Recent Development of Aminoacyl-tRNA Synthetase Inhibitors for Human Diseases: A Future Perspective



Abstract
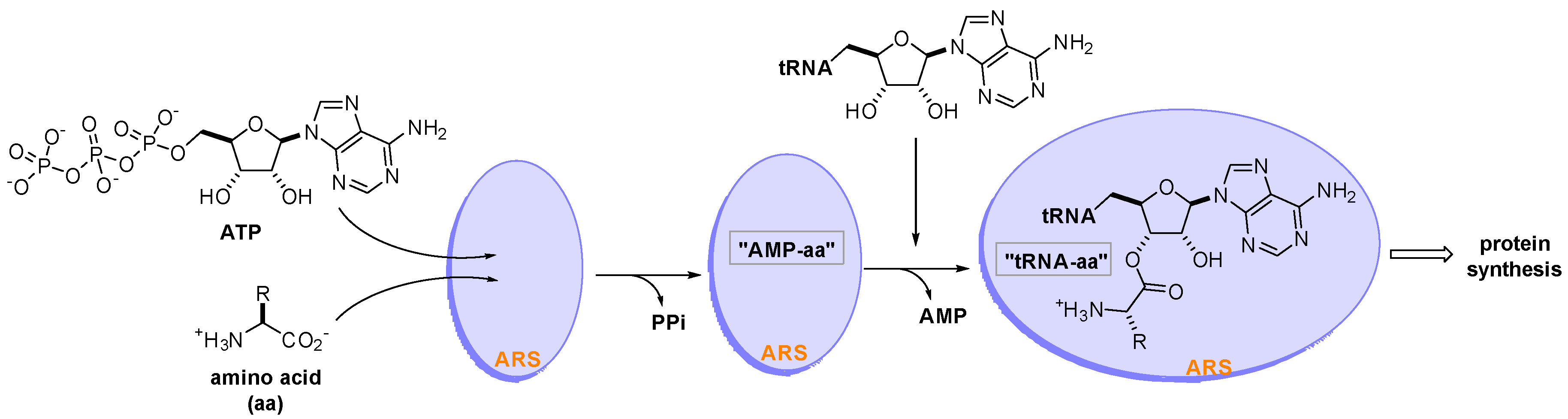
:1. Introduction
2. Structural Class of ARS Inhibitors
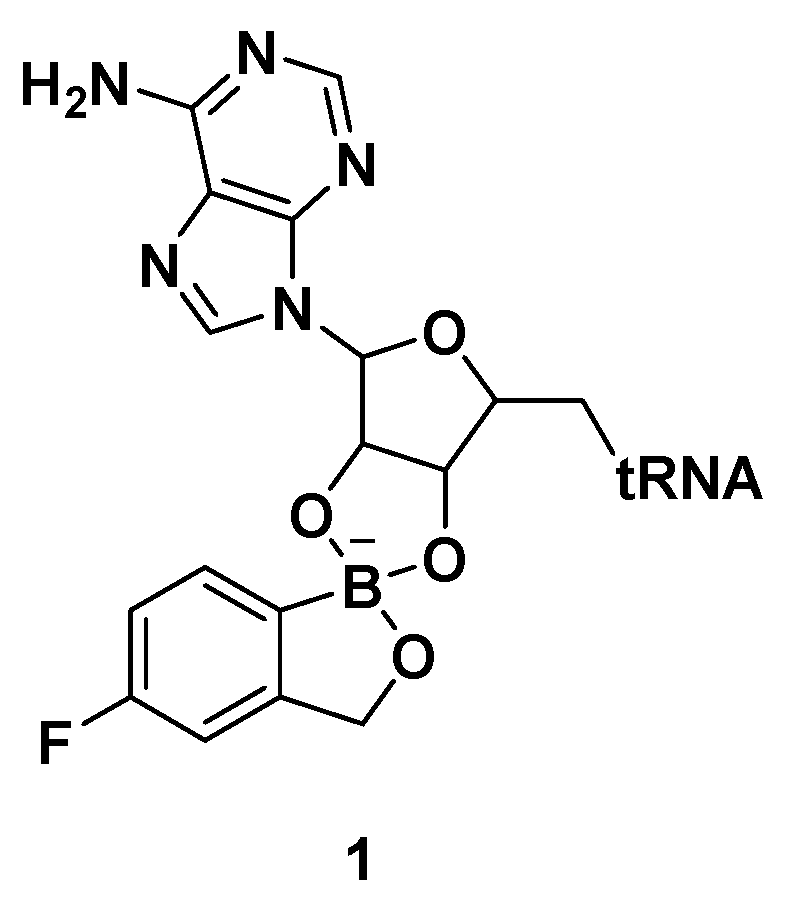
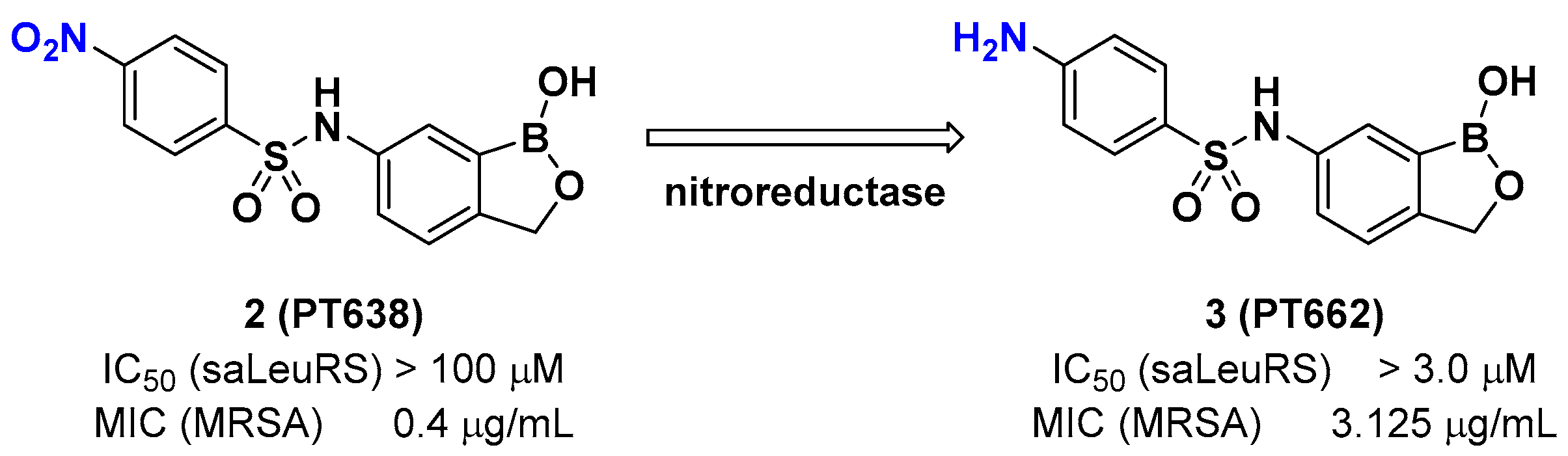
2.1. Benzoxaboroles
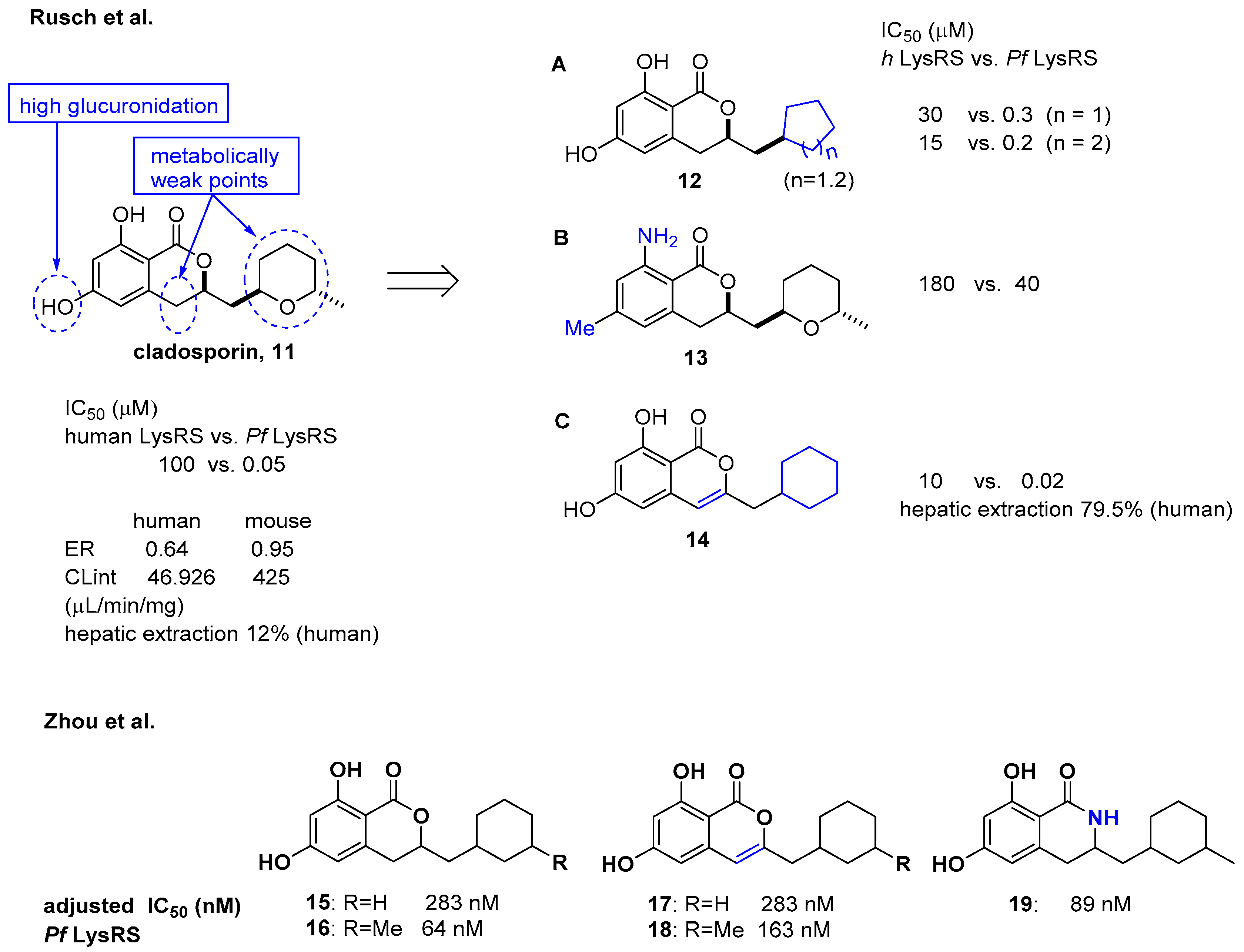
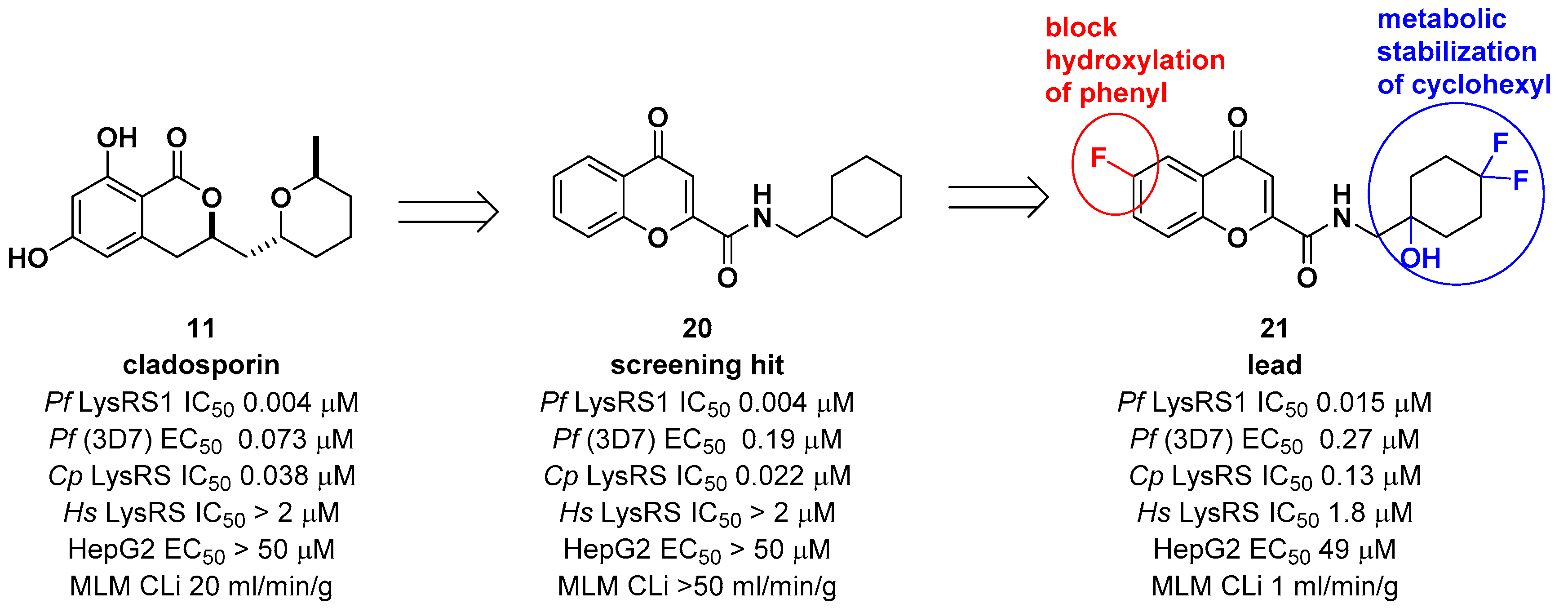
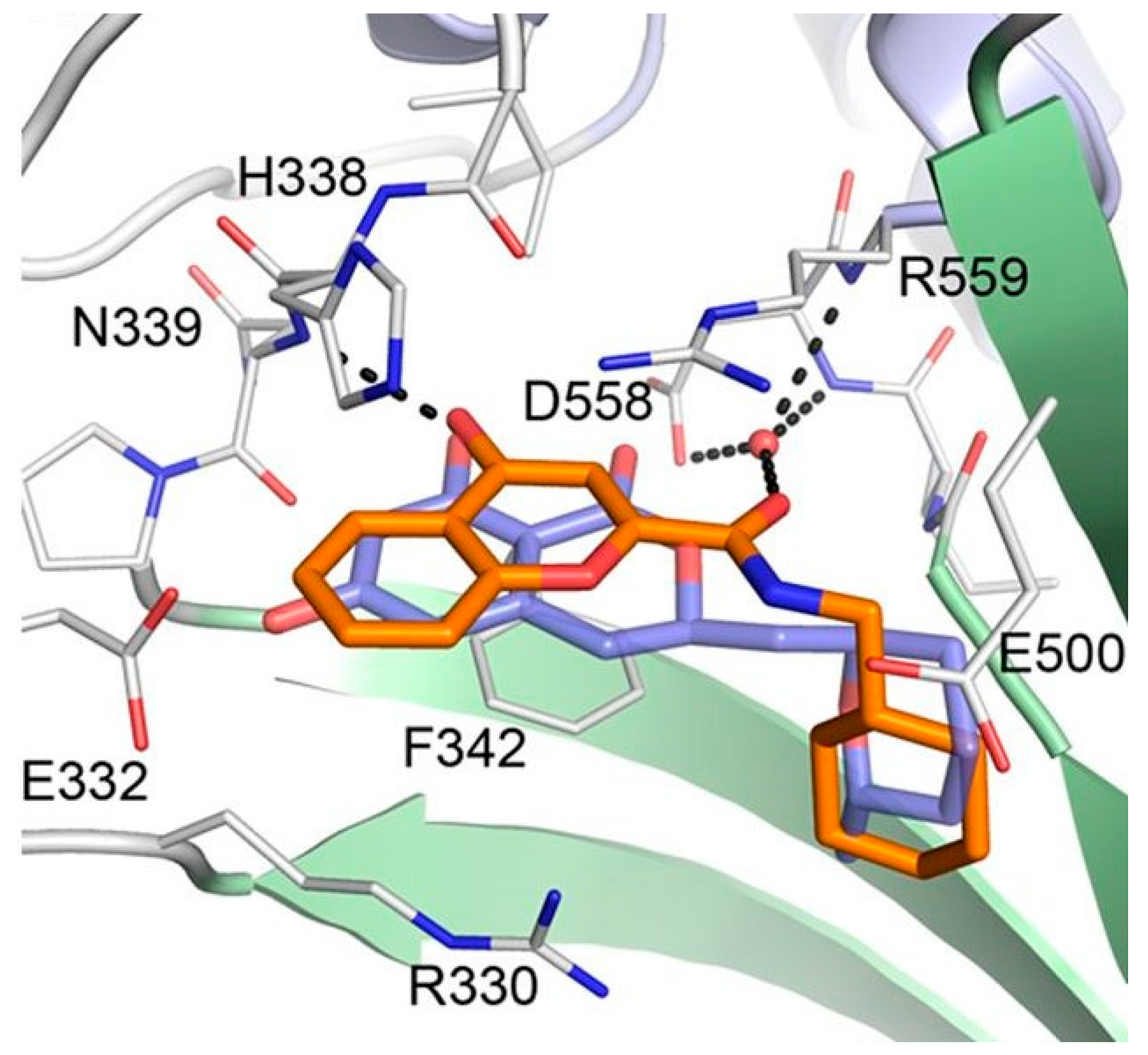
2.2. Cladosporin
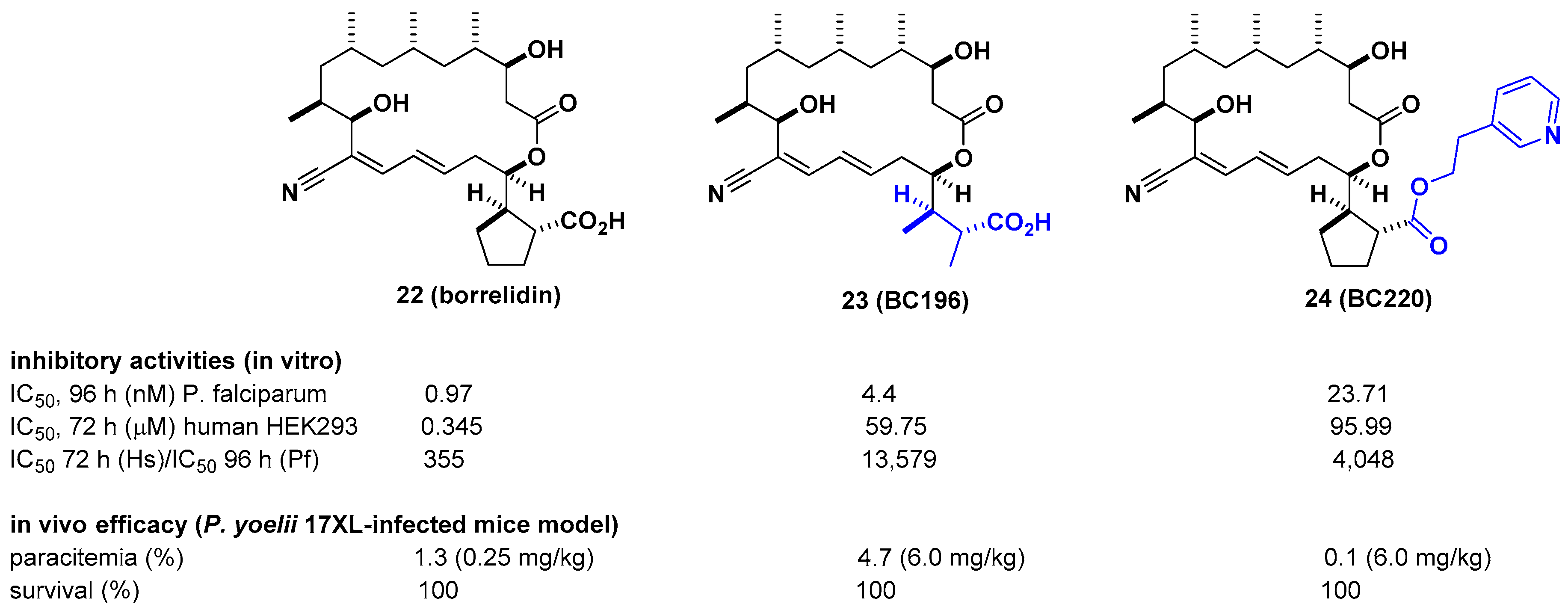
2.3. Borrelidin
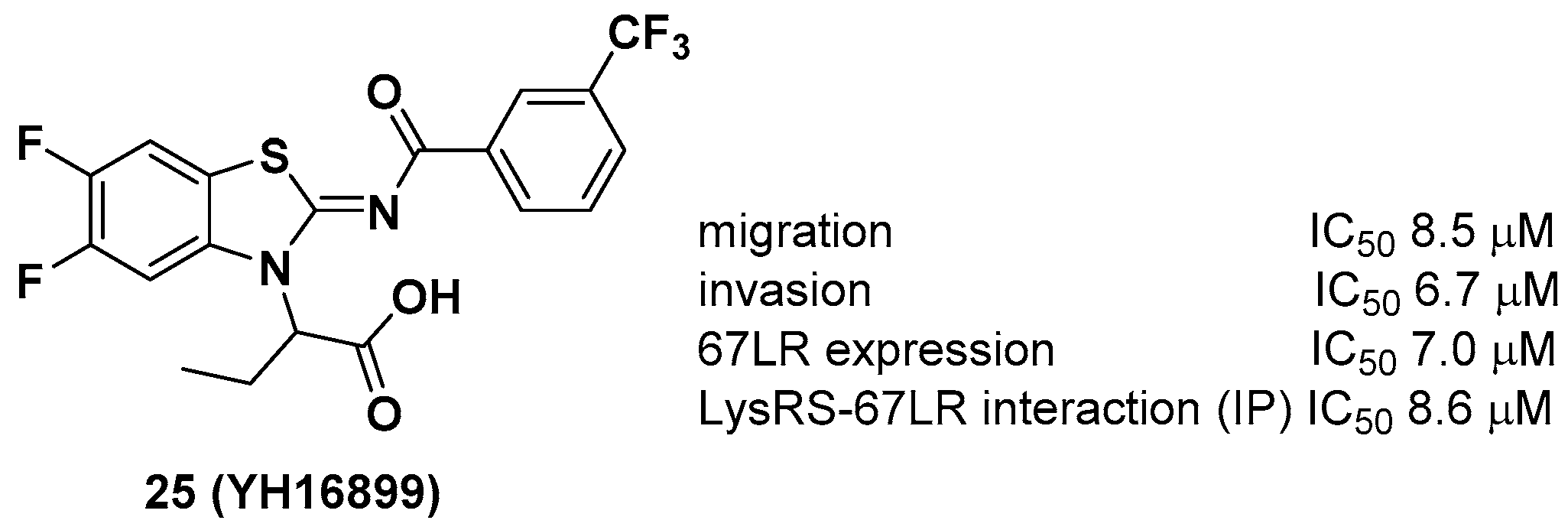
2.4. Benzothiazoles
2.5. Halofuginone
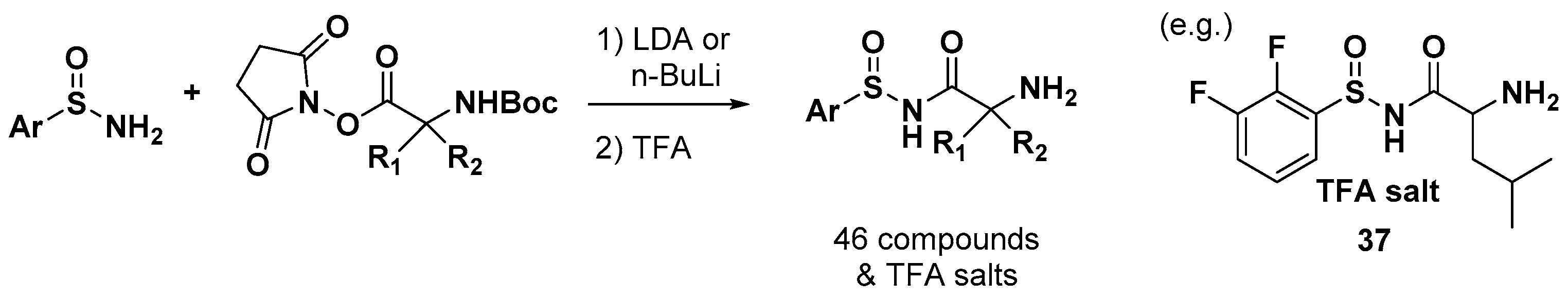
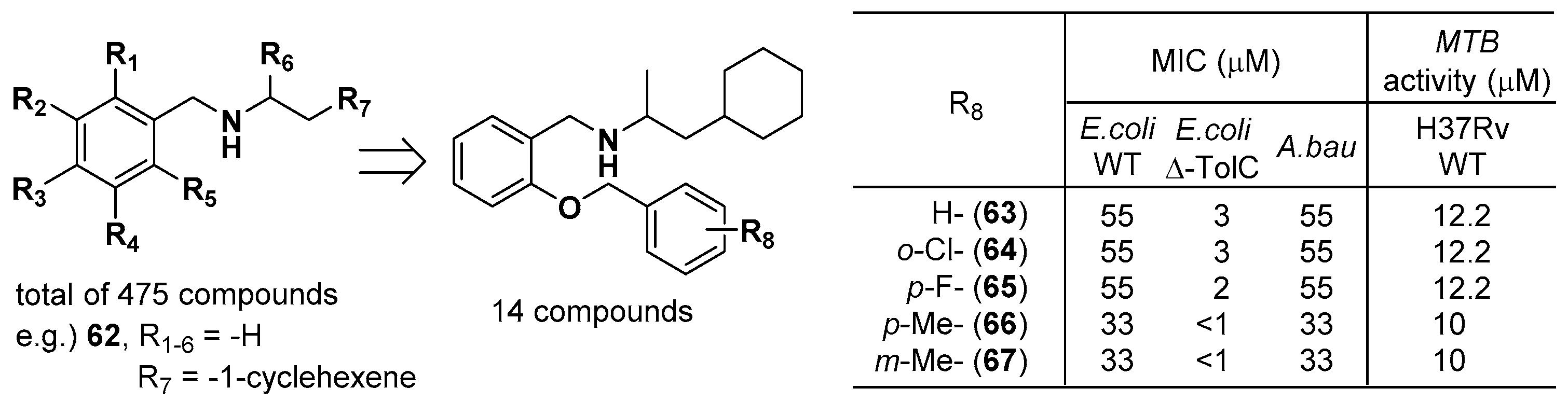
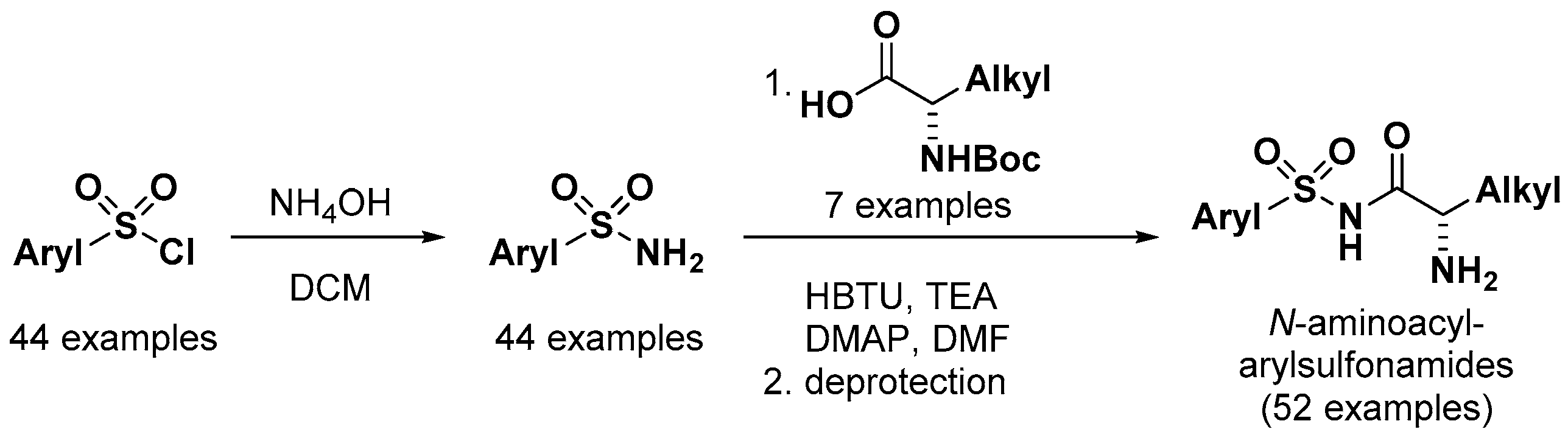
2.6. Sulfonamido Propanamides and 2-Aminopyrimidines
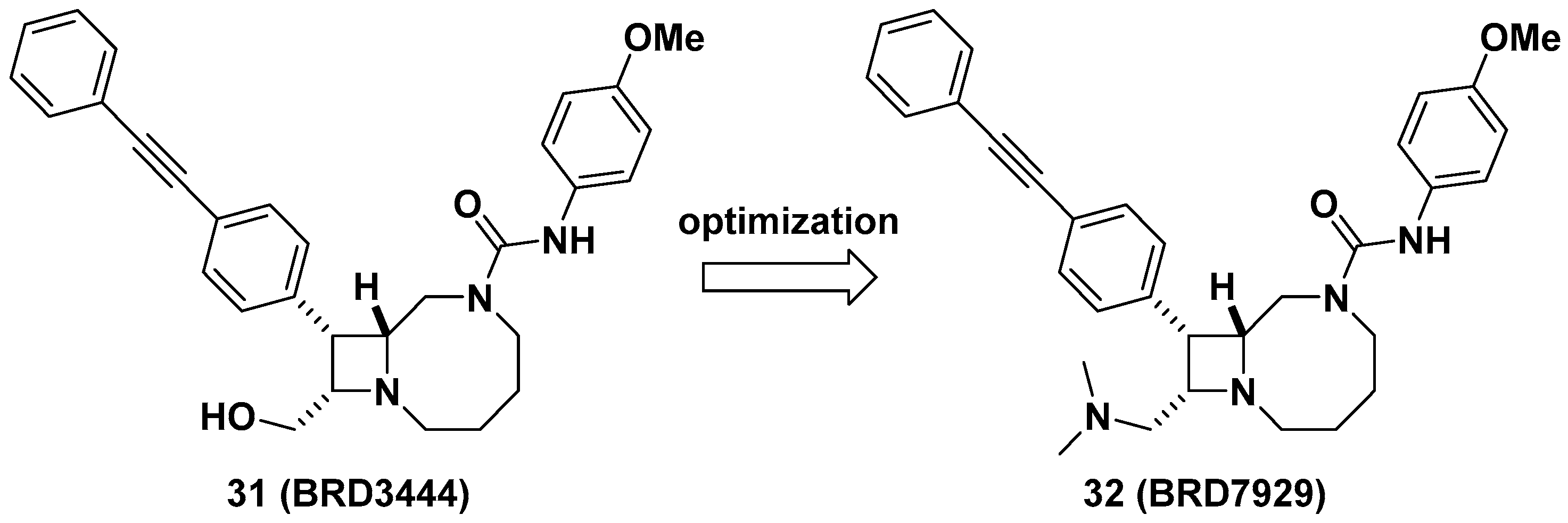
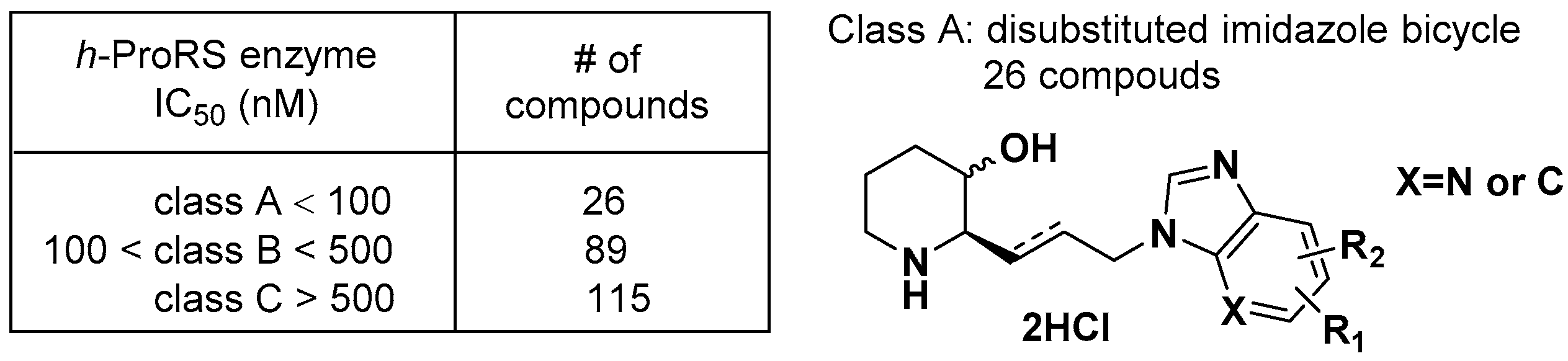
2.7. Bicylic Azetidines
3. Aminoacyl-tRNA Synthetase (ARS) Therapeutics in Patents
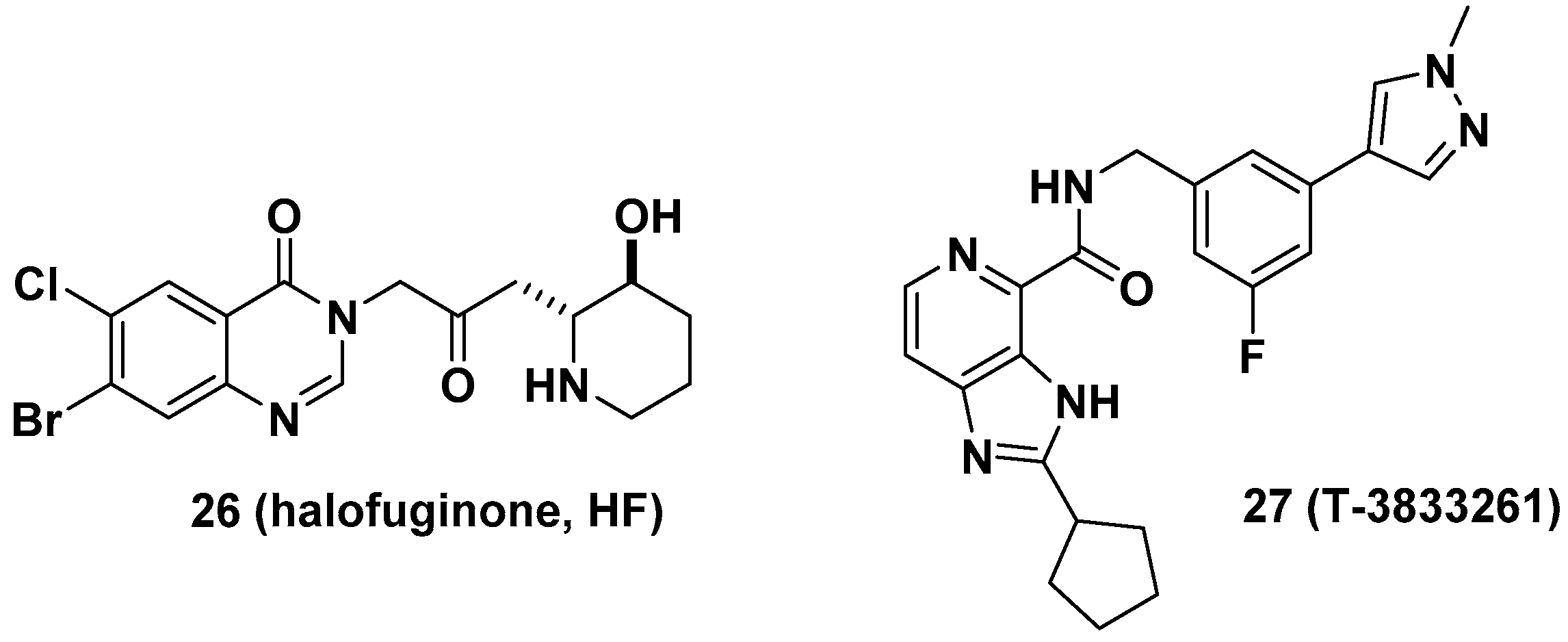
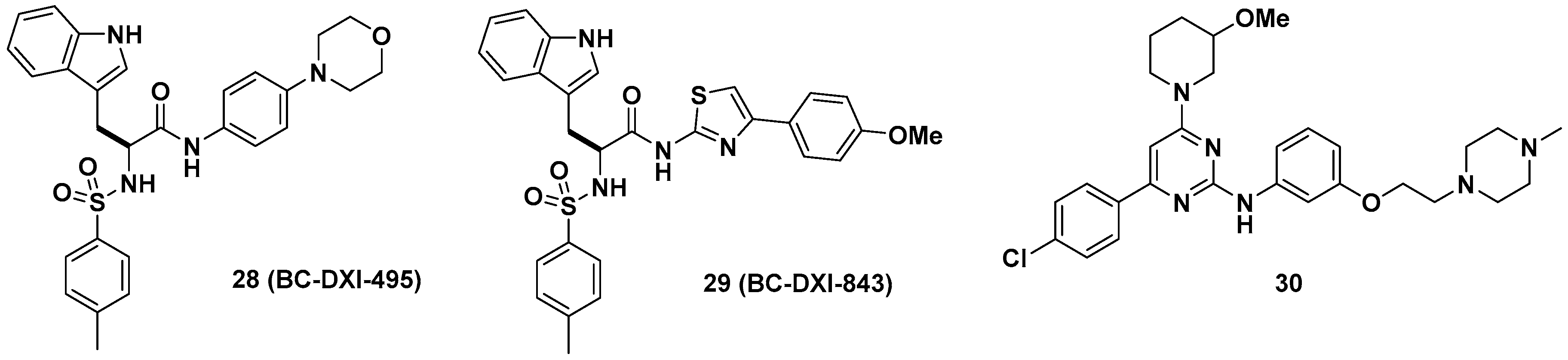
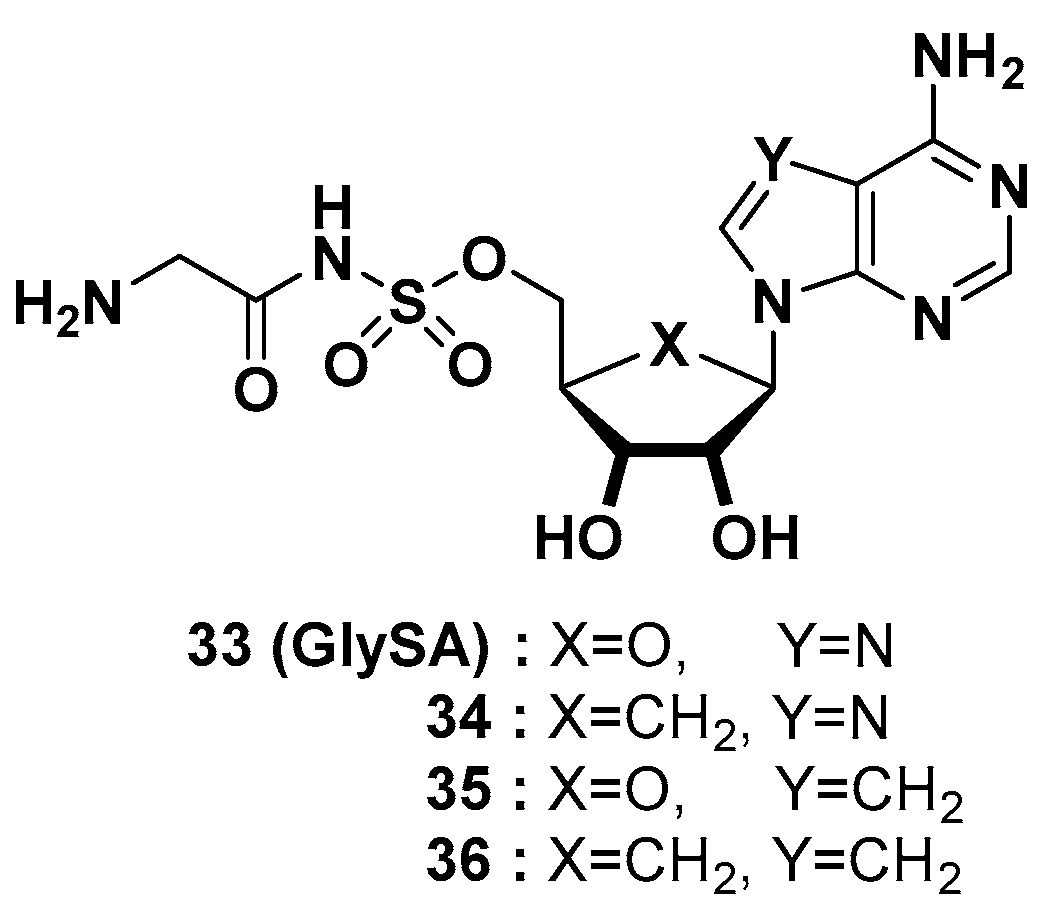
3.1. Glycyl-tRNA Synthetase (GlyRS) Inhibitors
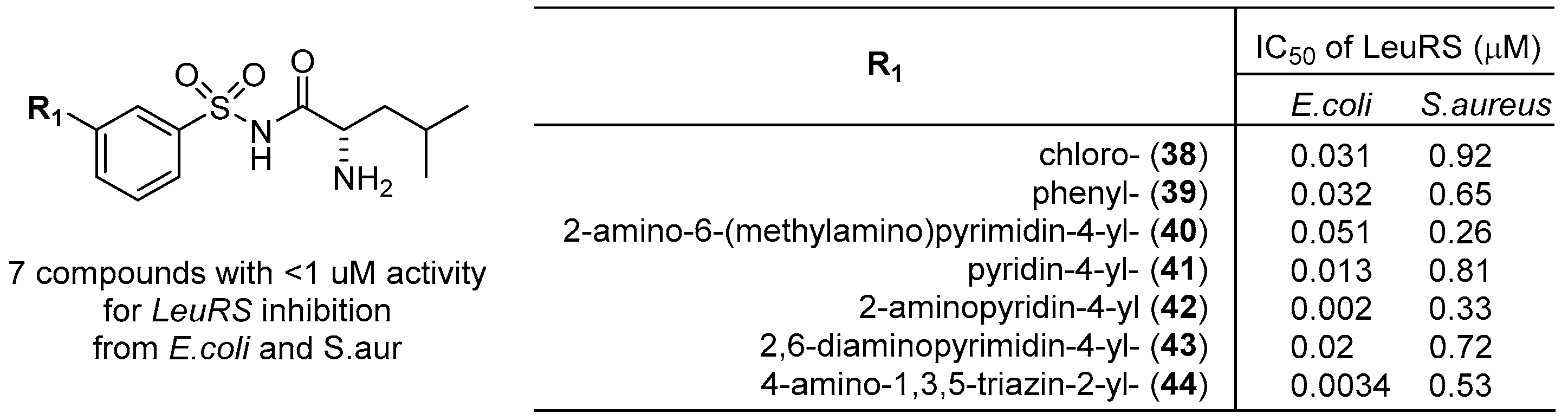
3.2. Leucyl-tRNA Synthetase (LeuRS) Inhibitors
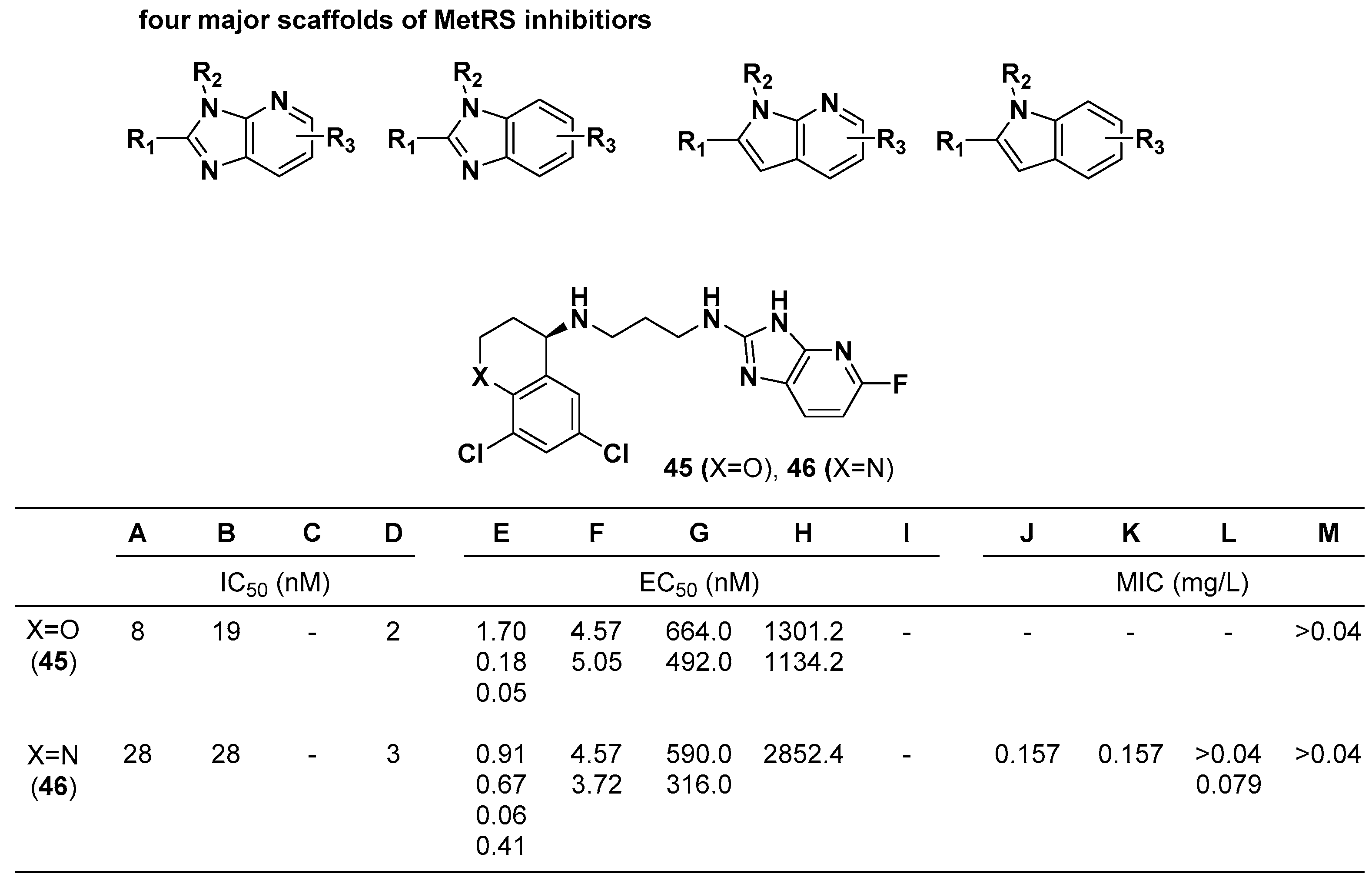
3.3. Methionyl-tRNA Synthetase (MetRS) Inhibitors
3.4. Phenylalanyl-tRNA Synthetase (PheRS) Inhibitors
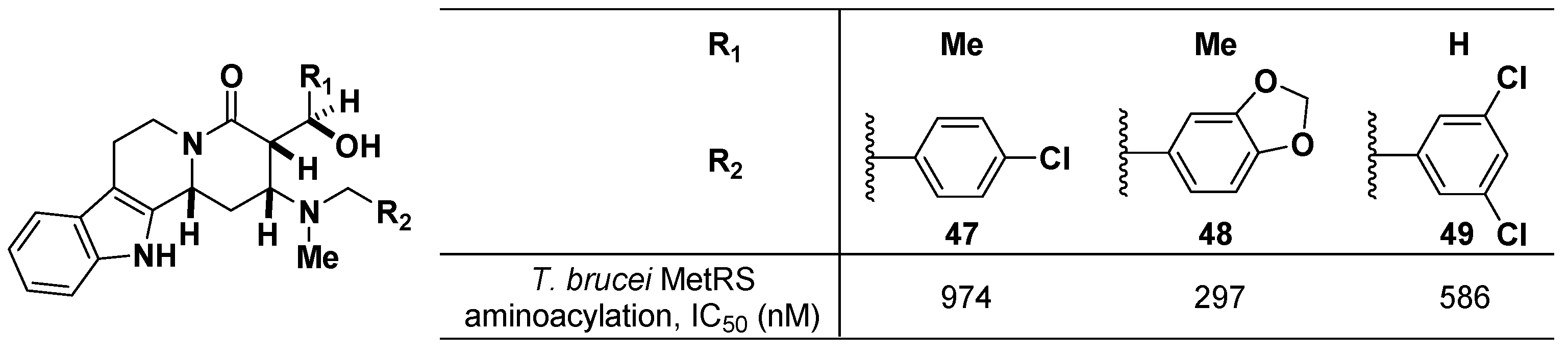
3.5. Prolyl-tRNA Synthetase (ProRS) Inhibitors
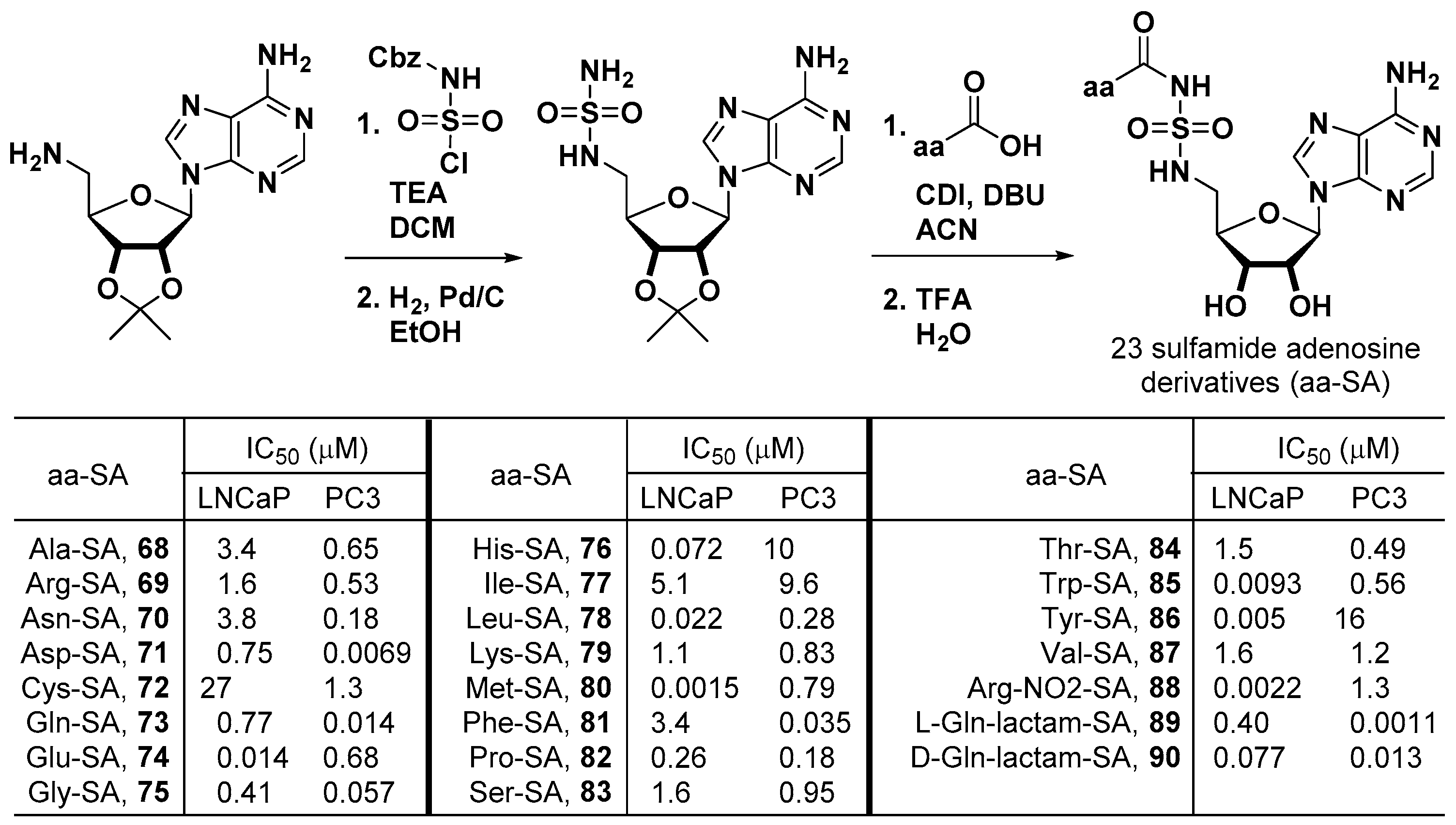
3.6. Multi-tRNA Synthetase Inhibitors
4. Aminoacyl-tRNA Synthetase (ARS) Therapeutics in Clinical Trials
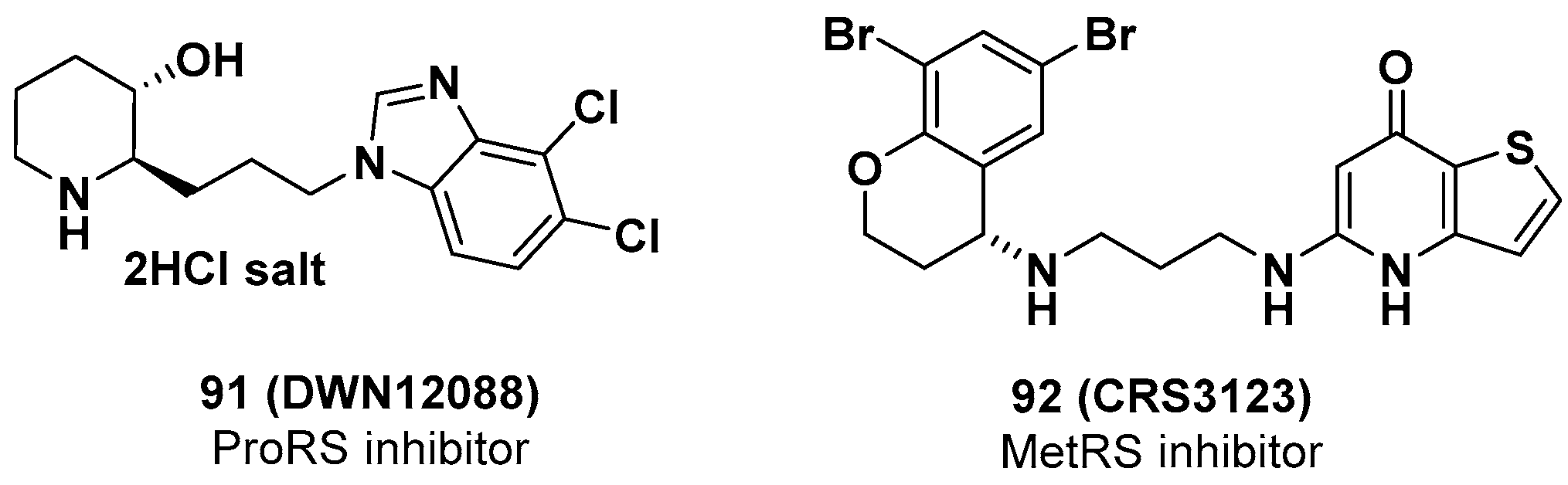
4.1. DWN12088, Eukaryotic Prolyl-tRNA Synthetase (eProRS) Inhibitor
4.2. CRS3123, Prokaryotic Methionyl-tRNA Synthetase (pMetRS) Inhibitor
4.2.1. Phase I Trial of a Single Dose of CRS3123 (NCT01551004)
4.2.2. Phase I Trial to Determine the Safety and Pharmacokinetics of CRS3123 (NCT02106338)
4.3. GSK3036656, Prokaryotic Leucyl-tRNA Synthetase (pLeuRS) Inhibitor
4.3.1. First-Time-in-Human (FTIH) Safety and Pharmacokinetics (PK) Study of GSK3036656 in Healthy Subjects (NCT03075410)
4.3.2. An Early Bactericidal Activity, Safety, and Tolerability Study of GSK3036656 in Subjects with Drug-sensitive Pulmonary Tuberculosis (NCT03557281)
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ibba, M.; Soll, D. Aminoacyl-tRNA Synthesis. Annu. Rev. Biochem. 2000, 69, 617–650. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.; Fox, P.L. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol. Med. 2013, 5, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Guo, M. Evolutionary limitation and opportunities for developing tRNA synthetase inhibitors with 5-binding-mode classification. Life 2015, 5, 1703–1725. [Google Scholar] [CrossRef]
- Hurdle, J.G.; O’Neill, A.J.; Chopra, I. Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob. Agents Chemother. 2005, 49, 4821–4833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwalab, V.; Nair, S.K. Aminoacyl tRNA synthetases as targets for antibiotic development. Med. Chem. Commun. 2012, 3, 887–898. [Google Scholar] [CrossRef]
- Lv, P.C.; Zhu, H.L. Aminoacyl-tRNA synthetase inhibitors as potent antibacterials. Curr. Med. Chem. 2012, 19, 3550–3563. [Google Scholar] [CrossRef] [PubMed]
- Gadakh, B.; Aerschot, A.V. Aminoacyl-tRNA synthetase inhibitors as antimicrobial agents: A patent review from 2006 till present. Expert Opin. Ther. Pat. 2012, 22, 1453–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vondenhoff, G.H.M.; Aerschot, A.V. Aminoacyl-tRNA synthetase inhibitors as potential antibiotics. Eur. J. Med. Chem. 2011, 46, 5227–5236. [Google Scholar] [CrossRef]
- Lee, E.Y.; Kim, S.; Kim, M.H. Aminoacyl-tRNA synthetases, therapeutic targets for infectious diseases. Biochem. Pharmacol. 2018, 154, 424–434. [Google Scholar] [CrossRef]
- Keller, T.L.; Zocco, D.; Sundrud, M.S.; Hendrick, M.; Edenius, M.; Yum, J.; Kim, Y.J.; Lee, H.K.; Cortese, J.F.; Wirth, D.F.; et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat. Chem. Biol. 2012, 8, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Shibata, A.; Kuno, M.; Adachi, R.; Sato, Y.; Hattori, H.; Matsuda, A.; Okuzono, Y.; Igaki, K.; Tominari, Y.; Takagi, T.; et al. Discovery and pharmacological characterization of a new class of prolyl-tRNA synthetase inhibitor for anti-fibrosis therapy. PLoS ONE 2017, 12, e0186587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamura, T.; Liu, D.; Towle, M.J.; Kageyama, R.; Tsukahara, N.; Wakabayashi, T.; Littlefield, B.A. Anti-angiogenesis effects of borrelidin are mediated through distinct pathways: Threonyl-tRNA synthetase and caspases are independently involved in suppression of proliferation and induction of apoptosis in endothelial cells. J. Antibiot. 2003, 56, 709–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otani, A.; Otani, A.; Slike, B.M.; Dorrell, M.I.; Hood, J.; Kinder, K.; Ewalt, K.L.; Cheresh, D.; Schimmel, P.; Friedlander, M. A fragment of human TrpRS as a potent antagonist of ocular angiogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Kwon, N.H.; Fox, P.L.; Kim, S. Aminoacyl-tRNA synthetases as therapeutic targets. Nat. Rev. Drug Discov. 2019, 18, 629–650. [Google Scholar] [CrossRef]
- Si, Y.; Basak, S.; Li, Y.; Merino, J.; Iuliano, J.N.; Walker, S.G.; Peter, J.T. Antibacterial Activity and Mode of Action of a Sulfonamide-Based Class of Oxaborole Leucyl-tRNA-Synthetase Inhibitors. ACS Infect. Dis. 2019, 5, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Francklyn, C.S.; Mullen, P. Progress and challenges in aminoacyl-tRNA synthetase-based therapeutics. J. Biol. Chem. 2019, 294, 5365–5385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rock, F.L.; Mao, W.; Yaremchuk, A.; Tukalo, M.; Crepin, T.; Zhou, H.; Zhang, Y.K.; Hernandez, V.; Akama, T.; Baker, S.J.; et al. An antifungal agent inhibits an aminoacyl tRNA synthetase by trapping tRNA in the editing site. Science 2007, 316, 1759–1761. [Google Scholar] [CrossRef]
- Dowlut, M.; Hall, D.G. An improved class of sugarbinding boronic acids, soluble and capable of complexing glycosides in neutral water. J. Am. Chem. Soc. 2006, 128, 4226–4227. [Google Scholar] [CrossRef]
- Berube, M.; Dowlut, M.; Hall, D.G. Benzoboroxoles as efficient glycopyranoside-binding agents in physiological conditions: Structure and selectivity of complex formation. J. Org. Chem. 2008, 73, 6471–6479. [Google Scholar] [CrossRef]
- Palencia, A.; Li, X.; Bu, W.; Choi, W.; Ding, C.Z.; Easom, E.E.; Feng, L.; Hernandez, V.; Houston, P.; Liu, L.; et al. Discovery of novel oral protein synthesis inhibitors of Mycobacterium tuberculosis that target leucyl-tRNA synthetase. Antimicrob. Agents Chemother. 2016, 60, 6271–6280. [Google Scholar] [CrossRef] [Green Version]
- Xianfeng, L.; Vincent, H.; Fernando, L.R.; Wai, C.; Yvonne, S.L.; Mak, M.M.; Weimin, M.; Yasheen, Z.; Eric, E.E.; Jacob, J.; et al. Discovery of a Potent and Specific M. tuberculosis Leucyl-tRNA Synthetase Inhibitor: (S)-3-(Aminomethyl)-4-chloro-7-(2-hydroxyethoxy)benzo[c][1,2]oxaborol-1(3H)-ol (GSK656). J. Med. Chem. 2017, 60, 8011–8026. [Google Scholar]
- David, T.; Geo, D.; Alex, C.; John, T.; Matt, D.; Simon, C.; Stephanie, G.; Alison, G.; Adeep, P.; Morris, M.; et al. First-Time-in-Human Study and Prediction of Early Bactericidal Activity for GSK3036656, a Potent Leucyl-tRNA Synthetase Inhibitor for Tuberculosis Treatment. Antimicrob. Agents Chemother. 2019, 63, e00240-19. [Google Scholar]
- Hoepfner, D.; McNamara, C.W.; Lim, C.S.; Studer, C.; Riedl, R.; Aust, T.; McCormack, S.L.; Plouffe, D.M.; Meister, S.; Schuierer, S.; et al. Selective and specific inhibition of the Plasmodium falciparum lysyl-tRNA synthetase by the fungal secondary metabolite cladosporin. Cell Host Microbe 2012, 11, 654–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Huang, X.; Li, H.; Tawa, G.; Fisher, E.; Tanaka, T.Q.; Shinn, P.; Huang, W.; Williamson, K.C.; Zheng, W. Novel lead structures with both Plasmodium falciparum gametocytocidal and asexual blood stage activity identified from high throughput compound screening. Malar. J. 2017, 16, 147. [Google Scholar] [CrossRef]
- Wampfler, R.; Hofmann, N.E.; Karl, S.; Betuela, I.; Kinboro, B.; Lorry, L.; Silkey, M.; Robinson, L.J.; Mueller, I.; Felger, I. Effects of liver-stage clearance by Primaquine on gametocyte carriage of Plasmodium vivax and P. falciparum. PLoS Negl. Trop. Dis. 2017, 11, e0005753. [Google Scholar] [CrossRef] [Green Version]
- Rusch, M.; Thevenon, A.; Hoepfner, D.; Aust, T.; Studer, C.; Patoor, M.; Rollin, P.; Livendahl, M.; Ranieri, B.; Schmitt, E.; et al. Design and synthesis of metabolically stable tRNA synthetase inhibitors derived from cladosporin. ChemBioChem 2019, 20, 644–649. [Google Scholar] [CrossRef] [Green Version]
- Fang, P.; Han, H.; Wang, J.; Chen, K.; Chen, X.; Guo, M. Structural Basis for Specific Inhibition of tRNA Synthetase by an ATP Competitive Inhibitor. Chem. Biol. 2015, 22, 734–744. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zheng, L.; Hei, Z.; Li, W.; Wang, J.; Yu, B.; Fang, P. Atomic Resolution Analyses of Isocoumarin Derivatives for Inhibition of Lysyl-tRNA Synthetase. ACS Chem. Biol. 2020, 15, 1016–1025. [Google Scholar] [CrossRef]
- Baragaña, B.; Forte, B.; Choi, R.; Hewitt, S.N.; Bueren-Calabuig, J.A.; Pisco, J.P.; Peet, C.; Dranow, D.M.; Robinson, D.A.; Jansen, C.; et al. Lysyl-tRNA synthetase as a drug target in malaria and cryptosporidiosis. Proc. Natl. Acad. Sci. USA 2019, 116, 7015–7020. [Google Scholar] [CrossRef] [Green Version]
- Novoa, E.M.; Camacho, N.; Tor, A.; Wilkinson, B.; Moss, S.; Marín-García, P.; Azcárate, I.G.; Bautista, J.M.; Mirando, A.C.; Francklyn, C.S.; et al. Analogs of natural aminoacyl-tRNA synthetase inhibitors clear malaria in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E5508–E5517. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.G.; Lee, J.Y.; Kwon, N.H.; Fang, P.; Zhang, Q.; Wang, J.; Young, N.L.; Guo, M.; Cho, H.Y.; Mushtaq, A.U.; et al. Chemical inhibition of prometastatic lysyl-tRNA synthetase–laminin receptor interaction. Nat. Chem. Biol. 2014, 10, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.G.; Choi, J.W.; Lee, J.Y.; Kim, H.; Oh, Y.S.; Lee, J.W.; Tak, Y.K.; Song, J.M.; Razin, E.; Yun, S.H.; et al. Interaction of two translational components, lysyl-tRNA synthetase and p40/37LRP, in plasma membrane promotes laminin-dependent cell migration. FASEB J. 2012, 26, 4142–4159. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.D.; Rice, D.P.; Ribacke, U.; Silterra, J.; Deik, A.A.; Moss, E.L.; Broadbent, K.M.; Neafsey, D.E.; Desai, M.M.; Clish, C.B.; et al. A genomic and evolutionary approach reveals non-genetic drug resistance in malaria. Genome Biol. 2014, 15, 511. [Google Scholar] [CrossRef] [Green Version]
- Kershenobich, D.; Fierro, F.J.; Rojkind, M. The relationship between the free pool of proline and collagen content in human liver cirrhosis. J. Clin. Investig. 1970, 49, 2246–2249. [Google Scholar] [CrossRef] [Green Version]
- Mendes, M.I.; Gutierrez Salazar, M.; Guerrero, K.; Thiffault, I.; Salomons, G.S.; Gauquelin, L.; Tran, L.T.; Forget, D.; Gauthier, M.S.; Waisfisz, Q.; et al. Bi-allelic mutations in EPRS, encoding the glutamyl-prolyl-aminoacyl-tRNA synthetase, cause a hypomyelinating leukodystrophy. Am. J. Hum. Genet. 2018, 102, 676–684. [Google Scholar] [CrossRef] [Green Version]
- US National Library of Medicine. Clinical Trials Related to Halogifunone. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=halofuginone&cntry=&state=&city=&dist= (accessed on 19 October 2020).
- US National Library of Medicine. NCT01847573, Safety, Tolerability, and Pharmacokinetics of Single and Multiple Doses of HT-100 in Duchenne Muscular Dystrophy. Available online: https://clinicaltrials.gov/ct2/show/NCT01847573 (accessed on 19 October 2020).
- US National Library of Medicine. NCT02525302, HT-100 Long-Term Study in DMD Patients Who Completed HALO-DMD-02. Available online: https://clinicaltrials.gov/ct2/show/NCT02525302 (accessed on 19 October 2020).
- US National Library of Medicine. NCT01978366, Open Label Extension Study of HT-100 in Patients with DMD. Available online: https://clinicaltrials.gov/ct2/show/NCT01978366 (accessed on 19 October 2020).
- Wu, J.; Subbaiah, K.C.V.; Xie, L.H.; Jiang, F.; Khor, E.S.; Mickelsen, D.; Myers, J.R.; Tang, W.H.W.; Yao, P. Glutamyl-prolyl-tRNA synthetase regulates proline-rich pro-fibrotic protein synthesis during cardiac fibrosis. Circ. Res. 2020, 127, 827–846. [Google Scholar] [CrossRef]
- Kim, D.G.; Lee, J.Y.; Lee, J.H.; Cho, H.Y.; Kang, B.S.; Jang, S.Y.; Kim, M.H.; Guo, M.; Han, J.M.; Kim, S.J.; et al. Oncogenic mutation of AIMP2/p38 inhibits its tumorsuppressive interaction with Smurf2. Cancer Res. 2016, 76, 3422–3436. [Google Scholar] [CrossRef] [Green Version]
- Yum, M.K.; Kang, J.S.; Lee, A.E.; Jo, Y.W.; Seo, J.Y.; Kim, H.A.; Kim, Y.Y.; Seong, J.; Lee, E.B.; Kim, J.H.; et al. AIMP2 controls intestinal stem cell compartments and tumorigenesis by modulating Wnt/β-catenin signaling. Cancer Res. 2016, 76, 4559–4568. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.W.; Lee, J.W.; Kim, J.K.; Jeon, H.K.; Choi, J.J.; Kim, D.G.; Kim, B.G.; Nam, D.H.; Kim, H.J.; Yun, S.H.; et al. Splicing variant of AIMP2 as an effective target against chemoresistant ovarian cancer. J. Mol. Cell Biol. 2012, 4, 164–173. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Cho, H.Y.; Kim, D.G.; Roh, Y.; Son, S.Y.; Mushtaq, A.U.; Kim, M.; Bhattarai, D.; Sivaraman, A.; Lee, Y.; et al. Targeting the interaction of AIMP2-DX2 with HSP70 suppresses cancer development. Nat. Chem. Biol. 2020, 16, 31–41. [Google Scholar] [CrossRef]
- Sivaraman, A.; Kim, D.G.; Bhattarai, D.; Kim, M.; Lee, H.Y.; Lim, S.; Kong, J.; Goo, J.I.; Shim, S.; Lee, S.; et al. Synthesis and structure−activity relationships of arylsulfonamides as AIMP2-DX2 inhibitors for the development of a novel anticancer therapy. J. Med. Chem. 2020, 63, 5139–5158. [Google Scholar] [CrossRef]
- Lee, S.; Kim, D.G.; Kim, K.; Kim, T.; Lim, S.; Kong, H.; Kim, S.; Suh, Y.G. 2-Aminophenylpyrimidines as novel inhibitors of aminoacyl-tRNA synthetase interacting multifunctional protein 2 (AIMP2)-DX2 for lung cancer treatment. J. Med. Chem. 2020, 63, 3908–3914. [Google Scholar] [CrossRef]
- Kato, N.; Comer, E.; Sakata-Kato, T.; Sharma, A.; Sharma, M.; Maetani1, M.; Bastien, J.; Brancucci, N.M.; Bittker, J.A.; Corey, V.; et al. Diversity-oriented synthesis yields novel multistage antimalarial inhibitors. Nature 2016, 538, 344–349. [Google Scholar] [CrossRef]
- Vinayak, S.; Jumani, R.S.; Miller, P.; Hasan, M.M.; McLeod, B.I.; Tandel, J.; Stebbins, E.E.; Teixeira, J.E.; Borrel, J.; Gonse, A.; et al. Bicyclic azetidines kill the diarrheal pathogen Cryptosporidium in mice by inhibiting parasite phenylalanyl-tRNA synthetase. Sci. Transl. Med. 2020, 12, eaba8412. [Google Scholar] [CrossRef]
- Yang, X.L.; Mo, Z.; Schimmel, P. Inhibition of Neddylation Using Glycyl-tRNA Synthetase (GlyRS) Inhibitors for Cancer Therapy. International Patent WO 2017066459 A1, 20 April 2017. [Google Scholar]
- US National Library of Medicine. A Representative Example of the Development of MLN4924 for Refractory Acute Myeloid Leukemia or Myelodysplastic Syndrome. Available online: https://clinicaltrials.gov/ct2/show/NCT03772925 (accessed on 20 November 2020).
- Finn, P.W.; Charlton, M.; Edmund, G.; Jirgensons, A.; Loza, E. 2-Amino-N-(Arylsulfinyl)-Acetamide Compounds as Inhibitors of Bacterial Aminoacyl-tRNA Synthetase and Their Preparation. International Patent WO 2018065611 A1, 12 April 2018. [Google Scholar]
- Jirgensons, A.; Loza, E.; Charlton, M.; Finn, P.W.; Ribas de Pouplana, L.; Saint-Leger, A. Preparation of Novel N-Acyl-Arylsulfonamide Derivatives as aaRS Inhibitors. International Patent WO 2016129983 A1; Japanese Patent JP6740256B2, 18 August 2016. [Google Scholar]
- Charlton, M.H.; Aleksis, R.; Saint-Leger, A.; Gupta, A.; Loza, E.; Ribas de Pouplana, L.; Kaula, I.; Gustina, D.; Madre, M.; Lola, D.; et al. N-leucinyl benzenesulfonamides as structurally simplified leucyl-tRNA synthetase inhibitors. ACS Med. Chem. Lett. 2018, 9, 84–88. [Google Scholar] [CrossRef]
- Buckner, F.S.; Alvarez, X.B.; Fan, E.; Gillespie, J.R.; Hol, W.G.J.; Huang, W.; Koh, C.Y.; Ranade, R.M.; Shibata, S.; Verlinde, C.L.M.; et al. Preparation of Substituted Imidazopyridines and Analogs as Specific Inhibitors of Methionyl-tRNA Synthetase. International Patent WO 2016029146 A1, 25 February 2016. [Google Scholar]
- Zhang, Z.; Koh, C.Y.; Ranade, R.M.; Shibata, S.; Gillespie, J.R.; Hulverson, M.A.; Huang, W.; Nguyen, J.; Pendem, N.; Gelb, M.H.; et al. 5-Fluoroimidazo[4,5-b]pyridine is a privileged fragment that conveys bioavailability to potent trypanosomal methionyl-tRNA synthetase inhibitors. ACS Infect. Dis. 2016, 2, 399–404. [Google Scholar] [CrossRef]
- Martin, S.F.; Sahn, J.J.; Meis, A.R.; Lepovitz, L.T. Novel Inhibitors of Trypanosoma Brucei Methionyl t-RNA Synthetase for the Treatment of African Trypanosomiasis. International Patent WO 2016004297 A1, 7 January 2016. [Google Scholar]
- Lepovitz, L.T.; Meis, A.R.; Thomas, S.M.; Wiedeman, J.; Pham, A.; Mensa-Wilmot, K.; Martin, S.F. Design, synthesis, and evaluation of novel anti-trypanosomal compounds. Tetrahedron 2020, 76, 131086. [Google Scholar] [CrossRef]
- Kahne, D.E.; Baidin, V. Preparation of Secondary Amine Compounds as tRNA Synthetase Inhibitors Useful in Killing Gram-Negative Bacteria. International Patent WO 2019140265 A1, 18 July 2019. [Google Scholar]
- Lee, B.Y.; Cho, M.J.; Lee, H.G.; Jung, M.G.; Oh, Y. Heterocyclic Compounds, Preparation Methods, Pharmaceutical Compositions, and Use as Prolyl-tRNA Synthetase Inhibitors. International Patent WO 2018147626 A1; Korean Patent KR102084772B1; Australian Patent AU2018218965B2, 16 August 2018. [Google Scholar]
- Qi, X.; Khaybullin, R.; Liang, X. Preparation of Sulfamide Adenosine Derivatives as Antitumor, Antiinflammatory, Antibacterial, Antifungal, and Parasiticide Agents. International Patent WO 2016201374 A1, 15 December 2016. [Google Scholar]
- Lee, C.H.; Cho, M.; Kim, J.M.; Lee, J.H.; Kim, D.-W.; Park, M.Y.; Kim, Y.K.; Han, J.; Park, J.S. A First-in-Class PRS Inhibitor, DWN12088, as a Novel Therapeutic Agent for Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, A2786. [Google Scholar]
- 2020 Clarivate Analytics, The CortellisTM Full Report of DWN12088. pp. 1–14. Available online: https://www.cortellis.com/intelligence/report/ci/nextgendrugall/108457 (accessed on 8 October 2020).
- Australian New Zealand Clinical Trials Registry. Available online: http://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=377846 (accessed on 3 October 2020).
- 2020 Clarivate Analytics, The CortellisTM Full Report of CRS3123. pp. 1–12. Available online: https://www.cortellis.com/intelligence/report/ci/nextgendrugall/58117 (accessed on 8 October 2020).
- Critchley, I.A.; Green, L.S.; Young, C.L.; Bullard, J.M.; Evans, R.J.; Price, M.; Jarvis, T.C.; Guiles, J.W.; Janjic, N.; Ochsner, U.A. Spectrum of activity and mode of action of REP3123, a new antibiotic to treat Clostridium difficile infections. J. Antimicrob. Chemother. 2009, 63, 954–963. [Google Scholar] [CrossRef] [Green Version]
- Ochsner, U.A.; Bell, S.J.; O’Leary, A.L.; Hoang, T.; Stone, K.C.; Young, C.L.; Critchley, I.A.; Janjic, N. Inhibitory effect of REP3123 on toxin and spore formation in Clostridium difficile, and in vivo efficacy in a hamster gastrointestinal infection model. J. Antimicrob. Chemother. 2009, 63, 964–971. [Google Scholar] [CrossRef] [Green Version]
- Citron, D.M.; Warren, Y.A.; Tyrrell, K.L.; Merriam, V.; Goldstein, E.J.C. Comparative in vitro activity of REP3123 against Clostridium difficile and other anaerobic intestinal bacteria. J. Antimicrob. Chemother. 2009, 63, 972–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomeli, B.K.; Galbraith, H.; Schettler, J.; Saviolakis, G.A.; El-Amin, W.; Osborn, B.; Ravel, J.; Hazleton, K.; Lozupone, C.A.; Evans, R.J.; et al. Multiple-Ascending-Dose Phase 1 Clinical Study of the Safety, Tolerability, and Pharmacokinetics of CRS3123, a Narrow-Spectrum Agent with Minimal Disruption of Normal Gut Microbiota. Antimicrob. Agents Chemother. 2020, 64, e01395-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US National Library of Medicine. Randomized, Double-Blind, Placebo-Controlled, Single Ascending Dose Phase I Trial to Determine the Safety and Pharmacokinetics of CRS3123 Administered Orally to Healthy Adults. Available online: https://clinicaltrials.gov/ct2/show/NCT01551004 (accessed on 4 October 2020).
- Nayak, S.U.; Griffiss, J.M.; Blumer, J.; O’Riordan, M.A.; Gray, W.; McKenzie, R.; Jurao, R.A.; An, A.T.; Le, M.; Bell, S.J.; et al. Safety, Tolerability, Systemic Exposure, and Metabolism of CRS3123, a Methionyl-tRNA Synthetase Inhibitor Developed for Treatment of Clostridium difficile, in a Phase 1 Study. Antimicrob. Agents Chemother. 2017, 61, e02760-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US National Library of Medicine. Randomized, Double-Blind, Placebo-Controlled, Multiple Ascending Dose Phase I Trial to Determine the Safety and Pharmacokinetics of CRS3123 Administered Orally to Healthy Adults. Available online: https://clinicaltrials.gov/ct2/show/NCT02106338 (accessed on 5 October 2020).
- 2020 Clarivate Analytics, The CortellisTM Full Report of GSK3036656. pp. 1–13. Available online: https://www.cortellis.com/intelligence/report/ci/nextgendrugall/70581 (accessed on 8 October 2020).
- US National Library of Medicine. A Double Blind, Placebo-Controlled First Time in Human Study to Evaluate the Safety, Tolerability and Pharmacokinetics of Single and Repeat Doses of GSK3036656 in Healthy Adult Volunteers. Available online: https://clinicaltrials.gov/ct2/show/NCT03075410 (accessed on 8 October 2020).
- US National Library of Medicine. A Phase IIa Open-label Trial to Investigate the Early Bactericidal Activity, Safety and Tolerability of GSK3036656 in Participants with Drug-sensitive Pulmonary Tuberculosis. Available online: https://clinicaltrials.gov/ct2/show/record/NCT03557281 (accessed on 9 October 2020).
- Nie, A.; Sun, B.; Fu, Z.; Yu, D. Roles of aminoacyl-tRNA synthetases in immune regulation and immune diseases. Cell Death Dis. 2019, 10, 901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahler, M.; Miller, F.W.; Fritzler, M.J. Idiopathic inflammatory myopathies and the anti-synthetase syndrome: A comprehensive review. Autoimmun. Rev. 2014, 13, 367–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lega, J.C.; Fabien, N.; Reynaud, Q.; Durieu, I.; Durupt, S.; Dutertre, M.; Cordier, J.F.; Cottin, V. The clinical phenotype associated with myositis-specific and associated autoantibodies: A meta-analysis revisiting the so-called antisynthetase syndrome. Autoimmun. Rev. 2014, 13, 883–891. [Google Scholar] [CrossRef]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Huapaya, J.A.; Albayda, J.; Paik, J.J.; Johnson, C.; Silhan, L.; Christopher-Stine, L.; Mammen, A.L.; Danoff, S.K. A longitudinal cohort study of the anti-synthetase syndrome: Increased severity of interstitial lung disease in black patients and patients with anti-PL7 and anti-PL12 autoantibodies. Rheumatology 2017, 56, 999–1007. [Google Scholar] [CrossRef] [Green Version]
- Koch, M.W.; Ilnytskyy, Y.; Golubov, A.; Metz, L.M.; Yong, V.W.; Kovalchuk, O. Global transcriptome profiling of mild relapsing-remitting versus primary progressive multiple sclerosis. Eur. J. Neurol. 2018, 25, 651–658. [Google Scholar] [CrossRef]
- Chen, J.; Jun, L.; Shiyong, C.; Li, H.; Zhu, M.; Shen, B. Increased TTS expression in patients with rheumatoid arthritis. Clin. Exp. Med. 2015, 15, 25–30. [Google Scholar] [CrossRef]
- Wang, C.Y.; Shi, Y.; Min, Y.N.; Zhu, X.J.; Guo, C.S.; Peng, J.; Dong, X.Y.; Qin, P.; Sun, J.Z.; Hou, M. Decreased IDO activity and increased TTS expression break immune tolerance in patients with immune thrombocytopenia. J. Clin. Immunol. 2011, 31, 643–649. [Google Scholar] [CrossRef]
- Zhang, Q.; Yin, X.; Wang, H.; Wu, X.; Li, X.; Li, Y.; Zhang, X.; Fu, C.; Li, H.; Qiu, Y. Fecal metabolomics and potential biomarkers for systemic lupus erythematosus. Front. Immunol. 2019, 10, 976. [Google Scholar] [CrossRef] [PubMed]
- Boasso, A.; Herbeuval, J.P.; Hardy, A.W.; Winkler, C.; Shearer, G.M. Regulation of indoleamine 2,3-dioxygenase and tryptophanyl-tRNA-synthetase by CTLA-4-Fc in human CD4+ T cells. Blood 2005, 105, 1574–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Mao, C.; Zhao, Z.; Gu, Q.; Jin, M.; Xiao, Y.; Jiang, X.; Zhao, Y.; Zhang, Y.; Ning, G. Increased TTS abrogates IDO-mediated CD4(+) T cells suppression in patients with Graves’ disease. Endocrine 2009, 36, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Wellman, T.L.; Eckenstein, M.; Wong, C.; Rincon, M.; Ashikaga, T.; Mount, S.L.; Francklyn, C.S.; Lounsbury, K.M. Threonyl-tRNA synthetase overexpression correlates with angiogenic markers and progression of human ovarian cancer. BMC Cancer 2014, 14, 620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, I.; Dewi, D.L. Upregulation of tryptophanyl-tRNA synthethase adapts human cancer cells to nutritional stress caused by tryptophan degradation. Oncoimmunology 2018, 7, e1486353. [Google Scholar] [CrossRef] [Green Version]
- Boczonadi, V.; Jenning, M.J.; Horvath, R. The role of tRNA synthetases in neurological and neuromuscular disorders. FEBS Lett. 2018, 592, 703–717. [Google Scholar] [CrossRef] [Green Version]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| ID | R1 | R2 | n | % Inhib. (10 μM) |
|---|---|---|---|---|
| 50 |  |  | 1 | 97 |
| 51 |  | 93 | ||
| 52 |  | 98 | ||
| 53 |  | 92 | ||
| 54 |  | 98 | ||
| 55 |  | 94 | ||
| 56 |  |  | 1 | 93 |
| 57 |  | 85 | ||
| 58 |  |  | 1 | 95 |
| 59 |  | 0 | 99 | |
| 60 | 2 | 96 | ||
| 61 (5-MeO -indole) | 1 | 98 (~60% @ 1 μM |
| A Panel of Cancer Cell Lines | IC50 (μM) L-Gln-lactam-SA (89) | |
|---|---|---|
| MOLT-4 | (human acute lymphoblastic leukemia) | 2.44 |
| HL60 | (human acute promyelocytic leukemia) | 0.46 |
| MDA231 | (breast cancer) | 11.03 |
| A549 | (lung cancer) | 7.93 |
| HCT116 | (colon cancer) | 1.48 |
| HeLa | (cervical cancer) | 25.24 |
| MCF-7 | (breast cancer) | 0.19 |
| Drug Name. | Company | Target | Indications | Status | Clinical Trial # (Country) |
|---|---|---|---|---|---|
| DWN12088 | Daewoong Pharma | eProRS | IPF; Scleroderma | Phase I | ACTRN12619001239156 (AU) |
| CRS3123 | Crestone | pMetRS | CDI; HPI | Phase I | NCT01551004 (US)NCT02106338 (US) |
| GSK3036656 | GSK | pLeuRS | MTBI | Phase I | NCT03075410 (UK)NCT03557281 (SA) |
| Mupirocin bioadhesive gel | Laboratorios Ojer Pharma | pIleRS | BSI; Impetigo | Phase III | Not specified (SP) |
| DWP17011 | Daewoong Pharma | eProRS | Fibrosis | Phase I | Not specified (KR) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-H.; Bae, S.; Song, M. Recent Development of Aminoacyl-tRNA Synthetase Inhibitors for Human Diseases: A Future Perspective. Biomolecules 2020, 10, 1625. https://doi.org/10.3390/biom10121625
Kim S-H, Bae S, Song M. Recent Development of Aminoacyl-tRNA Synthetase Inhibitors for Human Diseases: A Future Perspective. Biomolecules. 2020; 10(12):1625. https://doi.org/10.3390/biom10121625
Chicago/Turabian StyleKim, Soong-Hyun, Seri Bae, and Minsoo Song. 2020. "Recent Development of Aminoacyl-tRNA Synthetase Inhibitors for Human Diseases: A Future Perspective" Biomolecules 10, no. 12: 1625. https://doi.org/10.3390/biom10121625
APA StyleKim, S. -H., Bae, S., & Song, M. (2020). Recent Development of Aminoacyl-tRNA Synthetase Inhibitors for Human Diseases: A Future Perspective. Biomolecules, 10(12), 1625. https://doi.org/10.3390/biom10121625