Hyaluronan: Metabolism and Function


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
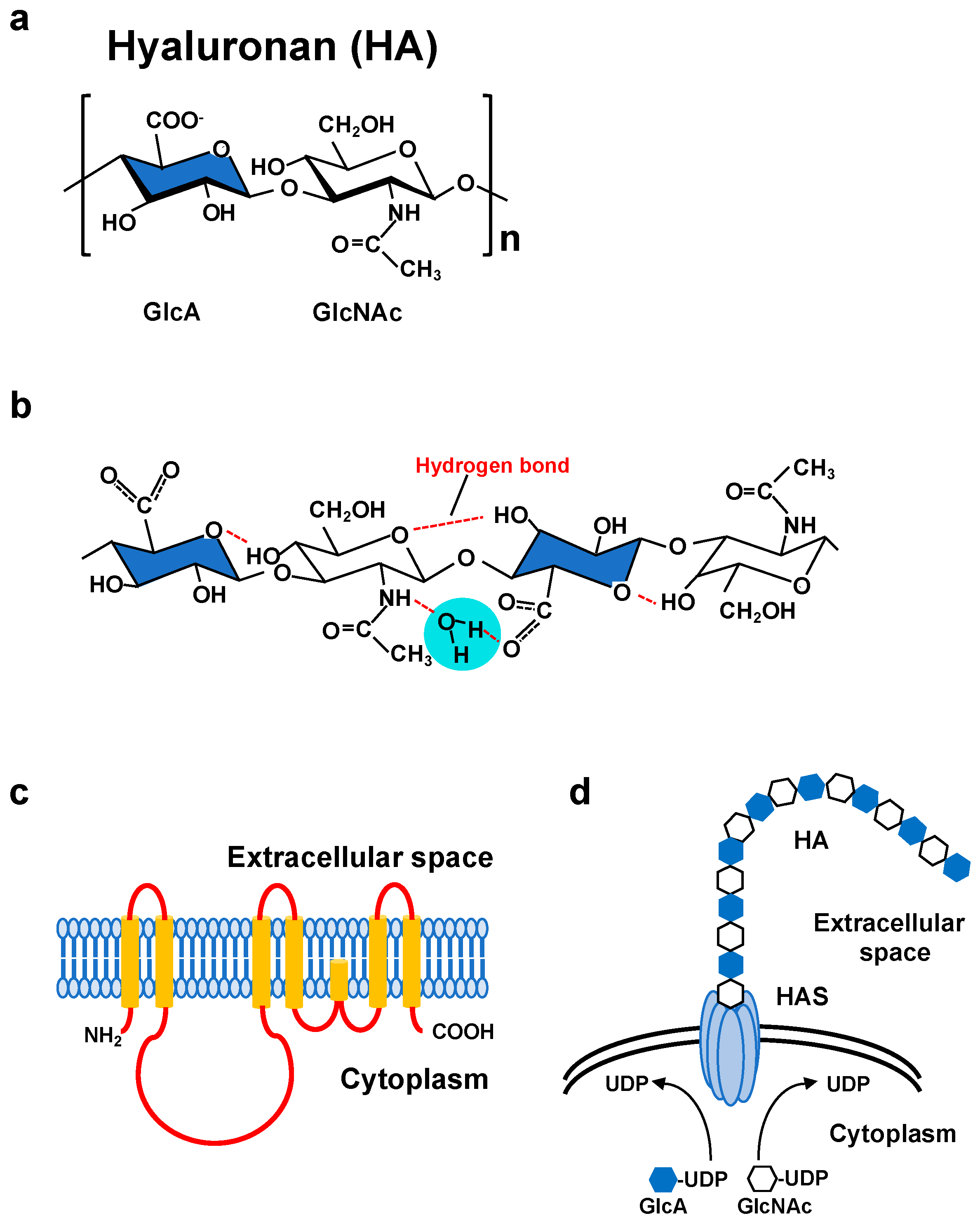
2. HA Biosynthesis
3. HA Catabolism
4. Close Associations between HA Metabolism and Functions
4.1. HA Properties
4.2. Importance of HA Metabolism in Morphogenesis and Wound Healing
4.2.1. Cardiovascular Development
4.2.2. Skeletal Development
4.2.3. Intestinal Development
4.2.4. Wound Healing
4.3. Altered HA Metabolism in Inflammatory Diseases
4.3.1. Synovial Fluid and Arthritis
4.3.2. Atherosclerosis
4.3.3. Obesity and Metabolic Disorder
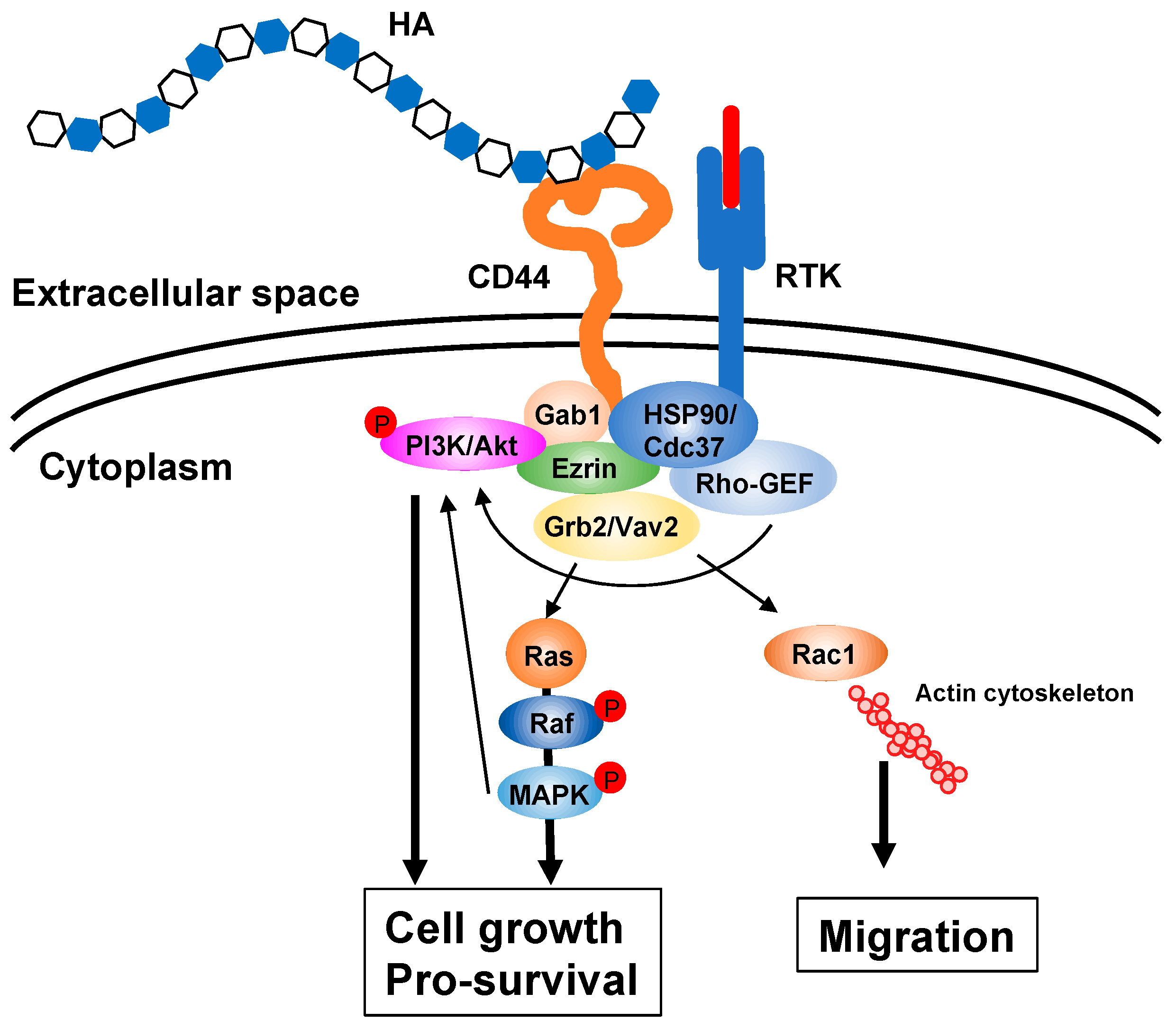
4.4. HA Metabolism and Cancer
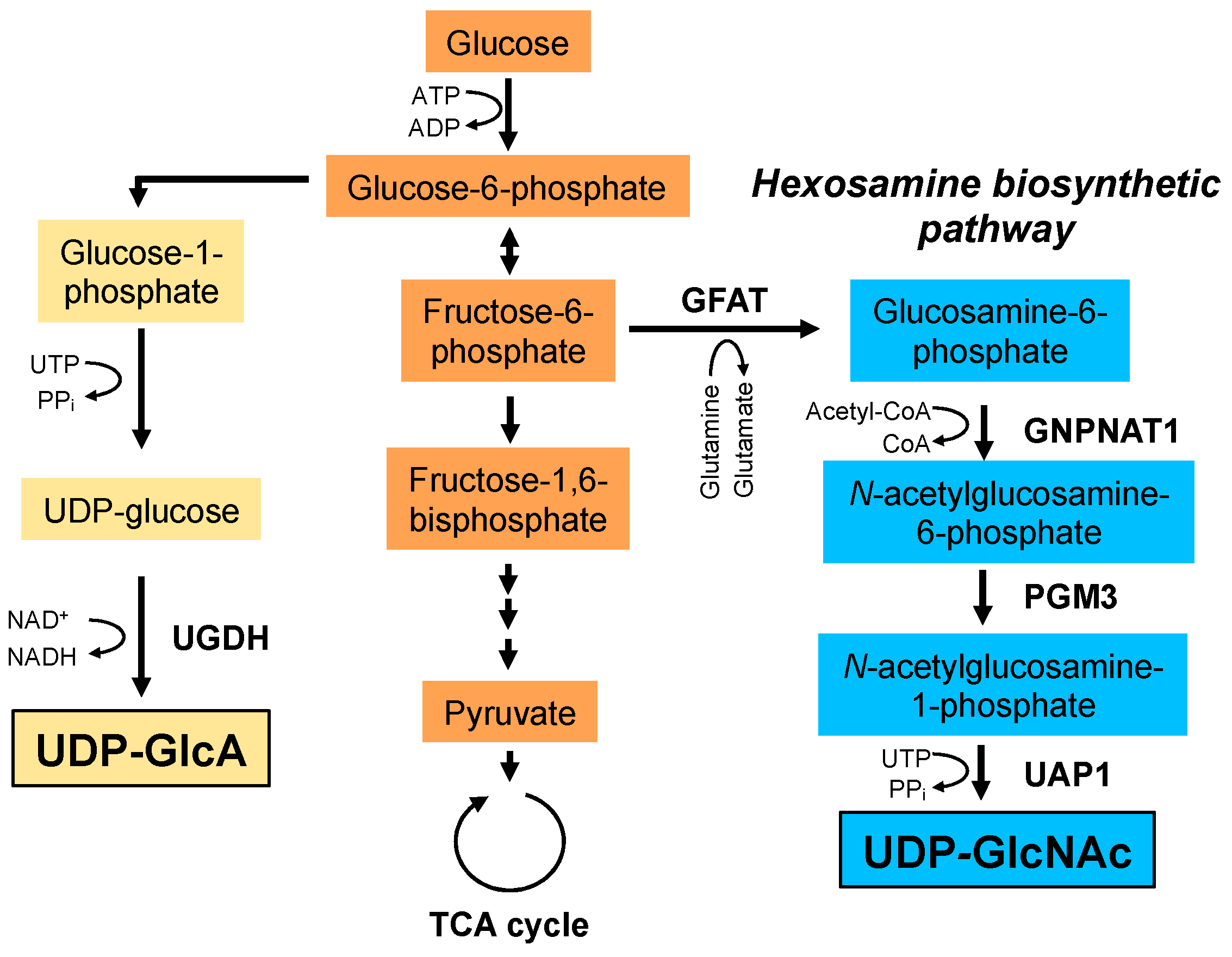
5. Metabolic Reprogramming Coupled with HA Production
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Meyer, K.; Palmer, J.W. The polysaccharide of the vitreous humor. J. Biol. Chem. 1934, 107, 629–634. [Google Scholar]
- Weissmann, B.; Meyer, K. The structure of hyalobiuronic acid and of hyaluronic acid from umbilical cord1,2. J. Am. Chem. Soc. 1954, 76, 1753–1757. [Google Scholar] [CrossRef]
- Holmes, M.W.; Bayliss, M.T.; Muir, H. Hyaluronic acid in human articular cartilage. Age-related changes in content and size. Biochem. J. 1988, 250, 435–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowman, M.K.; Lee, H.G.; Schwertfeger, K.L.; McCarthy, J.B.; Turley, E.A. The content and size of hyaluronan in biological fluids and tissues. Front. Immunol. 2015, 6, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hascall, V.; Esko, J.D. Hyaluronan. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 197–206. [Google Scholar] [CrossRef]
- Itano, N.; Sawai, T.; Yoshida, M.; Lenas, P.; Yamada, Y.; Imagawa, M.; Shinomura, T.; Hamaguchi, M.; Yoshida, Y.; Ohnuki, Y.; et al. Three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. J. Biol. Chem. 1999, 274, 25085–25092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csoka, A.B.; Frost, G.I.; Stern, R. The six hyaluronidase-like genes in the human and mouse genomes. Matrix Biol. 2001, 20, 499–508. [Google Scholar] [CrossRef]
- Yoshida, H.; Nagaoka, A.; Kusaka-Kikushima, A.; Tobiishi, M.; Kawabata, K.; Sayo, T.; Sakai, S.; Sugiyama, Y.; Enomoto, H.; Okada, Y.; et al. KIAA1199, a deafness gene of unknown function, is a new hyaluronan binding protein involved in hyaluronan depolymerization. Proc. Natl. Acad. Sci. USA 2013, 110, 5612–5617. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Tobisawa, Y.; Inubushi, T.; Irie, F.; Ohyama, C.; Yamaguchi, Y. A mammalian homolog of the zebrafish transmembrane protein 2 (TMEM2) is the long-sought-after cell-surface hyaluronidase. J. Biol. Chem. 2017, 292, 7304–7313. [Google Scholar] [CrossRef] [Green Version]
- Gribbon, P.; Heng, B.C.; Hardingham, T.E. The analysis of intermolecular interactions in concentrated hyaluronan solutions suggest no evidence for chain-chain association. Biochem. J. 2000, 350 Pt 1, 329–335. [Google Scholar]
- Toole, B.P. Hyaluronan and its binding proteins, the hyaladherins. Curr. Opin. Cell Biol. 1990, 2, 839–844. [Google Scholar] [CrossRef]
- Hascall, V.C.; Majors, A.K.; De La Motte, C.A.; Evanko, S.P.; Wang, A.; Drazba, J.A.; Strong, S.A.; Wight, T.N. Intracellular hyaluronan: A new frontier for inflammation? Biochim. Biophys. Acta 2004, 1673, 3–12. [Google Scholar] [CrossRef]
- Tighe, R.M.; Garantziotis, S. Hyaluronan interactions with innate immunity in lung biology. Matrix Biol. 2019, 78, 84–99. [Google Scholar] [CrossRef]
- Tavianatou, A.G.; Caon, I.; Franchi, M.; Piperigkou, Z.; Galesso, D.; Karamanos, N.K. Hyaluronan: Molecular size-dependent signaling and biological functions in inflammation and cancer. FEBS J. 2019, 286, 2883–2908. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Hascall, V.C.; Markwald, R.R.; Ghatak, S. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front. Immunol. 2015, 6, 201. [Google Scholar] [CrossRef] [Green Version]
- Toole, B.P. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Chanmee, T.; Ontong, P.; Itano, N. Hyaluronan: A modulator of the tumor microenvironment. Cancer Lett. 2016, 375, 20–30. [Google Scholar] [CrossRef]
- West, D.C.; Kumar, S. Hyaluronan and angiogenesis. Ciba Found. Symp. 1989, 143, 187–207. [Google Scholar] [CrossRef]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Izumikawa, T.; Higashide, M.; Mochizuki, N.; Chokchaitaweesuk, C.; Khansai, M.; Nakajima, K.; Kakizaki, I.; Kongtawelert, P.; et al. Hyaluronan production regulates metabolic and cancer stem-like properties of breast cancer cells via hexosamine biosynthetic pathway-coupled HIF-1 signaling. J. Biol. Chem. 2016, 291, 24105–24120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terabe, K.; Ohashi, Y.; Tsuchiya, S.; Ishizuka, S.; Knudson, C.B.; Knudson, W. Chondroprotective effects of 4-methylumbelliferone and hyaluronan synthase-2 overexpression involve changes in chondrocyte energy metabolism. J. Biol. Chem. 2019, 294, 17799–17817. [Google Scholar] [CrossRef]
- Itano, N.; Kimata, K. Mammalian hyaluronan synthases. IUBMB Life 2002, 54, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Weigel, P.H. Hyaluronan synthase: The mechanism of initiation at the reducing end and a pendulum model for polysaccharide translocation to the cell exterior. Int. J. Cell Biol. 2015, 2015, 367579. [Google Scholar] [CrossRef] [Green Version]
- Goentzel, B.J.; Weigel, P.H.; Steinberg, R.A. Recombinant human hyaluronan synthase 3 is phosphorylated in mammalian cells. Biochem. J. 2006, 396, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Vigetti, D.; Clerici, M.; Deleonibus, S.; Karousou, E.; Viola, M.; Moretto, P.; Heldin, P.; Hascall, V.C.; De Luca, G.; Passi, A. Hyaluronan synthesis is inhibited by adenosine monophosphate-activated protein kinase through the regulation of HAS2 activity in human aortic smooth muscle cells. J. Biol. Chem. 2011, 286, 7917–7924. [Google Scholar] [CrossRef] [Green Version]
- Vigetti, D.; Deleonibus, S.; Moretto, P.; Karousou, E.; Viola, M.; Bartolini, B.; Hascall, V.C.; Tammi, M.; De Luca, G.; Passi, A. Role of UDP-N-acetylglucosamine (GlcNAc) and O-GlcNAcylation of hyaluronan synthase 2 in the control of chondroitin sulfate and hyaluronan synthesis. J. Biol. Chem. 2012, 287, 35544–35555. [Google Scholar] [CrossRef] [Green Version]
- Karousou, E.; Kamiryo, M.; Skandalis, S.S.; Ruusala, A.; Asteriou, T.; Passi, A.; Yamashita, H.; Hellman, U.; Heldin, C.H.; Heldin, P. The activity of hyaluronan synthase 2 is regulated by dimerization and ubiquitination. J. Biol. Chem. 2010, 285, 23647–23654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigetti, D.; Deleonibus, S.; Moretto, P.; Bowen, T.; Fischer, J.W.; Grandoch, M.; Oberhuber, A.; Love, D.C.; Hanover, J.A.; Cinquetti, R.; et al. Natural antisense transcript for hyaluronan synthase 2 (HAS2-AS1) induces transcription of HAS2 via protein O-GlcNAcylation. J. Biol. Chem. 2014, 289, 28816–28826. [Google Scholar] [CrossRef] [Green Version]
- Kakizaki, I.; Kojima, K.; Takagaki, K.; Endo, M.; Kannagi, R.; Ito, M.; Maruo, Y.; Sato, H.; Yasuda, T.; Mita, S.; et al. A novel mechanism for the inhibition of hyaluronan biosynthesis by 4-methylumbelliferone. J. Biol. Chem. 2004, 279, 33281–33289. [Google Scholar] [CrossRef] [Green Version]
- Jokela, T.A.; Makkonen, K.M.; Oikari, S.; Karna, R.; Koli, E.; Hart, G.W.; Tammi, R.H.; Carlberg, C.; Tammi, M.I. Cellular content of UDP-N-acetylhexosamines controls hyaluronan synthase 2 expression and correlates with O-linked N-acetylglucosamine modification of transcription factors YY1 and SP1. J. Biol. Chem. 2011, 286, 33632–33640. [Google Scholar] [CrossRef] [Green Version]
- Grandoch, M.; Flogel, U.; Virtue, S.; Maier, J.K.; Jelenik, T.; Kohlmorgen, C.; Feldmann, K.; Ostendorf, Y.; Castaneda, T.R.; Zhou, Z.; et al. 4-Methylumbelliferone improves the thermogenic capacity of brown adipose tissue. Nat. Metab. 2019, 1, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Stern, R.; Asari, A.A.; Sugahara, K.N. Hyaluronan fragments: An information-rich system. Eur. J. Cell Biol. 2006, 85, 699–715. [Google Scholar] [CrossRef]
- Harada, H.; Takahashi, M. CD44-dependent intracellular and extracellular catabolism of hyaluronic acid by hyaluronidase-1 and -2. J. Biol. Chem. 2007, 282, 5597–5607. [Google Scholar] [CrossRef] [Green Version]
- Atmuri, V.; Martin, D.C.; Hemming, R.; Gutsol, A.; Byers, S.; Sahebjam, S.; Thliveris, J.A.; Mort, J.S.; Carmona, E.; Anderson, J.E.; et al. Hyaluronidase 3 (HYAL3) knockout mice do not display evidence of hyaluronan accumulation. Matrix Biol. 2008, 27, 653–660. [Google Scholar] [CrossRef]
- Kaneiwa, T.; Mizumoto, S.; Sugahara, K.; Yamada, S. Identification of human hyaluronidase-4 as a novel chondroitin sulfate hydrolase that preferentially cleaves the galactosaminidic linkage in the trisulfated tetrasaccharide sequence. Glycobiology 2010, 20, 300–309. [Google Scholar] [CrossRef]
- Martin-Deleon, P.A. Germ-cell hyaluronidases: Their roles in sperm function. Int. J. Androl. 2011, 34, e306–e318. [Google Scholar] [CrossRef]
- Culty, M.; Nguyen, H.A.; Underhill, C.B. The hyaluronan receptor (CD44) participates in the uptake and degradation of hyaluronan. J. Cell Biol. 1992, 116, 1055–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Q.; Knudson, C.B.; Knudson, W. Internalization of hyaluronan by chondrocytes occurs via receptor-mediated endocytosis. J. Cell Sci. 1993, 106 Pt 1, 365–375. [Google Scholar]
- Afify, A.M.; Stern, M.; Guntenhoner, M.; Stern, R. Purification and characterization of human serum hyaluronidase. Arch. Biochem. Biophys. 1993, 305, 434–441. [Google Scholar] [CrossRef]
- Rai, S.K.; Duh, F.M.; Vigdorovich, V.; Danilkovitch-Miagkova, A.; Lerman, M.I.; Miller, A.D. Candidate tumor suppressor HYAL2 is a glycosylphosphatidylinositol (GPI)-anchored cell-surface receptor for jaagsiekte sheep retrovirus, the envelope protein of which mediates oncogenic transformation. Proc. Natl. Acad. Sci. USA 2001, 98, 4443–4448. [Google Scholar] [CrossRef] [Green Version]
- Jadin, L.; Wu, X.; Ding, H.; Frost, G.I.; Onclinx, C.; Triggs-Raine, B.; Flamion, B. Skeletal and hematological anomalies in HYAL2-deficient mice: A second type of mucopolysaccharidosis IX? FASEB J. 2008, 22, 4316–4326. [Google Scholar] [CrossRef]
- Fraser, J.R.; Laurent, T.C.; Pertoft, H.; Baxter, E. Plasma clearance, tissue distribution and metabolism of hyaluronic acid injected intravenously in the rabbit. Biochem. J. 1981, 200, 415–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, J.R.; Appelgren, L.E.; Laurent, T.C. Tissue uptake of circulating hyaluronic acid. A whole body autoradiographic study. Cell Tissue Res. 1983, 233, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Oka, J.A.; Singh, A.; Weigel, P.H. Purification and subunit characterization of the rat liver endocytic hyaluronan receptor. J. Biol. Chem. 1999, 274, 33831–33834. [Google Scholar] [CrossRef] [Green Version]
- McGary, C.T.; Raja, R.H.; Weigel, P.H. Endocytosis of hyaluronic acid by rat liver endothelial cells. Evidence for receptor recycling. Biochem. J. 1989, 257, 875–884. [Google Scholar] [CrossRef] [PubMed]
- McGary, C.T.; Yannariello-Brown, J.; Kim, D.W.; Stinson, T.C.; Weigel, P.H. Degradation and intracellular accumulation of a residualizing hyaluronan derivative by liver endothelial cells. Hepatology 1993, 18, 1465–1476. [Google Scholar] [CrossRef]
- Scott, J.E.; Cummings, C.; Brass, A.; Chen, Y. Secondary and tertiary structures of hyaluronan in aqueous solution, investigated by rotary shadowing-electron microscopy and computer simulation. Hyaluronan is a very efficient network-forming polymer. Biochem. J. 1991, 274 Pt 3, 699–705. [Google Scholar] [CrossRef] [Green Version]
- Laurent, T.C.; Laurent, U.B.; Fraser, J.R. The structure and function of hyaluronan: An overview. Immunol. Cell Biol. 1996, 74, A1–A7. [Google Scholar] [CrossRef]
- Cowman, M.K.; Schmidt, T.A.; Raghavan, P.; Stecco, A. Viscoelastic properties of hyaluronan in physiological conditions. F1000Res 2015, 4, 622. [Google Scholar] [CrossRef] [Green Version]
- Comper, W.D.; Laurent, T.C. Physiological function of connective tissue polysaccharides. Physiol. Rev. 1978, 58, 255–315. [Google Scholar] [CrossRef]
- Gribbon, P.; Heng, B.C.; Hardingham, T.E. The molecular basis of the solution properties of hyaluronan investigated by confocal fluorescence recovery after photobleaching. Biophys. J. 1999, 77, 2210–2216. [Google Scholar] [CrossRef] [Green Version]
- Slevin, M.; Kumar, S.; Gaffney, J. Angiogenic oligosaccharides of hyaluronan induce multiple signaling pathways affecting vascular endothelial cell mitogenic and wound healing responses. J. Biol. Chem. 2002, 277, 41046–41059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, D.; Liang, J.; Noble, P.W. Hyaluronan as an immune regulator in human diseases. Physiol. Rev. 2011, 91, 221–264. [Google Scholar] [CrossRef] [Green Version]
- Campo, G.M.; Avenoso, A.; Campo, S.; D’Ascola, A.; Nastasi, G.; Calatroni, A. Small hyaluronan oligosaccharides induce inflammation by engaging both toll-like-4 and CD44 receptors in human chondrocytes. Biochem. Pharmacol. 2010, 80, 480–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowman, M.K.; Spagnoli, C.; Kudasheva, D.; Li, M.; Dyal, A.; Kanai, S.; Balazs, E.A. Extended, relaxed, and condensed conformations of hyaluronan observed by atomic force microscopy. Biophys. J. 2005, 88, 590–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toole, B.P. Hyaluronan in morphogenesis. Semin. Cell Dev. Biol. 2001, 12, 79–87. [Google Scholar] [CrossRef]
- Camenisch, T.D.; Spicer, A.P.; Brehm-Gibson, T.; Biesterfeldt, J.; Augustine, M.L.; Calabro, A., Jr.; Kubalak, S.; Klewer, S.E.; McDonald, J.A. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J. Clin. Investig. 2000, 106, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Camenisch, T.D.; Schroeder, J.A.; Bradley, J.; Klewer, S.E.; McDonald, J.A. Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of ErbB2-ErbB3 receptors. Nat. Med. 2002, 8, 850–855. [Google Scholar] [CrossRef]
- Chowdhury, B.; Hemming, R.; Hombach-Klonisch, S.; Flamion, B.; Triggs-Raine, B. Murine hyaluronidase 2 deficiency results in extracellular hyaluronan accumulation and severe cardiopulmonary dysfunction. J. Biol. Chem. 2013, 288, 520–528. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, B.; Xiang, B.; Muggenthaler, M.; Dolinsky, V.W.; Triggs-Raine, B. Hyaluronidase 2 deficiency is a molecular cause of cor triatriatum sinister in mice. Int. J. Cardiol. 2016, 209, 281–283. [Google Scholar] [CrossRef]
- Chowdhury, B.; Xiang, B.; Liu, M.; Hemming, R.; Dolinsky, V.W.; Triggs-Raine, B. Hyaluronidase 2 deficiency causes increased mesenchymal cells, congenital heart defects, and heart failure. Circ. Cardiovasc. Genet. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, K.; Li, Y.; Jakuba, C.; Sugiyama, Y.; Sayo, T.; Okuno, M.; Dealy, C.N.; Toole, B.P.; Takeda, J.; Yamaguchi, Y.; et al. Conditional inactivation of Has2 reveals a crucial role for hyaluronan in skeletal growth, patterning, chondrocyte maturation and joint formation in the developing limb. Development 2009, 136, 2825–2835. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Askew, E.B.; Knudson, C.B.; Knudson, W. CRISPR/Cas9 knockout of HAS2 in rat chondrosarcoma chondrocytes demonstrates the requirement of hyaluronan for aggrecan retention. Matrix Biol. 2016, 56, 74–94. [Google Scholar] [CrossRef] [Green Version]
- Shimoda, M.; Yoshida, H.; Mizuno, S.; Hirozane, T.; Horiuchi, K.; Yoshino, Y.; Hara, H.; Kanai, Y.; Inoue, S.; Ishijima, M.; et al. Hyaluronan-binding protein involved in hyaluronan depolymerization controls endochondral ossification through hyaluronan metabolism. Am. J. Pathol. 2017, 187, 1162–1176. [Google Scholar] [CrossRef] [Green Version]
- Sivakumar, A.; Mahadevan, A.; Lauer, M.E.; Narvaez, R.J.; Ramesh, S.; Demler, C.M.; Souchet, N.R.; Hascall, V.C.; Midura, R.J.; Garantziotis, S.; et al. Midgut laterality is driven by hyaluronan on the right. Dev. Cell 2018, 46, 533–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aya, K.L.; Stern, R. Hyaluronan in wound healing: Rediscovering a major player. Wound Repair Regen. 2014, 22, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Mack, J.A.; Feldman, R.J.; Itano, N.; Kimata, K.; Lauer, M.; Hascall, V.C.; Maytin, E.V. Enhanced inflammation and accelerated wound closure following tetraphorbol ester application or full-thickness wounding in mice lacking hyaluronan synthases Has1 and Has3. J. Investig. Dermatol. 2012, 132, 198–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fronza, M.; Caetano, G.F.; Leite, M.N.; Bitencourt, C.S.; Paula-Silva, F.W.; Andrade, T.A.; Frade, M.A.; Merfort, I.; Faccioli, L.H. Hyaluronidase modulates inflammatory response and accelerates the cutaneous wound healing. PLoS ONE 2014, 9, e112297. [Google Scholar] [CrossRef]
- Buhren, B.; Schrumpf, H.; Gorges, K.; Reiners, O.; Boelke, E.; Fischer, J.; Homey, B.; Gerber, A. Dose- and time-dependent effects of hyaluronidase on structural cells and the extracellular matrix of the skin. Eur. J. Med. Res. 2020. under review. [Google Scholar] [CrossRef]
- Dong, Y.; Arif, A.; Olsson, M.; Cali, V.; Hardman, B.; Dosanjh, M.; Lauer, M.; Midura, R.J.; Hascall, V.C.; Brown, K.L.; et al. Endotoxin free hyaluronan and hyaluronan fragments do not stimulate TNF-alpha, interleukin-12 or upregulate co-stimulatory molecules in dendritic cells or macrophages. Sci. Rep. 2016, 6, 36928. [Google Scholar] [CrossRef]
- Petrey, A.C.; de la Motte, C.A. Hyaluronan, a crucial regulator of inflammation. Front. Immunol. 2014, 5, 101. [Google Scholar] [CrossRef] [Green Version]
- de la Motte, C.A.; Hascall, V.C.; Drazba, J.; Bandyopadhyay, S.K.; Strong, S.A. Mononuclear leukocytes bind to specific hyaluronan structures on colon mucosal smooth muscle cells treated with polyinosinic acid:polycytidylic acid: Inter-alpha-trypsin inhibitor is crucial to structure and function. Am. J. Pathol. 2003, 163, 121–133. [Google Scholar] [CrossRef]
- Kang, I.; Harten, I.A.; Chang, M.Y.; Braun, K.R.; Sheih, A.; Nivison, M.P.; Johnson, P.Y.; Workman, G.; Kaber, G.; Evanko, S.P.; et al. Versican Deficiency Significantly Reduces Lung Inflammatory Response Induced by Polyinosine-Polycytidylic Acid Stimulation. J. Biol. Chem. 2017, 292, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Day, A.J.; de la Motte, C.A. Hyaluronan cross-linking: A protective mechanism in inflammation? Trends Immunol. 2005, 26, 637–643. [Google Scholar] [CrossRef]
- Tamer, T.M. Hyaluronan and synovial joint: Function, distribution and healing. Interdiscip. Toxicol. 2013, 6, 111–125. [Google Scholar] [CrossRef]
- Band, P.A.; Heeter, J.; Wisniewski, H.G.; Liublinska, V.; Pattanayak, C.W.; Karia, R.J.; Stabler, T.; Balazs, E.A.; Kraus, V.B. Hyaluronan molecular weight distribution is associated with the risk of knee osteoarthritis progression. Osteoarthr. Cartil. 2015, 23, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Shiozawa, J.; de Vega, S.; Cilek, M.Z.; Yoshinaga, C.; Nakamura, T.; Kasamatsu, S.; Yoshida, H.; Kaneko, H.; Ishijima, M.; Kaneko, K.; et al. Implication of HYBID (Hyaluronan-binding protein involved in hyaluronan depolymerization) in hyaluronan degradation by synovial fibroblasts in patients with knee osteoarthritis. Am. J. Pathol. 2020, 190, 1046–1058. [Google Scholar] [CrossRef]
- Yoshida, M.; Sai, S.; Marumo, K.; Tanaka, T.; Itano, N.; Kimata, K.; Fujii, K. Expression analysis of three isoforms of hyaluronan synthase and hyaluronidase in the synovium of knees in osteoarthritis and rheumatoid arthritis by quantitative real-time reverse transcriptase polymerase chain reaction. Arthritis Res. Ther. 2004, 6, R514–R520. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, Y.; Kozawa, E.; Urakawa, H.; Arai, E.; Futamura, N.; Zhuo, L.; Kimata, K.; Ishiguro, N.; Nishida, Y. Suppression of hyaluronan synthesis alleviates inflammatory responses in murine arthritis and in human rheumatoid synovial fibroblasts. Arthritis Rheum. 2013, 65, 1160–1170. [Google Scholar] [CrossRef]
- Papakonstantinou, E.; Roth, M.; Block, L.H.; Mirtsou-Fidani, V.; Argiriadis, P.; Karakiulakis, G. The differential distribution of hyaluronic acid in the layers of human atheromatic aortas is associated with vascular smooth muscle cell proliferation and migration. Atherosclerosis 1998, 138, 79–89. [Google Scholar] [CrossRef]
- Sussmann, M.; Sarbia, M.; Meyer-Kirchrath, J.; Nusing, R.M.; Schror, K.; Fischer, J.W. Induction of hyaluronic acid synthase 2 (HAS2) in human vascular smooth muscle cells by vasodilatory prostaglandins. Circ. Res. 2004, 94, 592–600. [Google Scholar] [CrossRef] [Green Version]
- Chai, S.; Chai, Q.; Danielsen, C.C.; Hjorth, P.; Nyengaard, J.R.; Ledet, T.; Yamaguchi, Y.; Rasmussen, L.M.; Wogensen, L. Overexpression of hyaluronan in the tunica media promotes the development of atherosclerosis. Circ. Res. 2005, 96, 583–591. [Google Scholar] [CrossRef] [Green Version]
- Homann, S.; Grandoch, M.; Kiene, L.S.; Podsvyadek, Y.; Feldmann, K.; Rabausch, B.; Nagy, N.; Lehr, S.; Kretschmer, I.; Oberhuber, A.; et al. Hyaluronan synthase 3 promotes plaque inflammation and atheroprogression. Matrix Biol. 2018, 66, 67–80. [Google Scholar] [CrossRef]
- Nieuwdorp, M.; Holleman, F.; de Groot, E.; Vink, H.; Gort, J.; Kontush, A.; Chapman, M.J.; Hutten, B.A.; Brouwer, C.B.; Hoekstra, J.B.; et al. Perturbation of hyaluronan metabolism predisposes patients with type 1 diabetes mellitus to atherosclerosis. Diabetologia 2007, 50, 1288–1293. [Google Scholar] [CrossRef] [Green Version]
- Auvinen, P.; Tammi, R.; Parkkinen, J.; Tammi, M.; Agren, U.; Johansson, R.; Hirvikoski, P.; Eskelinen, M.; Kosma, V.M. Hyaluronan in peritumoral stroma and malignant cells associates with breast cancer spreading and predicts survival. Am. J. Pathol. 2000, 156, 529–536. [Google Scholar] [CrossRef]
- Josefsson, A.; Adamo, H.; Hammarsten, P.; Granfors, T.; Stattin, P.; Egevad, L.; Laurent, A.E.; Wikstrom, P.; Bergh, A. Prostate cancer increases hyaluronan in surrounding nonmalignant stroma, and this response is associated with tumor growth and an unfavorable outcome. Am. J. Pathol. 2011, 179, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Lipponen, P.; Aaltomaa, S.; Tammi, R.; Tammi, M.; Agren, U.; Kosma, V.M. High stromal hyaluronan level is associated with poor differentiation and metastasis in prostate cancer. Eur. J. Cancer 2001, 37, 849–856. [Google Scholar] [CrossRef]
- Anttila, M.A.; Tammi, R.H.; Tammi, M.I.; Syrjanen, K.J.; Saarikoski, S.V.; Kosma, V.M. High levels of stromal hyaluronan predict poor disease outcome in epithelial ovarian cancer. Cancer Res. 2000, 60, 150–155. [Google Scholar] [PubMed]
- Pirinen, R.; Tammi, R.; Tammi, M.; Hirvikoski, P.; Parkkinen, J.J.; Johansson, R.; Bohm, J.; Hollmen, S.; Kosma, V.M. Prognostic value of hyaluronan expression in non-small-cell lung cancer: Increased stromal expression indicates unfavorable outcome in patients with adenocarcinoma. Int. J. Cancer 2001, 95, 12–17. [Google Scholar] [CrossRef]
- Auvinen, P.; Rilla, K.; Tumelius, R.; Tammi, M.; Sironen, R.; Soini, Y.; Kosma, V.M.; Mannermaa, A.; Viikari, J.; Tammi, R. Hyaluronan synthases (HAS1-3) in stromal and malignant cells correlate with breast cancer grade and predict patient survival. Breast Cancer Res. Treat. 2014, 143, 277–286. [Google Scholar] [CrossRef]
- Itano, N.; Sawai, T.; Miyaishi, O.; Kimata, K. Relationship between hyaluronan production and metastatic potential of mouse mammary carcinoma cells. Cancer Res. 1999, 59, 2499–2504. [Google Scholar] [PubMed]
- Liu, N.; Gao, F.; Han, Z.; Xu, X.; Underhill, C.B.; Zhang, L. Hyaluronan synthase 3 overexpression promotes the growth of TSU prostate cancer cells. Cancer Res. 2001, 61, 5207–5214. [Google Scholar]
- Kosaki, R.; Watanabe, K.; Yamaguchi, Y. Overproduction of hyaluronan by expression of the hyaluronan synthase Has2 enhances anchorage-independent growth and tumorigenicity. Cancer Res. 1999, 59, 1141–1145. [Google Scholar] [PubMed]
- Bernert, B.; Porsch, H.; Heldin, P. Hyaluronan synthase 2 (HAS2) promotes breast cancer cell invasion by suppression of tissue metalloproteinase inhibitor 1 (TIMP-1). J. Biol. Chem. 2011, 286, 42349–42359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosunen, A.; Ropponen, K.; Kellokoski, J.; Pukkila, M.; Virtaniemi, J.; Valtonen, H.; Kumpulainen, E.; Johansson, R.; Tammi, R.; Tammi, M.; et al. Reduced expression of hyaluronan is a strong indicator of poor survival in oral squamous cell carcinoma. Oral Oncol. 2004, 40, 257–263. [Google Scholar] [CrossRef]
- Bharadwaj, A.G.; Rector, K.; Simpson, M.A. Inducible hyaluronan production reveals differential effects on prostate tumor cell growth and tumor angiogenesis. J. Biol. Chem. 2007, 282, 20561–20572. [Google Scholar] [CrossRef] [Green Version]
- Schmaus, A.; Klusmeier, S.; Rothley, M.; Dimmler, A.; Sipos, B.; Faller, G.; Thiele, W.; Allgayer, H.; Hohenberger, P.; Post, S.; et al. Accumulation of small hyaluronan oligosaccharides in tumour interstitial fluid correlates with lymphatic invasion and lymph node metastasis. Br. J. Cancer 2014, 111, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, K.N.; Murai, T.; Nishinakamura, H.; Kawashima, H.; Saya, H.; Miyasaka, M. Hyaluronan oligosaccharides induce CD44 cleavage and promote cell migration in CD44-expressing tumor cells. J. Biol. Chem. 2003, 278, 32259–32265. [Google Scholar] [CrossRef] [Green Version]
- Delpech, B.; Laquerriere, A.; Maingonnat, C.; Bertrand, P.; Freger, P. Hyaluronidase is more elevated in human brain metastases than in primary brain tumours. Anticancer Res. 2002, 22, 2423–2427. [Google Scholar]
- Victor, R.; Chauzy, C.; Girard, N.; Gioanni, J.; d’Anjou, J.; Stora De Novion, H.; Delpech, B. Human breast-cancer metastasis formation in a nude-mouse model: Studies of hyaluronidase, hyaluronan and hyaluronan-binding sites in metastatic cells. Int. J. Cancer 1999, 82, 77–83. [Google Scholar] [CrossRef]
- Siiskonen, H.; Poukka, M.; Tyynela-Korhonen, K.; Sironen, R.; Pasonen-Seppanen, S. Inverse expression of hyaluronidase 2 and hyaluronan synthases 1-3 is associated with reduced hyaluronan content in malignant cutaneous melanoma. BMC Cancer 2013, 13, 181. [Google Scholar] [CrossRef] [Green Version]
- Lokeshwar, V.B.; Young, M.J.; Goudarzi, G.; Iida, N.; Yudin, A.I.; Cherr, G.N.; Selzer, M.G. Identification of bladder tumor-derived hyaluronidase: Its similarity to HYAL1. Cancer Res. 1999, 59, 4464–4470. [Google Scholar]
- Morera, D.S.; Hennig, M.S.; Talukder, A.; Lokeshwar, S.D.; Wang, J.; Garcia-Roig, M.; Ortiz, N.; Yates, T.J.; Lopez, L.E.; Kallifatidis, G.; et al. Hyaluronic acid family in bladder cancer: Potential prognostic biomarkers and therapeutic targets. Br. J. Cancer 2017, 117, 1507–1517. [Google Scholar] [CrossRef]
- Posey, J.T.; Soloway, M.S.; Ekici, S.; Sofer, M.; Civantos, F.; Duncan, R.C.; Lokeshwar, V.B. Evaluation of the prognostic potential of hyaluronic acid and hyaluronidase (HYAL1) for prostate cancer. Cancer Res. 2003, 63, 2638–2644. [Google Scholar]
- Kuscu, C.; Evensen, N.; Kim, D.; Hu, Y.J.; Zucker, S.; Cao, J. Transcriptional and epigenetic regulation of KIAA1199 gene expression in human breast cancer. PLoS ONE 2012, 7, e44661. [Google Scholar] [CrossRef]
- Koga, A.; Sato, N.; Kohi, S.; Yabuki, K.; Cheng, X.B.; Hisaoka, M.; Hirata, K. KIAA1199/CEMIP/HYBID overexpression predicts poor prognosis in pancreatic ductal adenocarcinoma. Pancreatology 2017, 17, 115–122. [Google Scholar] [CrossRef]
- Deng, F.; Lei, J.; Zhang, X.; Huang, W.; Li, Y.; Wu, D. Overexpression of KIAA1199: An independent prognostic marker in nonsmall cell lung cancer. J. Cancer Res. Ther. 2017, 13, 664–668. [Google Scholar] [CrossRef]
- Fink, S.P.; Myeroff, L.L.; Kariv, R.; Platzer, P.; Xin, B.; Mikkola, D.; Lawrence, E.; Morris, N.; Nosrati, A.; Willson, J.K.; et al. Induction of KIAA1199/CEMIP is associated with colon cancer phenotype and poor patient survival. Oncotarget 2015, 6, 30500–30515. [Google Scholar] [CrossRef] [Green Version]
- Jia, S.; Qu, T.; Wang, X.; Feng, M.; Yang, Y.; Feng, X.; Ma, R.; Li, W.; Hu, Y.; Feng, Y.; et al. KIAA1199 promotes migration and invasion by Wnt/beta-catenin pathway and MMPs mediated EMT progression and serves as a poor prognosis marker in gastric cancer. PLoS ONE 2017, 12, e0175058. [Google Scholar] [CrossRef]
- Evensen, N.A.; Kuscu, C.; Nguyen, H.L.; Zarrabi, K.; Dufour, A.; Kadam, P.; Hu, Y.J.; Pulkoski-Gross, A.; Bahou, W.F.; Zucker, S.; et al. Unraveling the role of KIAA1199, a novel endoplasmic reticulum protein, in cancer cell migration. J. Natl. Cancer Inst. 2013, 105, 1402–1416. [Google Scholar] [CrossRef] [Green Version]
- Jami, M.S.; Hou, J.; Liu, M.; Varney, M.L.; Hassan, H.; Dong, J.; Geng, L.; Wang, J.; Yu, F.; Huang, X.; et al. Functional proteomic analysis reveals the involvement of KIAA1199 in breast cancer growth, motility and invasiveness. BMC Cancer 2014, 14, 194. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Goodarzi, H.; Tavazoie, S.F.; Alarcon, C.R. TMEM2 Is a SOX4-regulated gene that mediates metastatic migration and invasion in breast cancer. Cancer Res. 2016, 76, 4994–5005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nykopp, T.K.; Rilla, K.; Sironen, R.; Tammi, M.I.; Tammi, R.H.; Hamalainen, K.; Heikkinen, A.M.; Komulainen, M.; Kosma, V.M.; Anttila, M. Expression of hyaluronan synthases (HAS1-3) and hyaluronidases (HYAL1-2) in serous ovarian carcinomas: Inverse correlation between HYAL1 and hyaluronan content. BMC Cancer 2009, 9, 143. [Google Scholar] [CrossRef] [Green Version]
- Nykopp, T.K.; Pasonen-Seppanen, S.; Tammi, M.I.; Tammi, R.H.; Kosma, V.M.; Anttila, M.; Sironen, R. Decreased hyaluronidase 1 expression is associated with early disease recurrence in human endometrial cancer. Gynecol. Oncol. 2015, 137, 152–159. [Google Scholar] [CrossRef]
- Cheng, X.B.; Sato, N.; Kohi, S.; Yamaguchi, K. Prognostic impact of hyaluronan and its regulators in pancreatic ductal adenocarcinoma. PLoS ONE 2013, 8, e80765. [Google Scholar] [CrossRef] [Green Version]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, R.; Su, X.; Pan, Y.; Han, X.; Shao, C.; Shi, Y. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol. Cancer 2019, 18, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Guo, L.; Yang, C.; Liu, Y.; He, Y.; Du, Y.; Wang, W.; Gao, F. A novel role of breast cancer-derived hyaluronan on inducement of M2-like tumor-associated macrophages formation. Oncoimmunology 2016, 5, e1172154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, D.M.; Wu, Y.; Chen, N.; Cheng, J.; Zhuang, S.M.; Zheng, L. Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood 2007, 110, 587–595. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Powell, J.D. Metabolism of immune cells in cancer. Nat. Rev. Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Shiratori, R.; Furuichi, K.; Yamaguchi, M.; Miyazaki, N.; Aoki, H.; Chibana, H.; Ito, K.; Aoki, S. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci. Rep. 2019, 9, 18699. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci Adv 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Boedtkjer, E.; Pedersen, S.F. The acidic tumor microenvironment as a driver of cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Shepard, H.M.; Cowell, J.A.; Zhao, C.; Osgood, R.J.; Rosengren, S.; Blouw, B.; Garrovillo, S.A.; Pagel, M.D.; Whatcott, C.J.; et al. Parallel accumulation of tumor hyaluronan, collagen, and other drivers of tumor progression. Clin. Cancer Res. 2018, 24, 4798–4807. [Google Scholar] [CrossRef] [Green Version]
- Payen, V.L.; Mina, E.; Van Hee, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol Metab 2020, 33, 48–66. [Google Scholar] [CrossRef]
- Slomiany, M.G.; Grass, G.D.; Robertson, A.D.; Yang, X.Y.; Maria, B.L.; Beeson, C.; Toole, B.P. Hyaluronan, CD44, and emmprin regulate lactate efflux and membrane localization of monocarboxylate transporters in human breast carcinoma cells. Cancer Res. 2009, 69, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.; Chen, H.; Madigan, M.C.; Cozzi, P.J.; Beretov, J.; Xiao, W.; Delprado, W.J.; Russell, P.J.; Li, Y. Co-expression of CD147 (EMMPRIN), CD44v3-10, MDR1 and monocarboxylate transporters is associated with prostate cancer drug resistance and progression. Br. J. Cancer 2010, 103, 1008–1018. [Google Scholar] [CrossRef] [Green Version]
- Itkonen, H.M.; Engedal, N.; Babaie, E.; Luhr, M.; Guldvik, I.J.; Minner, S.; Hohloch, J.; Tsourlakis, M.C.; Schlomm, T.; Mills, I.G. UAP1 is overexpressed in prostate cancer and is protective against inhibitors of N-linked glycosylation. Oncogene 2015, 34, 3744–3750. [Google Scholar] [CrossRef]
- Oikari, S.; Kettunen, T.; Tiainen, S.; Hayrinen, J.; Masarwah, A.; Sudah, M.; Sutela, A.; Vanninen, R.; Tammi, M.; Auvinen, P. UDP-sugar accumulation drives hyaluronan synthesis in breast cancer. Matrix Biol. 2018, 67, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Chokchaitaweesuk, C.; Kobayashi, T.; Izumikawa, T.; Itano, N. Enhanced hexosamine metabolism drives metabolic and signaling networks involving hyaluronan production and O-GlcNAcylation to exacerbate breast cancer. Cell Death Dis. 2019, 10, 803. [Google Scholar] [CrossRef]
- Ngo, H.; Tortorella, S.M.; Ververis, K.; Karagiannis, T.C. The Warburg effect: Molecular aspects and therapeutic possibilities. Mol. Biol. Rep. 2015, 42, 825–834. [Google Scholar] [CrossRef]
- Ferrer, C.M.; Lynch, T.P.; Sodi, V.L.; Falcone, J.N.; Schwab, L.P.; Peacock, D.L.; Vocadlo, D.J.; Seagroves, T.N.; Reginato, M.J. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol. Cell 2014, 54, 820–831. [Google Scholar] [CrossRef] [Green Version]
- Ciavardelli, D.; Rossi, C.; Barcaroli, D.; Volpe, S.; Consalvo, A.; Zucchelli, M.; De Cola, A.; Scavo, E.; Carollo, R.; D’Agostino, D.; et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. 2014, 5, e1336. [Google Scholar] [CrossRef] [Green Version]
- Peng, F.; Wang, J.H.; Fan, W.J.; Meng, Y.T.; Li, M.M.; Li, T.T.; Cui, B.; Wang, H.F.; Zhao, Y.; An, F.; et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2018, 37, 1062–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshmukh, A.; Deshpande, K.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Cancer stem cell metabolism: A potential target for cancer therapy. Mol. Cancer 2016, 15, 69. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobayashi, T.; Chanmee, T.; Itano, N. Hyaluronan: Metabolism and Function. Biomolecules 2020, 10, 1525. https://doi.org/10.3390/biom10111525
Kobayashi T, Chanmee T, Itano N. Hyaluronan: Metabolism and Function. Biomolecules. 2020; 10(11):1525. https://doi.org/10.3390/biom10111525
Chicago/Turabian StyleKobayashi, Takashi, Theerawut Chanmee, and Naoki Itano. 2020. "Hyaluronan: Metabolism and Function" Biomolecules 10, no. 11: 1525. https://doi.org/10.3390/biom10111525
APA StyleKobayashi, T., Chanmee, T., & Itano, N. (2020). Hyaluronan: Metabolism and Function. Biomolecules, 10(11), 1525. https://doi.org/10.3390/biom10111525