In Vivo Quantitative Estimation of DNA-Dependent Interaction of Sox2 and Oct4 Using BirA-Catalyzed Site-Specific Biotinylation



Abstract
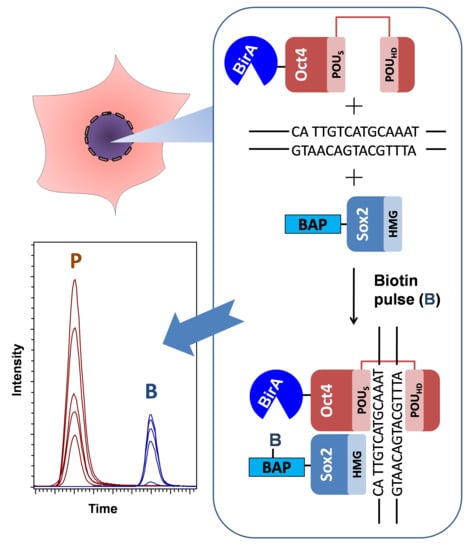
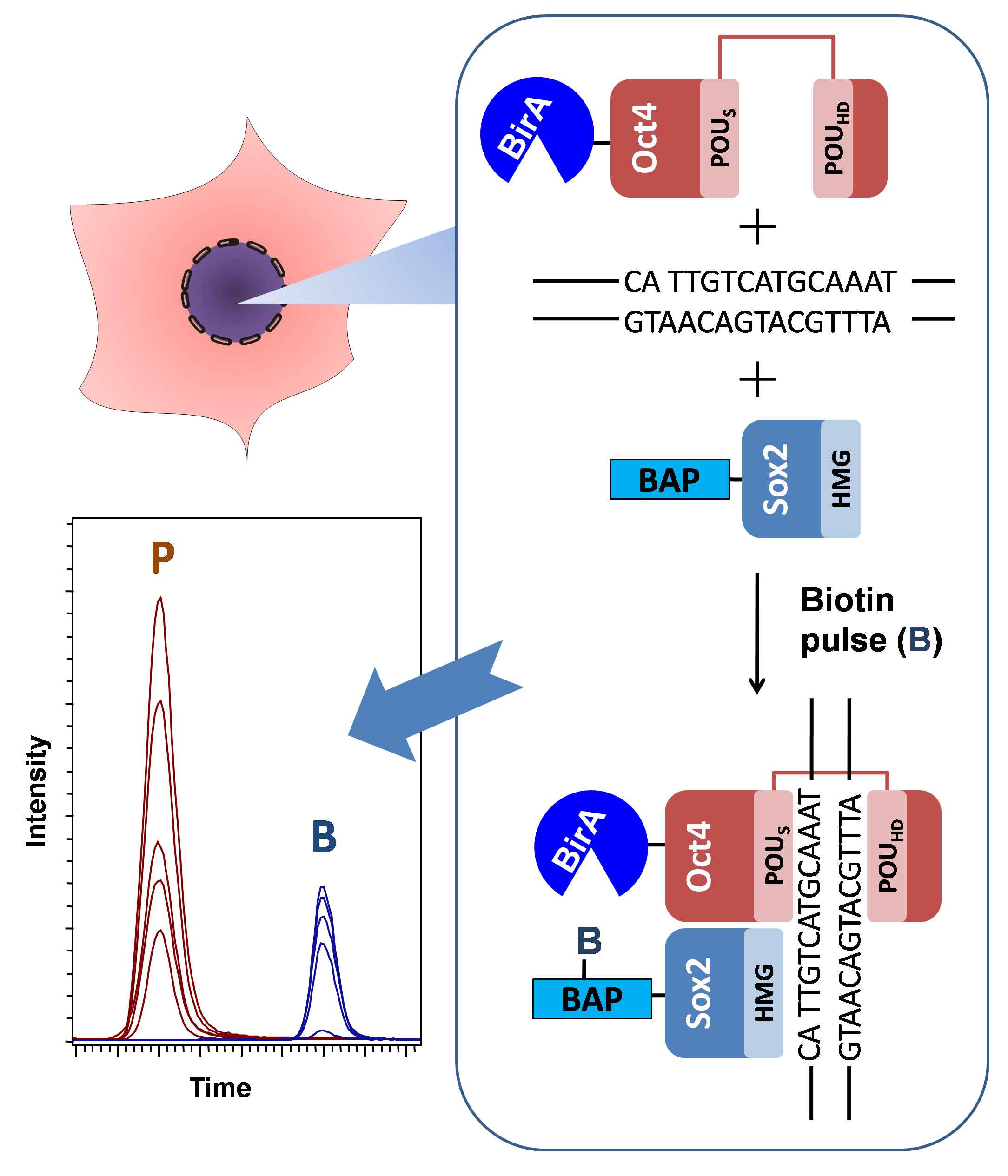
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
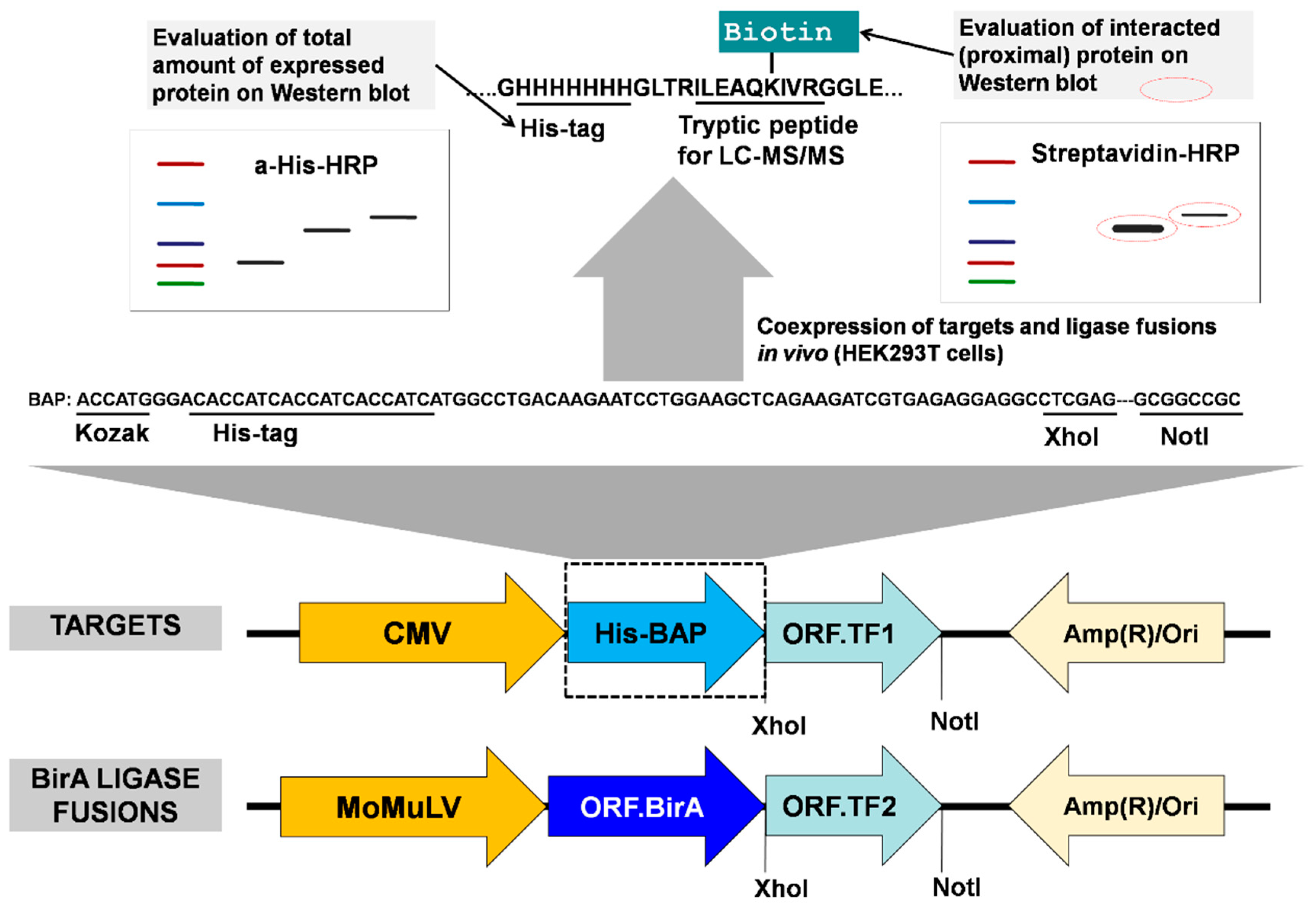
2.1. Design and Preparation of Constructs for Transient Expression
- Primer 1 Sox2_FD: CACACACACTCGAGGGCATGTACAACATGATGGAGACGGA
- Primer 2 Sox2_RS: TGTGTGTGGCGGCCGCTCACATGTGTGAGAGGGGCAGTGT
- Primer 3 Oct4_FD: CACACACAGTCGACGGCATGGCGGGACACCTGGCTTCGGA
- Primer 4 Oct4_RS: TGTGTGTGGCGGCCGCTCAGTTTGAATGCATGGGAGAGCC
2.2. Cell Culture, Transient Transfection, and Biotin Labeling In Vivo
2.2.1. Materials
2.2.2. Methods
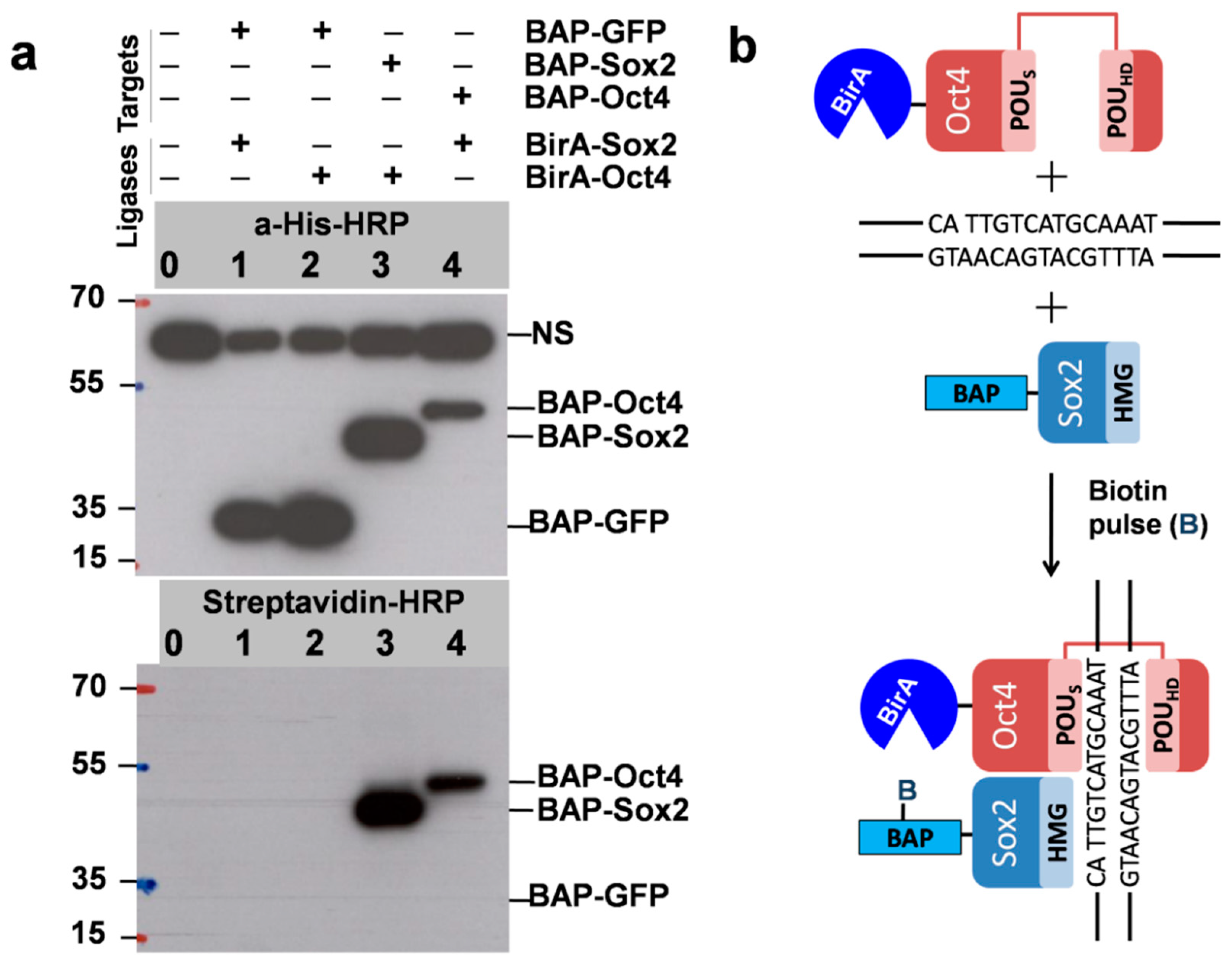
- pcDNA3-BAP-GFP, 3.0 µg; pcDNA3-BAP-Sox2, 5.0 µg; pOz-BirA-Oct4, 2.0 µg.
- 0—Control (No plasmid)
- 1—pcDNA3-BAP-GFP + pOz-BirA-Oct4 (for experiment with biotin pulse of 9 h)
- 2—pcDNA3-BAP-Sox2 + pOz-BirA-Oct4 (for experiment with biotin pulse of 9 h)
- 3—pcDNA3-BAP-GFP + pOz-BirA-Oct4 (for experiment with biotin pulse of 3 h)
- 4—pcDNA3-BAP-Sox2 + pOz-BirA-Oct4 (for experiment with biotin pulse of 3 h)
2.3. Cell Lysis and Sample Preparation
2.3.1. Materials
2.3.2. Methods
2.4. Ni-Sepharose Protein Binding, Purification, Propionylation, and On-Bead Digestion
2.4.1. Materials
2.4.2. Methods
2.5. Desalting of Tryptic Peptide Mixturesby Ziptip
2.5.1. Materials
2.5.2. Methods
2.6. LC–MS/MS Analysis
2.6.1. Materials
2.6.2. Methods
2.7. Creating the Multiple Reaction Monitoring Method
- In the otofControl 4.0 program (Bruker Daltonik GmbH, Germany), the method named “Targeted protein quantification middle-band CID-MRM.m” was selected, and then for global settings, the spectra rate was set to1.0 Hz;
- The mass range of the MS scan was set to extend from m/z 200 to 1300 in positive ion polarity mode;
- In the Source page of the system configuration pane, nanoBooster box was selected, and 1300 V for the capillary, 3.0 L/min for dry gas, and 150 °C for dry temperature were chosen;
- In the MRM subpage of the MS/MS page, the m/z values of propionylated BAP (563.2) with collision energy 27.0 eV and biotinylated BAP (648.8) with collision energy 33.0 eV were added. Mass width was set to3.00;
3. Results and Discussions
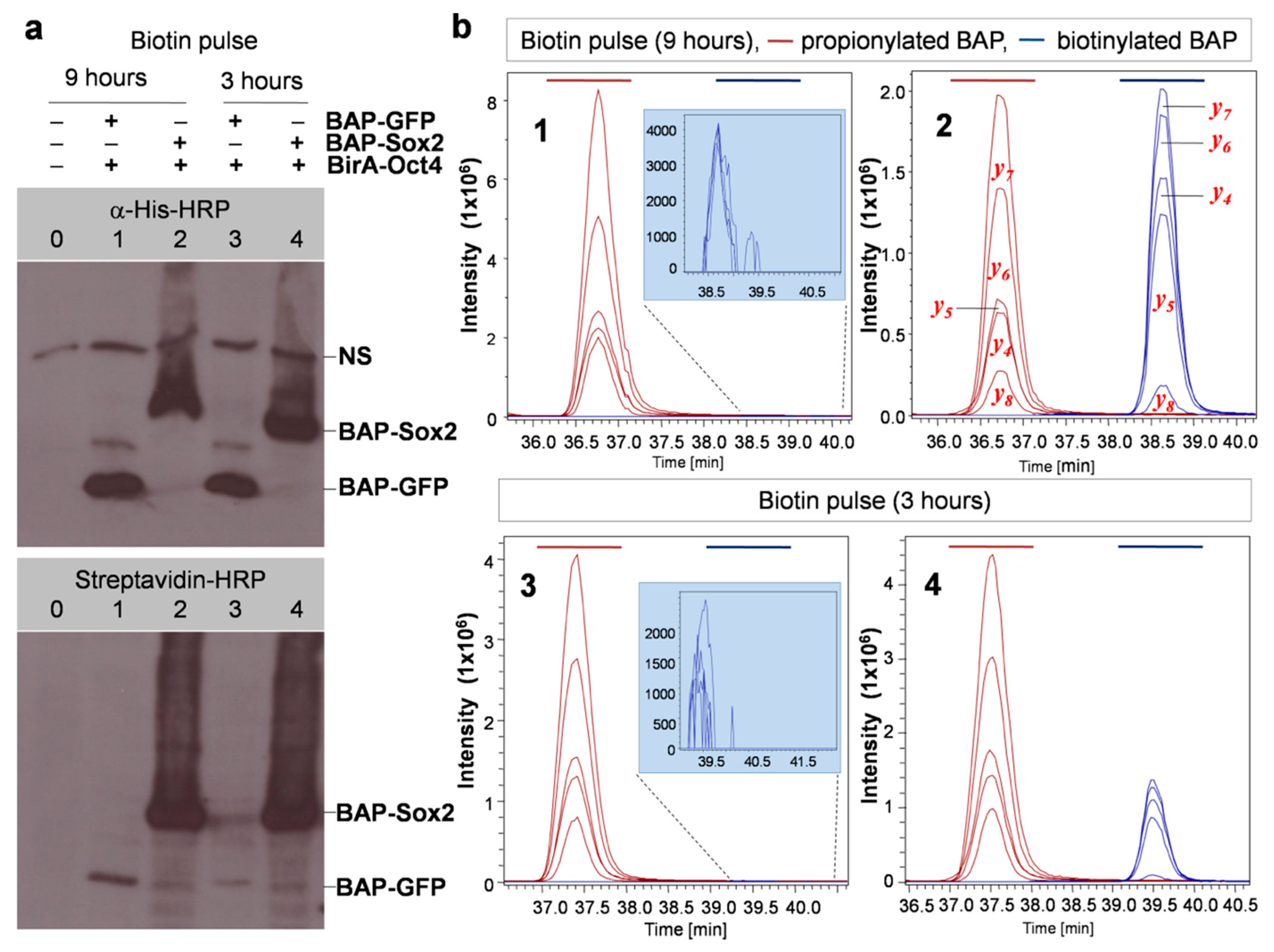
3.1. Overview of the Technique
3.2. Experimental Design
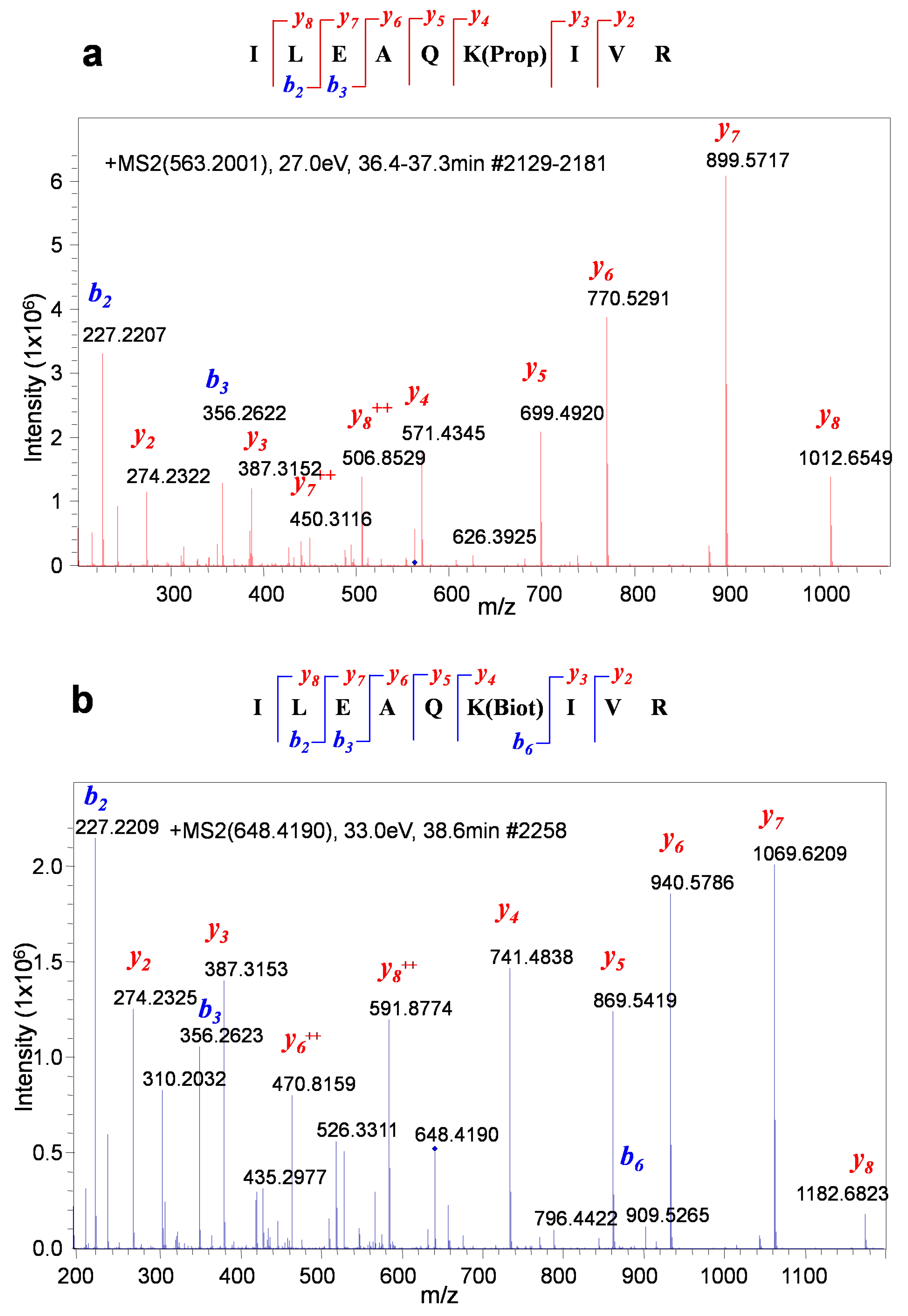
3.3. Development of the Protocol
3.4. Quantitative Evaluation of DNA-Dependant Interactions of Sox2 and Oct4
3.5. Applications, Advantages, and Limitations of the Method
- The design of BAP allows using both His-tag and streptavidin beads for the purification of the target proteins from non-specifically bound proteins, even under harsh conditions (high ionic strength of the solvent, presence of detergents, chaotropic agents).
- A wide range of commercially available reagents can be used for the detection and purification of His-tagged and biotin-labeled target proteins.
- Inside mammalian cells, the bacterial BirA enzyme does not biotinylate any endogenous protein, and conversely, the BAP is not recognized by the mammalian biotin ligase [32].
- A vector is used that generates a BAP peptide with a wide temporal dynamic range of biotinylation linearity, which results in the isolation of a large number of even weakly interacting proteins. Alternative methods like BICON use Avitag, which gives a higher background biotinylation [38].
- The generation of a permanent covalent mark on one of the proteins of interest will allow one to bypass the limitations imposed by the extraction and purification procedures. Thus, the method should prove useful for the study of interactions that are otherwise difficult to detect by the Co-IP and tandem affinity purification (TAP) methods [30,39].
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Krallinger, M.; Leitner, F.; Rodriguez-Penagos, C.; Valencia, A. Overview of the protein-protein interaction annotation extraction task of BioCreative II. Genome Biol. 2008, 9 (Suppl. 2), S4. [Google Scholar] [CrossRef] [Green Version]
- Keskin, O.; Tuncbag, N.; Gursoy, A. Predicting Protein-Protein Interactions from the Molecular to the Proteome Level. Chem. Rev. 2016, 116, 4884–4909. [Google Scholar] [CrossRef]
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of protein-protein interactions: What are the preferred ways for proteins to interact? Chem. Rev. 2008, 108, 1225–1244. [Google Scholar] [CrossRef]
- Ivanov, A.A.; Khuri, F.R.; Fu, H. Targeting protein-protein interactions as an anticancer strategy. Trends Pharmacol. Sci. 2013, 34, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.J. Inhibition of protein-protein interactions using designed molecules. Chem. Soc. Rev. 2009, 38, 3289–3300. [Google Scholar] [CrossRef] [Green Version]
- Westermarck, J.; Ivaska, J.; Corthals, G.L. Identification of protein interactions involved in cellular signaling. Mol. Cell. Proteom. 2013, 12, 1752–1763. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Sawyer, N.; Regan, L. Protein-protein interactions: General trends in the relationship between binding affinity and interfacial buried surface area. Protein Sci. 2013, 22, 510–515. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, J. Inhibiting transient protein-protein interactions: Lessons from the Cdc25 protein tyrosine phosphatases. Nat. Rev. Cancer 2007, 7, 202–211. [Google Scholar] [CrossRef]
- Karsenti, E. Self-organization in cell biology: A brief history. Nat. Rev. Mol. Cell Biol. 2008, 9, 255–262. [Google Scholar] [CrossRef]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016, 17, 183–193. [Google Scholar] [CrossRef]
- Yamanaka, S. Induced pluripotent stem cells: Past, present, and future. Cell Stem Cell 2012, 10, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, S.; Blau, H.M. Nuclear reprogramming to a pluripotent state by three approaches. Nature 2010, 465, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Chambers, I.; Tomlinson, S.R. The transcriptional foundation of pluripotency. Development 2009, 136, 2311–2322. [Google Scholar] [CrossRef] [Green Version]
- Esch, D.; Vahokoski, J.; Groves, M.R.; Pogenberg, V.; Cojocaru, V.; Vom Bruch, H.; Han, D.; Drexler, H.C.; Arauzo-Bravo, M.J.; Ng, C.K.; et al. A unique Oct4 interface is crucial for reprogramming to pluripotency. Nat. Cell Biol. 2013, 15, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Merino, F.; Ng, C.K.L.; Veerapandian, V.; Scholer, H.R.; Jauch, R.; Cojocaru, V. Structural basis for the SOX-dependent genomic redistribution of OCT4 in stem cell differentiation. Structure 2014, 22, 1274–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapia, N.; MacCarthy, C.; Esch, D.; Gabriele Marthaler, A.; Tiemann, U.; Arauzo-Bravo, M.J.; Jauch, R.; Cojocaru, V.; Scholer, H.R. Dissecting the role of distinct OCT4-SOX2 heterodimer configurations in pluripotency. Sci. Rep. 2015, 5, 13533. [Google Scholar] [CrossRef]
- White, M.D.; Angiolini, J.F.; Alvarez, Y.D.; Kaur, G.; Zhao, Z.W.; Mocskos, E.; Bruno, L.; Bissiere, S.; Levi, V.; Plachta, N. Long-Lived Binding of Sox2 to DNA Predicts Cell Fate in the Four-Cell Mouse Embryo. Cell 2016, 165, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Goolam, M.; Scialdone, A.; Graham, S.J.L.; Macaulay, I.C.; Jedrusik, A.; Hupalowska, A.; Voet, T.; Marioni, J.C.; Zernicka-Goetz, M. Heterogeneity in Oct4 and Sox2 Targets Biases Cell Fate in 4-Cell Mouse Embryos. Cell 2016, 165, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Thomson, M.; Liu, S.J.; Zou, L.N.; Smith, Z.; Meissner, A.; Ramanathan, S. Pluripotency factors in embryonic stem cells regulate differentiation into germ layers. Cell 2011, 145, 875–889. [Google Scholar] [CrossRef] [Green Version]
- Kulyyassov, A.; Shoaib, M.; Pichugin, A.; Kannouche, P.; Ramanculov, E.; Lipinski, M.; Ogryzko, V. PUB-MS: A Mass Spectrometry-based Method to Monitor Protein-Protein Proximity in vivo. J. Proteome Res. 2011, 10, 4416–4427. [Google Scholar] [CrossRef]
- Viens, A.; Mechold, U.; Lehrmann, H.; Harel-Bellan, A.; Ogryzko, V. Use of protein biotinylation in vivo for chromatin immunoprecipitation. Anal. Biochem. 2004, 325, 68–76. [Google Scholar] [CrossRef]
- Mechold, U.; Gilbert, C.; Ogryzko, V. Codon optimization of the BirA enzyme gene leads to higher expression and an improved efficiency of biotinylation of target proteins in mammalian cells. J. Biotechnol. 2005, 116, 245–249. [Google Scholar] [CrossRef]
- Betancourt, L.H.; Sanchez, A.; Pla, I.; Kuras, M.; Zhou, Q.M.; Andersson, R.; Marko-Varga, G. Quantitative Assessment of Urea in-Solution Lys-C/Trypsin Digestions Reveals Superior Performance at Room Temperature over Traditional Proteolysis at 37 degrees C. J. Proteome Res. 2018, 17, 2556–2561. [Google Scholar] [CrossRef]
- Villar-Garea, A.; Imhof, A. The analysis of histone modifications. BBA-Proteins Proteom. 2006, 1764, 1932–1939. [Google Scholar] [CrossRef]
- Robin, P.; Fritsch, L.; Philipot, O.; Svinarchuk, F.; Ait-Si-Ali, S. Post-translational modifications of histones H3 and H4 associated with the histone methyltransferases Suv39h1 and G9a. Genome Biol. 2007, 8, R270. [Google Scholar] [CrossRef] [Green Version]
- Shoaib, M.; Kulyyassov, A.; Robin, C.; Winczura, K.; Tarlykov, P.; Despas, E.; Kannouche, P.; Ramanculov, E.; Lipinski, M.; Ogryzko, V. PUB-NChIP-“in vivo biotinylation” approach to study chromatin in proximity to a protein of interest. Genome Res. 2013, 23, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Rao, S.; Chu, J.; Shen, X.; Levasseur, D.N.; Theunissen, T.W.; Orkin, S.H. A protein interaction network for pluripotency of embryonic stem cells. Nature 2006, 444, 364–368. [Google Scholar] [CrossRef]
- Slavoff, S.A.; Liu, D.S.; Cohen, J.D.; Ting, A.Y. Imaging protein-protein interactions inside living cells via interaction-dependent fluorophore ligation. J. Am. Chem. Soc. 2011, 133, 19769–19776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Suarez, M.; Chen, T.S.; Ting, A.Y. Protein-protein interaction detection in vitro and in cells by proximity biotinylation. J. Am. Chem. Soc. 2008, 130, 9251–9253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, I.; Howarth, M.; Lin, W.; Ting, A.Y. Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat. Methods 2005, 2, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Hymer, W.C.; Kuff, E.L. Isolation of nuclei from mammalian tissues through the use of triton X-100. J. Histochem. Cytochem. 1964, 12, 359–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mir, R.A.; Bele, A.; Mirza, S.; Srivastava, S.; Olou, A.A.; Ammons, S.A.; Kim, J.H.; Gurumurthy, C.B.; Qiu, F.; Band, H.; et al. A Novel Interaction of Ecdysoneless (ECD) Protein with R2TP Complex Component RUVBL1 Is Required for the Functional Role of ECD in Cell Cycle Progression. Mol. Cell. Biol. 2016, 36, 886–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, T.; Wendler, P.; Sigala, B.; Saibil, H.; Tsaneva, I.R. Dodecameric structure and ATPase activity of the human TIP48/TIP49 complex. J. Mol. Biol. 2007, 366, 179–192. [Google Scholar] [CrossRef]
- Cabantous, S.; Nguyen, H.B.; Pedelacq, J.D.; Koraichi, F.; Chaudhary, A.; Ganguly, K.; Lockard, M.A.; Favre, G.; Terwilliger, T.C.; Waldo, G.S. A New Protein-Protein Interaction Sensor Based on Tripartite Split-GFP Association. Sci. Rep. UK 2013, 3, 2854. [Google Scholar] [CrossRef]
- Lau, P.N.; Cheung, P. Elucidating combinatorial histone modifications and crosstalks by coupling histone-modifying enzyme with biotin ligase activity. Nucleic Acids Res. 2013, 41, e49. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.F. The tandem affinity purification technology: An overview. Biotechnol. Lett. 2011, 33, 1487–1499. [Google Scholar] [CrossRef]
- Weaver, L.H.; Kwon, K.; Beckett, D.; Matthews, B.W. Corepressor-induced organization and assembly of the biotin repressor: A model for allosteric activation of a transcriptional regulator. Proc. Natl. Acad. Sci. USA 2001, 98, 6045–6050. [Google Scholar] [CrossRef] [Green Version]
- Kulyyassov, A.; Ramanculov, E. Application of Impact II high resolution quadrupole Time of-flight (QTOF) instrumentation in shotgun proteomics. Eurasian J. Appl. Biotechnol. 2018, 3, 3–18. [Google Scholar]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulyyassov, A.; Ogryzko, V. In Vivo Quantitative Estimation of DNA-Dependent Interaction of Sox2 and Oct4 Using BirA-Catalyzed Site-Specific Biotinylation. Biomolecules 2020, 10, 142. https://doi.org/10.3390/biom10010142
Kulyyassov A, Ogryzko V. In Vivo Quantitative Estimation of DNA-Dependent Interaction of Sox2 and Oct4 Using BirA-Catalyzed Site-Specific Biotinylation. Biomolecules. 2020; 10(1):142. https://doi.org/10.3390/biom10010142
Chicago/Turabian StyleKulyyassov, Arman, and Vasily Ogryzko. 2020. "In Vivo Quantitative Estimation of DNA-Dependent Interaction of Sox2 and Oct4 Using BirA-Catalyzed Site-Specific Biotinylation" Biomolecules 10, no. 1: 142. https://doi.org/10.3390/biom10010142
APA StyleKulyyassov, A., & Ogryzko, V. (2020). In Vivo Quantitative Estimation of DNA-Dependent Interaction of Sox2 and Oct4 Using BirA-Catalyzed Site-Specific Biotinylation. Biomolecules, 10(1), 142. https://doi.org/10.3390/biom10010142