1. Introduction and Calculations
Scattering by single-electron systems is always of interest because the wave function of the target is known exactly. Also, scattering from atoms helps us to understand their static and dynamic properties. It also has applications in astrophysics and in the investigation of controlled thermonuclear devices. Different approaches have been applied to study scattering of electrons and positrons from various targets, to infer the feasibility of various calculations and the accuracy of their results. In other words, this is an ideal problem for testing various theories. A simple method is to assume that the wave function of the incident electron on hydrogen atom is given by
, where
is the ground state of the hydrogen atom and
is the function whose asymptotic limit determines phase shift of the scattering. Morse and Allis [
1] introduced exchange between electrons, writing the wave function in the form:
The ground state function is given by:
The upper sign in Equation (1) corresponds to the singlet state and the lower sign corresponds to triplet state of the system. The equation for the scattering function
u(r) (letting
r1 = r) is obtained by the Kohn variation of the functional:
where
E =
k2 −
Z2 and
k2 is the kinetic energy of the incident electron, and
Z is the charge of the nucleus. Considering the proton stationary during the interaction, the Hamiltonian
H of the system in Rydberg units is given by:
In the above equation,
. Using Equation (3), we get the integro-differential equation:
The exchange terms with
,
, and
are the nonlocal potentials and are functions of u(r):
and
The singlet and triplet
S-wave phase shifts in the exchange approximation are given in
Table 1. Among the different methods used is the variational principle of Kohn [
2]. Schwartz [
3] used this principle to calculate phase shifts of electrons and positrons scattering from hydrogen atoms. There are no bounds on phase shifts in this principle except at incident energy
k = 0. There are also singularities in this calculation. In spite of singularities, the results obtained in [
3] are fairly accurate and have stood the test of time. Another extensively used method is the close-coupling approximation [
4], in which the total wave function is expanded in eigenstates of the hydrogen atom. Accurate results have been obtained using the
R-matrix formulation [
5] in which most of the complications of interactions are taken into account within the inner region of a certain radius and outside this radius plane waves or Coulomb waves are used for continuum functions.
It has been emphasized by Wigner [
8] that long range forces determine the nature of the scattering parameters at threshold. A method in which the longest-range potential proportional to −1/r
4 is taken into account is the method of polarized orbital [
6,
9]. This method includes the essential physics of the problem and has been widely used not only to calculate scattering parameters for electrons and positrons but also to calculate photoabsorption cross sections. Using the first-order perturbation theory and the dipole part of the perturbed wave function, Temkin [
9] showed that the effective target wave function in the presence of the incident electron can be written as
where
Now, the function in Equation (1) is written as:
The equation for the scattering function
can be derived from:
The equation for the scattering function
for all momenta has been given by Sloan [
10]:
where
The scattering equation has the well-known attractive potential which for large
r is equal to
,
α = 4.5
being the asymptotic polarizability for a hydrogen atom. Temkin and Lamkin [
6], using this method of polarized orbitals, calculated
S-wave phase shifts, as well as for higher partial waves, for scattering of an electron from a hydrogen atom. Phase shifts are determined by the asymptotic limit of the function
:
The Coulomb phase in the above equation is given by:
S-wave phase shift obtained by this method are given in
Table 1 and they differ from those obtained in the exchange approximation, showing the importance of polarization of the target resulting in the long-range potential −1/r
4. However, this method does not provide variational bounds to the exact phase shifts.
The short-range correlations can be important in addition to the long-range correlations. These short-range correlations can be included using the projection-operator formalism of Feshbach [
11]:
The wave function in Equation (1) is augmented by a correlation function
:
where
The radial functions are of Hylleraas type and
D’s are rotational harmonics [
12].
The projections are defined by:
Using the projection operators
P and
Q, we get the equation for the scattering function
, having an optical potential [
7]. The equation for the scattering function
is given by the following equation:
where
and
are the direct and nonlocal exchange potentials of the ‘exchange approximation’ [
1].
is the optical potential which is negative definite and therefore attractive. The optical potential acting on
is given by:
We find
S-wave phase shifts, given in
Table 1, increase compared to the exchange approximation when short-range correlations are included but not the long-range correlations at the same time. However, the phase shifts obtained by this method have lower bounds to the exact phase shifts.
Is it possible to consider the long-range and short-range correlations at the same time? The projection-operator formalism, when both type of correlations are included, does not work because it is not possible to construct projection operators when
instead of
is used in the definitions given in Equation (21). However, we can write a wave function which has a long-range and a short-range parts at the same time and we call this formulation hybrid theory because it includes both type of correlations: short-range as well as long-range at the same time. We describe this formalism by confining ourselves to the e-H partial wave (denoted by
L) problem. The total spatial wave function is written as:
The functions and are given in Equations (9) and (20), respectively. This approach is variationally correct and phase shifts obtained have lower bounds to the exact phase shifts. This has been achieved by introducing separate correlation functions and then amalgamating them into the scattering problem, via an optical potential, in order to replace the many-particle Schrödinger equation with a single-particle Schrödinger equation.
We can derive the equation for the scattering function u(r) from (letting r
1 =
r) from the functional:
For
S-waves, we can write:
Clmn in Equation (26) are coefficients which are determined when eigenvalues are calculated using the Ritz variational principle. For
= 1, the functional can be written as:
where
and
We can determine
C1 by:
which implies that:
We can generalize the above treatment to any number of unknown coefficients in Equation (24). Now all the quantities in Equation (24) are known except
. The equation for the scattering function
can be derived from:
The resulting equation is fairly complicated and the various quantities are given in [
13]. We give a few of them below. We can generalize the above treatment to any number of unknown coefficients in Equation (24). We write the wave function in the form:
We have replaced the step function
by:
The exponent n is found to be 3 or 4. Now, polarization takes place for any value of r1, the coordinate of the incident electron. In the method of polarized orbitals, the orbital of the target was perturbed only when the incident electron was outside the target. Now perturbation takes place whether the incident electron is outside or inside of the target.
In addition to the nonlinear parameters in the correlation function,
β is another nonlinear parameter and it is a function of the incident momentum
k. This further helps in getting improved results. There is another form of the smooth cutoff function given by Shertzer and Temkin [
14]:
All the quantities are now known in the wave function given in Equation (24). We can derive the equation for the scattering function using Equation (35). The equation for the scattering function is again is of the same form as Equation (22), but with more terms:
As stated earlier, most of the quantities are given in [
13]. Some of these quantities occurring in the direct potential are given below:
The direct potentials are given by:
We repeat here the exchange terms already indicated in Equation (5):
The last term in
has:
For
,
has a term
which is the dipole polarizability of the target with nuclear charge
Z.
= optical potential which is attractive, but does not depend on projection operators, and is given by:
The function
is the function given in Equation (24) without the correlation terms. Phase shifts, given in
Table 2, are calculated using the asymptotic limit of the function
given in Equation (16).
We see that the method of polarized orbitals over estimates phase shifts. In the variational formulation, phase shifts have lower bounds to the exact phase shifts, that is, they can be higher than those given in
Table 2 when the number of correlations terms in Equation (24) is increased. The phase shifts obtained using hybrid theory agree well with those obtained by Schwartz [
3] and also with those obtained by Scholz et al. [
15] using the close-coupling approximation. Similar calculations have been carried out for
S-wave electron scattering from He
+ and Li
2+ [
16]. These results are shown in
Table 3 and they agree with those obtained by Gien [
17,
18] using the Harris-Nesbet variational method.
Calculations have been carried out for partial wave
L = 1 for electron scattering from H, He
+, and Li
2+ [
19,
20]. The
P-wave phase shifts for electron-H scattering are given in
Table 4. They are compared with those obtained using the
R-matrix method [
15] and by the finite-element method [
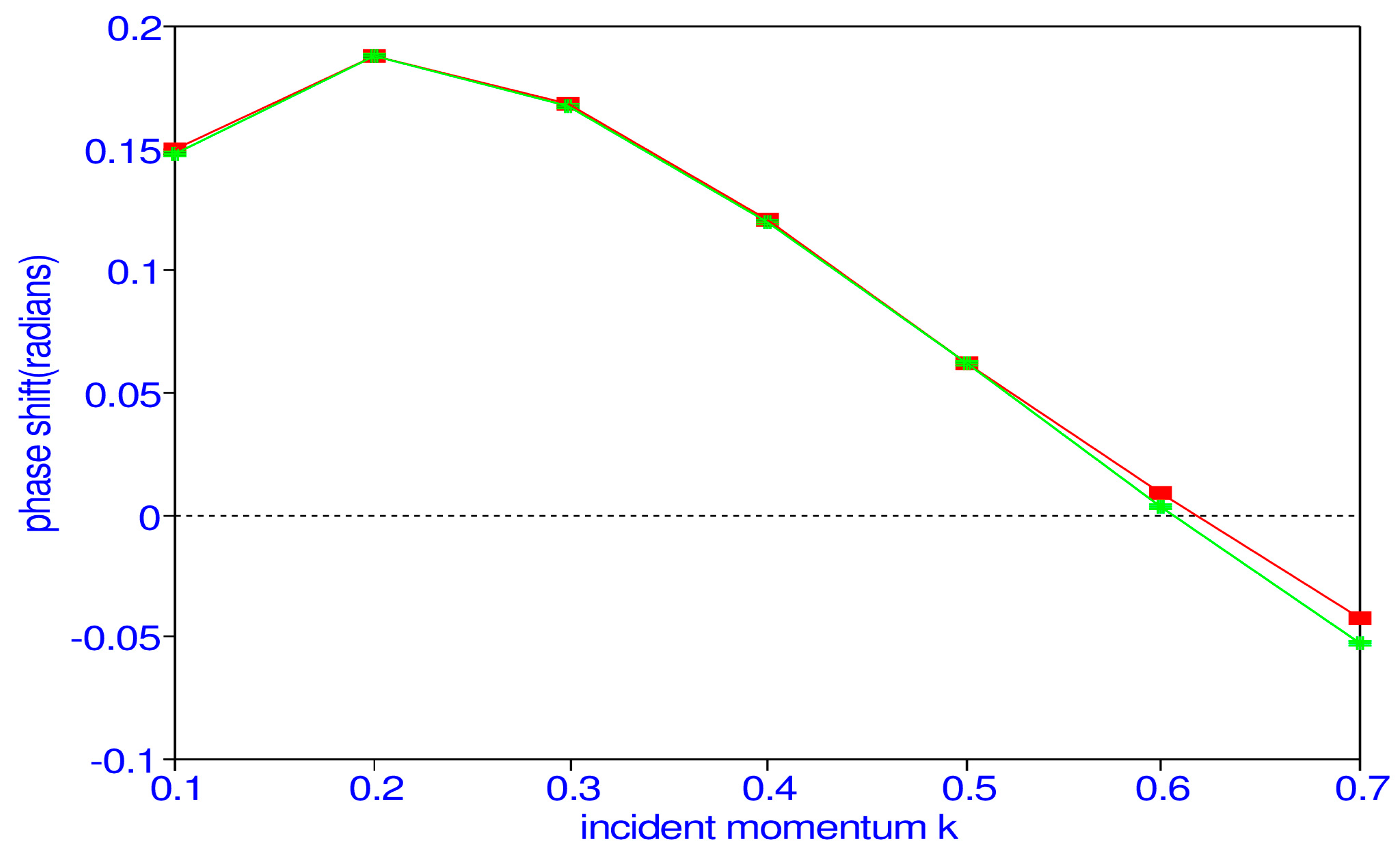
21]. We see that the singlet
P phase shifts have a peak at
k = 0.3, while the triplet
P phase shifts increase continuously.
Calculations have also been carried for partial wave
L = 2 for electron scattering from H, He
+, and Li
2+ and photoabsorption when the final state is a 2
p state [
22].
5. Photoabsorption
It was pointed out by Wildt [
26] that the opacity in the sun is due to the absorption of photons:
The photoabsorption or photodetachment cross section (in length form and in units of (
) for a transition from an initial state ‘
i’ to the final state ‘
f’ is given by:
where
α is the fine structure constant,
k is the momentum of the outgoing electron, and
ω is the energy of the incident photon:
where
is the ionization of the system absorbing the photon and
k2 is the energy of the ejected electron. The Hylleraas-type normalized function
is the ground-state wave function of the two-electron systems and it is given by:
The upper sign refers to the singlet states and the lower sign refers to the triplet states. In the
Table 6, the photodetachment cross sections of
[
27] are given for a few values of
k, momentum of the ejected electron. These results have been obtained without the short-range correlations. They are compared in
Table 6 with those obtained by Bell and Kingston [
28] who used the method of polarized orbitals. Their results are higher than those obtained using the variational approach [
27]. Addition of short-range correlations in the final state wave function gave improved results for cross sections, given in
Table 7. However, we find that at low energies the cross sections do not change considerably, showing that at low-energies the long-range correlations are more important than the short-range correlations.
Ohmura and Ohmura [
29], using the effective range theory and the loosely bound structure of hydrogen ion, obtained:
where
γ = 0.2355883 and
ρ = 2.646 ± 0.0004,
γ is the square root of the binding energy of the electron and
ρ is the effective range. The cross sections obtained using the effective-range theory are higher than those obtained in [
27]. However, at low energies they are close to each other. Photodetachment cross sections for
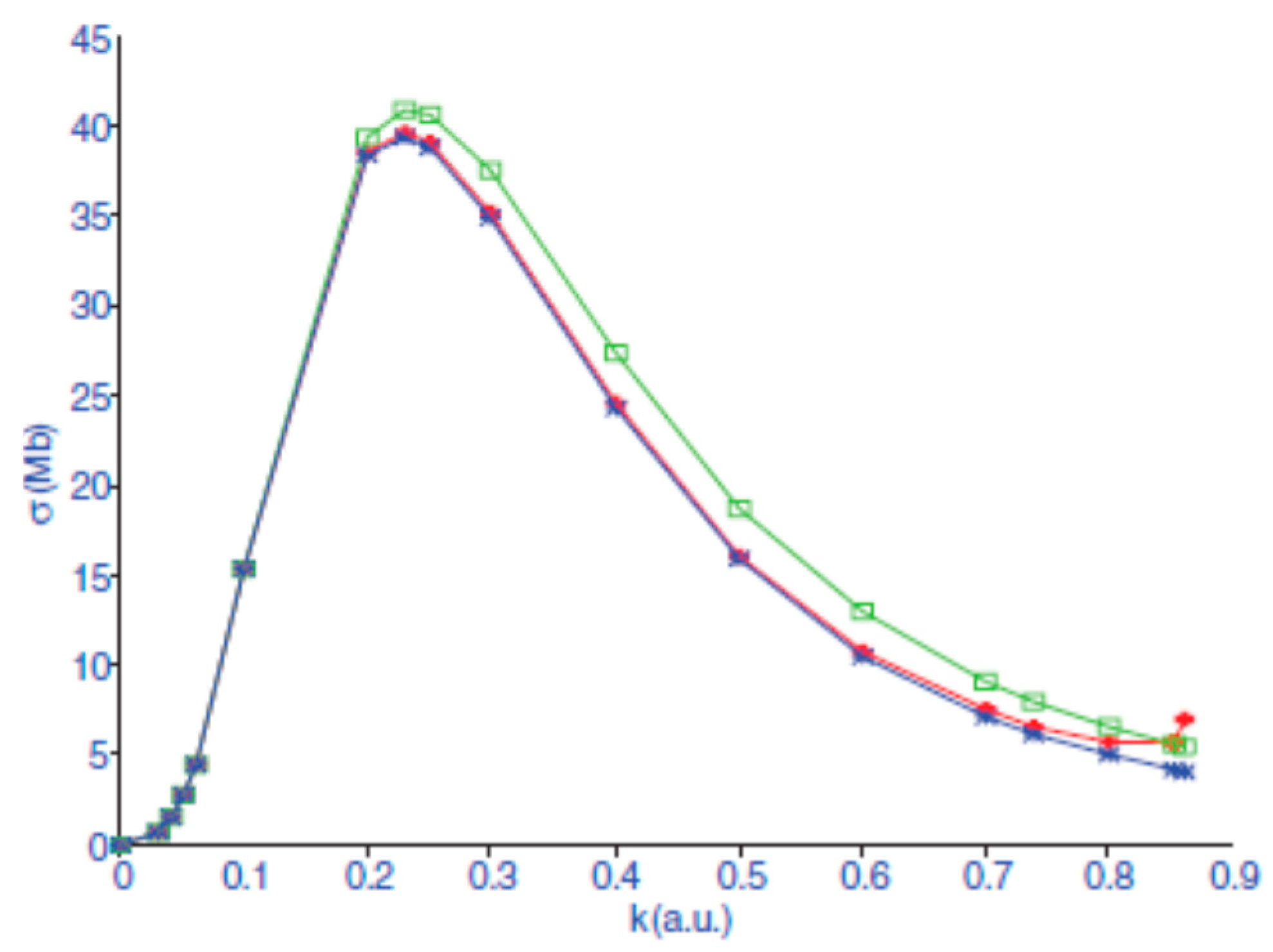
are shown in
Figure 1 from Ref. [
27].
A similar calculation, including short-range and long-range correlations, has been carried out for the photoionization of He [
27]. The results, given in
Table 8, agree well with those obtained using the
R-matrix approach [
30] and the experimental results of Samson et al. [
31]. Photoionization cross sections for Li ion have also been calculated and they are also given in [
27].
We notice that in the limit , the photodetachment cross section for H− goes to zero because the plane wave is normalized as sin(kr)/kr which is equal to 1 for , while the photoionization cross section of He goes to a finite value because of the final state Coulomb wave, which is proportional to for .
7. Excitation of the 2S State
Cross section for excitation of the 2
S state of atomic hydrogen at low incident electron energies has been carried out using different methods. We mention a few: Burke et al. [
32] used close-coupling approximation retaining 1
s, 2
s, and 2
p eigenstates. Callaway et al. [
33] carried out calculations in the range 12 to 54 eV using 11-state expansion including seven pseudostates. A similar calculation in the above energy range has been carried by Callaway et al. [
34] using six-state close-coupling expansion including an optical potential. The results of these calculations are essentially based on the close-coupling approximation. Scott el al. [
35] used the standard
R-matrix close-coupling method using nine-state basis, consisting of three eigenstates and six pseudostates. Poet [
36] carried out calculations by assuming spherically symmetric wave functions. Lloyd and McDowell [
37] used the method of polarized orbitals for the first three partial waves and Bessel functions for the higher partial waves. All these results are different from each other. Even though the hydrogen atom is the simplest system, it seems it is not easy to get exact cross sections for excitation. Excitation cross sections have been calculated using the variational method of polarized orbitals [
13]. The excitation cross section [
38] from state ‘
i’ to state ‘
f’ is given by:
where
ki and
kf are the initial and final momenta and
Tfi is a matrix element given by:
The interaction potential is:
The initial state wave function
, in principle, is an exact solution of the Schrödinger equation [
38]. Using this method, excitation cross sections have been calculated in the energy range 10.30 to 54.5 eV by using nine partial waves to get convergence, which is good up to the fifth and sixth decimal places for low energies, while at high energies it is good up to the third decimal place [
38]. Some of the results of the various calculations are given in
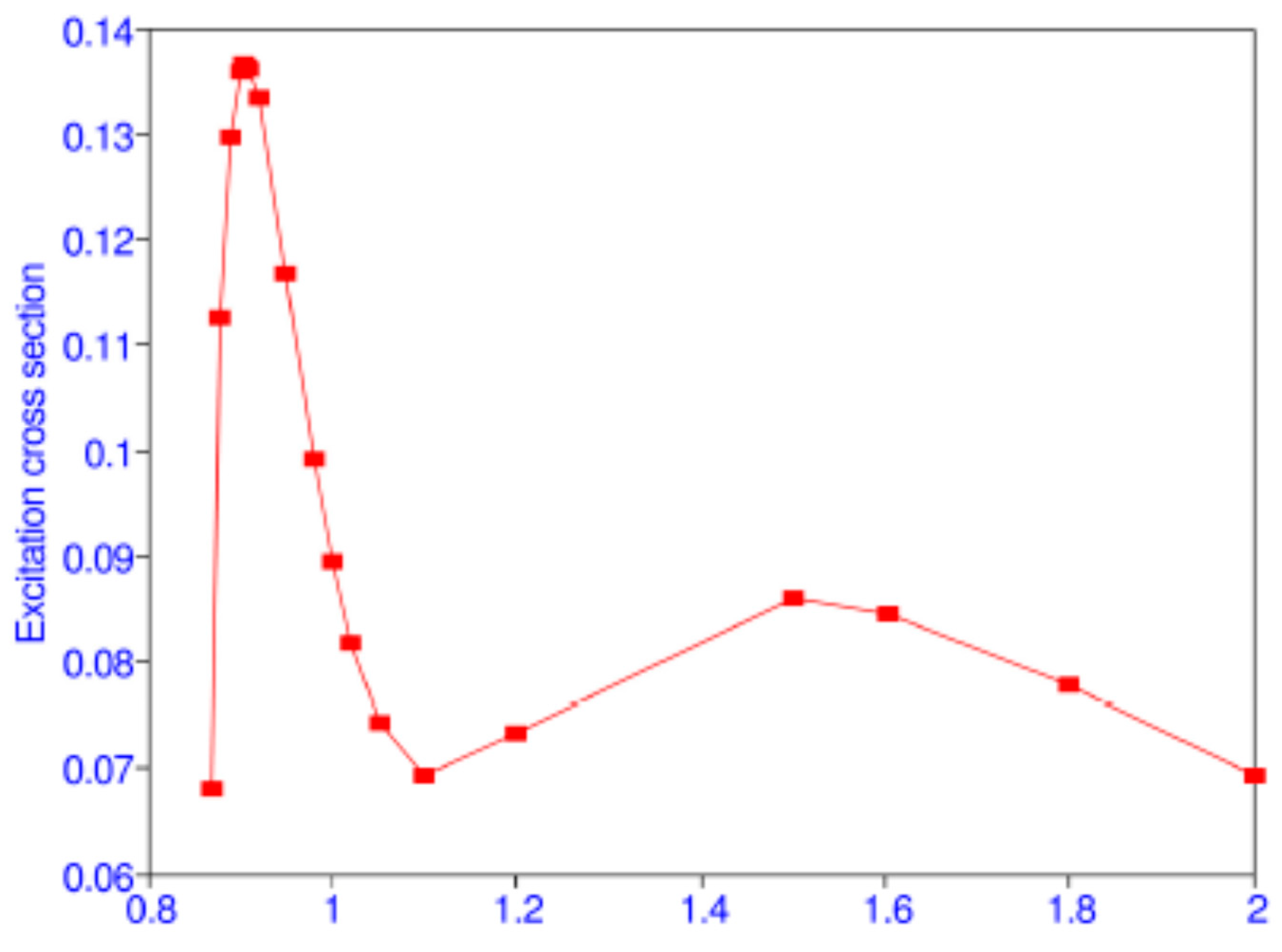
Table 11. The maximum of the cross section 0.137
at 11.4 eV which is close to the experimental result 0.163 ± 0.2
at 11.6 ± 0.2 eV of Kauppila et al. [
39]. There is a minimum of the cross section at 16.46 eV and another maximum at 34.82 eV, as shown in
Figure 2 from Ref. [
38]. Differential and spin cross sections have also been calculated [
38].





{kind=link}
{kind=link}
{kind=link}