
In Silico Studies on Selected Neutral Molecules, CGa2Ge2, CAlGaGe2, and CSiGa2Ge Containing Planar Tetracoordinate Carbon


Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
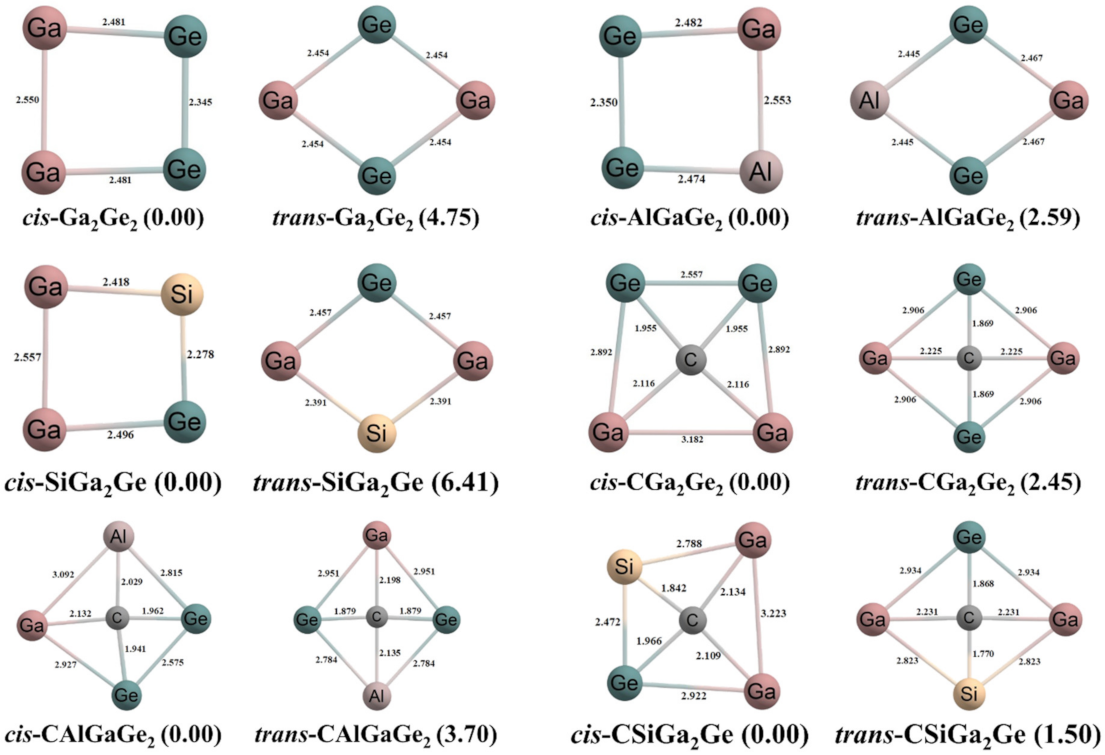
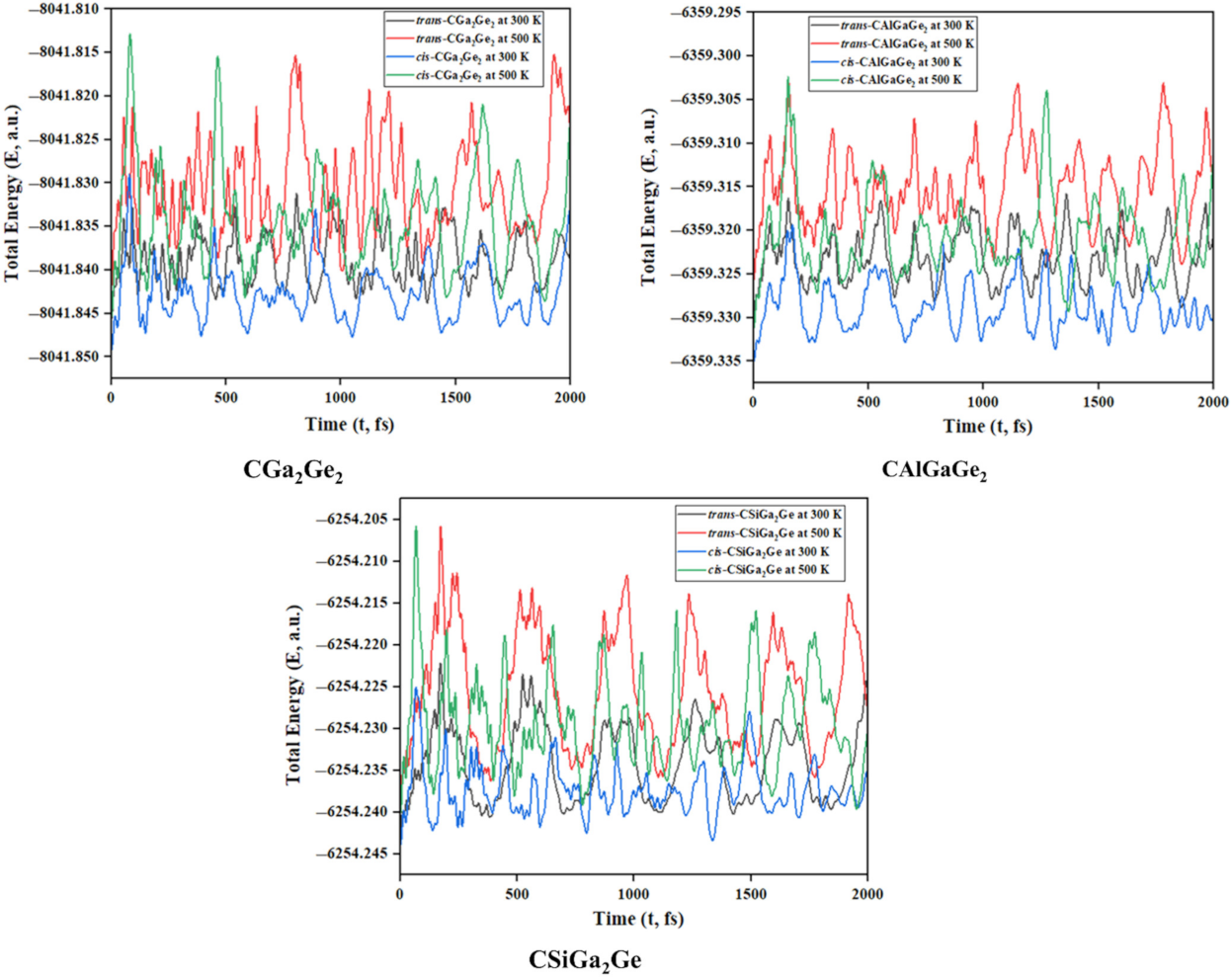
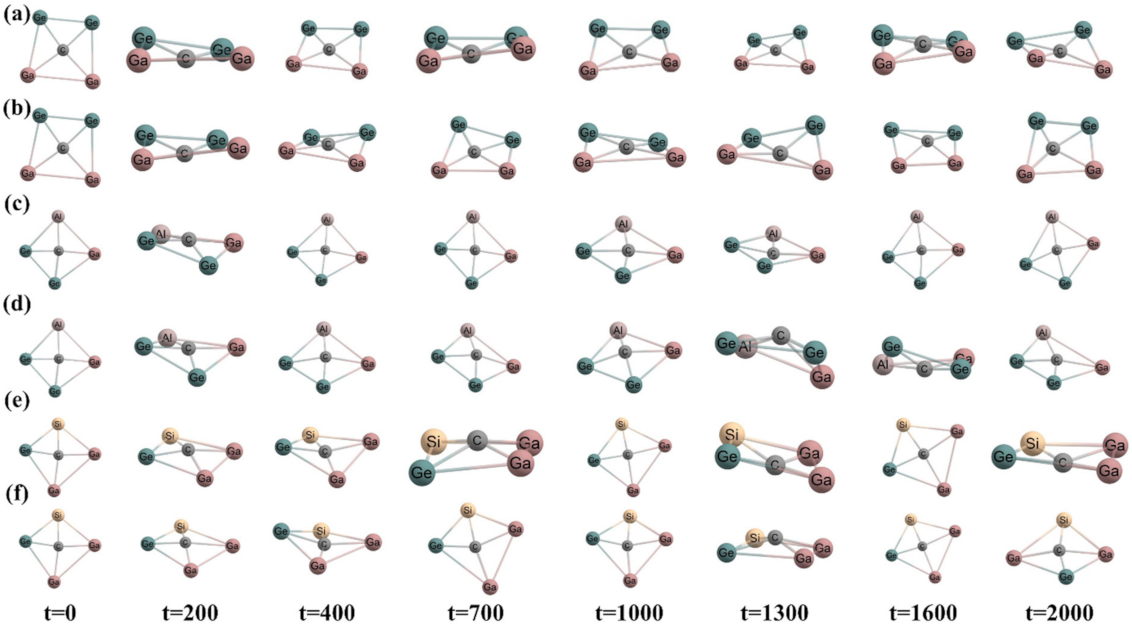
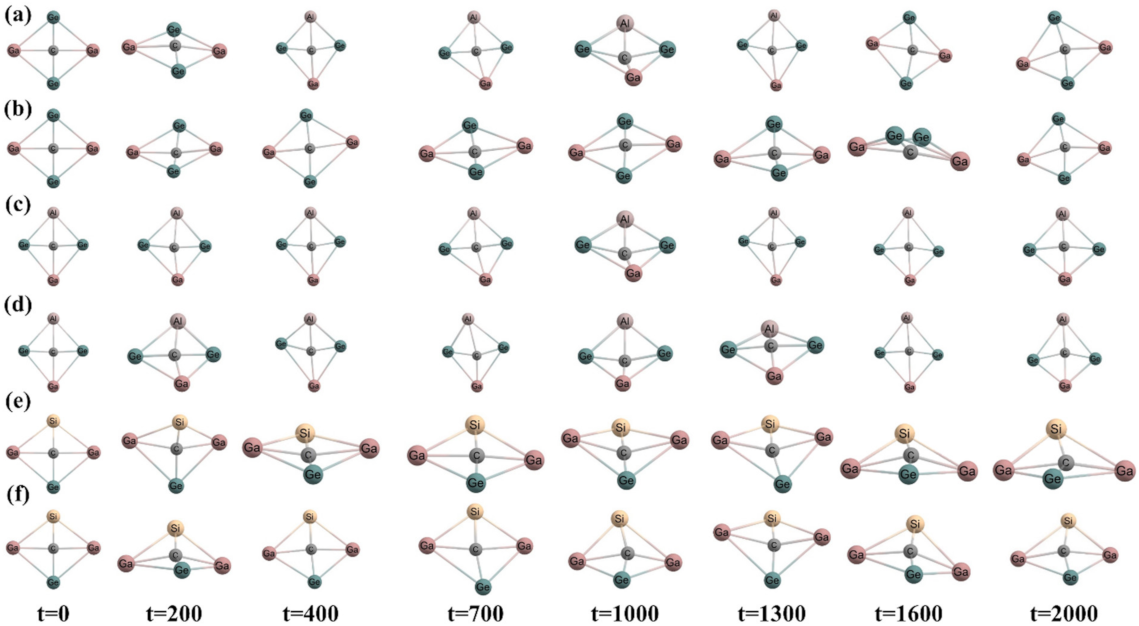
3.1. Geometries
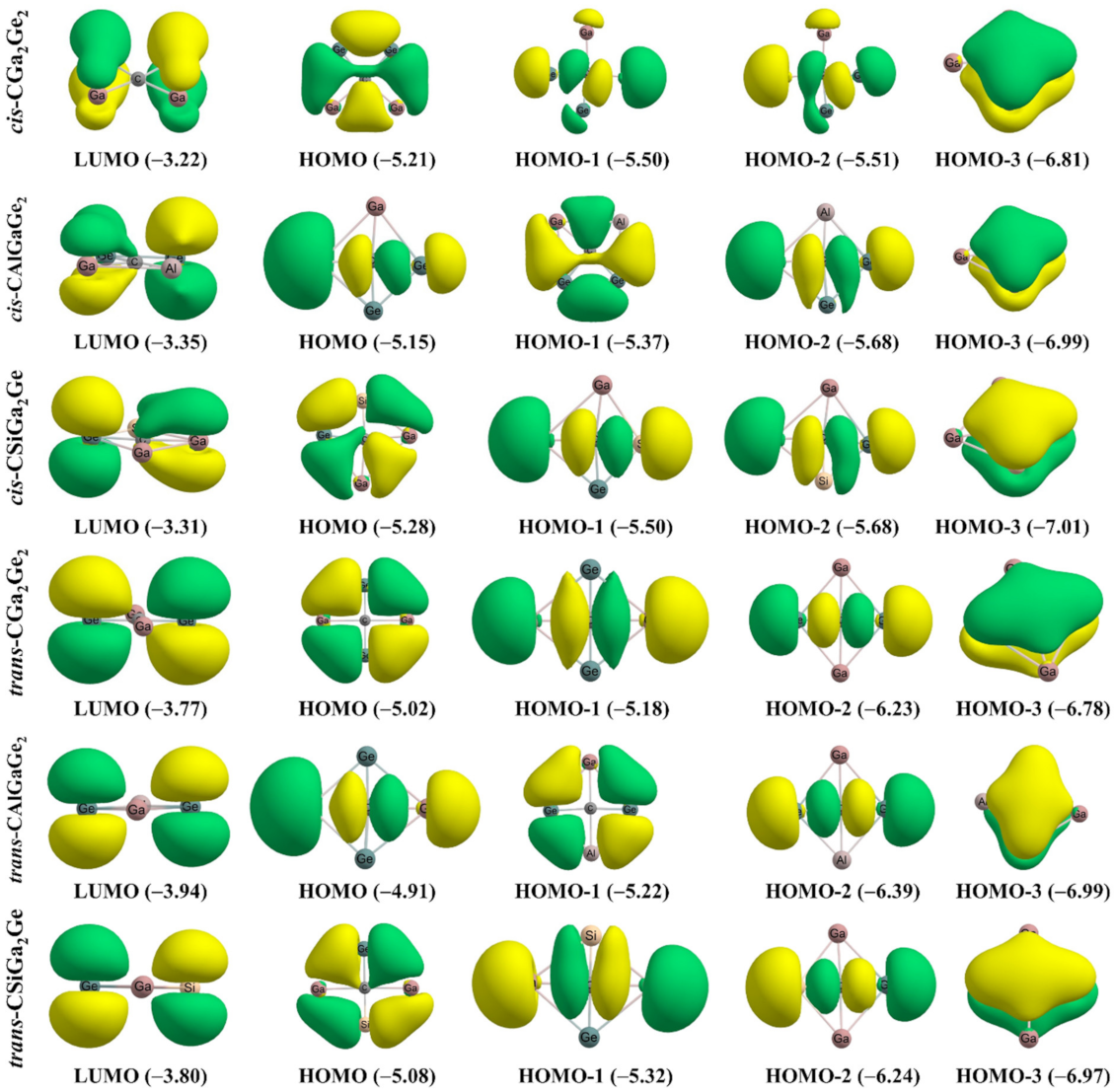
3.2. Molecular Orbitals
3.3. Natural Bond Orbital (NBO) Analysis
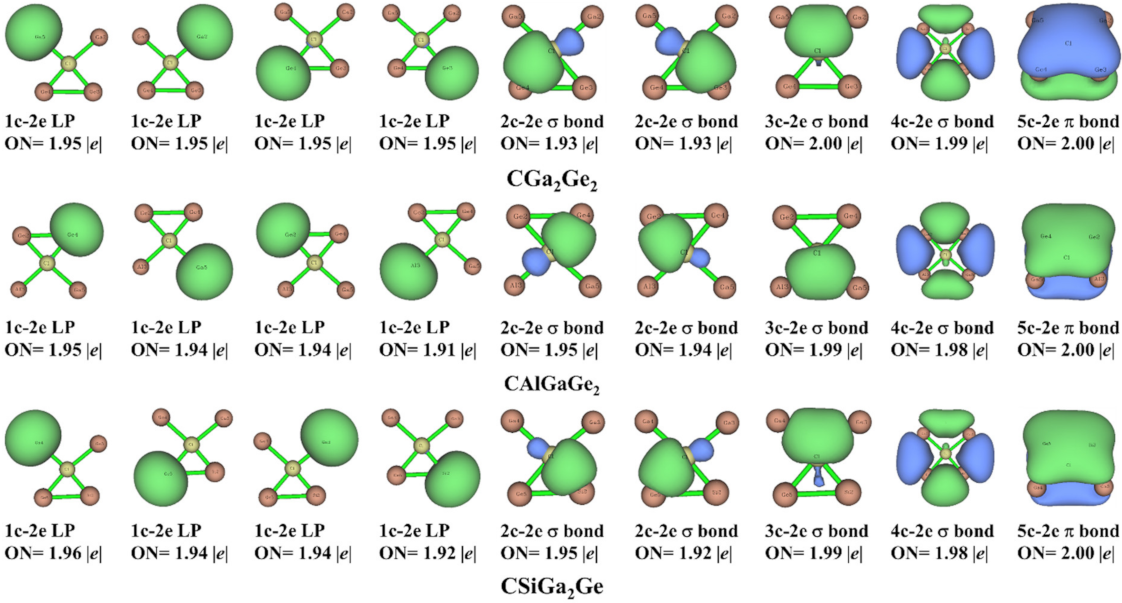
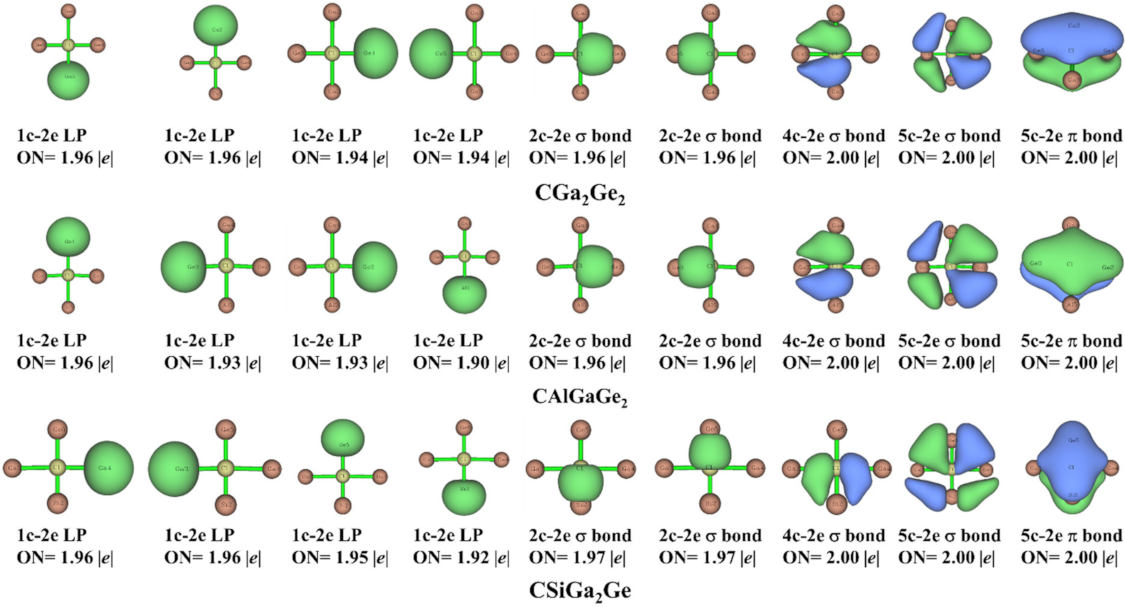
3.4. Adaptive Natural Density Partitioning (AdNDP) Analysis
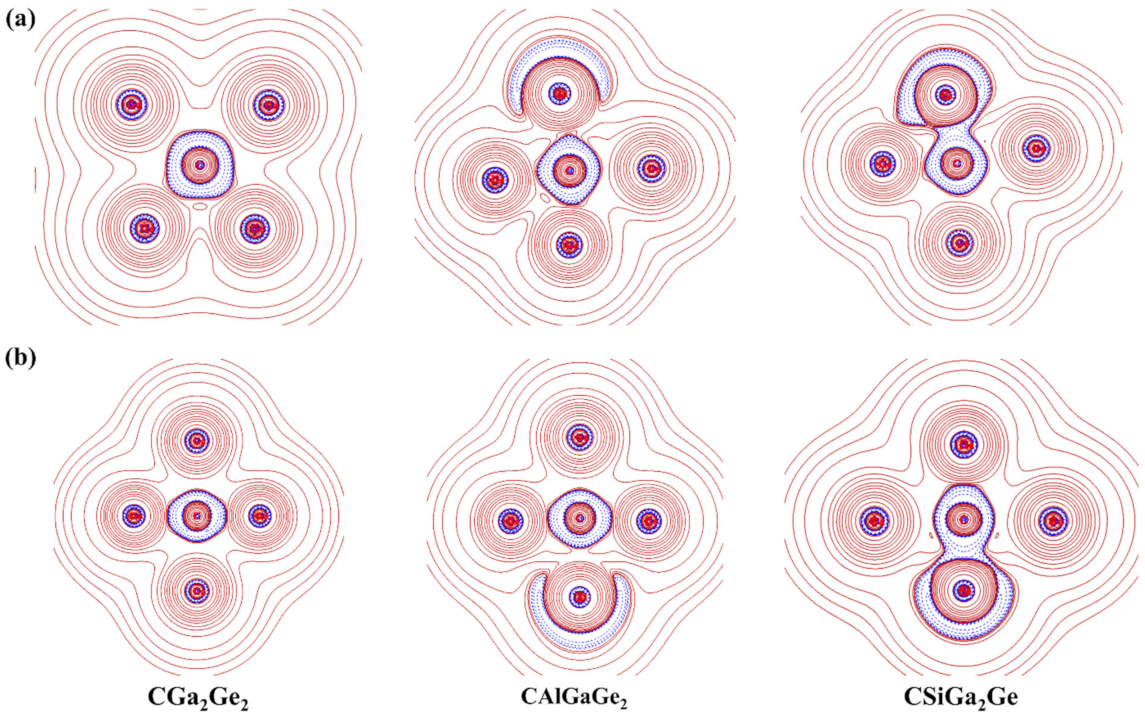
3.5. Atoms in Molecule (AIM) Analysis
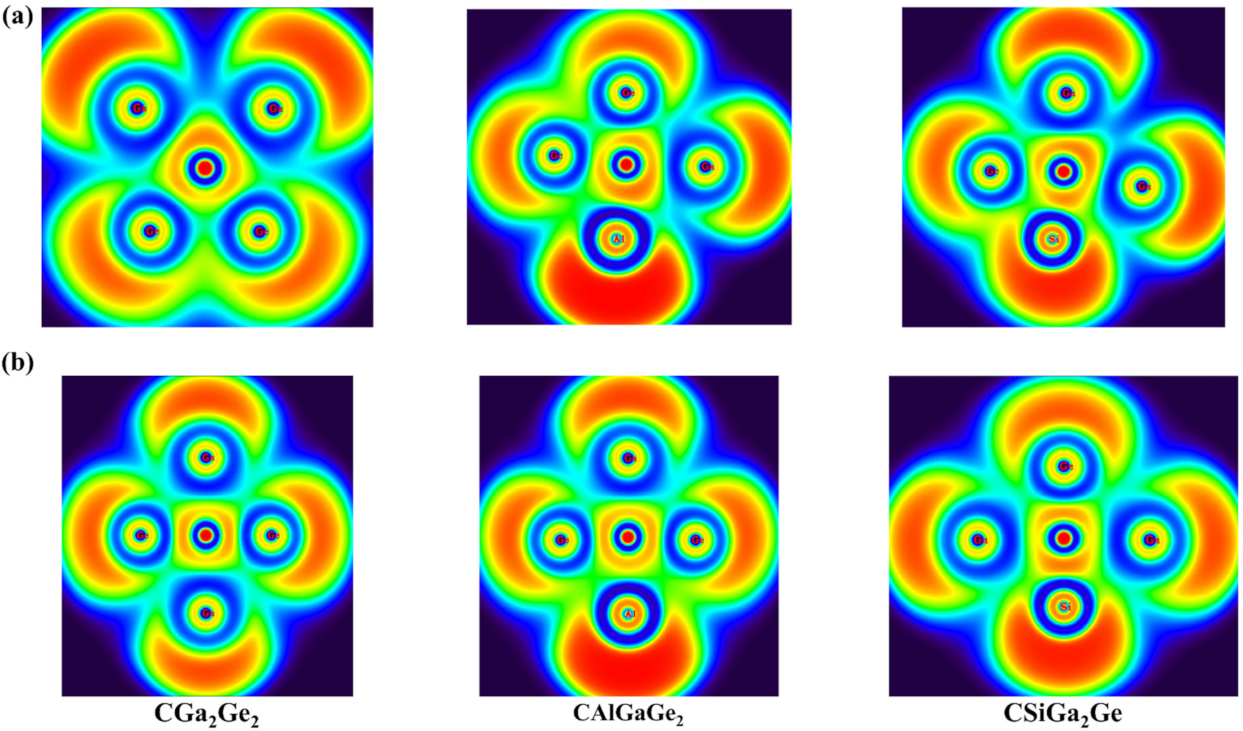
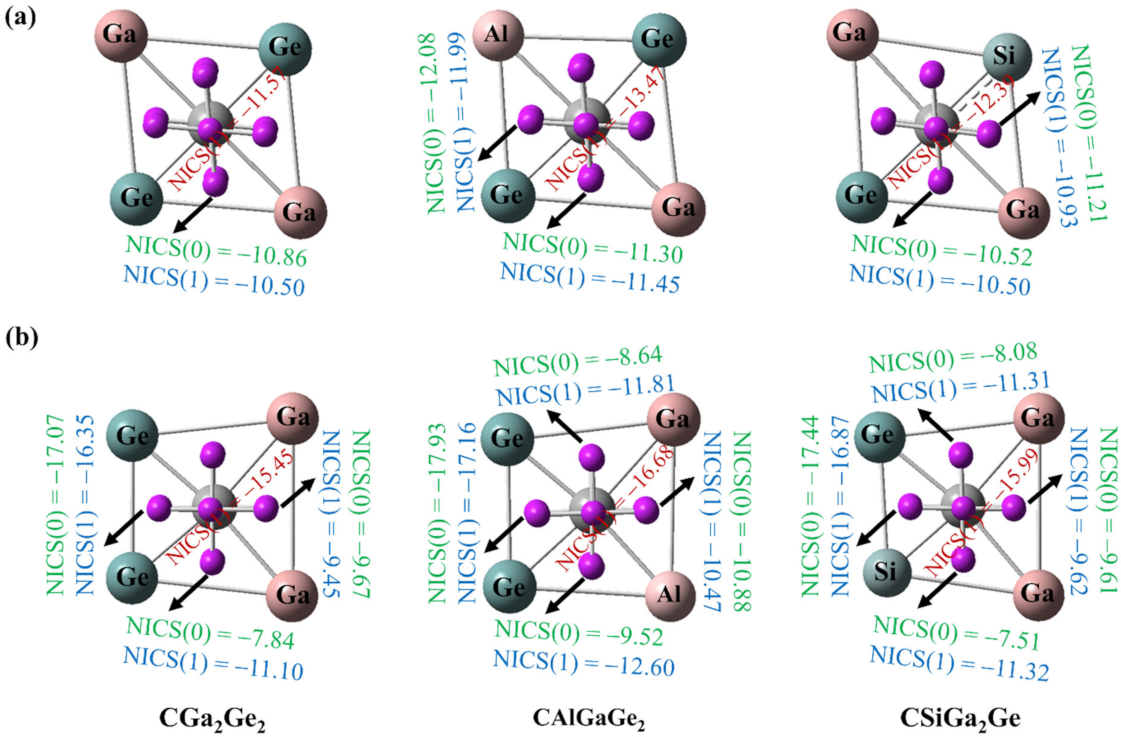
3.6. Aromaticity Analysis
4. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, X.; Wang, L.S.; Boldyrev, A.I.; Simons, J. Tetracoordinated Planar Carbon in the Al4C− Anion. A Combined Photoelectron Spectroscopy and ab Initio Study. J. Am. Chem. Soc. 1999, 121, 6033–6038. [Google Scholar] [CrossRef]
- Boldyrev, A.I.; Simons, J. Tetracoordinated Planar Carbon in Pentaatomic Molecules. J. Am. Chem. Soc. 1998, 120, 7967–7972. [Google Scholar] [CrossRef]
- Röttger, D.; Erker, G. Compounds Containing Planar-Tetracoordinate Carbon. Angew. Chem. Int. Ed. Engl. 1997, 36, 812–827. [Google Scholar] [CrossRef]
- Erker, G. Planar-Tetracoordinate Carbon: Making Stable Anti-van’t Hoff/Le Bel Compounds. Comment. Inorg. Chem. 1992, 13, 111–131. [Google Scholar] [CrossRef]
- Keese, R. Carbon Flatland: Planar Tetracoordinate Carbon and Fenestranes. Chem. Rev. 2006, 106, 4787–4808. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Yu, S.; Ding, Y.H.; Bowen, K.H. Identifying the Hydrogenated Planar Tetracoordinate Carbon: A Combined Experimental and Theoretical Study of CAl4H and CAl4H−. J. Phys. Chem. Lett. 2017, 8, 2263–2267. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, H.F.; Wang, L.S.; Geske, G.; Boldyrev, A. Pentaatomic Tetracoordinate Planar Carbon, [CAl4]2−: A New Structural Unit and Its Salt Complexes. Angew. Chem. Int. Ed. 2000, 39, 3630–3632. [Google Scholar] [CrossRef]
- Hoffmann, R.; Alder, R.W.; Wilcox, C.F. Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 1970, 92, 4992–4993. [Google Scholar] [CrossRef]
- Collins, J.B.; Dill, J.D.; Jemmis, E.D.; Apeloig, Y.; von Ragué Schleyer, P.; Seeger, R.; Pople, J.A. Stabilization of Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 1976, 98, 5419–5427. [Google Scholar] [CrossRef]
- Merino, G.; Méndez-Rojas, M.A.; Beltrán, H.I.; Corminboeuf, C.; Heine, T.; Vela, A. Theoretical Analysis of the Smallest Carbon Cluster Containing a Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 2004, 126, 16160–16169. [Google Scholar] [CrossRef]
- Yang, L.M.; Ganz, E.; Chen, Z.; Wang, Z.X.; von Ragué Schleyer, P. Four Decades of the Chemistry of Planar Hypercoordinate Compounds. Angew. Chem. Int. Ed. 2015, 54, 9468–9501. [Google Scholar] [CrossRef]
- Thirumoorthy, K.; Thimmakondu, V.S. Flat Crown Ethers with Planar Tetracoordinate Carbon Atoms. Int. J. Quantum Chem. 2021, 121, e26479. [Google Scholar] [CrossRef]
- Yañez, O.; Vásquez-Espinal, A.; Báez-Grez, R.; Rabanal-León, W.A.; Osorio, E.; Ruiz, L.; Tiznado, W. Carbon Rings Decorated with Group 14 Elements: New Aromatic Clusters Containing Planar Tetracoordinate Carbon. New J. Chem. 2019, 43, 6781–6785. [Google Scholar] [CrossRef]
- Thirumoorthy, K.; Karton, A.; Thimmakondu, V.S. From High-Energy C7H2 Isomers with A Planar Tetracoordinate Carbon Atom to an Experimentally Known Carbene. J. Phys. Chem. A 2018, 122, 9054–9064. [Google Scholar] [CrossRef] [Green Version]
- Suresh, C.H.; Frenking, G. Direct 1-3 Metal-Carbon Bonding and Planar Tetracoordinated Carbon in Group 6 Metallacyclobutadienes. Organometallics 2010, 29, 4766–4769. [Google Scholar] [CrossRef]
- van’t Hoff, J.H. A Suggestion Looking to the Extension into Space of the Structural Formulas at Present Used in Chemistry, and a Note upon the Relation between the Optical Activity and the Chemical Constitution of Organic Compounds. Arch. Neerl. Sci. Exactes Nat. 1874, 9, 445–454. [Google Scholar]
- Le-Bel, J.A. On the Relations Which Exist between the Atomic Formulas of Organic Compounds and the Rotatory Power of Their Solutions. Bull. Soc. Chim. Fr. 1874, 22, 337–347. [Google Scholar]
- Monkhorst, H.J. Activation Energy for Interconversion of Enantiomers Containing an Asymmetric Carbon Atom without Breaking Bonds. Chem. Commun. 1968, 11, 1111–1112. [Google Scholar] [CrossRef]
- Hoffmann, R. The theoretical design of novel stabilized systems. Pure Appl. Chem. 1971, 28, 181–194. [Google Scholar] [CrossRef]
- Sateesh, B.; Srinivas Reddy, A.; Narahari Sastry, G. Towards Design of the Smallest Planar Tetracoordinate Carbon and Boron Systems. J. Comput. Chem. 2007, 28, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Job, N.; Karton, A.; Thirumoorthy, K.; Cooksy, A.L.; Thimmakondu, V.S. Theoretical Studies of SiC4H2 Isomers Delineate Three Low-Lying Silylidenes are Missing in the Laboratory. J. Phys. Chem. A 2020, 124, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.C.; Feng, L.Y.; Dong, C.; Zhai, H.J. Ternary 12-electron CBe3X3+ (X = H, Li, Na, Cu, Ag) Clusters: Planar Tetracoordinate Carbons and Superalkali Cations. Phys. Chem. Chem. Phys. 2019, 21, 22048–22056. [Google Scholar] [CrossRef]
- Nandula, A.; Trinh, Q.T.; Saeys, M.; Alexandrova, A.N. Origin of Extraordinary Stability of Square-Planar Carbon Atoms in Surface Carbides of Cobalt and Nickel. Angew. Chem. Int. Ed. 2015, 54, 5312–5316. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.H.; Contreras, M.; Ding, Y.H.; Merino, G. Planar Tetracoordinate Carbon versus Planar Tetracoordinate Boron: The Case of CB4 and Its Cation. J. Am. Chem. Soc. 2011, 133, 13228–13231. [Google Scholar] [CrossRef]
- Thirumoorthy, K.; Cooksy, A.; Thimmakondu, V.S. Si2C5H2 Isomers–Search Algorithms Versus Chemical Intuition. Phys. Chem. Chem. Phys. 2020, 22, 5865–5872. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Halla, J.O.C.; Wu, Y.B.; Wang, Z.X.; Islas, R.; Heine, T.; Merino, G. CAl4Be and CAl3Be2−: Global Minima with a Planar Pentacoordinate Carbon Atom. Chem. Commun. 2010, 46, 8776–8778. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.H.; Ding, Y.H.; Cabellos, J.L.; Osorio, E.; Islas, R.; Restrepo, A.; Merino, G. Planar tetracoordinate carbons with a double bond in CAl3E clusters. Phys. Chem. Chem. Phys. 2015, 17, 8769–8775. [Google Scholar] [CrossRef] [PubMed]
- von Ragué Schleyer, P.; Boldyrev, A.I. A new, general strategy for achieving planar tetracoordinate geometries for carbon and other second row periodic elements. J. Chem. Soc. Chem. Commun. 1991, 21, 1536–1538. [Google Scholar] [CrossRef]
- Wu, X.-F.; Cheng, Y.-X.; Guo, J.-C. CLiAl2E and CLi2AlE (E = P, As, Sb, Bi): Planar Tetracoordinate Carbon Clusters with 16 and 14 Valence Electrons. ACS Omega 2019, 4, 21311–21318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Job, N.; Khatun, M.; Thirumoorthy, K.; CH, S.S.R.; Chandrasekaran, V.; Anoop, A.; Thimmakondu, V.S. CAl4Mg0/−: Global Minima with a Planar Tetracoordinate Carbon Atom. Atoms 2021, 9, 24. [Google Scholar] [CrossRef]
- Wang, L.-S.; Boldyrev, A.I.; Li, X.; Simons, J. Experimental Observation of Pentaatomic Tetracoordinate Planar Carbon-Containing Molecules. J. Am. Chem. Soc. 2000, 122, 7681–7687. [Google Scholar] [CrossRef]
- Boldyrev, A.I.; Wang, L.-S. Beyond Classical Stoichiometry: Experiment and Theory. J. Phys. Chem. A 2001, 105, 10759–10775. [Google Scholar] [CrossRef]
- Pei, Y.; An, W.; Ito, K.; von Ragué Schleyer, P.; Zeng, X.C. Planar Pentacoordinate Carbon in CAl5+: A Global Minimum. J. Am. Chem. Soc. 2008, 130, 10394–10400. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Cabellos, J.L.; Orozco, M.; Merino, G.; Zhao, L.; Chattaraj, P.K. Planar Pentacoordinate Carbon in CGa5+ Derivatives. Phys. Chem. Chem. Phys. 2018, 20, 12350–12355. [Google Scholar] [CrossRef] [PubMed]
- Vassilev-Galindo, V.; Pan, S.; Donald, J.K.; Merino, G. Planar Pentacoordinate Carbons. Nat. Chem. Rev. 2018, 2, 0114. [Google Scholar] [CrossRef]
- Exner, K.; von Ragué Schleyer, P. Planar Hexacoordinate Carbon: A Viable Possibility. Science 2000, 290, 1937–1940. [Google Scholar] [CrossRef]
- Averkiev, B.B.; Zubarev, D.Y.; Wang, L.M.; Huang, W.; Wang, L.S.; Boldyrev, A.I. Carbon Avoids Hypercoordination in CB6−, CB62−, and C2B5− Planar Carbon-Boron Clusters. J. Am. Chem. Soc. 2008, 130, 9248–9250. [Google Scholar] [CrossRef]
- Ito, K.; Chen, Z.; Corminboeuf, C.; Wannere, C.S.; Zhang, X.H.; Li, Q.S.; von Ragué Schleyer, P. Myriad Planar Hexacoordinate Carbon Molecules Inviting Synthesis. J. Am. Chem. Soc. 2007, 129, 1510–1511. [Google Scholar] [CrossRef]
- Zhang, C.F.; Han, S.J.; Wu, Y.B.; Lu, H.G.; Lu, G. Thermodynamic Stability versus Kinetic Stability: Is the Planar Hexacoordinate Carbon Species D3h CN3Mg3+ Viable? J. Phys. Chem. A 2014, 118, 3319–3325. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Metz, B.; Stoll, H.; Dolg, M. Small-core multiconfiguration-Dirac-Hartree-Fock-adjusted pseudopotentials for post-d main group elements: Application to PbH and PbO. J. Chem. Phys. 2000, 113, 2563–2569. [Google Scholar] [CrossRef]
- Peterson, K.A. Systematically convergent basis sets with relativistic pseudopotentials. I. Correlation consistent basis sets for the post-d group 13–15 elements. J. Chem. Phys. 2003, 119, 11099–11112. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revis on B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Schlegel, H.B.; Millam, J.M.; Iyengar, S.S.; Voth, G.A.; Daniels, A.D.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. J. Chem. Phys. 2001, 114, 9758–9763. [Google Scholar] [CrossRef]
- Iyengar, S.S.; Schlegel, H.B.; Millam, J.M.; Voth, G.A.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. II. Generalizations based on mass-weighting, idempotency, energy conservation and choice of initial conditions. J. Chem. Phys. 2001, 115, 10291–10302. [Google Scholar] [CrossRef]
- Schlegel, H.B.; Iyengar, S.S.; Li, X.; Millam, J.M.; Voth, G.A.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. III. Comparison with Born-Oppenheimer dynamics. J. Chem. Phys. 2002, 117, 8694–8704. [Google Scholar] [CrossRef] [Green Version]
- Wiberg, K.B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO 3.1 QCPE Bulletin. 1990, Volume 10, p. 58. Available online: https://www.scirp.org/(S(czeh2tfqw2orz553k1w0r45))/reference/referencespapers.aspx?referenceid=1595007 (accessed on 1 August 2021).
- Bader, R.F.W. Atoms in Molecules. In A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Parr, R.G.; Chattaraj, P.K. Principle of maximum hardness. J. Am. Chem. Soc. 1991, 113, 1854–1855. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Cedillo, A.; Parr, R.G.; Arnett, E.M. Appraisal of Chemical Bond Making, Bond Breaking, and Electron Transfer in Solution in the Light of the Principle of Maximum Hardness. J. Org. Chem. 1995, 60, 4707–4714. [Google Scholar] [CrossRef]
- Chakraborty, D.; Chattaraj, P.K. Conceptual Density Functional Theory based Electronic Structure Principles. Chem. Sci. 2021, 12, 6264–6279. [Google Scholar] [CrossRef]
- Zhou, K. Theoretical studies on the pentaatomic planar tetracoordinate carbon molecules CGa3Si and CGa3Si−. Comput. Theor. Chem. 2013, 1009, 30–34. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Developing Paradigms of Chemical Bonding: Adaptive Natural Density Partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Revealing Intuitively Assessable Chemical Bonding Patterns in Organic Aromatic Molecules via Adaptive Natural Density Partitioning. J. Org. Chem. 2008, 73, 9251–9258. [Google Scholar] [CrossRef]
- von Ragué Schleyer, P.; Jiao, H.; Hommes, N.v.E.; Malkin, V.G.; Malkina, O.L. An Evaluation of the Aromaticity of Inorganic Rings: Refined Evidence from Magnetic Properties. J. Am. Chem. Soc. 1997, 119, 12669–12670. [Google Scholar] [CrossRef]
- von Ragué Schleyer, P.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N.J.R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | qC | qAl | qSi | qGa | qGe | Valence Electronic Configuration of C |
|---|---|---|---|---|---|---|
| cis-Ga2Ge2 | - | - | - | 0.32 0.32 | −0.32 −0.32 | - |
| trans-Ga2Ge2 | - | - | - | 0.21 0.21 | −0.21 −0.21 | - |
| cis-CGa2Ge2 | −2.14 | - | - | 0.59 0.59 | 0.49 0.49 | 2s1.606 2px1.583 2py1.598 2pz1.315 |
| trans-CGa2Ge2 | −2.14 | - | - | 0.50 0.50 | 0.58 0.58 | 2s1.548 2px1.408 2py1.552 2pz1.596 |
| cis-AlGaGe2 | - | 0.47 | - | 0.36 | −0.42 −0.42 | - |
| trans-AlGaGe2 | - | 0.33 | - | 0.15 | −0.17 −0.30 | - |
| cis-CAlGaGe2 | −2.22 | 0.64 | - | 0.59 | 0.48 0.51 | 2s1.612 2px1.593 2py1.638 2pz1.341 |
| trans-CAlGaGe2 | −2.23 | 0.55 | - | 0.54 | 0.57 0.57 | 2s1.556 2px1.412 2py1.555 2pz1.661 |
| cis-SiGa2Ge | - | - | −0.41 | 0.34 0.34 | −0.27 | - |
| trans-SiGa2Ge | - | - | −0.28 | 0.22 0.25 | −0.19 | - |
| cis-CSiGa2Ge | −2.16 | - | 0.47 | 0.59 0.60 | 0.51 | 2s1.590 2px1.581 2py1.635 2pz1.312 |
| trans-CSiGa2Ge | −2.18 | - | 0.58 | 0.51 0.51 | 0.59 | 2s1.528 2px1.421 2py1.590 2pz1.593 |
| Complexes | WBI (C–Al) | WBI (C–Si) | WBI (C–Ga) | WBI (C–Ge) |
|---|---|---|---|---|
| cis-CGa2Ge2 | - | - | 0.37 0.37 | 1.04 1.04 |
| trans-CGa2Ge2 | - | - | 0.31 0.31 | 1.13 1.13 |
| cis-CAlGaGe2 | 0.33 | - | 0.34 | 1.02 1.03 |
| trans-CAlGaGe2 | 0.30 | - | 0.31 | 1.09 1.09 |
| cis-CSiGa2Ge | - | 1.09 | 0.34 0.36 | 0.99 |
| trans-CSiGa2Ge | - | 1.18 | 0.29 0.29 | 1.09 |
| Complexes | BCP | ρ(rc) | ∇2ρ(rc) | G(rc) | V(rc) | H(rc) | ELF | −G(rc)/V(rc) | G(rc)/ρ(rc) |
|---|---|---|---|---|---|---|---|---|---|
| cis-CGa2Ge2 | C-Ga | 0.078 | 0.131 | 0.060 | −0.088 | −0.028 | 0.311 | 0.682 | 0.769 |
| C-Ge | 0.115 | 0.139 | 0.092 | −0.148 | −0.057 | 0.420 | 0.622 | 0.800 | |
| trans-CGa2Ge2 | C-Ga | 0.063 | 0.098 | 0.044 | −0.063 | −0.019 | 0.295 | 0.698 | 0.698 |
| C-Ge | 0.134 | 0.207 | 0.124 | −0.196 | −0.072 | 0.399 | 0.633 | 0.925 | |
| cis-CAlGaGe2 | C-Al | 0.064 | 0.224 | 0.072 | −0.088 | −0.016 | 0.224 | 0.818 | 1.125 |
| C-Ga | 0.075 | 0.125 | 0.057 | −0.083 | −0.026 | 0.125 | 0.687 | 0.760 | |
| C-Ge | 0.112 | 0.137 | 0.089 | −0.144 | −0.055 | 0.417 | 0.618 | 0.795 | |
| trans-CAlGaGe2 | C-Al | 0.053 | 0.152 | 0.052 | −0.065 | −0.014 | 0.149 | 0.800 | 0.981 |
| C-Ga | 0.067 | 0.103 | 0.047 | −0.069 | −0.022 | 0.308 | 0.681 | 0.701 | |
| C-Ge | 0.131 | 0.198 | 0.119 | −0.188 | −0.069 | 0.399 | 0.633 | 0.908 | |
| cis-CSiGa2Ge | C-Si | 0.112 | 0.284 | 0.136 | −0.201 | −0.065 | 0.233 | 0.677 | 1.214 |
| C-Ga | 0.074 | 0.126 | 0.057 | −0.082 | −0.025 | 0.305 | 0.695 | 0.770 | |
| C-Ge | 0.111 | 0.139 | 0.088 | −0.142 | −0.054 | 0.139 | 0.620 | 0.793 | |
| trans-CSiGa2Ge | C-Si | 0.126 | 0.415 | 0.175 | −0.246 | −0.071 | 0.211 | 0.711 | 1.389 |
| C-Ga | 0.061 | 0.099 | 0.043 | −0.062 | −0.018 | 0.287 | 0.694 | 0.705 | |
| C-Ge | 0.135 | 0.209 | 0.126 | −0.199 | −0.073 | 0.400 | 0.633 | 0.933 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, P.; Chattaraj, P.K. In Silico Studies on Selected Neutral Molecules, CGa2Ge2, CAlGaGe2, and CSiGa2Ge Containing Planar Tetracoordinate Carbon. Atoms 2021, 9, 65. https://doi.org/10.3390/atoms9030065
Das P, Chattaraj PK. In Silico Studies on Selected Neutral Molecules, CGa2Ge2, CAlGaGe2, and CSiGa2Ge Containing Planar Tetracoordinate Carbon. Atoms. 2021; 9(3):65. https://doi.org/10.3390/atoms9030065
Chicago/Turabian StyleDas, Prasenjit, and Pratim Kumar Chattaraj. 2021. "In Silico Studies on Selected Neutral Molecules, CGa2Ge2, CAlGaGe2, and CSiGa2Ge Containing Planar Tetracoordinate Carbon" Atoms 9, no. 3: 65. https://doi.org/10.3390/atoms9030065
APA StyleDas, P., & Chattaraj, P. K. (2021). In Silico Studies on Selected Neutral Molecules, CGa2Ge2, CAlGaGe2, and CSiGa2Ge Containing Planar Tetracoordinate Carbon. Atoms, 9(3), 65. https://doi.org/10.3390/atoms9030065