Modulation of Mitochondrial Metabolic Reprogramming and Oxidative Stress to Overcome Chemoresistance in Cancer

 , , , and
, , , and
Abstract
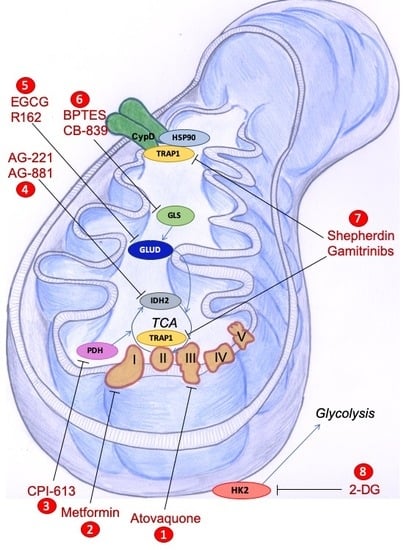
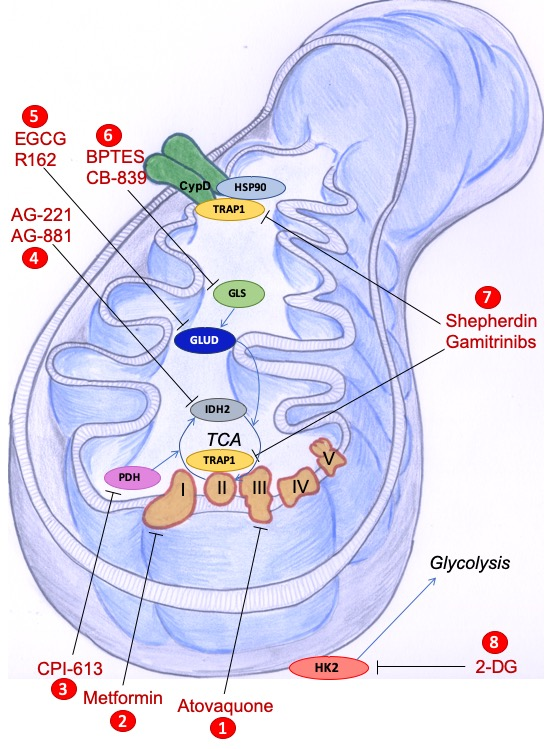
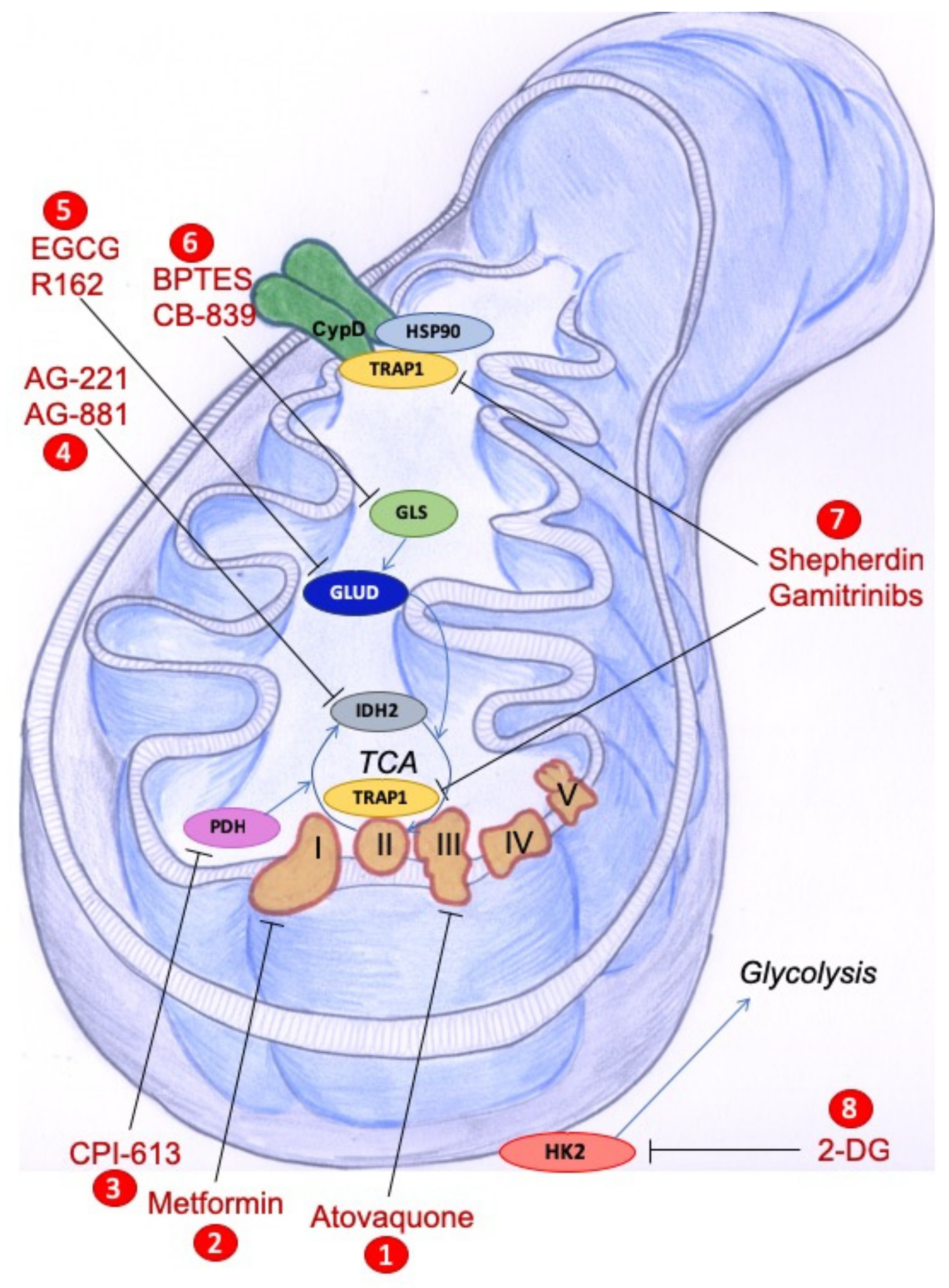
:
1. Introduction
2. The Role of Mitochondria in ATP Synthesis and Reactive Oxygen Species Production
3. The Antioxidant Response in Cancer
4. Metabolic Reprogramming, Oxidative Stress and Chemoresistance in Cancer
5. Metabolic Enzymes and Cancer
6. The Mitochondrial Chaperone Tumor Necrosis Factor Receptor-Associated Protein 1
Tumor Necrosis Factor receptor-Associated Protein 1 as Driver of Metabolic Rewiring
7. Mitochondria-Directed Therapeutic Strategies
7.1. Natural Compounds
7.2. Drugs Targeting Metabolic Pathways
7.3. Inhibition of Mitochondrial HSP90s as a Therapeutic Strategy
8. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 11, 59950–59964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentric, G.; Mieulet, V.; Mechta-Grigoriou, F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxid Redox Signal 2017, 26, 462–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amoroso, M.R.; Matassa, D.S.; Agliarulo, I.; Avolio, R.; Maddalena, F.; Condelli, V.; Landriscina, M.; Esposito, F. Stress-Adaptive Response in Ovarian Cancer Drug Resistance: Role of TRAP1 in Oxidative Metabolism-Driven Inflammation. Adv. Protein Chem. Struct. Biol. 2017, 108, 163–198. [Google Scholar] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Emmings, E.; Mullany, S.; Chang, Z.; Landen, C.N., Jr.; Linder, S.; Bazzaro, M. Targeting Mitochondria for Treatment of Chemoresistant Ovarian Cancer. Int. J. Mol. Sci. 2019, 20, 229. [Google Scholar] [CrossRef] [Green Version]
- Lang, B.F.; Gray, M.W.; Burger, G. Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 1999, 33, 351–397. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Miettinen, T.P.; Bjorklund, M. Mitochondrial Function and Cell Size: An Allometric Relationship. Trends Cell Biol. 2017, 27, 393–402. [Google Scholar] [CrossRef]
- Prasai, K. Regulation of mitochondrial structure and function by protein import: A current review. Pathophysiology 2017, 24, 107–122. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.U.; Fisher, P.R. Import of nuclear-encoded mitochondrial proteins: A cotranslational perspective. Int. Rev. Cell Mol. Biol. 2009, 273, 49–68. [Google Scholar] [PubMed]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Cho, U.; Kim, S.; Park, I.S.; Cho, J.H.; Dhanasekaran, D.N.; Song, Y.S. Tumor microenvironment on mitochondrial dynamics and chemoresistance in cancer. Free Radic. Res. 2018, 52, 1271–1287. [Google Scholar] [CrossRef] [PubMed]
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilization. Biochim. Biophys. Acta 2014, 1837, 418–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, B.E.; Chan, S.I. Structures and proton-pumping strategies of mitochondrial respiratory enzymes. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 23–65. [Google Scholar] [CrossRef] [Green Version]
- Sazanov, L.A. A giant molecular proton pump: Structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 2015, 16, 375–388. [Google Scholar] [CrossRef]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Moscow) 2005, 70, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, Oxidative Stress and Innate Immunity. Front. Physiol. 2018, 9, 1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scalcon, V.; Bindoli, A.; Rigobello, M.P. Significance of the mitochondrial thioredoxin reductase in cancer cells: An update on role, targets and inhibitors. Free Radic. Biol. Med. 2018, 127, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Thompson, M.A.; Tamayo, A.T.; Zuo, Z.; Lee, J.; Vega, F.; Ford, R.J.; Pham, L.V. Over-expression of Thioredoxin-1 mediates growth, survival, and chemoresistance and is a druggable target in diffuse large B-cell lymphoma. Oncotarget 2012, 3, 314–326. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Yu, A.Q. The functional role of peroxiredoxin 3 in reactive oxygen species, apoptosis, and chemoresistance of cancer cells. J. Cancer Res. Clin. Oncol. 2015, 141, 2071–2077. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.Y.; Cantley, L.C.; DeNicola, G.M. NRF2 Rewires Cellular Metabolism to Support the Antioxidant Response. In A Master Regulator of Oxidative Stress–The Transcription Factor Nrf2; IntechOpen: London, UK, 2016. [Google Scholar]
- Almeida, A.S.; Soares, N.L.; Sequeira, C.O.; Pereira, S.A.; Sonnewald, U.; Vieira, H.L. Improvement of neuronal differentiation by carbon monoxide: Role of pentose phosphate pathway. Redox Biol. 2018, 17, 338–347. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Bott, A.J.; Maimouni, S.; Zong, W. The Pleiotropic Effects of Glutamine Metabolism in Cancer. Cancers 2019, 11, 770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Le, A. Glutamine Metabolism in Cancer. In The Heterogeneity of Cancer Metabolism; Advances in Experimental Medicine and Biology; Le, A., Ed.; Springer: Cham, Switzerland, 2018; Volume 1063. [Google Scholar]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Abildgaard, C.; Guldberg, P. Molecular drivers of cellular metabolic reprogramming in melanoma. Trends Mol. Med. 2015, 21, 164–171. [Google Scholar] [CrossRef]
- Bidkhori, G.; Benfeitas, R.; Klevstig, M.; Zhang, C.; Nielsen, J.; Uhlen, M.; Boren, J.; Mardinoglu, A. Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumour subtypes. Proc. Natl. Acad. Sci. USA 2018, 115, E11874–E11883. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Ettinger, S.L.; Qu, S.; Xue, H.; Nabavi, N.; Choi, S.Y.C.; Bell, R.H.; Mo, F.; Haegert, A.M.; Gout, P.W.; et al. Metabolic heterogeneity signature of primary treatment-naive prostate cancer. Oncotarget 2017, 8, 25928–25941. [Google Scholar]
- Gentric, G.; Kieffer, Y.; Mieulet, V.; Goundiam, O.; Bonneau, C.; Nemati, F.; Hurbain, I.; Raposo, G.; Popova, T.; Stern, M.H.; et al. PML-Regulated Mitochondrial Metabolism Enhances Chemosensitivity in Human Ovarian Cancers. Cell Metab. 2019, 29, 156–173. [Google Scholar] [CrossRef] [Green Version]
- Matassa, D.S.; Amoroso, M.R.; Lu, H.; Avolio, R.; Arzeni, D.; Procaccini, C.; Faicchia, D.; Maddalena, F.; Simeon, V.; Agliarulo, I.; et al. Oxidative metabolism drives inflammation-induced platinum resistance in human ovarian cancer. Cell Death Differ. 2016, 23, 1542–1554. [Google Scholar] [CrossRef] [Green Version]
- Waghray, D.; Zhang, Q. Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef]
- Meijerman, I.; Beijnen, J.H.; Schellens, J.H. Combined action and regulation of phase II enzymes and multidrug resistance proteins in multidrug resistance in cancer. Cancer Treat. Rev. 2008, 34, 505–520. [Google Scholar] [CrossRef]
- Dar, S.; Chhina, J.; Mert, I.; Chitale, D.; Buekers, T.; Kaur, H.; Giri, S.; Munkarah, A.; Rattan, R. Bioenergetic Adaptations in Chemoresistant Ovarian Cancer Cells. Sci. Rep. 2017, 7, 8760. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Luo, C.; Vazquez, F.; Puigserver, P. Targeting mitochondrial oxidative metabolism in melanoma causes metabolic compensation through glucose and glutamine utilization. Cancer Res. 2014, 74, 3535–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Xu, H.; Chen, X.; Li, X.; Luo, B. Inhibition of mitochondrial respiration overcomes hepatocellular carcinoma chemoresistance. Biochem. Biophys. Res. Commun. 2019, 508, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Vellinga, T.T.; Borovski, T.; de Boer, V.C.; Fatrai, S.; van Schelven, S.; Trumpi, K.; Verheem, A.; Snoeren, N.; Emmink, B.L.; Koster, J.; et al. SIRT1/PGC1alpha-Dependent Increase in Oxidative Phosphorylation Supports Chemotherapy Resistance of Colon Cancer. Clin. Cancer Res. 2015, 15, 2870–2879. [Google Scholar] [CrossRef] [Green Version]
- Denise, C.; Paoli, P.; Calvani, M.; Taddei, M.; Giannoni, E.; Kopetz, S.; Kazmi, S.M.; Pia, M.M.; Pettazzoni, P.; Sacco, E.; et al. 5-Fluorouracil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem-like traits. Oncotarget 2015, 6, 41706–41721. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumourigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [Green Version]
- Nobrega-Pereira, S.; Caiado, F.; Carvalho, T.; Matias, I.; Graça, G.; Gonçalves, L.G.; Silva-Santos, B.; Norell, H.; Dias, S. VEGFR2-Mediated Reprogramming of Mitochondrial Metabolism Regulates the Sensitivity of Acute Myeloid Leukemia to Chemotherapy. Cancer Res. 2018, 78, 731–741. [Google Scholar] [CrossRef] [Green Version]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef] [Green Version]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumourigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Yang, T.; Ren, C.; Qiao, P.; Han, X.; Wang, L.; Lv, S.; Sun, Y.; Liu, Z.; Du, Y.; Yu, Z. PIM2-mediated phosphorylation of hexokinase 2 is critical for tumour growth and paclitaxel resistance in breast cancer. Oncogene 2018, 37, 5997–6009. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Wang, H.L.; Xu, J.; Tan, J.; Fu, L.N.; Wang, J.L.; Zou, T.H.; Sun, D.F.; Gao, Q.Y.; Chen, Y.X.; et al. Sirtuin5 contributes to colorectal carcinogenesis by enhancing glutaminolysis in a deglutarylation-dependent manner. Nat. Commun. 2018, 9, 545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Cao, Y.; Meng, G.; Qian, L.; Xu, T.; Yan, C.; Luo, O.; Wang, S.; Wei, J.; Ding, Y.; et al. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine 2019, 39, 239–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesi, G.; Walbrecq, G.; Zimmer, A.; Kreis, S.; Haan, C. ROS production induced by BRAF inhibitor treatment rewires metabolic processes affecting cell growth of melanoma cells. Mol. Cancer 2017, 16, 102. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.W.; Kuo, W.H.; Chan, S.H.; Chan, H.L.; Chang, K.J.; Wang, L.H. Transketolase Regulates the Metabolic Switch to Control Breast Cancer Cell Metastasis via the alpha-Ketoglutarate Signaling Pathway. Cancer Res. 2018, 78, 2799–2812. [Google Scholar] [CrossRef] [Green Version]
- Amoroso, M.R.; Matassa, D.S.; Sisinni, L.; Lettini, G.; Landriscina, M.; Esposito, F. TRAP1 revisited: Novel localizations and functions of a ’next-generation’ biomarker (review). Int. J. Oncol. 2014, 45, 969–977. [Google Scholar] [CrossRef] [Green Version]
- Lettini, G.; Maddalena, F.; Sisinni, L.; Condelli, V.; Matassa, D.S.; Costi, M.P.; Simoni, D.; Esposito, F.; Landriscina, M. TRAP1: A viable therapeutic target for future cancer treatments? Expert Opin. Ther. Targets 2017, 21, 805–815. [Google Scholar] [CrossRef] [Green Version]
- Leskovar, A.; Wegele, H.; Werbeck, N.D.; Buchner, J.; Reinstein, J. The ATPase cycle of the mitochondrial Hsp90 analog Trap1. J. Biol. Chem. 2008, 283, 11677–11688. [Google Scholar] [CrossRef] [Green Version]
- Masgras, I.; Sanchez-Martin, C.; Colombo, G.; Rasola, A. The Chaperone TRAP1 as a Modulator of the Mitochondrial Adaptations in Cancer Cells. Front. Oncol. 2017, 7, 58. [Google Scholar] [CrossRef] [Green Version]
- Matassa, D.S.; Agliarulo, I.; Amoroso, M.R.; Maddalena, F.; Sepe, L.; Ferrari, M.C.; Sagar, V.; D’Amico, S.; Loreni, F.; Paolella, G.; et al. TRAP1-dependent regulation of p70S6K is involved in the attenuation of protein synthesis and cell migration: Relevance in human colorectal tumours. Mol. Oncol. 2014, 8, 1482–1494. [Google Scholar] [CrossRef] [Green Version]
- Amoroso, M.R.; Matassa, D.S.; Laudiero, G.; Egorova, A.V.; Polishchuk, R.S.; Maddalena, F.; Piscazzi, A.; Paladino, S.; Sarnataro, D.; Garbi, C.; et al. TRAP1 and the proteasome regulatory particle TBP7/Rpt3 interact in the endoplasmic reticulum and control cellular ubiquitination of specific mitochondrial proteins. Cell Death Differ. 2012, 19, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of tumour cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 2007, 131, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Costantino, E.; Maddalena, F.; Calise, S.; Piscazzi, A.; Tirino, V.; Fersini, A.; Ambrosi, A.; Neri, V.; Esposito, F.; Landriscina, M. TRAP1, a novel mitochondrial chaperone responsible for multi-drug resistance and protection from apoptotis in human colorectal carcinoma cells. Cancer Lett. 2009, 279, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Leav, I.; Plescia, J.; Goel, H.L.; Li, J.; Jiang, Z.; Cohen, R.J.; Languino, L.R.; Altieri, D.C. Cytoprotective mitochondrial chaperone TRAP-1 as a novel molecular target in localized and metastatic prostate cancer. Am. J. Pathol. 2010, 176, 393–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, G.; Zhang, Q.; Fan, Z. Heat shock protein 75 (TRAP1) antagonizes reactive oxygen species generation and protects cells from granzyme M-mediated apoptosis. J. Biol. Chem. 2007, 282, 20553–20560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montesano Gesualdi, N.; Pirozzi, G.; Costantino, E.; Landriscina, M.; Esposito, F. Tumour necrosis factor-associated protein 1 (TRAP-1) protects cells from oxidative stress and apoptosis. Stress 2007, 10, 342–350. [Google Scholar] [CrossRef]
- Larsen, S.B.; Hanss, Z.; Kruger, R. The genetic architecture of mitochondrial dysfunction in Parkinson’s disease. Cell Tissue Res. 2018, 373, 21–37. [Google Scholar] [CrossRef] [Green Version]
- Altieri, D.C.; Stein, G.S.; Lian, J.B.; Languino, L.R. TRAP-1, the mitochondrial Hsp90. Biochim. Biophys. Acta 2012, 1823, 767–773. [Google Scholar] [CrossRef] [Green Version]
- Im, C.N.; Lee, J.S.; Zheng, Y.; Seo, J.S. Iron chelation study in a normal human hepatocyte cell line suggests that tumour necrosis factor receptor-associated protein 1 (TRAP1) regulates production of reactive oxygen species. J. Cell Biochem. 2007, 100, 474–486. [Google Scholar] [CrossRef]
- Masuda, Y.; Shima, G.; Aiuchi, T.; Horie, M.; Hori, K.; Nakajo, S.; Kajimoto, S.; Shibayama-Imazu, T.; Nakaya, K. Involvement of tumour necrosis factor receptor-associated protein 1 (TRAP1) in apoptosis induced by beta-hydroxyisovalerylshikonin. J. Biol. Chem. 2004, 279, 42503–42515. [Google Scholar] [CrossRef] [Green Version]
- Landriscina, M.; Laudiero, G.; Maddalena, F.; Amoroso, M.R.; Piscazzi, A.; Cozzolino, F.; Monti, M.; Garbi, C.; Fersini, A.; Pucci, P.; et al. Mitochondrial chaperone Trap1 and the calcium binding protein Sorcin interact and protect cells against apoptosis induced by antiblastic agents. Cancer Res. 2010, 70, 6577–6586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matassa, D.S.; Agliarulo, I.; Avolio, R.; Landriscina, M.; Esposito, F. TRAP1 Regulation of Cancer Metabolism: Dual Role as Oncogene or Tumour Suppressor. Genes 2018, 9, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciacovelli, M.; Guzzo, G.; Morello, V.; Frezza, C.; Zheng, L.; Nannini, N.; Calabrese, F.; Laudiero, G.; Esposito, F.; Landriscina, M.; et al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. 2013, 17, 988–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizza, S.; Montagna, C.; Cardaci, S.; Maiani, E.; Di Giacomo, G.; Sanchez-Quiles, V.; Blagoev, B.; Rasola, A.; De Zio, D.; Stamler, J.S.; et al. S-nitrosylation of the Mitochondrial Chaperone TRAP1 Sensitizes Hepatocellular Carcinoma Cells to Inhibitors of Succinate Dehydrogenase. Cancer Res. 2016, 76, 4170–4182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, S.; Tsutsumi, S.; Muhlebach, G.; Sourbier, C.; Lee, M.J.; Lee, S.; Vartholomaiou, E.; Tatokoro, M.; Beebe, K.; Miyajima, N.; et al. Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis. Proc. Natl. Acad. Sci. USA 2013, 110, E1604–E1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.K.; Hong, J.H.; Oh, Y.T.; Kim, S.S.; Yin, J.; Lee, A.J.; Chae, Y.C.; Kim, J.H.; Park, S.H.; Park, C.K.; et al. Interplay between TRAP1 and Sirtuin-3 Modulates Mitochondrial Respiration and Oxidative Stress to Maintain Stemness of Glioma Stem Cells. Cancer Res. 2019, 79, 1369–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, Y.C.; Angelin, A.; Lisanti, S.; Kossenkov, A.V.; Speicher, K.D.; Wang, H.; Powers, J.F.; Tischler, A.S.; Pacak, K.; Fliedner, S.; et al. Landscape of the mitochondrial Hsp90 metabolome in tumours. Nat. Commun. 2013, 4, 2139. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, E.; Altman, B.J.; Seo, J.H.; Ghosh, J.C.; Kossenkov, A.V.; Tang, H.Y.; Krishn, S.R.; Languino, L.R.; Gabrilovich, D.I.; Speicher, D.W.; et al. Myc-mediated transcriptional regulation of the mitochondrial chaperone TRAP1 controls primary and metastatic tumour growth. J. Biol. Chem. 2019, 294, 10407–10414. [Google Scholar] [CrossRef] [Green Version]
- Lisanti, S.; Tavecchio, M.; Chae, Y.C.; Liu, Q.; Brice, A.K.; Thakur, M.L.; Languino, L.R.; Altieri, D.C. Deletion of the mitochondrial chaperone TRAP-1 uncovers global reprogramming of metabolic networks. Cell Rep. 2014, 8, 671–677. [Google Scholar] [CrossRef]
- Yu, L.; Lu, M.; Jia, D.; Ma, J.; Ben-Jacob, E.; Levine, H.; Kaipparettu, B.A.; Onuchic, J.N. Modeling the Genetic Regulation of Cancer Metabolism: Interplay between Glycolysis and Oxidative Phosphorylation. Cancer Res. 2017, 77, 1564–1574. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Wang, F.; Trachootham, D.; Huang, P. Preferential killing of cancer cells with mitochondrial dysfunction by natural compounds. Mitochondrion 2010, 10, 614–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Hao, Y.; Wu, L.; Dong, X.; Jiang, N.; Cong, B.; Liu, J.; Zhang, W.; Tang, D.; De Perrot, M.; et al. Curcumin induces apoptosis and inhibits angiogenesis in murine malignant mesothelioma. Int. J. Oncol. 2018, 53, 2531–2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghtaderi, H.; Sepehri, H.; Delphi, L.; Attari, F. Gallic acid and curcumin induce cytotoxicity and apoptosis in human breast cancer cell MDA-MB-231. Bioimpacts 2018, 8, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H. ROS-mediated activation of AMPK plays a critical role in sulforaphane-induced apoptosis and mitotic arrest in AGS human gastric cancer cells. Gen. Physiol. Biophys. 2018, 37, 129–140. [Google Scholar] [CrossRef]
- Liang, W.; Cai, A.; Chen, G.; Xi, H.; Wu, X.; Cui, J.; Zhang, K.; Zhao, X.; Yu, J.; Wei, B.; et al. Shikonin induces mitochondria-mediated apoptosis and enhances chemotherapeutic sensitivity of gastric cancer through reactive oxygen species. Sci. Rep. 2016, 6, 38267. [Google Scholar] [CrossRef]
- Zhou, J.; Gong, J.; Ding, C.; Chen, G. Quercetin induces the apoptosis of human ovarian carcinoma cells by upregulating the expression of microRNA-145. Mol. Med. Rep. 2015, 12, 3127–3131. [Google Scholar] [CrossRef] [Green Version]
- Brummer, C.; Faerber, S.; Bruss, C.; Blank, C.; Lacroix, R.; Haferkamp, S.; Herr, W.; Kreutz, M.; Renner, K. Metabolic targeting synergizes with MAPK inhibition and delays drug resistance in melanoma. Cancer Lett. 2019, 442, 453–463. [Google Scholar] [CrossRef]
- Lucantoni, F.; Dussmann, H.; Llorente-Folch, I.; Prehn, J.H.M. BCL2 and BCL(X)L selective inhibitors decrease mitochondrial ATP production in breast cancer cells and are synthetically lethal when combined with 2-deoxy-D-glucose. Oncotarget 2018, 9, 26046–26063. [Google Scholar] [CrossRef] [Green Version]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Meena, A.S.; Bhat, M.K. Targeting metabolic flexibility by simultaneously inhibiting respiratory complex I and lactate generation retards melanoma progression. Oncotarget 2015, 35, 37281–37299. [Google Scholar] [CrossRef] [Green Version]
- Zaafar, D.K.; Zaitone, S.A.; Moustafa, Y.M. Role of metformin in suppressing 1, 2-dimethylhydrazine-induced colon cancer in diabetic and non-diabetic mice: Effect on tumor angiogenesis and cell proliferation. PLoS ONE 2014, 9, e100562. [Google Scholar] [CrossRef]
- Dallaglio, K.; Bruno, A.; Cantelmo, A.R.; Esposito, A.I.; Ruggiero, L.; Orecchioni, S.; Calleri, A.; Bertolini, F.; Pfeffer, U.; Noonan, D.M.; et al. Paradoxic effects of metformin on endothelial cells and angiogenesis. Carcinogenesis 2014, 35, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Kurelac, I.; Umesh Ganesh, N.; Iorio, M.; Porcelli, A.M.; Gasparre, G. The multifaceted effects of metformin on tumor microenvironment. Semin. Cell Dev. Biol. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Alistar, A.; Morris, B.B.; Desnoyer, R.; Klepin, H.D.; Hosseinzadeh, K.; Clark, C.; Cameron, A.; Leyendecker, J.; D’Agostino, R., Jr.; Topaloglu, U.; et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: A single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017, 18, 770–778. [Google Scholar] [CrossRef]
- Boddu, P.; Borthakur, G. Therapeutic targeting of isocitrate dehydrogenase mutant AML. Expert Opin. Investig. Drugs 2017, 26, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.K.; Park, K.G. Targeting Glutamine Metabolism for Cancer Treatment. Biomol. Ther. (Seoul) 2018, 1, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.H.; Plescia, J.; Song, H.Y.; Meli, M.; Colombo, G.; Beebe, K.; Scroggins, B.; Neckers, L.; Altieri, D.C. Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J. Clin. Investig. 2009, 119, 454–464. [Google Scholar] [CrossRef] [Green Version]
- Neckers, L.; Kern, A.; Tsutsumi, S. Hsp90 inhibitors disrupt mitochondrial homeostasis in cancer cells. Chem. Biol. 2007, 14, 1204–1206. [Google Scholar] [CrossRef] [Green Version]
- Plescia, J.; Salz, W.; Xia, F.; Pennati, M.; Zaffaroni, N.; Daidone, M.G.; Meli, M.; Dohi, T.; Fortugno, P.; Nefedova, Y.; et al. Rational design of shepherdin, a novel anticancer agent. Cancer Cell 2005, 7, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Siegelin, M.D.; Dohi, T.; Raskett, C.M.; Orlowski, G.M.; Powers, C.M.; Gilbert, C.A.; Ross, A.H.; Plescia, J.; Altieri, D.C. Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells. J. Clin. Investig. 2011, 121, 1349–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, Y.C.; Caino, M.C.; Lisanti, S.; Ghosh, J.C.; Dohi, T.; Danial, N.N.; Villanueva, J.; Ferrero, S.; Vaira, V.; Santambrogio, L.; et al. Control of tumour bioenergetics and survival stress signaling by mitochondrial HSP90s. Cancer Cell 2012, 22, 331–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rondanin, R.; Lettini, G.; Oliva, P.; Baruchello, R.; Costantini, C.; Trapella, C.; Simoni, D.; Bernardi, T.; Sisinni, L.; Pietrafesa, M.; et al. New TRAP1 and Hsp90 chaperone inhibitors with cationic components: Preliminary studies on mitochondrial targeting. Bioorg. Med. Chem. Lett. 2018, 28, 2289–2293. [Google Scholar] [CrossRef] [PubMed]
- D’Annessa, I.; Sattin, S.; Tao, J.; Pennati, M.; Sànchez-Martìn, C.; Moroni, E.; Rasola, A.; Zaffaroni, N.; Agard, D.A.; Bernardi, A.; et al. Design of Allosteric Stimulators of the Hsp90 ATPase as New Anticancer Leads. Chemistry 2017, 23, 5188–5192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffel, J.; Bhattacharyya, A.K.; Gallegos, A.; Cui, H.; Einspahr, J.G.; Alberts, D.S.; Powis, G. Increased expression of thioredoxin-1 in human colorectal cancer is associated with decreased patient survival. J. Lab. Clin. Med. 2003, 142, 46–51. [Google Scholar] [CrossRef]
- Choi, J.H.; Kim, T.N.; Kim, S.; Baek, S.H.; Kim, J.H.; Lee, S.R.; Kim, J.R. Overexpression of mitochondrial thioredoxin reductase and peroxiredoxin III in hepatocellular carcinomas. Anticancer Res. 2002, 22, 3331–3335. [Google Scholar] [PubMed]
- Topkas, E.; Cai, N.; Cumming, A.; Hazar-Rethinam, M.; Gannon, O.M.; Burgess, M.; Saunders, N.A.; Endo-Munoz, L. Auranofin is a potent suppressor of osteosarcoma metastasis. Oncotarget 2016, 7, 831–844. [Google Scholar] [CrossRef]
- Bu, L.; Li, W.; Ming, Z.; Shi, J.; Fang, P.; Yang, S. Inhibition of TrxR2 suppressed NSCLC cell proliferation, metabolism and induced cell apoptosis through decreasing antioxidant activity. Life Sci. 2017, 178, 35–41. [Google Scholar] [CrossRef]
- Chua, P.J.; Lee, E.H.; Yu, Y.; Yip, G.W.; Tan, P.H.; Bay, B.H. Silencing the Peroxiredoxin III gene inhibits cell proliferation in breast cancer. Int. J. Oncol. 2010, 26, 359–364. [Google Scholar]
- Qiao, B.; Wang, J.; Xie, J.; Niu, Y.; Ye, S.; Wan, Q.; Ye, Q. Detection and identification of peroxiredoxin 3 as a biomarker in hepatocellular carcinoma by a proteomic approach. Int. J. Mol. Med. 2012, 29, 832–840. [Google Scholar]
- Kim, Y.S.; Lee, H.L.; Lee, K.B.; Park, J.H.; Chung, W.Y.; Lee, K.S.; Sheen, S.S.; Park, K.J.; Hwang, S.C. Nuclear factor E2-related factor 2 dependent overexpression of sulfiredoxin and peroxiredoxin III in human lung cancer. Korean J. Int. Med. 2011, 26, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Su, S.K.; Ki, T.K.; Kang, M.S.; Jeong, D.H.; Lee, D.S.; Jung, E.J.; Kim, Y.N.; Han, J.; Song, I.S.; et al. Overexpression of peroxiredoxin-3 and -5 is a potential biomarker for prognosis in endometrial cancer. Oncol. Lett. 2018, 15, 5111–5118. [Google Scholar]
- Whitaker, H.C.; Patel, D.; Howat, W.J.; Warren, A.Y.; Kay, J.D.; Sangan, T.; Marioni, J.C.; Mitchell, J.; Aldridge, S.; Luxton, H.J.; et al. Peroxiredoxin-3 is overexpressed in prostate cancer and promotes cancer cell survival by protecting cells from oxidative stress. Br. J. Cancer 2013, 109, 983–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.F.; Jiang, S.J.; Pan, Z.P.; Cheng, W.D.; Zhang, W.J.; Yao, X.K.; Li, Y.C.; Lun, Y.Z. Expression and clinical significance of Sirt1 in colorectal cancer. Oncol. Lett. 2016, 11, 1167–1172. [Google Scholar] [CrossRef] [Green Version]
- Suh, D.H.; Kim, M.A.; Kim, H.; Kim, M.K.; Kim, H.S.; Chung, H.H.; Kim, Y.B.; Song, Y.S. Association of overexpression of hexokinase II with chemoresistance in epithelial ovarian cancer. Clin. Exp. Med. 2014, 14, 345–353. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Zhang, M.; Cong, Q.; Zhang, M.X.; Zhang, M.Y.; Lu, Y.Y.; Xu, C.J. Hexokinase 2 confers resistance to cisplatin in ovarian cancer cells by enhancing cisplatin-induced autophagy. Int. J. Biochem. Cell Biol. 2018, 95, 9–16. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Protein | Function | Tumor Type | References |
|---|---|---|---|
| Thioredoxin-1 (Trx1) | Redox protein member of thioredoxin system which plays a crucial role in the cellular redox homeostasis | DLBCL | [24] |
| CRC | [107] | ||
| Thioredoxin reductase-2 (TrxR2) | Member of the family of pyridine nucleotide-disulfide oxidoreductases, component of the antioxidant thioredoxin system | HCC | [108] |
| OS | [109] | ||
| LC | [110] | ||
| Peroxiredoxin 3 (Prx3) | Antioxidant enzyme with mitochondrial localization, member of the thioredoxin system | BC | [111] |
| HCC | [112] | ||
| MESO | [113] | ||
| EC | [114] | ||
| PC | [115] | ||
| SIRT1 | NAD+-dependent protein deacetylase, plays key roles in DNA damage response and metabolic adaptation to energy stress. Along with PGC1α, is involved in chemotherapy-induced shift to OXPHOS in CRC cells | CRC | [47,116] |
| SIRT5 | NAD-dependent protein lysine demalonylase, desuccinylase and deglutarylase able to remove malonyl, succinyl, and glutaryl groups from the lysine residues of proteins | CRC | [54] |
| Hexokinase II (HK2) | Catalyzes the phosphorylation of glucose to generate glucose-6-phosphate in the first step of glycolysis. It promotes chemoresistance by enhancing cisplatin-induced autophagy | OC | [117,118] |
| Transketolase (TKT) | Ezyme catalyzing important reaction both in the Calvin cycle and in pentose phosphate pathway | BC | [57] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avolio, R.; Matassa, D.S.; Criscuolo, D.; Landriscina, M.; Esposito, F. Modulation of Mitochondrial Metabolic Reprogramming and Oxidative Stress to Overcome Chemoresistance in Cancer. Biomolecules 2020, 10, 135. https://doi.org/10.3390/biom10010135
Avolio R, Matassa DS, Criscuolo D, Landriscina M, Esposito F. Modulation of Mitochondrial Metabolic Reprogramming and Oxidative Stress to Overcome Chemoresistance in Cancer. Biomolecules. 2020; 10(1):135. https://doi.org/10.3390/biom10010135
Chicago/Turabian StyleAvolio, Rosario, Danilo Swann Matassa, Daniela Criscuolo, Matteo Landriscina, and Franca Esposito. 2020. "Modulation of Mitochondrial Metabolic Reprogramming and Oxidative Stress to Overcome Chemoresistance in Cancer" Biomolecules 10, no. 1: 135. https://doi.org/10.3390/biom10010135
APA StyleAvolio, R., Matassa, D. S., Criscuolo, D., Landriscina, M., & Esposito, F. (2020). Modulation of Mitochondrial Metabolic Reprogramming and Oxidative Stress to Overcome Chemoresistance in Cancer. Biomolecules, 10(1), 135. https://doi.org/10.3390/biom10010135