
Cross Sections and Rate Coefficients for Vibrational Excitation of H2O by Electron Impact

 ,
, 
Abstract
:1. Introduction
2. Theoretical Approach
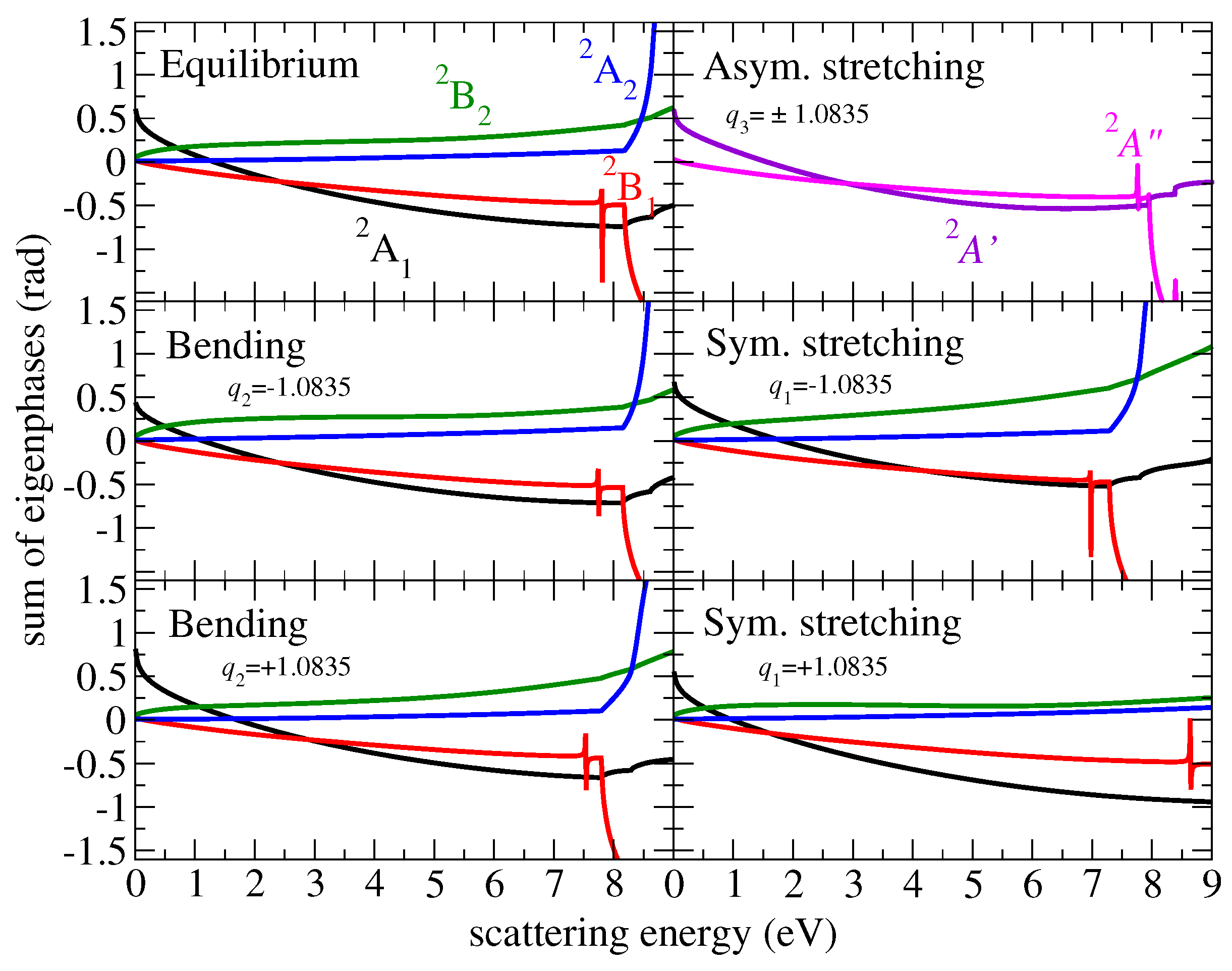
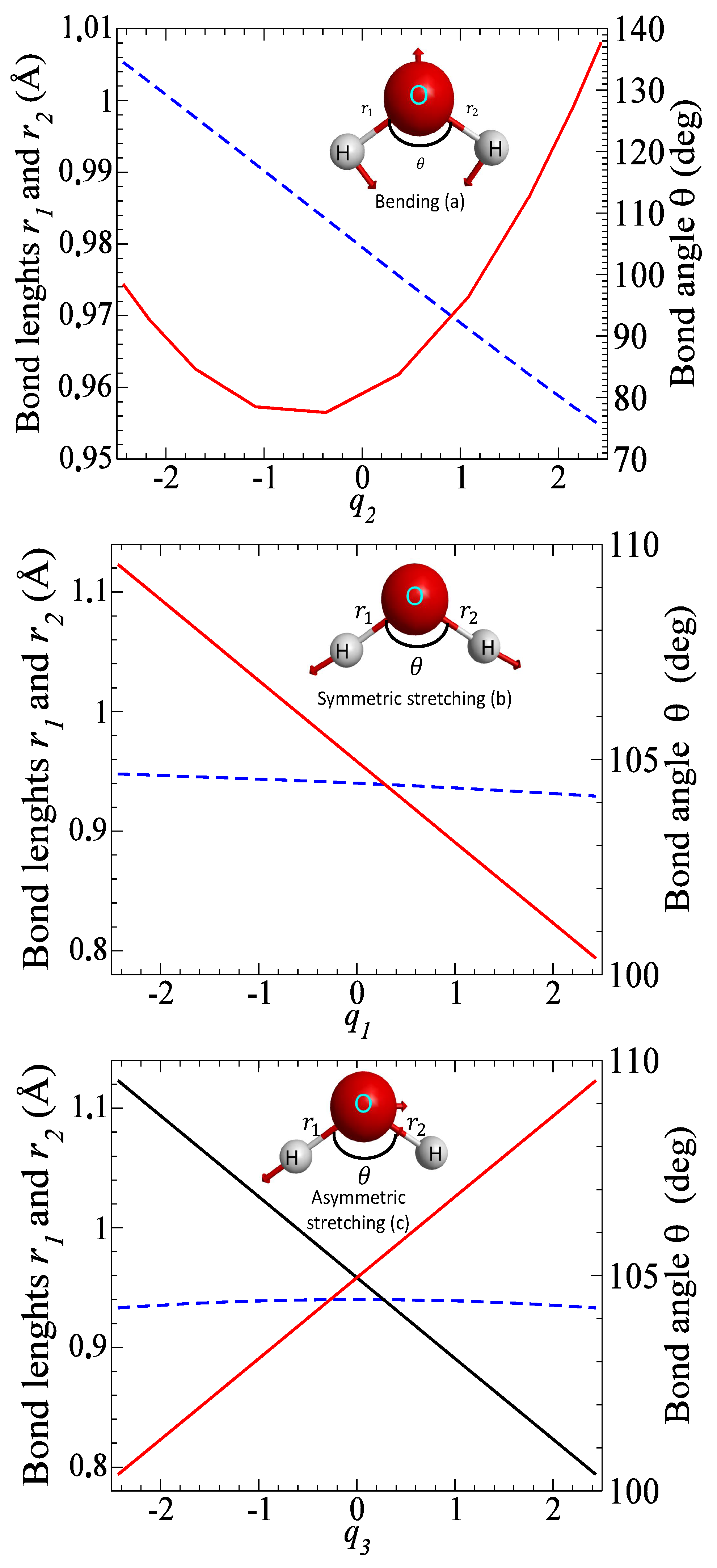
2.1. Ab Initio Calculations
2.2. Cross-Sections for Vibrational Excitation
3. Results
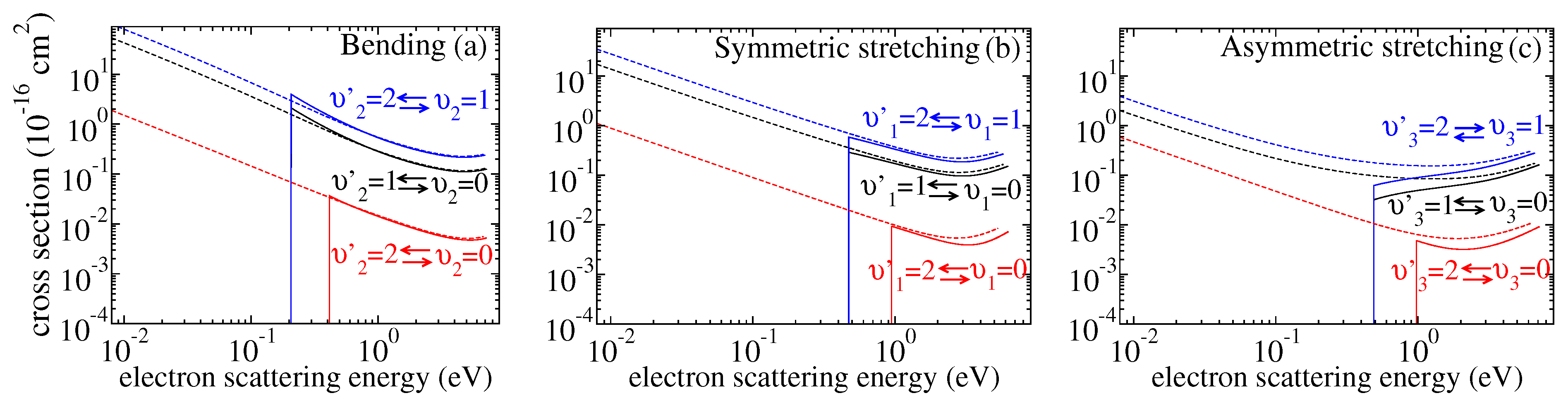
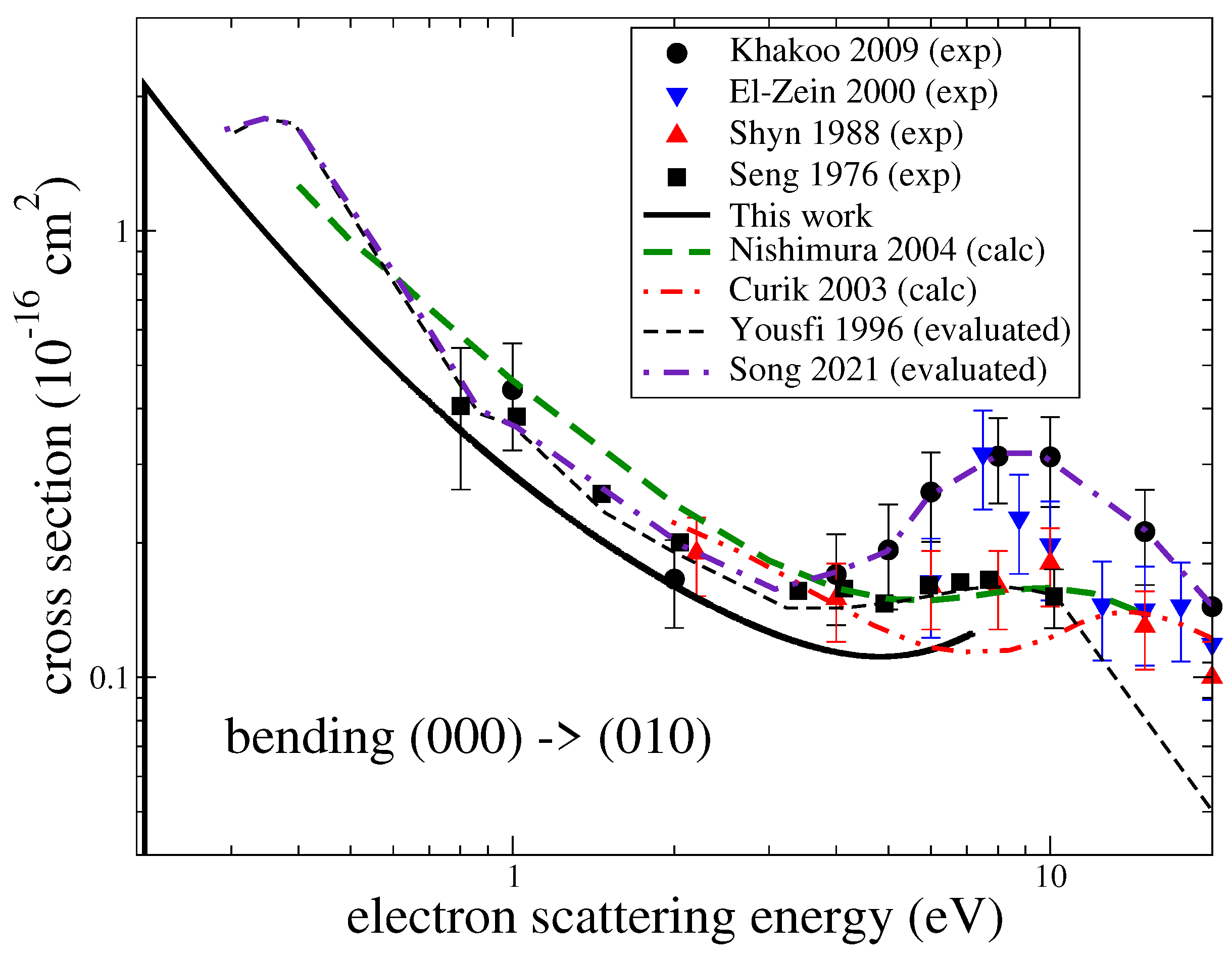
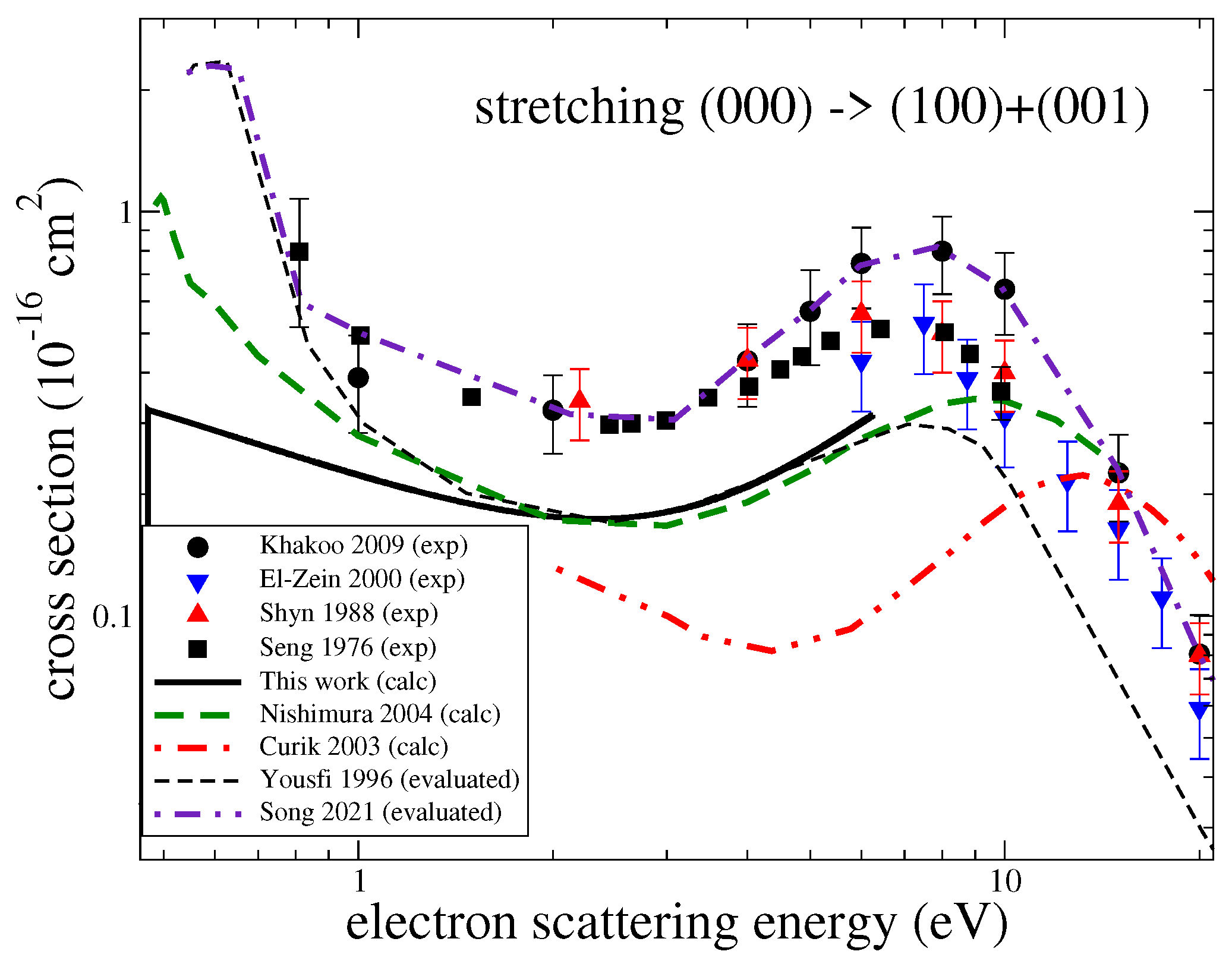
3.1. Cross Sections
3.2. Rate Coefficients
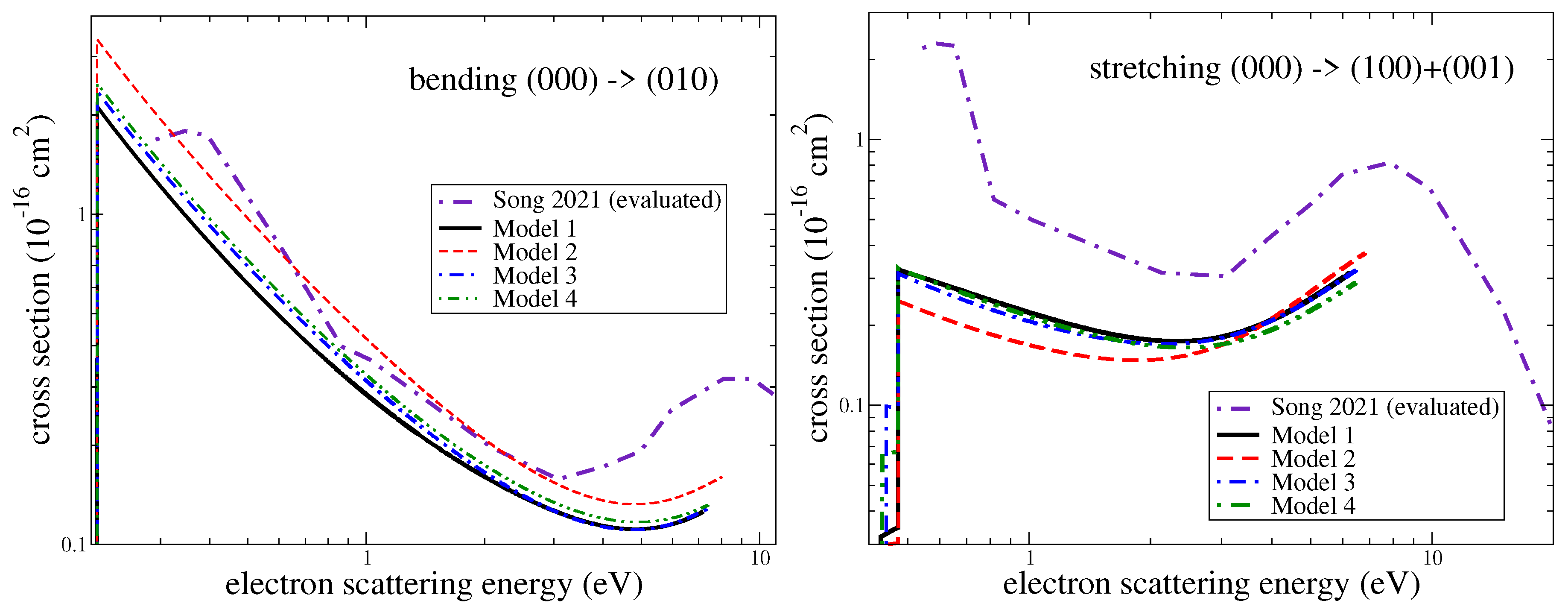
3.3. Assessment of Uncertainties
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Van Dishoeck, E.F.; Herbst, E.; Neufeld, D.A. Interstellar Water Chemistry: From Laboratory to Observations. Chem. Rev. 2013, 113, 9043–9085. [Google Scholar] [CrossRef] [Green Version]
- Song, M.Y.; Cho, H.; Karwasz, G.P.; Kokoouline, V.; Nakamura, Y.; Tennyson, J.; Faure, A.; Mason, N.J.; Itikawa, Y. Cross Sections for Electron Collisions with H2O. J. Phys. Chem. Ref. Data 2021, 50, 023103. [Google Scholar] [CrossRef]
- Machado, L.E.; Brescansin, L.M.; Iga, I.; Lee, M.T. Elastic and rotational excitation cross-sections for electron-water collisions in the low- and intermediate-energy ranges. Eur. Phys. J. D 2005, 33, 193–199. [Google Scholar] [CrossRef]
- Faure, A.; Gorfinkiel, J.D.; Tennyson, J. Low-energy electron collisions with water: Elastic and rotationally inelastic scattering. J. Phys. B At. Mol. Phys. 2004, 37, 801–807. [Google Scholar] [CrossRef] [Green Version]
- Itikawa, Y.; Mason, N. Cross Sections for Electron Collisions with Water Molecules. J. Phys. Chem. Ref. Data 2005, 34, 1–22. [Google Scholar] [CrossRef]
- Khakoo, M.; Winstead, C.; McKoy, V. Vibrational excitation of water by electron impact. Phys. Rev. A 2009, 79, 052711. [Google Scholar] [CrossRef] [Green Version]
- Seng, G.; Linder, F. Vibrational excitation of polar molecules by electron impact. II. Direct and resonant excitation in H2O. J. Phys. B At. Mol. Phys. 1976, 9, 2539–2551. [Google Scholar] [CrossRef]
- Nishimura, T.; Gianturco, F. Vibrational excitation of water by low-energy electron scattering: Calculations and experiments. Europhys. Lett. 2004, 65, 179. [Google Scholar] [CrossRef] [Green Version]
- Faure, A.; Josselin, E. Collisional excitation of water in warm astrophysical media. I. Rate coefficients for rovibrationally excited states. Astron. Astrophys. 2008, 492, 257–264. [Google Scholar] [CrossRef]
- Baudry, A.; Humphreys, E.M.L.; Herpin, F.; Torstensson, K.; Vlemmings, W.H.T.; Richards, A.M.S.; Gray, M.D.; De Breuck, C.; Olberg, M. Vibrationally excited water emission at 658 GHz from evolved stars. Astron. Astrophys. 2018, 609, A25. [Google Scholar] [CrossRef] [Green Version]
- Tenenbaum, E.D.; Dodd, J.L.; Milam, S.N.; Woolf, N.J.; Ziurys, L.M. Comparative Spectra of Oxygen-rich Versus Carbon-rich Circumstellar Shells: VY Canis Majoris and IRC +10216 at 215–285 GHz. Astrophys. J. Lett. 2010, 720, L102–L107. [Google Scholar] [CrossRef] [Green Version]
- Stoecklin, T.; Denis-Alpizar, O.; Clergerie, A.; Halvick, P.; Faure, A.; Scribano, Y. Rigid-Bender Close-Coupling Treatment of the Inelastic Collisions of H2O with para-H2. J. Phys. Chem. A 2019, 123, 5704–5712. [Google Scholar] [CrossRef] [PubMed]
- Stoecklin, T.; Cabrera-González, L.D.; Denis-Alpizar, O.; Páez-Hernández, D. A close coupling study of the bending relaxation of H2O by collision with He. J. Chem. Phys. 2021, 154, 144307. [Google Scholar] [CrossRef]
- Ayouz, M.; Kokoouline, V. Cross Sections and Rate Coefficients for Vibrational Excitation of HeH+ Molecule by Electron Impact. Atoms 2016, 4, 30. [Google Scholar] [CrossRef] [Green Version]
- Ayouz, M.; Kokoouline, V. Cross Sections and Rate Coefficients for Rovibrational Excitation of HeH+ Isotopologues by Electron Impact. Atoms 2019, 7, 67. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; dos Santos, S.F.; Yuen, C.H.; Cortona, P.; Kokoouline, V.; Ayouz, M. Theoretical study of electron-induced vibrational excitation of NO2. Plasma Sources Sci. Technol. 2019, 28, 105017. [Google Scholar] [CrossRef]
- Liu, H.; Santos, S.F.d.; Yuen, C.H.; Cortona, P.; Ayouz, M.; Kokoouline, V. Vibrational excitation of N2O by an electron impact and the role of the Renner-Teller effect in the process. Phys. Rev. A 2020, 102, 032808. [Google Scholar] [CrossRef]
- Tennyson, J. Electron–molecule collision calculations using the R-matrix method. Phys. Rep. 2010, 491, 29–76. [Google Scholar] [CrossRef]
- Carr, J.; Galiatsatos, P.; Gorfinkiel, J.; Harvey, A.; Lysaght, M.; Madden, D.; Mašín, Z.; Plummer, M.; Tennyson, J.; Varambhia, H. UKRmol: A low-energy electron- and positron-molecule scattering suite. Eur. Phys. J. D 2012, 66, 58. [Google Scholar] [CrossRef]
- Tennyson, J.; Brown, D.B.; Munro, J.J.; Rozum, I.; Varambhia, H.N.; Vinci, N. Quantemol-N: An expert system for performing electron molecule collision calculations using the R-matrix method. J. Phys. Conf. Ser. 2007, 86, 012001. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Functions for Use in Molecular Calculations. I. Contraction of (9s5p) Atomic Basis Sets for the First-Row Atoms. J. Chem. Phys. 1970, 53, 2823–2833. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A general-purpose quantum chemistry program package. WIREs Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
- Johnson, R.D. NIST Computational Chemistry Comparison and Benchmark Database; NIST Standard Reference Database Number 101; NIST: Gaithersburg, MD, USA, 2010.
- Császár, A.G.; Czako, G.; Furtenbacher, T.; Tennyson, J.; Szalay, V.; Shirin, S.V.; Zobov, N.F.; Polyansky, O.L. On equilibrium structures of the water molecule. J. Chem. Phys. 2005, 122, 214305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorn, P.; Campbell, L.; Brunger, M. Electron excitation and energy transfer rates for H2O in the upper atmosphere. PMC Phys. B 2009, 2, 1. [Google Scholar] [CrossRef]
- El-Zein, A.; Brunger, M.; Newell, W. Excitation of vibrational quanta in water by electron impact. J. Phys. B At. Mol. Opt. Phys. 2000, 33, 5033. [Google Scholar] [CrossRef]
- Shyn, T.; Cho, S.; Cravens, T. Vibrational-excitation cross sections of water molecules by electron impact. Phys. Rev. A 1988, 38, 678. [Google Scholar] [CrossRef] [PubMed]
- Curik, R.; Carsky, P. Vibrationally inelastic electron scattering on polyatomic molecules by the discrete momentum representation (DMR) method. J. Phys. B At. Mol. Opt. Phys. 2003, 36, 2165. [Google Scholar] [CrossRef]
- Yousfi, M.; Benabdessadok, M.D. Boltzmann equation analysis of electron-molecule collision cross sections in water vapor and ammonia. J. Appl. Phys. 1996, 80, 6619–6630. [Google Scholar] [CrossRef]
- Laporta, V.; Cassidy, C.; Tennyson, J.; Celiberto, R. Electron-impact resonant vibration excitation cross sections and rate coefficients for carbon monoxide. Plasma Sources Sci. Technol. 2012, 21, 045005. [Google Scholar] [CrossRef]
- Jones, M.; Tennyson, J. On the use of pseudostates to calculate molecular polarizabilities. J. Phys. B 2010, 43, 045101. [Google Scholar] [CrossRef]
- Kokoouline, V.; Faure, A.; Tennyson, J.; Greene, C.H. Calculation of rate constants for vibrational and rotational excitation of the H3+ ion by electron impact. Mon. Not. R. Astron. Soc. 2010, 405, 1195–1202. [Google Scholar]
- Jiang, X.; Yuen, C.H.; Cortona, P.; Ayouz, M.; Kokoouline, V. Cross sections for vibronic excitation of CH+ by low-energy electron impact. Phys. Rev. A 2019, 100, 062711. [Google Scholar] [CrossRef]
- Cooper, B.; Tudorovskaya, M.; Mohr, S.; O’Hare, A.; Hanicinec, M.; Dzarasova, A.; Gorfinkiel, J.; Benda, J.; Mašín, Z.; Al-Refaie, A.; et al. Quantemol Electron Collision: An expert system for performing UKRmol+ electron molecule collision calculations. Atoms 2019, 7, 97. [Google Scholar] [CrossRef] [Green Version]
- Mašín, Z.; Benda, J.; Gorfinkiel, J.D.; Harvey, A.G.; Tennyson, J. UKRmol+: A suite for modelling of electronic processes in molecules interacting with electrons, positrons and photons using the R-matrix method. Comput. Phys. Comms. 2020, 249, 107092. [Google Scholar] [CrossRef] [Green Version]
- Fonseca dos Santos, S.; Douguet, N.; Kokoouline, V.; Orel, A. Scattering matrix approach to the dissociative recombination of HCO+ and N2H+. J. Chem. Phys. 2014, 140, 164308. [Google Scholar] [CrossRef] [Green Version]
- Douguet, N.; Fonseca dos Santos, S.; Kokoouline, V.; Orel, A. Simplified model to describe the dissociative recombination of linear polyatomic ions of astrophysical interest. EPJ Web Conf. 2015, 84, 07003. [Google Scholar] [CrossRef] [Green Version]
- Kokoouline, V.; Ayouz, M.; Mezei, J.Z.; Hassouni, K.; Schneider, I.F. Theoretical study of dissociative recombination and vibrational excitation of the BF2+ ion by an electron impact. Plasma Sources Sci. Technol. 2018, 27, 115007. [Google Scholar] [CrossRef]
- Ayouz, M.A.; Yuen, C.H.; Balucani, N.; Ceccarelli, C.; Schneider, I.F.; Kokoouline, V. Dissociative electron recombination of NH2CHOH+ and implications for interstellar formamide abundance. Mon. Not. R. Astron. Soc. 2019, 490, 1325–1331. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode | This Study | Exp. [24,25] |
|---|---|---|
| Bending (010) | 0.207 | 0.198 |
| Symmetric stretching (100) | 0.472 | 0.453 |
| Asymmetric stretching (001) | 0.488 | 0.466 |
| Bond lengths , (Å) | 0.958 | 0.958 |
| Bond angle (Degrees) | 104.44 | 104.50 |
| (K) | 2403 | 4807 | 0 | 2403 | 0 | 0 |
| 5.76 | 2.23 | 2.63 | 1.08 | 8.33 | 3.99 | |
| −6.32 | −3.05 | 2.24 | −1.19 | 7.07 | 6.83 | |
| 2.76 | 1.95 | −2.97 | 5.30 | −5.27 | −7.34 |
| (K) | 5489 | 10978 | 0 | 5488 | 0 | 0 |
| 2.92 | 1.01 | 1.10 | 5.69 | 6.62 | 2.32 | |
| −5.68 | −1.06 | −8.08 | −1.07 | −1.82 | −1.79 | |
| 4.29 | 7.76 | 1.09 | 8.00 | 2.39 | 2.16 |
| (K) | 5673 | 11345 | 0 | 5672 | 0 | 0 |
| 3.56 | 1.66 | 4.27 | 6.64 | 4.49 | 8.57 | |
| −9.80 | −3.59 | −1.51 | −1.82 | −5.01 | −2.94 | |
| 7.05 | 2.45 | 1.66 | 1.31 | 5.15 | 3.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayouz, M.; Faure, A.; Tennyson, J.; Tudorovskaya, M.; Kokoouline, V. Cross Sections and Rate Coefficients for Vibrational Excitation of H2O by Electron Impact. Atoms 2021, 9, 62. https://doi.org/10.3390/atoms9030062
Ayouz M, Faure A, Tennyson J, Tudorovskaya M, Kokoouline V. Cross Sections and Rate Coefficients for Vibrational Excitation of H2O by Electron Impact. Atoms. 2021; 9(3):62. https://doi.org/10.3390/atoms9030062
Chicago/Turabian StyleAyouz, Mehdi, Alexandre Faure, Jonathan Tennyson, Maria Tudorovskaya, and Viatcheslav Kokoouline. 2021. "Cross Sections and Rate Coefficients for Vibrational Excitation of H2O by Electron Impact" Atoms 9, no. 3: 62. https://doi.org/10.3390/atoms9030062
APA StyleAyouz, M., Faure, A., Tennyson, J., Tudorovskaya, M., & Kokoouline, V. (2021). Cross Sections and Rate Coefficients for Vibrational Excitation of H2O by Electron Impact. Atoms, 9(3), 62. https://doi.org/10.3390/atoms9030062