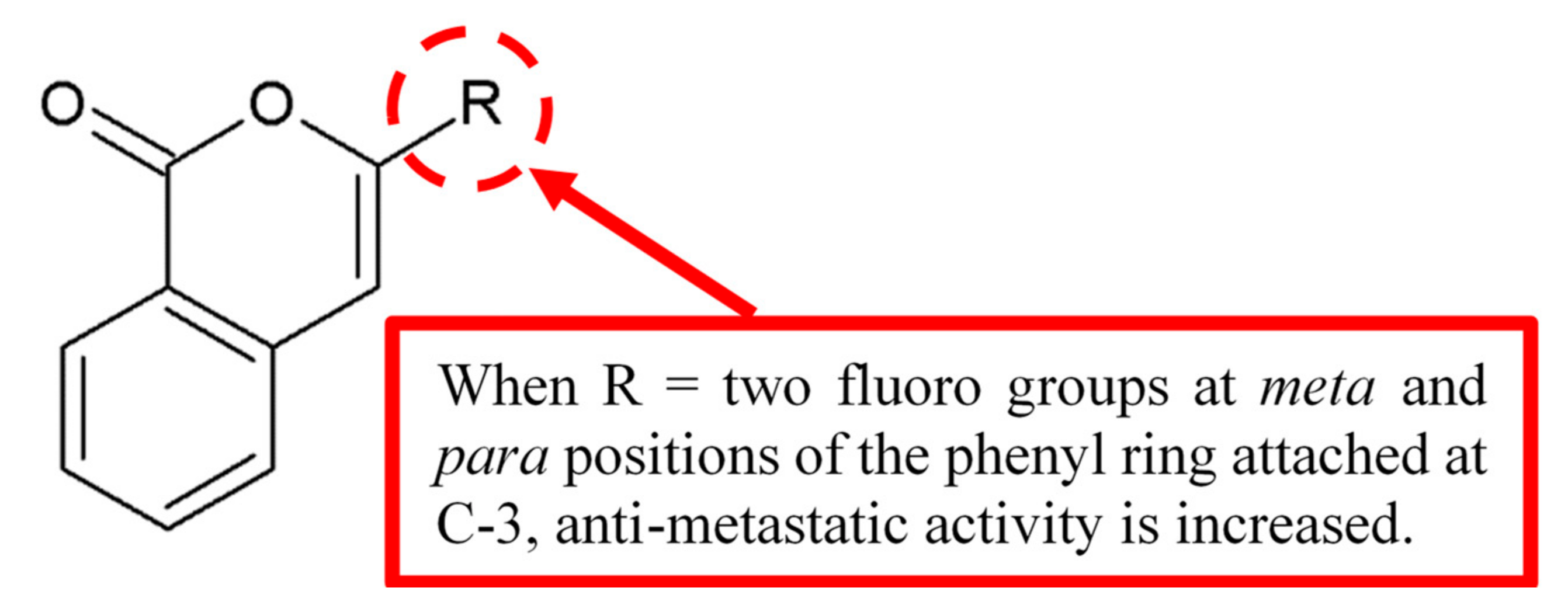
Figure 1.
Structure–activity relationship (SAR) study of isocoumarin derivative 1.
Figure 1.
Structure–activity relationship (SAR) study of isocoumarin derivative 1.
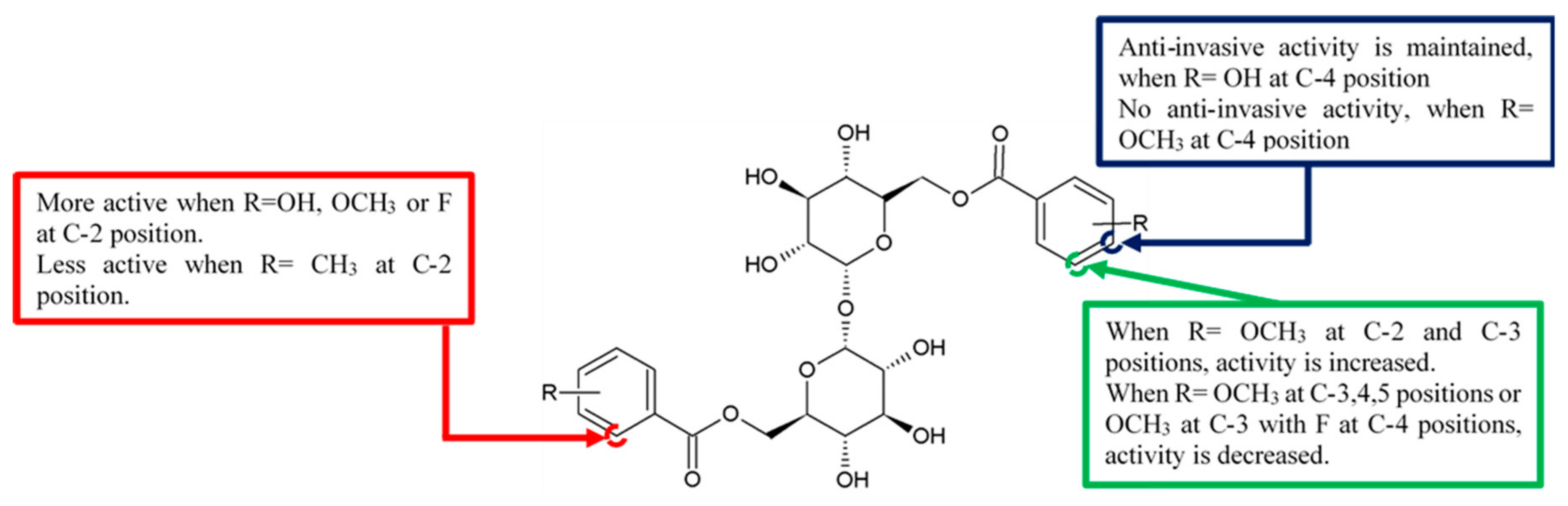
Figure 2.
SAR study of brartemicin derivatives.
Figure 2.
SAR study of brartemicin derivatives.
Figure 3.
SAR study of 1,3,4-oxadiazole derivatives.
Figure 3.
SAR study of 1,3,4-oxadiazole derivatives.
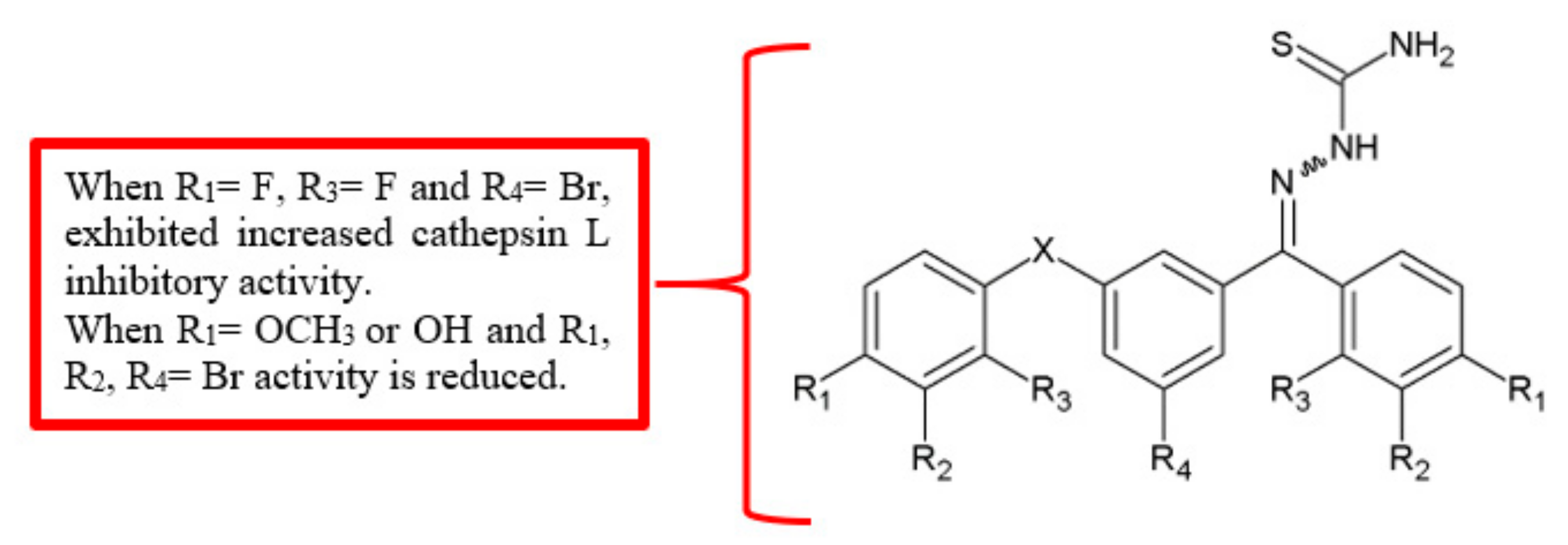
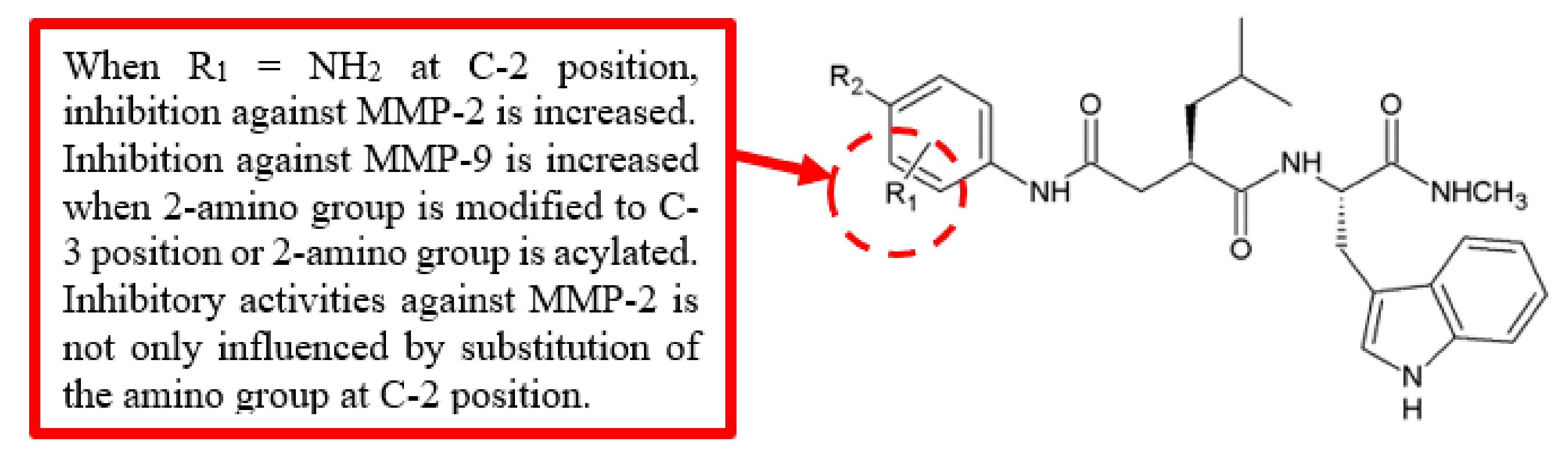
Figure 4.
SAR study of benzoylbenzophenone thiosemicarbazone derivatives.
Figure 4.
SAR study of benzoylbenzophenone thiosemicarbazone derivatives.
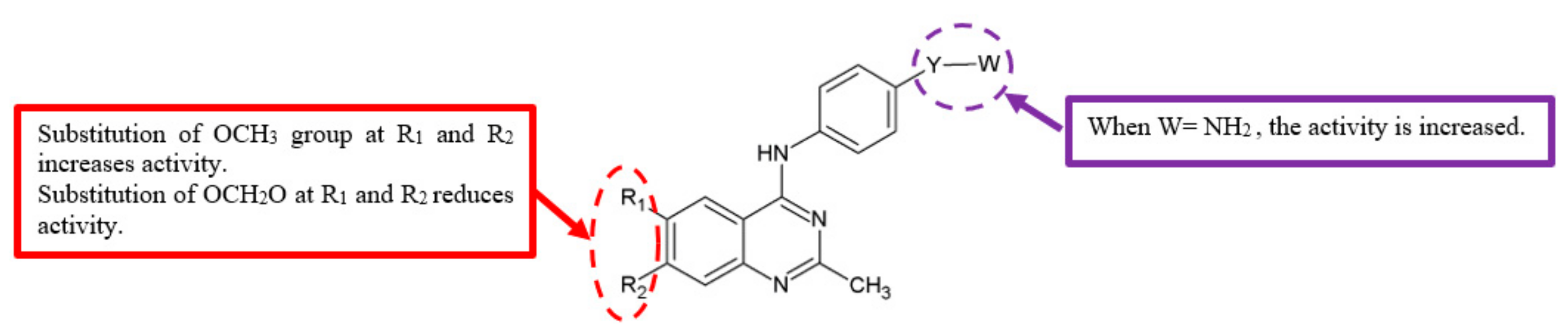
Figure 5.
SAR study of 4-anilino-quinazoline derivatives.
Figure 5.
SAR study of 4-anilino-quinazoline derivatives.
Figure 6.
SAR study of 2-furanylvinylquinoline derivative 22.
Figure 6.
SAR study of 2-furanylvinylquinoline derivative 22.
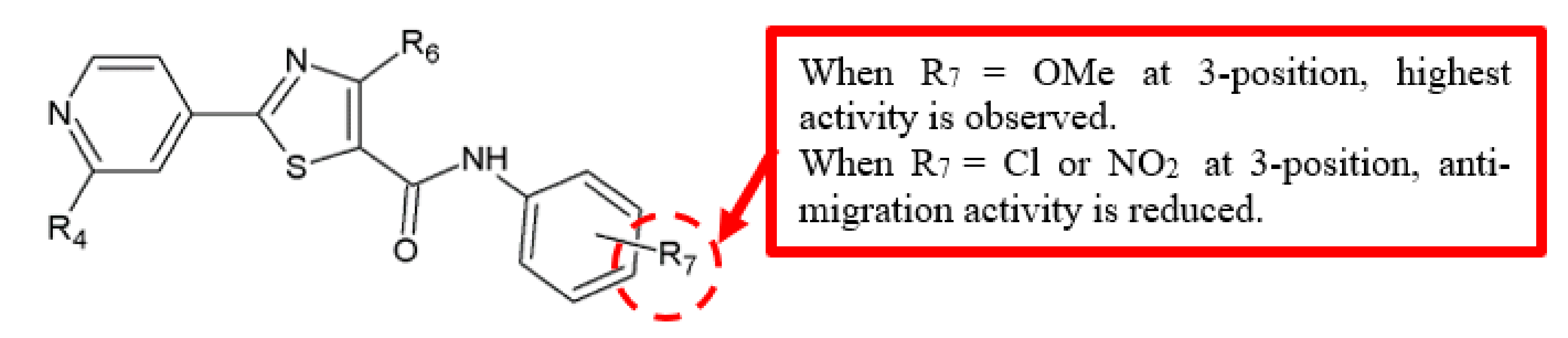
Figure 7.
SAR study of 4-methyl-2-(4-pyridinyl)thiazole-5-carboxamide derivatives.
Figure 7.
SAR study of 4-methyl-2-(4-pyridinyl)thiazole-5-carboxamide derivatives.
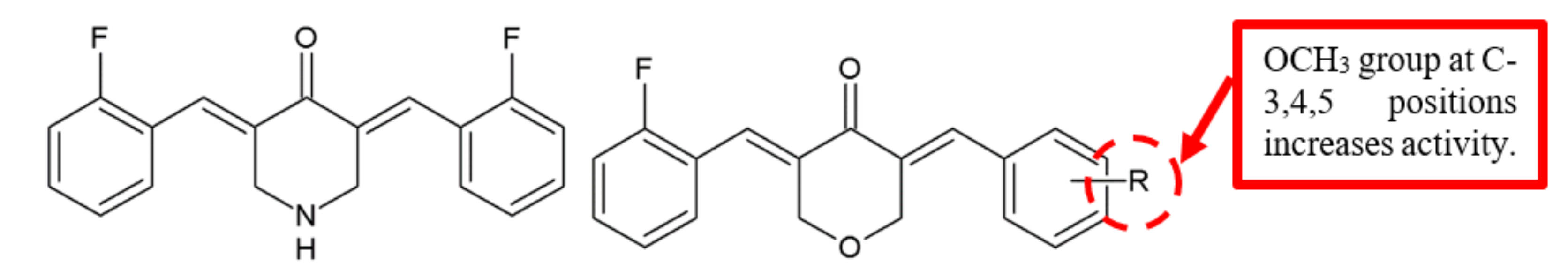
Figure 8.
SAR study of EF24 derivatives.
Figure 8.
SAR study of EF24 derivatives.
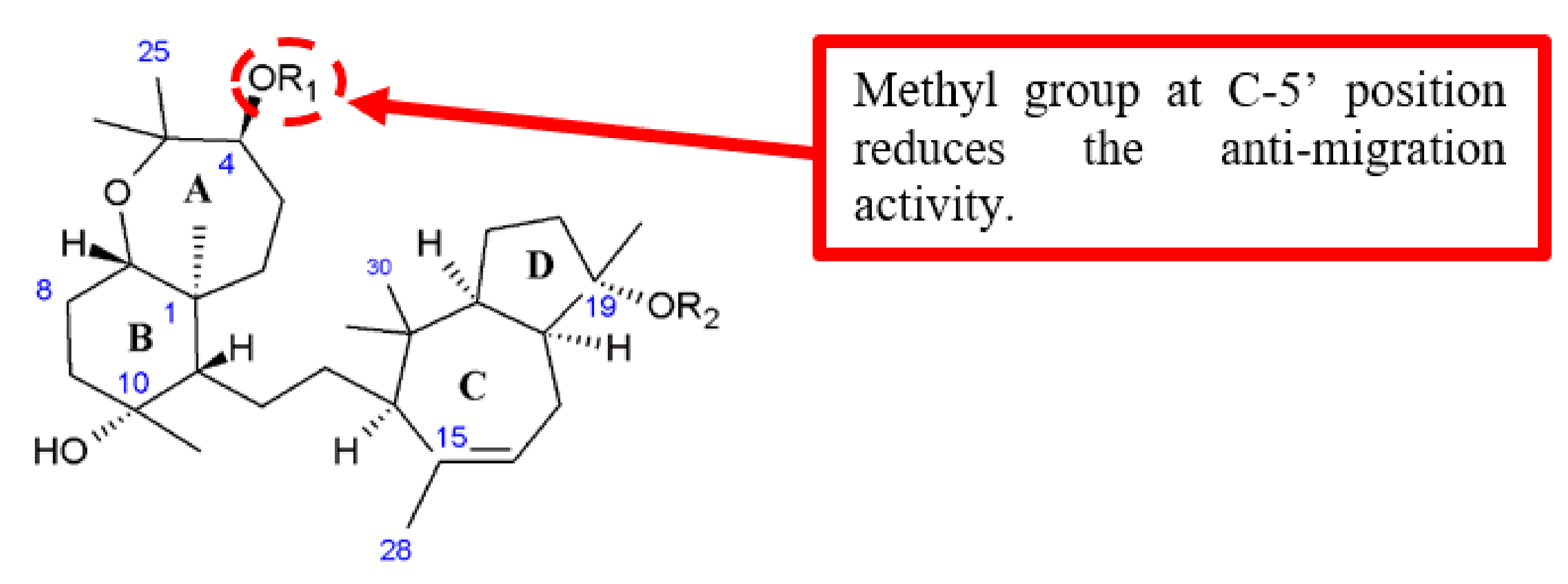
Figure 9.
SAR study of BU-4664L derivative 27.
Figure 9.
SAR study of BU-4664L derivative 27.
Figure 10.
SAR study of isomalyngamide A derivatives.
Figure 10.
SAR study of isomalyngamide A derivatives.
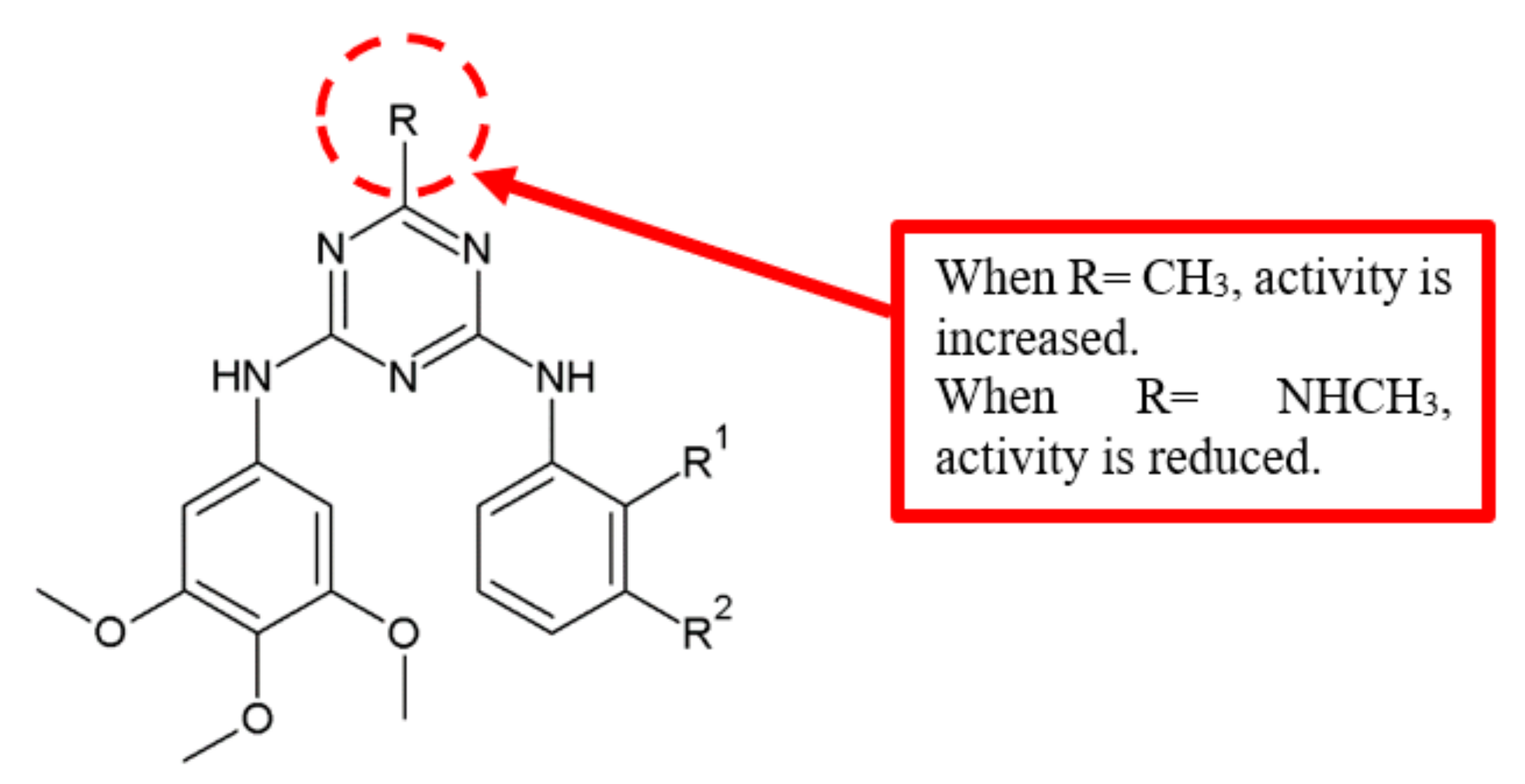
Figure 11.
SAR study of diarylamino-1,3,5-triazine derivatives.
Figure 11.
SAR study of diarylamino-1,3,5-triazine derivatives.
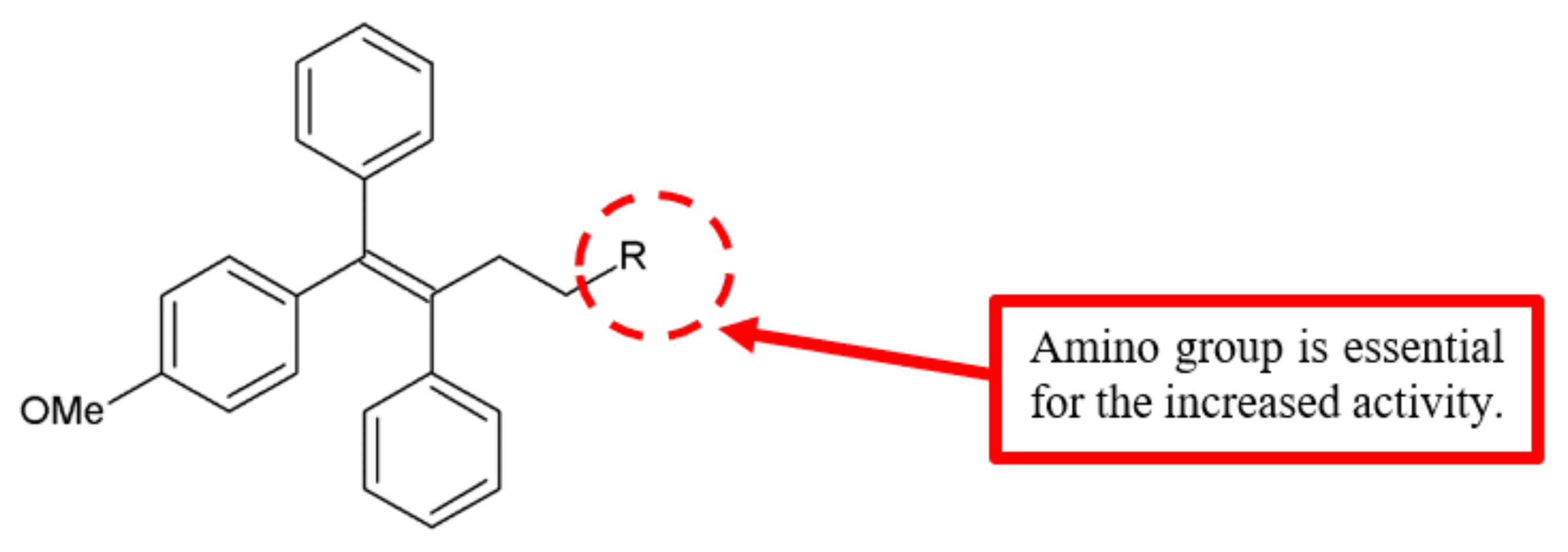
Figure 12.
SAR study of triarylethylene derivatives.
Figure 12.
SAR study of triarylethylene derivatives.
Figure 13.
SAR study of benzamide Ilomastat derivatives.
Figure 13.
SAR study of benzamide Ilomastat derivatives.
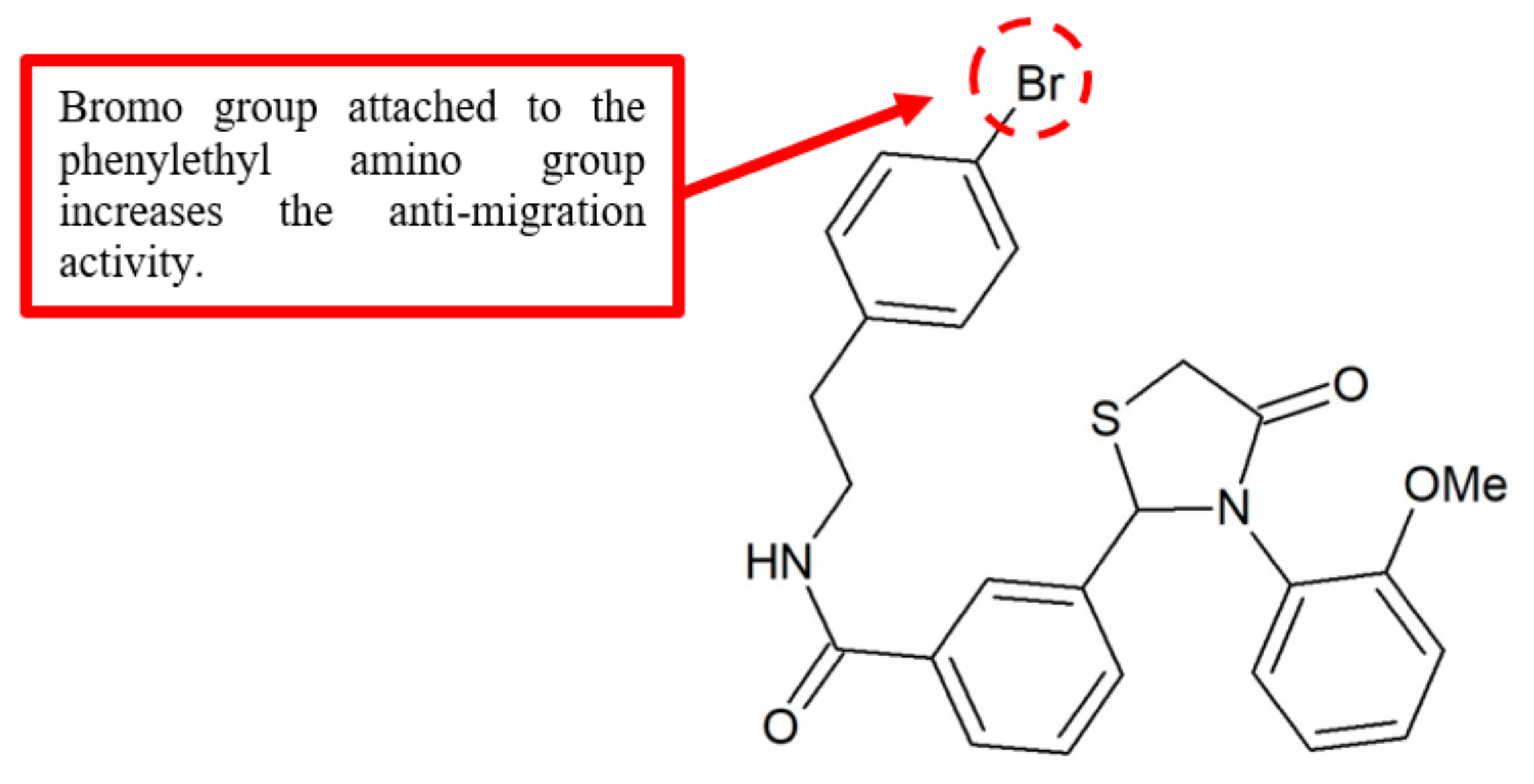
Figure 14.
SAR study of 2,3-diaryl-4-thiazolidinone derivative 36.
Figure 14.
SAR study of 2,3-diaryl-4-thiazolidinone derivative 36.
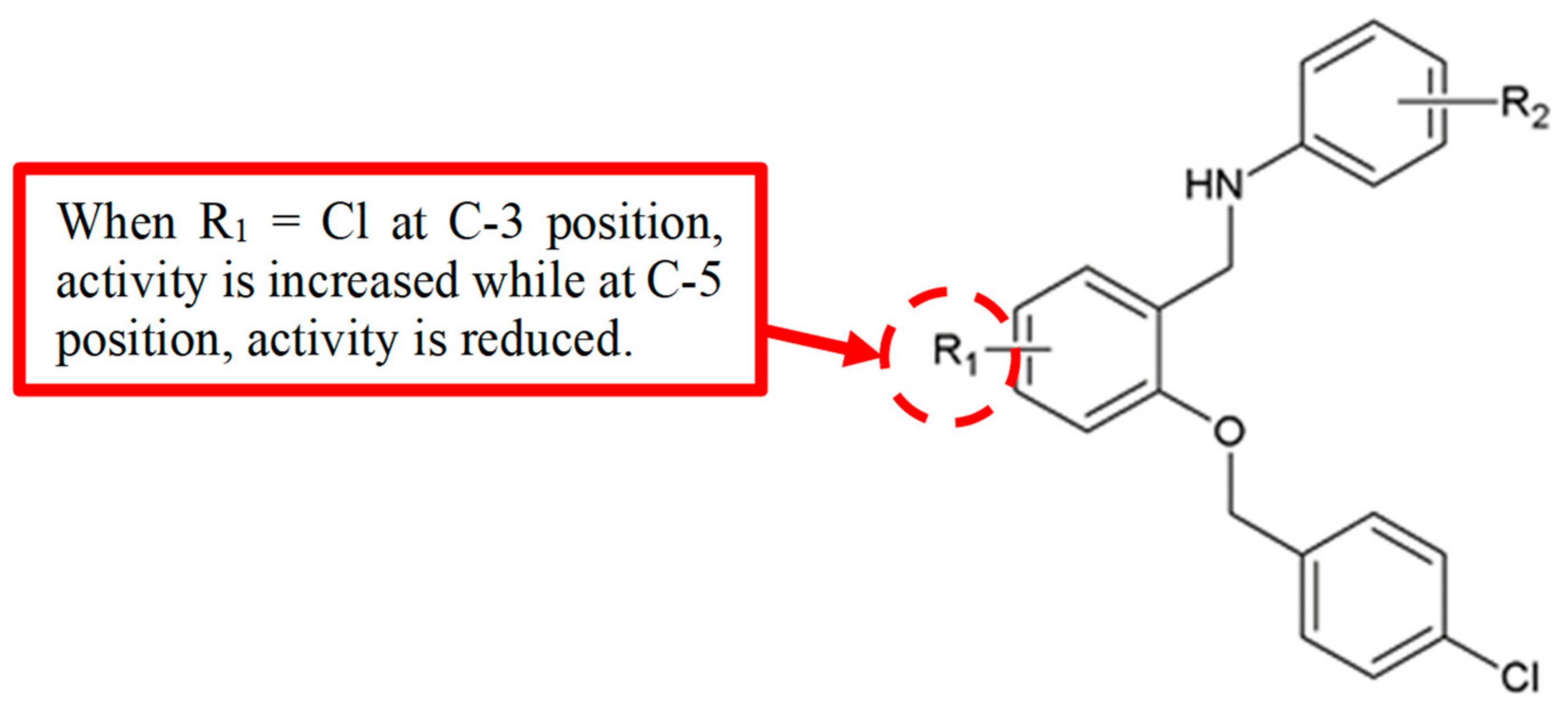
Figure 15.
SAR study of benzyloxyphenylmethylaminophenol derivatives.
Figure 15.
SAR study of benzyloxyphenylmethylaminophenol derivatives.
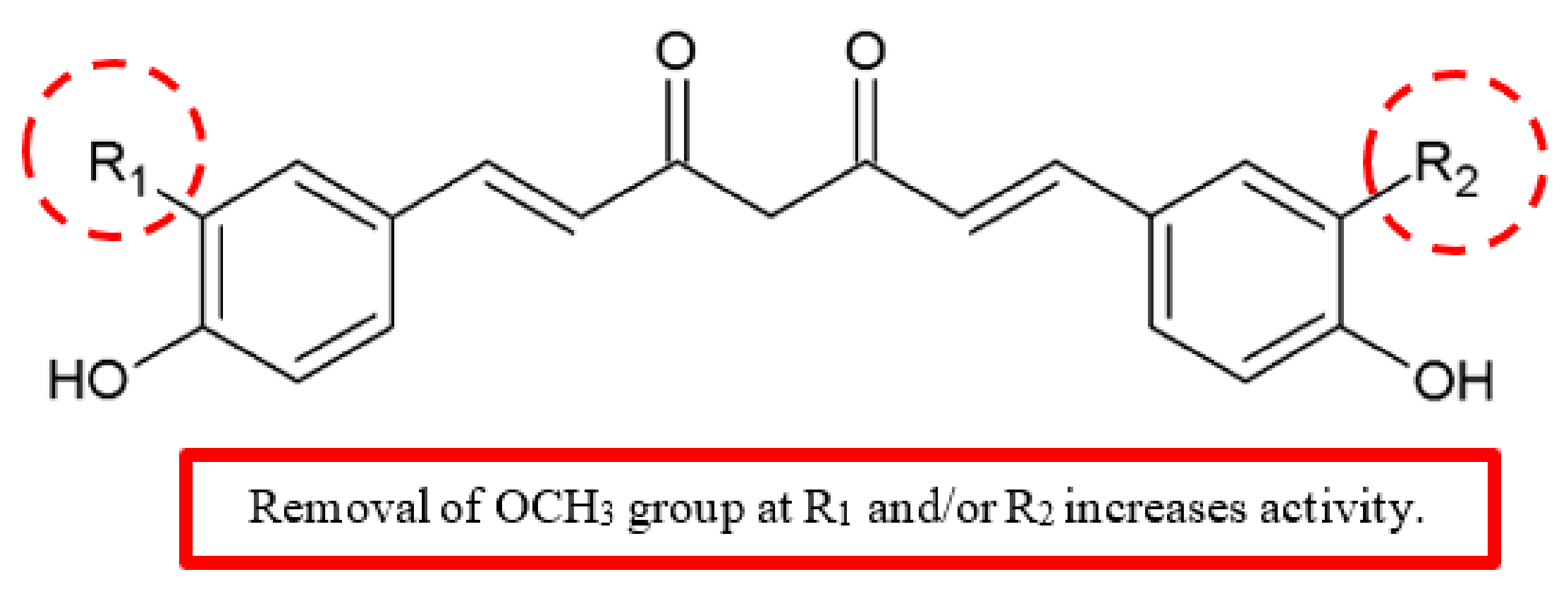
Figure 16.
SAR study of curcumin derivatives.
Figure 16.
SAR study of curcumin derivatives.
Figure 17.
SAR study of sipholenol A derivatives.
Figure 17.
SAR study of sipholenol A derivatives.
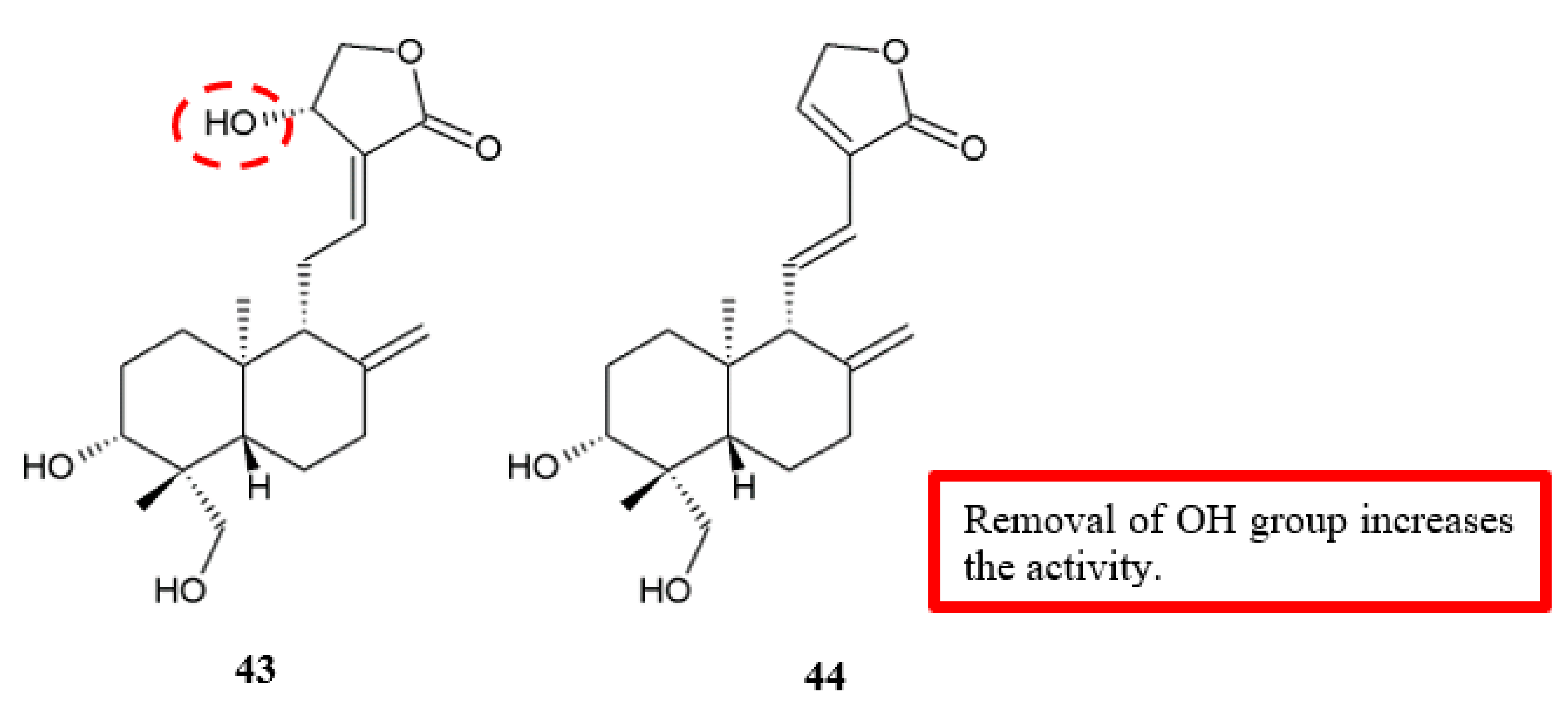
Figure 18.
SAR study of andrographolide derivatives.
Figure 18.
SAR study of andrographolide derivatives.
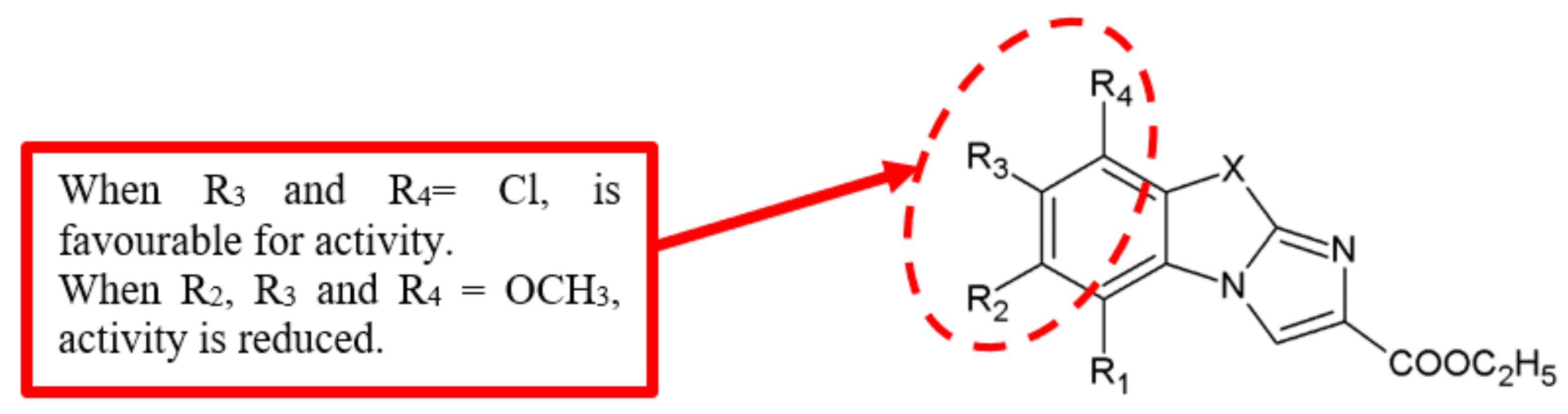
Figure 19.
SAR study of imidazobenzothiazole derivatives.
Figure 19.
SAR study of imidazobenzothiazole derivatives.
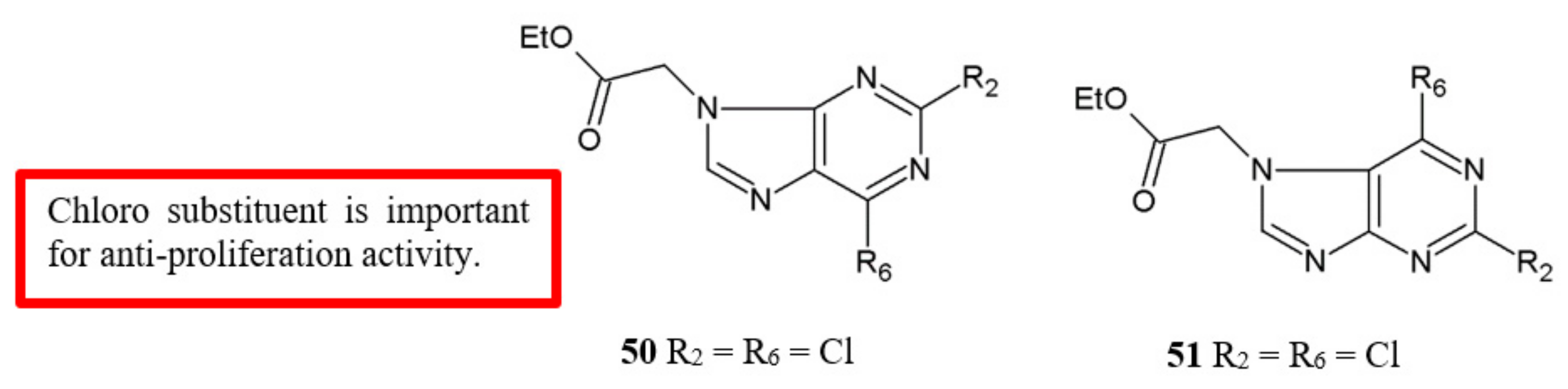
Figure 20.
SAR study of purine derivatives.
Figure 20.
SAR study of purine derivatives.
Figure 21.
SAR study of 8-hydroxyquinoline derivatives.
Figure 21.
SAR study of 8-hydroxyquinoline derivatives.
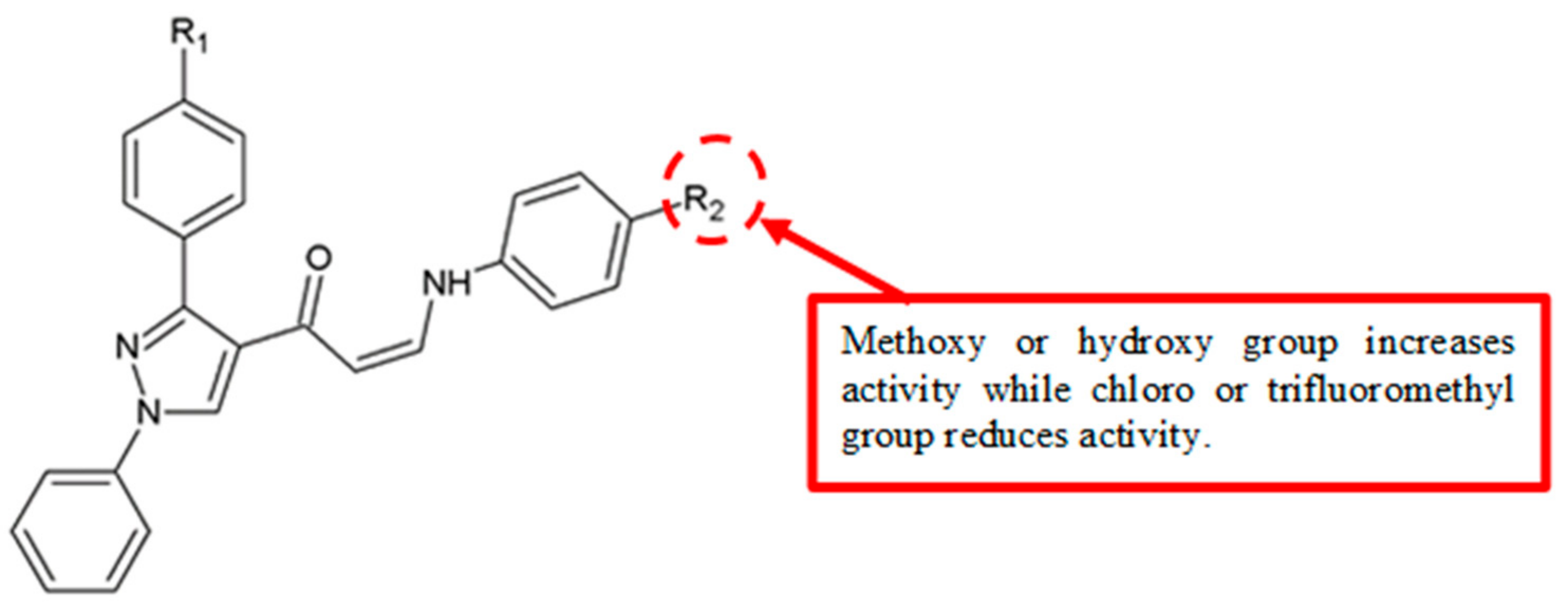
Figure 22.
SAR study of (Z)-1-(1,3-diphenyl-1H-pyrazol-4-yl)-3-(phenylamino)prop-2-en-1-one derivatives.
Figure 22.
SAR study of (Z)-1-(1,3-diphenyl-1H-pyrazol-4-yl)-3-(phenylamino)prop-2-en-1-one derivatives.
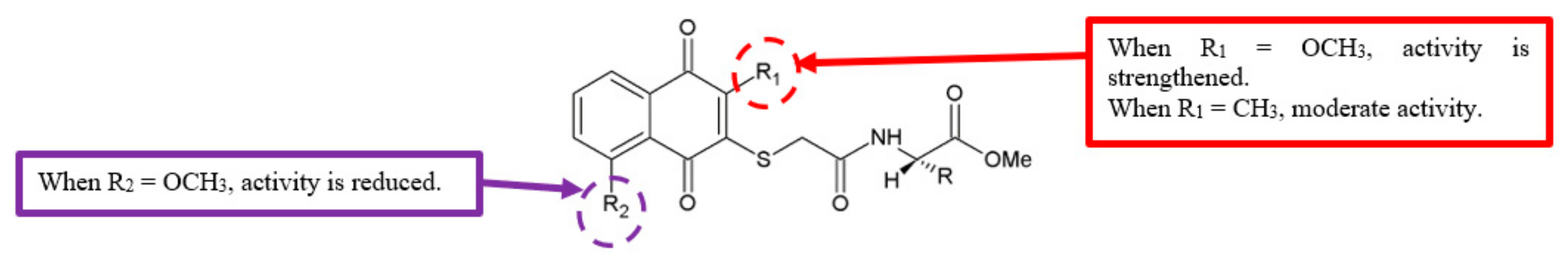
Figure 23.
SAR study of naphthoquinone amide derivatives.
Figure 23.
SAR study of naphthoquinone amide derivatives.
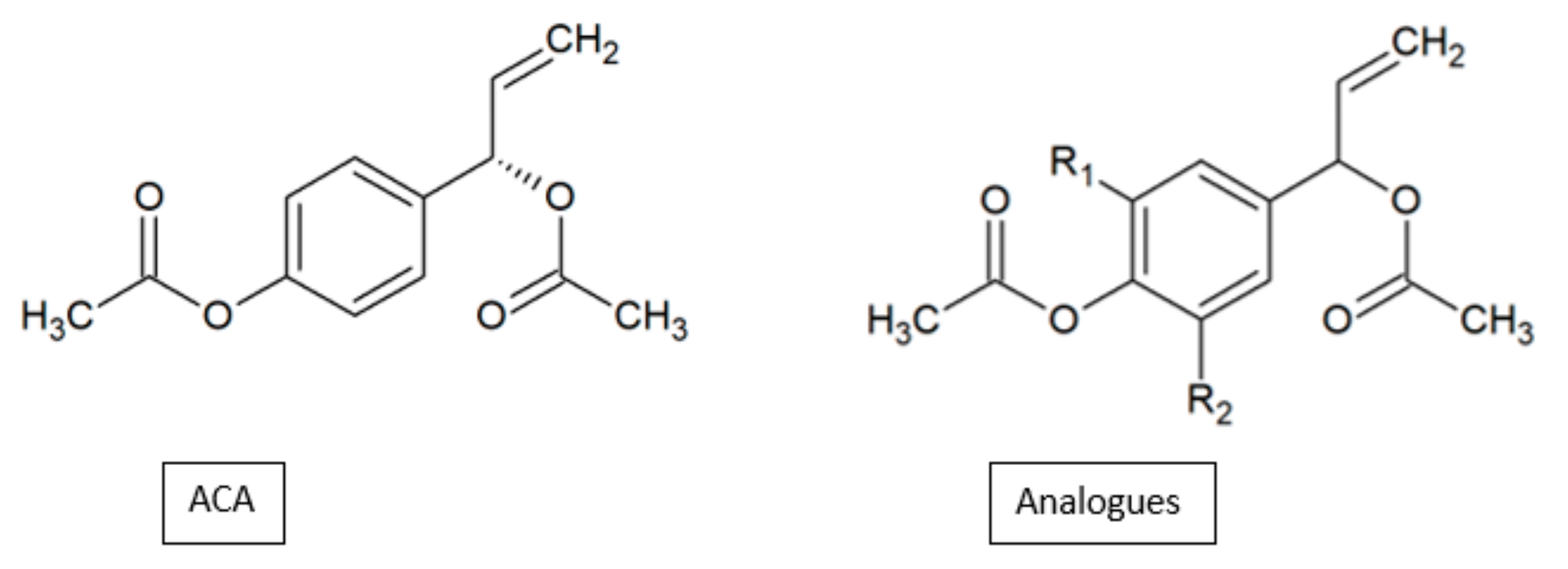
Figure 24.
SAR study of 1’S-1’-acetoxychavicol acetate (ACA) derivatives.
Figure 24.
SAR study of 1’S-1’-acetoxychavicol acetate (ACA) derivatives.
Figure 25.
SAR study of 6,7-disubstituted-4-phenoxyquinoline derivatives.
Figure 25.
SAR study of 6,7-disubstituted-4-phenoxyquinoline derivatives.
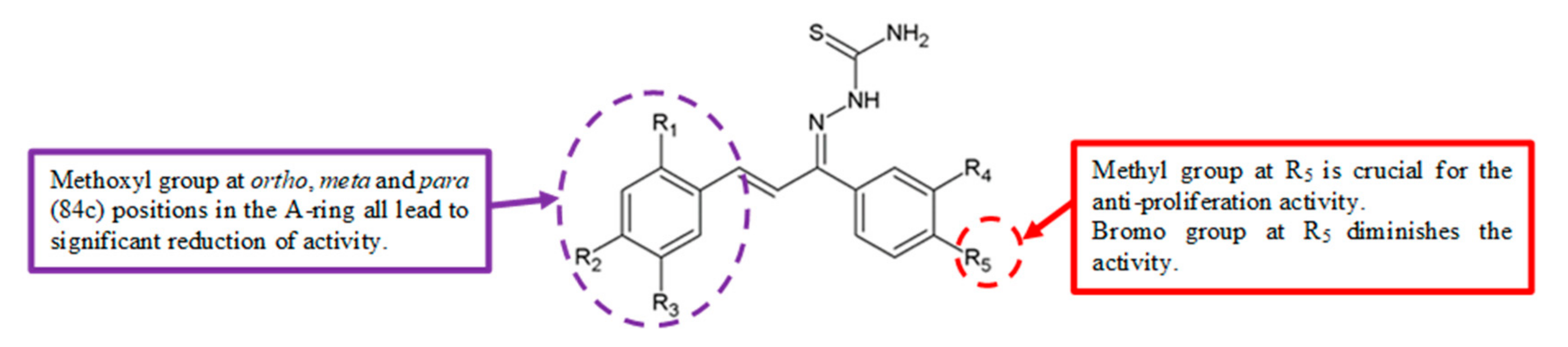
Figure 26.
SAR study of chalcone thiosemicarbazide derivatives.
Figure 26.
SAR study of chalcone thiosemicarbazide derivatives.
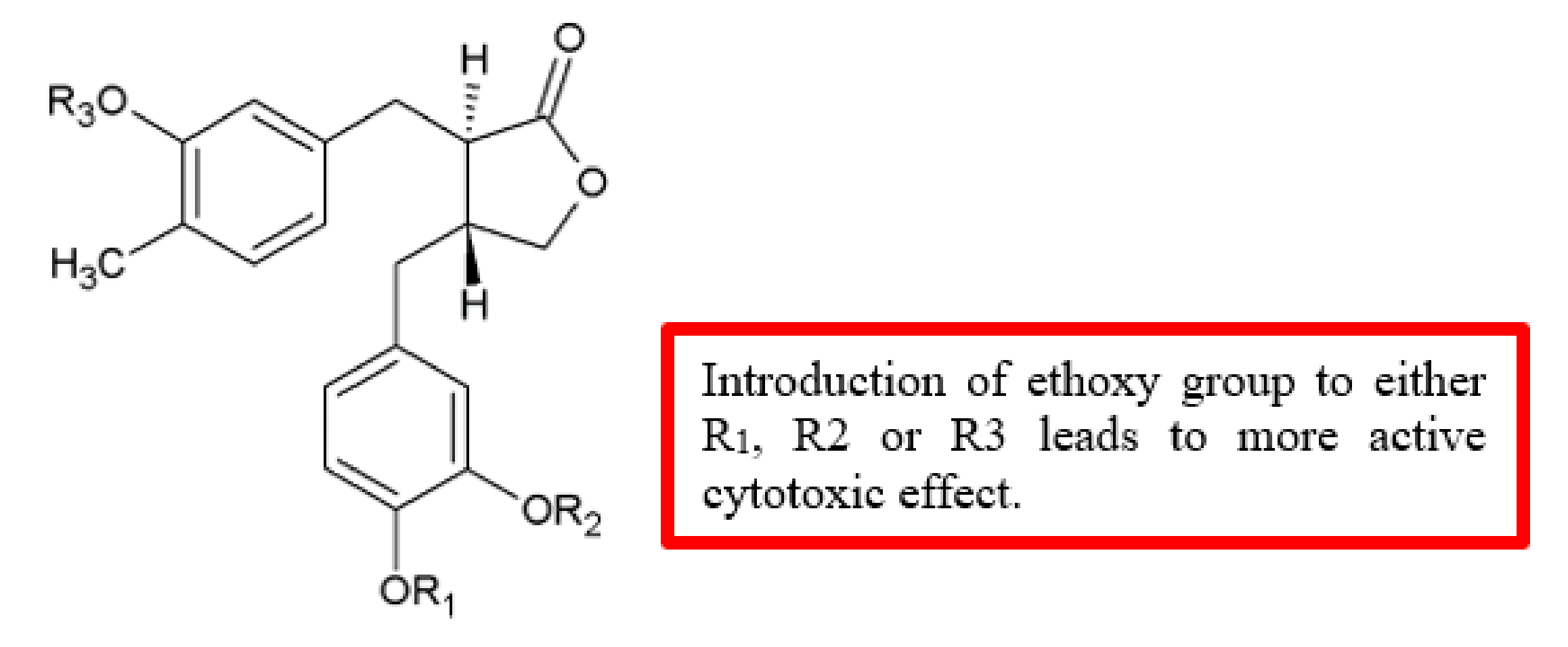
Figure 27.
SAR study of (‒)-arctigenin derivatives.
Figure 27.
SAR study of (‒)-arctigenin derivatives.
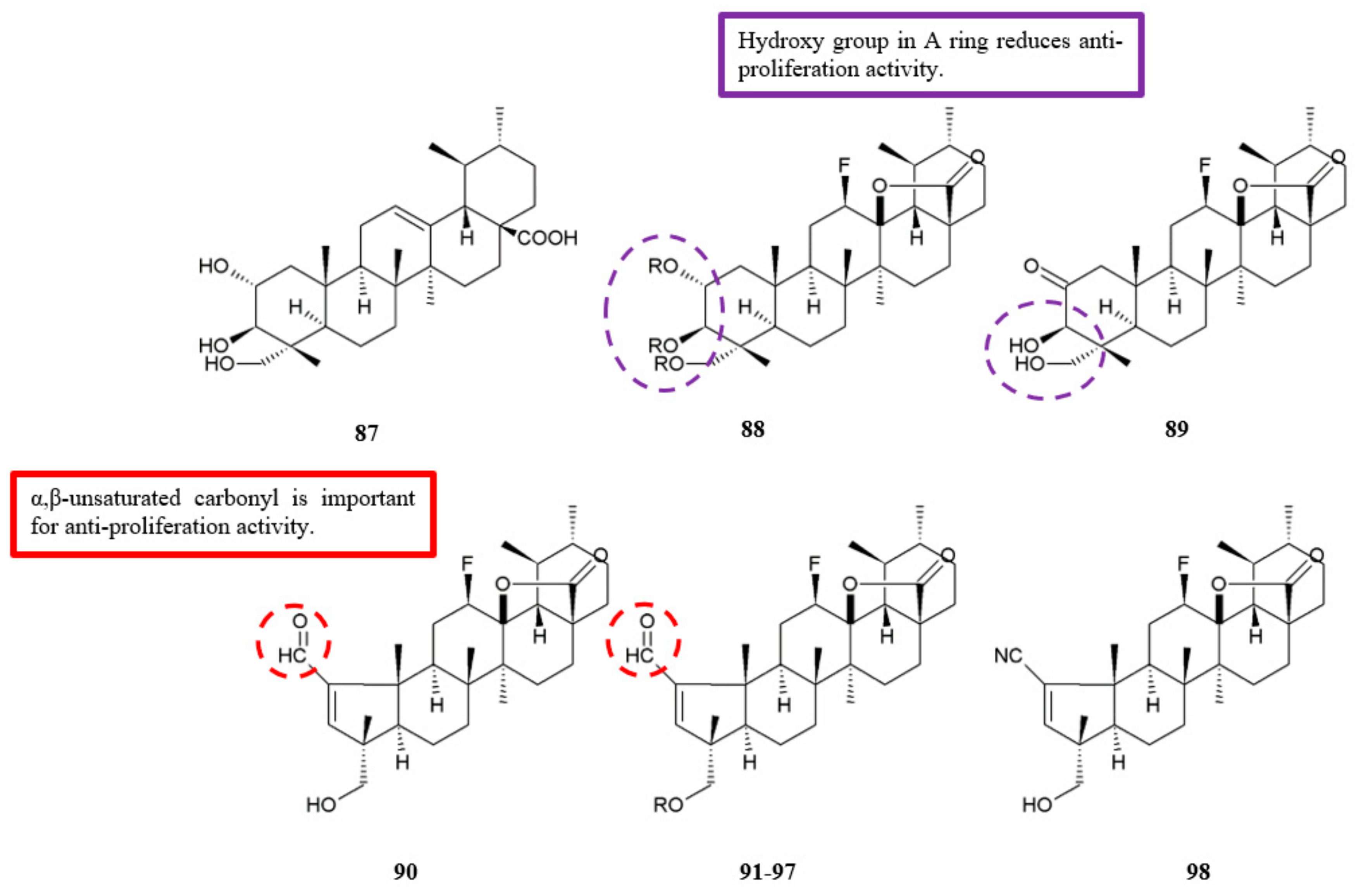
Figure 28.
SAR study of asiatic acid derivatives.
Figure 28.
SAR study of asiatic acid derivatives.
Table 1.
SAR study of brartemicin derivatives.
Table 1.
SAR study of brartemicin derivatives.
| Compounds | R | IC50 (μg/mL) |
|---|
| Anti-Invasive Activity In Vitro |
|---|
| Brartemicin, 2 | 2,4-(OH)2-6-CH3 | 0.25 |
|---|
| 3a | 2-OCH3 | 1.0 |
| 3b | 2-CH3 | NA |
| 3c | 4-OCH3 | NA |
| 3d | 4-OH | 1.0 |
| 3e | 2,3-(OCH3)2 | 0.10 |
| 3f | 3,4,5-(OCH3)3 | NA |
| 3g | 3-OCH3-4-F | NA |
| 3h | 2,6-F2 | 1.0 |
| 4 | 2-OH | <1.0 |
| 5 | 2,3-(OH)2 | <1.0 |
Table 2.
SAR study of 1,3,4-oxadiazole derivatives.
Table 2.
SAR study of 1,3,4-oxadiazole derivatives.
| Compounds | R | Anti-Proliferation Activity (IC50, μg/mL) | FAK Inhibitory Activity (IC50, μM) |
|---|
| MCF-7 | HT29 |
|---|
| 6 | 2-F | 5.68 | 10.21 | 1.2 ± 0.3 |
| 7 | 2-CH3 | 18.89 | 26.81 | 12.1 ± 1.3 |
| 8 | 3-F | 8.25 | 15.47 | 7.1 ± 0.3 |
| 9 | 3-CH3 | 28.92 | 38.50 | 15.8 ± 1.1 |
| 10 | 4-F | 8.70 | 17.62 | 9.1 ± 0.5 |
| 11 | 4-CH3 | 30.23 | 42.30 | 33.8 ± 1.4 |
| Cisplatin | - | 11.20 | 15.83 | 8.6 ± 0.2 |
Table 3.
SAR study of benzoylbenzophenone thiosemicarbazone derivatives.
Table 3.
SAR study of benzoylbenzophenone thiosemicarbazone derivatives.
| Compounds | R1 | R2 | R3 | R4 | X | Cathepsin L Inhibitory Activity (IC50, nM) |
|---|
| 12 | H | H | H | H | C=O | 9.9 |
| 13 | F | H | H | H | C=O | 14.4 |
| 14 | Br | H | H | H | C=NHHC(S)NH2 | >10,000 |
| 15 | OCH3 | H | H | H | C=O | 5117 |
| 16 | OH | H | H | H | C=O | 340 |
| 17 | H | Br | H | OH | C=O | ~10,000 |
| 18 | H | H | F | Br | C=O | 8.1 |
| 19 | H | Br | H | Br | C=O | 10,347 |
Table 4.
SAR study of 4-anilino-quinazoline derivatives.
Table 4.
SAR study of 4-anilino-quinazoline derivatives.
| Compounds | R1, R2 | Y | W | Inhibitory Activity on EGFR (IC50, μM) | Inhibitory Activity on VEGFR-2 (IC50, μM) |
|---|
| 20 | OCH3, OCH3 | SO2 | ![Biomolecules 10 00138 i001]() | 9.70 | 7.79 |
| 21a | OCH2O | SO2 | ![Biomolecules 10 00138 i001]() | >100 | >100 |
| 21b | OCH3, OCH3 | SO2 | CH3 | 61.5 | >100 |
| 21c | OCH2O | SO2 | CH3 | >100 | >100 |
| 21d | OCH3, OCH3 | SO2 | NH2 | 2.37 | 1.02 |
| 21e | OCH3, OCH3 | - | N(CH3)2 | 36.0 | 39.3 |
| 21f | OCH2O | - | N(CH3)2 | >100 | >100 |
| 21g | OCH3, OCH3 | C=O | NH2 | 0.90 | 1.17 |
Table 5.
SAR study of 4-methyl-2-(4-pyridinyl)thiazole-5-carboxamide derivatives.
Table 5.
SAR study of 4-methyl-2-(4-pyridinyl)thiazole-5-carboxamide derivatives.
| Compounds | R4 | R6 | R7 | Anti-Migration Activity (IC50, μM) |
|---|
| HUVEC |
|---|
| 23 | - | - | - | 6.0 ± 1.6 |
| 24a | n-Pr | Me | 3’-OMe | 3.4 ± 0.2 |
| 24b | n-Pr | Me | 3’-Cl | 6.8 ± 2.3 |
| 24c | n-Pr | Me | 3’-NO2 | 8.5 ± 6.7 |
Table 6.
SAR study of EF24 derivatives.
Table 6.
SAR study of EF24 derivatives.
| Compounds | R | Anti-Proliferation Activity (IC50, μM) | Migration Rate at the Concentration of 20 µM (%) |
|---|
| A549 | LLC | H1650 | A549 |
|---|
| EF24, 25 | - | 7.1 ± 3.2 | 8.4 ± 3.0 | 14.6 ± 10 | - |
| 26 | 3,4,5-OCH3 | 6.3 ± 0.3 | 6.1 ± 0.9 | 6.8 ± 0.3 | 37.9 |
Table 7.
SAR study of BU-4664L derivative 27.
Table 7.
SAR study of BU-4664L derivative 27.
| Compounds | Side Chain | R | IC50 (μg/mL) |
|---|
| Anti-Invasive Activity | Anti-Angiogenic Activity | Inhibition of Cellular Motility |
|---|
| Colon 26-L5 | Renca | Colon 26-L5 | HUVEC |
|---|
| 27 | Saturated | Me | 1.0 | 0.78 | 0.11 | 0.67 | 0.0076 |
Table 8.
SAR study of isomalyngamide A derivatives.
Table 8.
SAR study of isomalyngamide A derivatives.
| Compounds | Percentage of Cell Proliferation Inhibition at 20 μM (%) | Anti-Migration Activity (IC50, μM) |
|---|
| MCF-7 | MDA-MB-231 | MDA-MB-231 |
|---|
| 28 | 7% | 0% | 22.7 ± 1.3 |
| 29 | 19% | 0% | 29.9 ± 0.6 |
Table 9.
SAR study of diarylamino-1,3,5-triazine derivatives.
Table 9.
SAR study of diarylamino-1,3,5-triazine derivatives.
| Compounds | R1 | R2 | R | FAK Inhibitory Activity—FRET (IC50, μM) | Anti-Proliferation Activity (IC50, μM) |
|---|
| HUVEC |
|---|
| 30 | NHSO2CH3 | H | Cl | 41.9 ± 4.6 | 9.5 ± 1.0 |
| 31 | NHSO2CH3 | H | NHCH3 | 65.9 ± 9.6 | 34.2 ± 7.6 |
| 32 | NHSO2CH3 | H | CH3 | 7.9 ± 0.9 | 8.5 ± 0.4 |
Table 10.
SAR study of triarylethylene derivatives.
Table 10.
SAR study of triarylethylene derivatives.
| Compounds | R | Anti-Proliferation Activity (IC50, μM) | Migration Rate at the Concentration of 1 µM (%) |
|---|
| MDA-MB-231 | MCF-7 | MDA-MB-231 |
|---|
| 33 | NH2 | 11.4 ± 4.2 | 16.9 ± 7.7 | 25 |
| Tamoxifen | - | >50 | 50 | - |
| Ospemifene | - | >50 | >50 | >70% |
Table 11.
SAR study of benzamide Ilomastat derivatives.
Table 11.
SAR study of benzamide Ilomastat derivatives.
| Compounds | R1 | R2 | Inhibitory Activity (IC50, nM) |
|---|
| MMP-2 | MMP-9 |
|---|
| Ilomastat, 34 | - | - | 0.94 | 0.55 |
| 35a | 2-NH2 | H | 0.19 | 1579.01 |
| 35b | 2-NH2 | F | 2.20 | 7.75 |
| 35c | 2-NH2 | CF3 | >104 | >104 |
| 35d | 2-NH2 | COPh | >104 | >104 |
| 35e | 2-NH2 | CH3 | >104 | 7297.04 |
| 35f | 2-NH2 | Br | 21.80 | 27.32 |
| 35g | 2-NHCOCH3 | H | >104 | 155.19 |
| 35h | 3-NH2 | H | 2.05 | 13.52 |
Table 12.
SAR study of 2,3-diaryl-4-thiazolidinone derivative 39.
Table 12.
SAR study of 2,3-diaryl-4-thiazolidinone derivative 39.
| Compounds | Anti-Proliferation Activity (IC50, μM) | Anti-Migration Activity (IC50, μM) |
|---|
| A549 | MDA-MB-231 | MDA-MB-231 |
|---|
| 36 | 0.21 | 0.23 | <0.05 |
Table 13.
SAR study of benzyloxyphenylmethylaminophenol derivatives.
Table 13.
SAR study of benzyloxyphenylmethylaminophenol derivatives.
| Compounds | R1 | R2 | Inhibitory Activity on STAT3 (IC50, μM) | Anti-Proliferation Activity (IC50, μM) |
|---|
| HepG2 | MDA-MB-468 |
|---|
| 37a | H | 4′-OH | 7.71 | 9.61 |
| 37b | 3-Cl | 4′-OH | 1.38 | 19.70 |
| 37c | 5-Cl | 4′-OH | 26.68 | 18.83 |
| 37d | 5-Cl | 4′-SO2NH2 | 35.67 | 24.34 |
Table 14.
SAR study of curcumin derivatives.
Table 14.
SAR study of curcumin derivatives.
| Compounds | R1, R2 | IC50 (μM) |
|---|
| Inhibition of Active-MMP-2 Secretion | Inhibition of Active-MMP-9 Secretion | Inhibition of uPA Secretion | Inhibition of Collagenase Activity | Inhibition of MMP-2 Activity |
|---|
| Curcumin, 38 | OCH3, OCH3 | 9.0 | >10.0 | 10.0 | 50.0 | >50.0 |
| Demethoxycurcumin, 39 | OCH3, - | 6.0 | 8.0 | 7.5 | 47.0 | 45.0 |
| Bisdemethoxycurcumin, 40 | -, - | 7.0 | >10.0 | 7.0 | >50.0 | 40.0 |
Table 15.
SAR study of sipholenol A derivatives.
Table 15.
SAR study of sipholenol A derivatives.
| Compounds | R1 | R2 | Anti-Proliferation Activity (IC50, μM) | Anti-Migration Activity (IC50, μM) |
|---|
| MCF-7 | MDA-MB-231 | MDA-MB-231 |
|---|
| 41 | ![Biomolecules 10 00138 i002]() | H | >50 | 33.4 | 11.8 |
| 42 | ![Biomolecules 10 00138 i003]() | H | 33.5 | 11.3 | 2.4 |
Table 16.
SAR study of andrographolide derivatives. Im (%) refers to the percentage of inhibition on cell migration at 10.0 μM, except for that of 5637 cells treated by compound 43 at 5 μM, the minimum effective concentration for cell migration. a No inhibitory activities against cell migration were observed at 10.0 μM.
Table 16.
SAR study of andrographolide derivatives. Im (%) refers to the percentage of inhibition on cell migration at 10.0 μM, except for that of 5637 cells treated by compound 43 at 5 μM, the minimum effective concentration for cell migration. a No inhibitory activities against cell migration were observed at 10.0 μM.
| Compounds | Anti-Migration Activity (Im, %) |
|---|
| 5637 | SGC-7901 | PC-3 |
|---|
| Andrographolide, 43 | 34.5% | 20.8% | N a |
| 44 | 44.7% | N a | N a |
Table 17.
SAR study of imidazobenzothiazole derivatives. a Log of molar concentration that inhibits 50% net cell growth, MG MID—mean graph midpoint.
Table 17.
SAR study of imidazobenzothiazole derivatives. a Log of molar concentration that inhibits 50% net cell growth, MG MID—mean graph midpoint.
| Compounds | X | R1 | R2 | R3 | R4 | log MG MID GI50 (M) a |
|---|
| 45 | S | H | H | Cl | H | −4.74 |
| 46 | S | H | H | OCH3 | H | −4.87 |
| 47 | S | H | OCH3 | OCH3 | H | −4.14 |
| 48 | S | H | H | Cl | Cl | −4.89 |
| 49 | S | H | OCH3 | OCH3 | OCH3 | −4.26 |
Table 18.
SAR study of purine derivatives.
Table 18.
SAR study of purine derivatives.
| Compounds | Anti-proliferation activity (IC50, μM) |
|---|
| MCF-7 | HCT-116 | A-375 | G-361 |
|---|
| 50 | 3.93 ± 0.04 | 6.20 ± 0.05 | 1.18 ± 0.03 | 3.06 ± 0.01 |
| 51 | 5.63 ± 0.03 | 6.36 ± 0.06 | 4.98 ± 0.07 | 5.67 ± 0.01 |
Table 19.
SAR study of 8-hydroxyquinoline derivatives. a Log of molar concentration of compound that inhibits cell growth by 50%, MG MID—mean graph midpoint.
Table 19.
SAR study of 8-hydroxyquinoline derivatives. a Log of molar concentration of compound that inhibits cell growth by 50%, MG MID—mean graph midpoint.
| Compounds | R | log MG MID GI50 (M) a |
|---|
| 52a | 4-I | 1.8 |
| 52b | 4-F | 1.6 |
| 52c | 4-Cl | 0.7 |
| 52d | 4-Br | 1.3 |
| 52e | 2,4,6-(Cl)3 | 0.7 |
Table 20.
SAR study of (Z)-1-(1,3-diphenyl-1H-pyrazol-4-yl)-3-(phenylamino)prop-2-en-1-one derivatives.
Table 20.
SAR study of (Z)-1-(1,3-diphenyl-1H-pyrazol-4-yl)-3-(phenylamino)prop-2-en-1-one derivatives.
| Compounds | R1 | R2 | Anti-proliferation activity (IC50, μM) |
|---|
| HT29 | PC3 | A549 | U87MG | HaCaT |
|---|
| 53 | H | Cl | 3.61 ± 0.56 | 5.2 ± 0.93 | 14.05 ± 0.76 | 11.4 ± 0.29 | 28.4 ± 4.1 |
| 54 | H | CF3 | >50 | >50 | >50 | >50 | >50 |
| 55 | H | OH | 5.0 ± 0.96 | 3.6 ± 0.65 | 3.21 ± 1.2 | 4.29 ± 0.89 | 44.6 ± 3.6 |
| 56 | H | OMe | 1.56 ± 0.32 | 6.4 ± 1.1 | 3.25 ± 0.19 | >50 | >50 |
| 57 | H | 3,4,5-OMe | 9.8 ± 1.3 | 5.93 ± 1.7 | 6.34 ± 0.83 | 2.35 ± 0.65 | 36.2 ± 1.8 |
| 58 | F | Cl | 4.6 ± 0.61 | 3.8 ± 0.56 | 6.7 ± 0.85 | 8.9 ± 1.3 | 21.3 ± 2.1 |
| 59 | F | CF3 | >50 | >50 | >50 | >50 | >50 |
| 60 | F | OH | 1.9 ± 0.21 | 2.6 ± 0.19 | 1.5 ± 0.45 | 4.7 ± 0.8 | 16.3 ± 1.2 |
| 61 | F | OMe | 3.2 ± 0.9 | 4.6 ± 0.7 | 8.9 ± 0.51 | 2.5 ± 0.61 | 19.6 ± 0.93 |
| 62 | F | 3,4,5-OMe | 7.77 ± 0.96 | 4.89 ± 1.35 | 9.35 ± 1.8 | 12.6 ± 2.1 | 32.8 ± 3.4 |
| 63 | Cl | Cl | 4.65 ± 0.63 | 3.89 ± 0.79 | 3.67 ± 0.3 | 13.12 ± 1.2 | 23.4 ± 3.7 |
| 64 | Cl | CF3 | >50 | >50 | >50 | >50 | >50 |
| 65 | Cl | OH | 2.5 ± 0.27 | 4.43 ± 1.3 | 1.91 ± 0.21 | 1.50 ± 0.43 | 22.6 ± 2.3 |
| 66 | Cl | OMe | 8.93 ± 1.4 | 10.65 ± 1.1 | 6.46 ± 2.7 | 6.89 ± 1.95 | 30.8 ± 2.9 |
| 67 | Cl | 3,4,5-OMe | 12.7 ± 2.6 | 9.98 ± 0.69 | 5.64 ± 0.56 | 17.8 ± 3.6 | 46.6 ± 5.2 |
| 68 | OMe | Cl | 4.76 ± 0.57 | 3.89 ± 0.33 | 2.97 ± 0.26 | 8.86 ± 0.3 | 20.9 ± 1.5 |
| 69 | OMe | CF3 | 9.87 ± 0.31 | >50 | >50 | >50 | >50 |
| 70 | OMe | OH | 2.46 ± 0.57 | 1.98 ± 0.16 | 2.77 ± 0.24 | 3.73 ± 0.66 | 34.6 ± 2.5 |
| 71 | OMe | OMe | 8.76 ± 0.98 | 13.4 ± 1.7 | 6.78 ± 3.4 | >50 | >50 |
| 72 | OMe | 3,4,5-OMe | 14.6 ± 1.7 | 18.9 ± 2.3 | 11.2 ± 1.65 | >50 | >50 |
| 73 | 3,4,5-OMe | Cl | 7.68 ± 0.92 | 11.2 ± 1.43 | 8.67 ± 0.75 | 3.21 ± 0.36 | 30.2 ± 2.8 |
| 74 | 3,4,5-OMe | CF3 | >50 | >50 | >50 | >50 | >50 |
| 75 | 3,4,5-OMe | OH | 2.5 ± 0.09 | 4.6 ± 0.78 | 3.16 ± 0.92 | 1.8 ± 0.57 | 17.6 ± 1.1 |
| 76 | 3,4,5-OMe | OMe | 5.78 ± 1.9 | 9.6 ± 1.7 | 4.78 ± 0.41 | 12.8 ± 2.3 | >50 |
| 77 | 3,4,5-OMe | 3,4,5-OMe | 8.4 ± 2.63 | 14.1 ± 1.94 | 7.98 ± 1.78 | 5.1 ± 0.93 | 20.9 ± 1.5 |
Table 21.
SAR study of naphthoquinone amide derivatives.
Table 21.
SAR study of naphthoquinone amide derivatives.
| Compounds | R | R1 | R2 | Anti-Proliferation Activity (IC50, μM) |
|---|
| HeLa | SAS |
|---|
| 78a | CH2Ph | CH3 | OCH3 | >100 | 56.5 |
| 78b | CH2·CH(CH3)2 | CH3 | OCH3 | >100 | 78.5 |
| 79a | H | OCH3 | H | 77.5 | 12.0 |
| 79b | CH3 | OCH3 | H | 39.0 | 14.0 |
| 79c | CH(CH3)2 | CH3 | H | 20.0 | 16.0 |
Table 22.
SAR study of 1’S-1’-acetoxychavicol acetate (ACA) derivatives.
Table 22.
SAR study of 1’S-1’-acetoxychavicol acetate (ACA) derivatives.
| Compounds | R1 | R2 | Anti-Proliferation Activity (IC50, μM) |
|---|
| ACA | - | - | 4.8 ± 0.4 |
| AEA | - | OCH3 | 9.5 ± 0.3 |
| AMCA | OCH3 | OCH3 | 29.6 ± 5.6 |
Table 23.
SAR study of 6,7-disubstituted-4-phenoxyquinoline derivatives.
Table 24.
SAR study of chalcone thiosemicarbazide derivatives.
Table 24.
SAR study of chalcone thiosemicarbazide derivatives.
| Compounds | R1 | R2 | R3 | R4 | R5 | Anti-Proliferation Activity (IC50, μM) |
|---|
| HepG2 |
|---|
| 84a | OMe | H | H | H | H | 20 ± 3 |
| 84b | H | H | OMe | H | H | 5.53 ± 0.3 |
| 84c | H | OMe | H | H | H | 10 ± 2 |
| 84d | H | H | H | H | Br | 6.35 ± 0.34 |
| 84e | H | H | H | H | Me | 0.78 ± 0.05 |
Table 25.
SAR study of (‒)-arctigenin derivatives.
Table 25.
SAR study of (‒)-arctigenin derivatives.
| Compounds | R1 | R2 | R3 | Preferential Cytotoxicity (PC50, μM) |
|---|
| Arctigenin, 85 | Me | Me | Me | 0.80 |
| 86a | Me | Et | Et | 0.66 |
| 86b | Et | Me | Me | 0.49 |
| 86c | Et | Et | Et | 0.78 |
Table 26.
SAR study of asiatic acid derivatives.
Table 27.
Summary of the antimigration and antiproliferation effects of different substituents.
Table 27.
Summary of the antimigration and antiproliferation effects of different substituents.
| Effective Functional Groups for Antimigration Effect |
| Functional Groups | Analogues of | Reference | Figure |
| Fluoro | Isocoumarin | [9] | Figure 1 |
| Brartemicin | [10] | Figure 2 |
| 1,3,4-oxadiazole | [12] | Figure 3 |
| Benzoylbenzophenone thiosemicarbazone | [15] | Figure 4 |
| Methoxy | 4-anilino-quinazoline | [23] | Figure 5 |
| 2-furanylvinylquinoline | [24] | Figure 6 |
| 4-methyl-2-(4-pyridinyl)thiazole-5-carboxamide | [27] | Figure 7 |
| EF24 | [28] | Figure 8 |
| Methyl | BU-4664L | [29] | Figure 9 |
| Isomalyngamide A | [31] | Figure 10 |
| Diarylamino-1,3,5-triazine | [32] | Figure 11 |
| Amino | 4-anilino-quinazoline | [23] | Figure 5 |
| Triarylethylene | [33] | Figure 12 |
| Benzamide Ilomastat | [38] | Figure 13 |
| Hydroxy | Brartemicin | [10] | Figure 2 |
| Nitro | 4-methyl-2-(4-pyridinyl)thiazole-5-carboxamide | [27] | Figure 7 |
| Bromo | 2,3-diaryl-4-thiazolidinone | [39] | Figure 14 |
| Chloro | Benzyloxyphenylmethylaminophenol | [44] | Figure 15 |
| Weak Functional Groups for Antimigration Effect |
| Functional Groups | Analogues of | Reference | Figure |
| Methoxy | Brartemicin | [10] | Figure 2 |
| Benzoylbenzophenone thiosemicarbazone | [15] | Figure 4 |
| Curcumin | [45] | Figure 16 |
| Methyl | Brartemicin | [10] | Figure 2 |
| 1,3,4-oxadiazole | [12] | Figure 3 |
| Sipholenol A | [46] | Figure 17 |
| Hydroxy | Benzoylbenzophenone thiosemicarbazone | [15] | Figure 4 |
| Andrographolide | [47] | Figure 18 |
| Bromo | Benzoylbenzophenone thiosemicarbazone | [15] | Figure 4 |
| Chloro | 4-methyl-2-(4-pyridinyl)thiazole-5-carboxamide | [27] | Figure 7 |
| Methylamino | Diarylamino-1,3,5-triazine | [32] | Figure 11 |
| Effective Functional Groups for Antiproliferation Effect |
| Functional Groups | Analogues of | Reference | Figure |
| Chloro | Imidazobenzothiazole | [48] | Figure 19 |
| Purine | [49] | Figure 20 |
| 8-hydroxyquinoline | [50] | Figure 21 |
| Methoxy | (Z)-1-(1,3-diphenyl-1H-pyrazol-4-yl)-3-(phenylamino)prop-2-en-1-one | [51] | Figure 22 |
| Naphthoquinone amide | [52] | Figure 23 |
| Fluoro | 1,3,4-oxadiazole | [12] | Figure 3 |
| 6,7-disubstituted-4-phenoxyquinoline | [54] | Figure 24 |
| Methyl | Naphthoquinone amide | [52] | Figure 2 |
| Thiosemicarbazide | [55] | Figure 25 |
| Hydroxy | (Z)-1-(1,3-diphenyl-1H-pyrazol-4-yl)-3-(phenylamino)prop-2-en-1-one | [51] | Figure 22 |
| Ethoxy | (‒)-arctigenin | [56] | Figure 26 |
| Carbonyl | Fluorinated asiatic acid | [57] | Figure 27 |
| Weak Functional Groups for Antiproliferation Effect |
| Functional Groups | Analogues of | Reference | Figure |
| Methoxy | Imidazobenzothiazole | [48] | Figure 19 |
| Thiosemicarbazide | [55] | Figure 25 |
| Bromo | 8-hydroxyquinoline | [50] | Figure 21 |
| Thiosemicarbazide | [55] | Figure 25 |
| Methyl | 1,3,4-oxadiazole | [12] | Figure 3 |
| Fluoro | 8-hydroxyquinoline | [50] | Figure 21 |
| Iodo | 8-hydroxyquinoline | [50] | Figure 21 |
| Chloro | (Z)-1-(1,3-diphenyl-1H-pyrazol-4-yl)-3-(phenylamino)prop-2-en-1-one | [51] | Figure 22 |
| Trifluoromethyl | (Z)-1-(1,3-diphenyl-1H-pyrazol-4-yl)-3-(phenylamino)prop-2-en-1-one | [51] | Figure 22 |
| Hydroxy | Fluorinated asiatic acid | [57] | Figure 27 |






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












