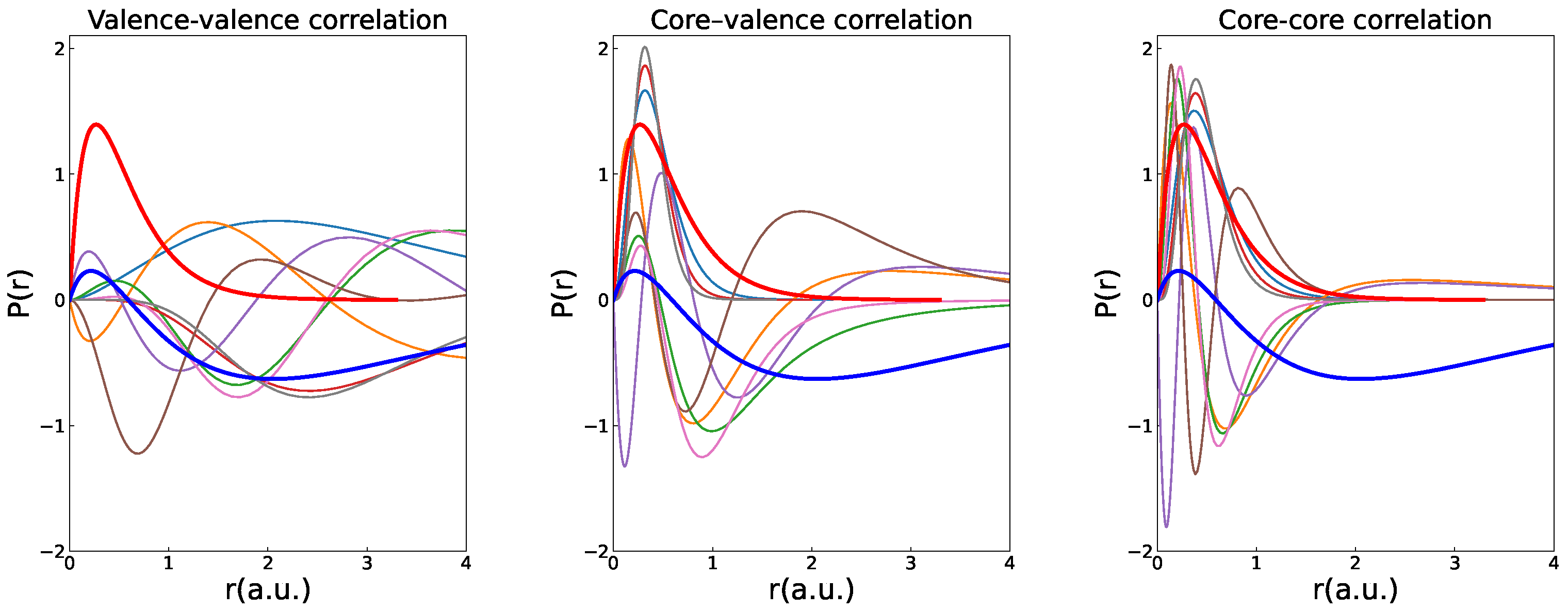
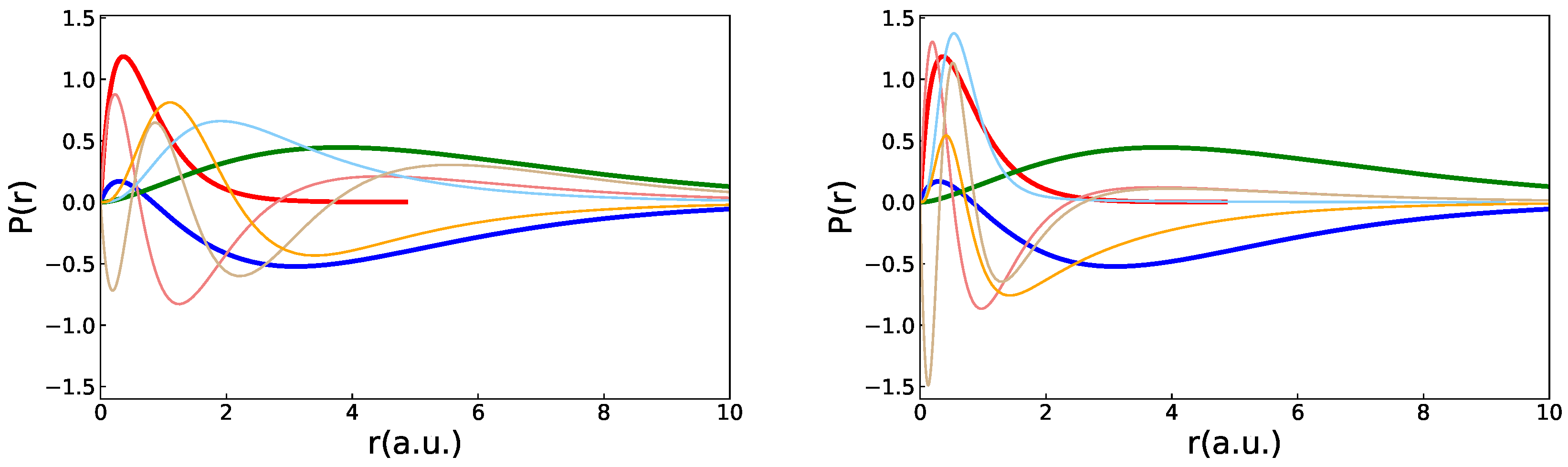
3.2. Polarization Effects
3.3. Localization of the Polarization Orbitals
- Perform a weighted average Dirac–Fock calculation for and ;
- Keep frozen and perform weighted average MCDHF calculations for and based on the CSF expansions formed by allowing S’s substitution from the reference configuration to a set of orbitals;
- Compute the hyperfine interaction constants and monitor the convergence as the set of orbitals is increased;
- Stop when the hyperfine interaction constants are not changing anymore.
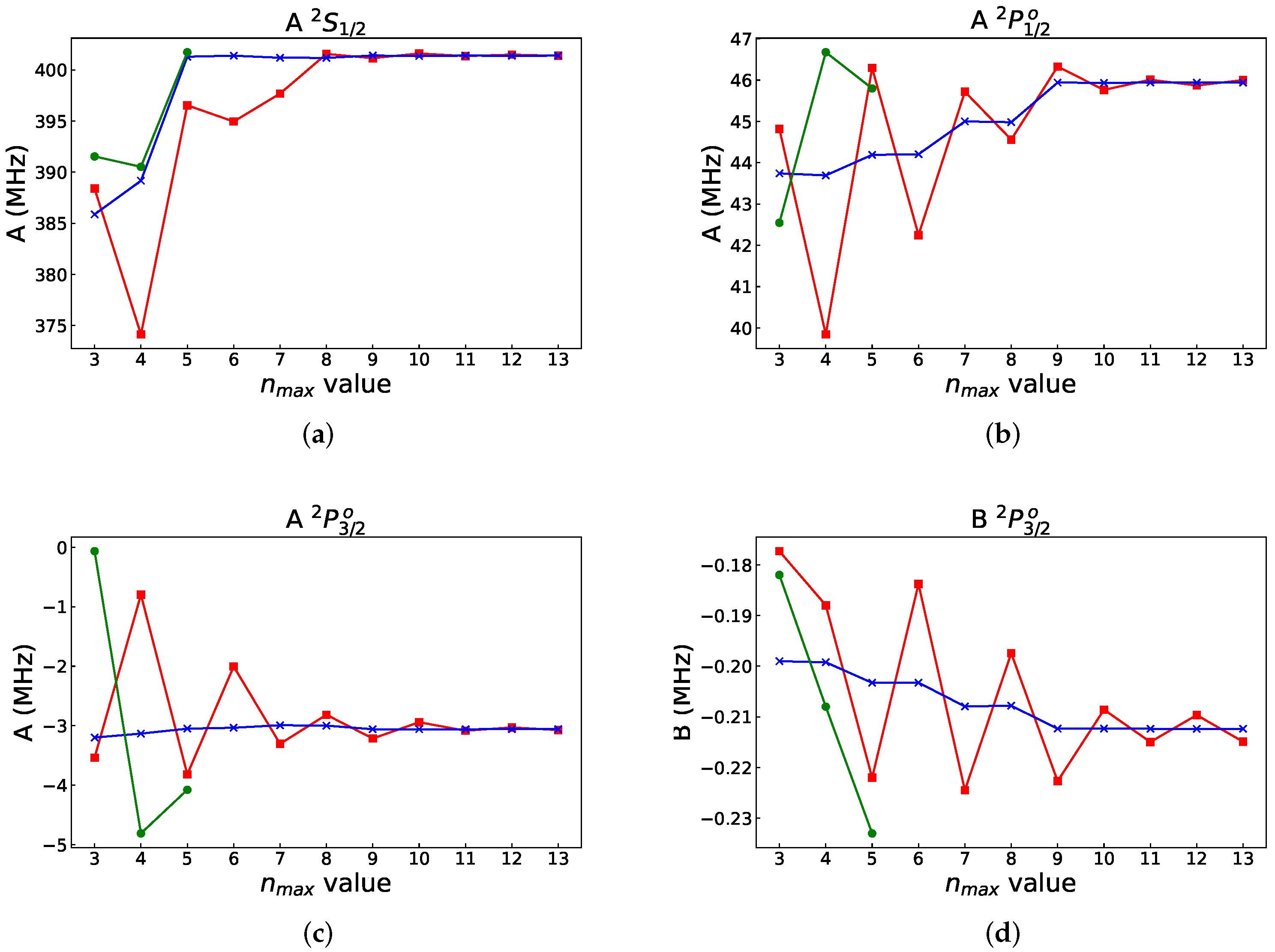
4. Hyperfine Interaction Constants in Different Orbital Bases
4.1. Orbital Basis from Energy-Driven Calculations
4.2. Polarization Orbitals Augmented to the Orbital Basis from Energy-Driven Calculations
5. Summary and Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Froese Fischer, C.; Gaigalas, G.; Jönsson, P.; Bieroń, J. GRASP2018—A Fortran 95 version of the general relativistic atomic structure package. Comput. Phys. Commun. 2019, 237, 184–187. [Google Scholar] [CrossRef]
- Jönsson, P.; Gaigalas, G.; Rynkun, P.; Radžiūtė, L.; Ekman, J.; Gustafsson, S.; Hartman, H.; Wang, K.; Godefroid, M.; Froese Fischer, C.; et al. Multiconfiguration Dirac-Hartree-Fock Calculations with Spectroscopic Accuracy: Applications to Astrophysics. Atoms 2017, 5, 16. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Wang, K.; Godefroid, M.; Jönsson, P.; Si, R.; Chen, C.Y. Benchmarking calculations with spectroscopic accuracy of excitation energies and wavelengths in sulfur-like tungsten. Phys. Rev. A 2020, 101, 032509. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.Y.; Wang, K.; Si, R.; Godefroid, M.; Jönsson, P.; Xiao, J.; Gu, M.F.; Chen, C.Y. Benchmarking calculations with spectroscopic accuracy of level energies and wavelengths in W LVII–W LXII tungsten ions. J. Quant. Spectrosc. Radiat. Transf. 2021, 269, 107650. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Li, J.Q.; Wang, K.; Si, R.; Godefroid, M.; Jönsson, P.; Xiao, J.; Gu, M.F.; Chen, C.Y. Benchmarking calculations of wavelengths and transition rates with spectroscopic accuracy for W xlviii through W lvi tungsten ions. Phys. Rev. A 2022, 105, 022817. [Google Scholar] [CrossRef]
- Andersson, M.; Grumer, J.; Ryde, N.; Blackwell-Whitehead, R.; Hutton, R.; Zou, Y.; Jönsson, P.; Brage, T. Hyperfine-dependent gf values of Mn I lines in the 1.49–180 μm H Band. Astrophys. J. Suppl. 2015, 216, 21. [Google Scholar] [CrossRef] [Green Version]
- Si, R.; Brage, T.; Li, W.; Grumer, J.; Li, M.; Hutton, R. A First Spectroscopic Measurement of the Magnetic-field Strength for an Active Region of the Solar Corona. Astrophys. J. Lett. 2020, 898, L34. [Google Scholar] [CrossRef]
- Filippin, L.; Bieroń, J.; Gaigalas, G.; Godefroid, M.; Jönsson, P. Multiconfiguration calculations of electronic isotope-shift factors in Zn I. Phys. Rev. A 2017, 96, 042502. [Google Scholar] [CrossRef] [Green Version]
- Ekman, J.; Jönsson, P.; Godefroid, M.; Nazé, C.; Gaigalas, G.; Bieroń, J. ris4: A program for relativistic isotope shift calculations. Comput. Phys. Commun. 2019, 235, 433–446. [Google Scholar] [CrossRef]
- Bieroń, J.; Froese Fischer, C.; Fritzsche, S.; Gaigalas, G.; Grant, I.P.; Indelicato, P.; Jönsson, P.; Pyykkö, P. Ab initio MCDHF calculations of electron-nucleus interactions. Phys. Scr. 2015, 90, 054011. [Google Scholar] [CrossRef]
- Papoulia, A.; Schiffmann, S.; Bieroń, J.; Gaigalas, G.; Godefroid, M.; Harman, Z.; Jönsson, P.; Oreshkina, N.S.; Pyykkö, P.; Tupitsyn, I.I. Ab initio electronic factors of the A and B hyperfine structure constants for the 5s25p6s 1,3P1o states in Sn I. Phys. Rev. A 2021, 103, 022815. [Google Scholar] [CrossRef]
- Li, J.; Gaigalas, G.; Bieroń, J.; Ekman, J.; Jönsson, P.; Godefroid, M.; Froese Fischer, C. Re-Evaluation of the Nuclear Magnetic Octupole Moment of 209Bi. Atoms 2022, 10, 132. [Google Scholar] [CrossRef]
- Barzakh, A.; Andreyev, A.N.; Raison, C.; Cubiss, J.G.; Van Duppen, P.; Péru, S.; Hilaire, S.; Goriely, S.; Andel, B.; Antalic, S.; et al. Large shape staggering in neutron-deficient Bi isotopes. Phys. Rev. Lett. 2021, 127, 192501. [Google Scholar] [CrossRef] [PubMed]
- Wraith, C.; Yang, X.; Xie, L.; Babcock, C.; Bieroń, J.; Billowes, J.; Bissell, M.; Blaum, K.; Cheal, B.; Filippin, L.; et al. Evolution of nuclear structure in neutron-rich odd-Zn isotopes and isomers. Phys. Lett. B 2017, 771, 385–391. [Google Scholar] [CrossRef]
- Barzakh, A.; Cubiss, J.; Andreyev, A.; Seliverstov, M.; Andel, B.; Antalic, S.; Ascher, P.; Atanasov, D.; Beck, D.; Bieroń, J.; et al. Inverse odd-even staggering in nuclear charge radii and possible octupole collectivity in 217,218,219At revealed by in-source laser spectroscopy. Phys. Rev. C 2019, 99, 054317. [Google Scholar] [CrossRef] [Green Version]
- Jönsson, P.; Godefroid, M.; Gaigalas, G.; Ekman, J.; Grumer, J.; Li, W.; Li, J.; Brage, T.; Grant, I.P.; Bieroń, J.; et al. An introduction to relativistic theory as implemented in GRASP. Atoms 2022, in press. [Google Scholar]
- Froese Fischer, C.; Brage, T.; Jönsson, P. Computational Atomic Structure; Institute of Physics Publishing (IoP): Bristol, UK, 1997. [Google Scholar]
- Godefroid, M.R.; Van Meulebeke, G.; Jönsson, P.; Froese Fischer, C. Large-scale MCHF calculations of hyperfine structures in nitrogen and oxygen. Z. Phys. D—Atoms Mol. Clust. 1997, 42, 193–201. [Google Scholar] [CrossRef]
- Papoulia, A.; Ekman, J.; Gaigalas, G.; Godefroid, M.; Gustafsson, S.; Hartman, H.; Li, W.; Radžiūtė, L.; Rynkun, P.; Schiffmann, S.; et al. Coulomb (Velocity) Gauge Recommended in Multiconfiguration Calculations of Transition Data Involving Rydberg Series. Atoms 2019, 7, 106. [Google Scholar] [CrossRef] [Green Version]
- Grant, I.P. Relativistic Quantum Theory of Atoms and Molecules: Theory and Computation; Springer Science and Business Media, LLC: New York, NY, USA, 2007. [Google Scholar]
- Froese Fischer, C.; Godefroid, M.; Brage, T.; Jönsson, P.; Gaigalas, G. Advanced multiconfiguration methods for complex atoms: I. Energies and wave functions. J. Phys. B At. Mol. Opt. Phys. 2016, 49, 182004. [Google Scholar] [CrossRef] [Green Version]
- Jönsson, P.; Godefroid, M.; Gaigalas, G.; Ekman, J.; Grumer, J.; Li, W.; Li, J.; Brage, T.; Grant, I.P.; Bieroń, J.; et al. GRASP Manual for Users. Atoms 2022, in press. [Google Scholar]
- Godefroid, M.R.; Jönsson, P.; Froese Fischer, C. Atomic structure variational calculations in spectroscopy. Phys. Scr. 1998, 1998, 33. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, C. Theory of hyperfine structure. Phys. Rev. 1955, 97, 380. [Google Scholar] [CrossRef]
- Lindgren, I.; Rosén, A. Relativistic self-consistent-field calculations with application to atomic hyperfine interaction. Case Stud. At. Phys. 1974, 3, 93–196. [Google Scholar]
- Lindgren, I. Effective operators in the atomic hyperfine interaction. Rep. Prog. Phys. 1984, 47, 345. [Google Scholar] [CrossRef]
- Beckmann, A.; Böklen, K.; Elke, D. Precision measurements of the nuclear magnetic dipole moments of 6Li, 7Li, 23Na, 39K and 41K. Z. Phys. 1974, 270, 173–186. [Google Scholar] [CrossRef]
- Orth, H.; Ackermann, H.; Otten, E. Fine and hyperfine structure of the 2 2P term of 7Li; determination of the nuclear quadrupole moment. Z. Phys. A Atoms Nucl. 1975, 273, 221–232. [Google Scholar] [CrossRef]
- Desclaux, J. A multiconfiguration relativistic DIRAC-FOCK program. Comput. Phys. Commun. 1975, 9, 31–45. [Google Scholar] [CrossRef]
- Indelicato, P. Projection operators in multiconfiguration Dirac-Fock calculations: Application to the ground state of heliumlike ions. Phys. Rev. A 1995, 51, 1132–1145. [Google Scholar] [CrossRef]
- Boucard, S.; Indelicato, P. Relativistic Many-Body and Qed Effects on the Hyperfine Structure of Lithium-Like Ions. Eur. Phys. J. A 2000, 8, 59–73. [Google Scholar] [CrossRef]
- Indelicato, P.; Lindroth, E.; Desclaux, J. Nonrelativistic Limit of Dirac-Fock Codes: The Role of Brillouin Configurations. Phys. Rev. Lett. 2005, 94, 013002. [Google Scholar] [CrossRef] [Green Version]
- Grant, I.P.; McKenzie, B.J.; Norrington, P.H.; Mayers, D.F.; Pyper, N.C. An atomic multiconfigurational Dirac-Fock package. Comput. Phys. Commun. 1980, 21, 207–231. [Google Scholar] [CrossRef]
- Dyall, K.G.; Grant, I.P.; Johnson, C.T.; Parpia, F.A.; Plummer, E.P. GRASP: A general-purpose relativistic atomic structure program. Comput. Phys. Commun. 1989, 55, 425–456. [Google Scholar] [CrossRef]
- Verdebout, S.; Rynkun, P.; Jönsson, P.; Gaigalas, G.; Froese Fischer, C.; Godefroid, M. A partitioned correlation function interaction approach for describing electron correlation in atoms. J. Phys. B At. Mol. Opt. Phys. 2013, 46, 085003. [Google Scholar] [CrossRef]
- Bieroń, J.; Jönsson, P.; Froese Fischer, C. Large-scale multiconfiguration Dirac-Fock calculations of the hyperfine-structure constants of the 2s 2S1/2, 2p 2P1/2, and 2p 2P3/2 states of lithium. Phys. Rev. A 1996, 53, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Puchalski, M.; Pachucki, K. Ground State Hyperfine Splitting in 6,7Li Atoms and the Nuclear Structure. Phys. Rev. Lett. 2013, 111, 243001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froese Fischer, C.; Verdebout, S.; Godefroid, M.; Rynkun, P.; Jönsson, P.; Gaigalas, G. Doublet-quartet energy separation in boron: A partitioned-correlation-function-interaction method. Phys. Rev. A 2013, 88, 062506. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.T.; Wang, K.; Si, R.; Godefroid, M.; Gaigalas, G.; Chen, C.Y.; Jönsson, P. Reducing the computational load—Atomic multiconfiguration calculations based on configuration state function generators. Comput. Phys. Commun. 2023, 283, 108562. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DF | 290.249 | 32.356 | −6.469 | |
| 374.047 | 44.741 | |||
| 380.692 | 42.461 | |||
| 380.341 | 42.610 | |||
| 380.342 | 42.611 | |||
| Exp. | 401.752043 | 45.914(25) | −3.055(14) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Jönsson, P.; Godefroid, M.; Gaigalas, G.; Bieroń, J.; Marques, J.P.; Indelicato, P.; Chen, C. Independently Optimized Orbital Sets in GRASP—The Case of Hyperfine Structure in Li I. Atoms 2023, 11, 4. https://doi.org/10.3390/atoms11010004
Li Y, Jönsson P, Godefroid M, Gaigalas G, Bieroń J, Marques JP, Indelicato P, Chen C. Independently Optimized Orbital Sets in GRASP—The Case of Hyperfine Structure in Li I. Atoms. 2023; 11(1):4. https://doi.org/10.3390/atoms11010004
Chicago/Turabian StyleLi, Yanting, Per Jönsson, Michel Godefroid, Gediminas Gaigalas, Jacek Bieroń, José Pires Marques, Paul Indelicato, and Chongyang Chen. 2023. "Independently Optimized Orbital Sets in GRASP—The Case of Hyperfine Structure in Li I" Atoms 11, no. 1: 4. https://doi.org/10.3390/atoms11010004
APA StyleLi, Y., Jönsson, P., Godefroid, M., Gaigalas, G., Bieroń, J., Marques, J. P., Indelicato, P., & Chen, C. (2023). Independently Optimized Orbital Sets in GRASP—The Case of Hyperfine Structure in Li I. Atoms, 11(1), 4. https://doi.org/10.3390/atoms11010004