S-adenosyl-l-homocysteine Hydrolase: A Structural Perspective on the Enzyme with Two Rossmann-Fold Domains


Abstract
:1. Introduction
2. Methods
Databases and Software Used
3. Results and Discussion
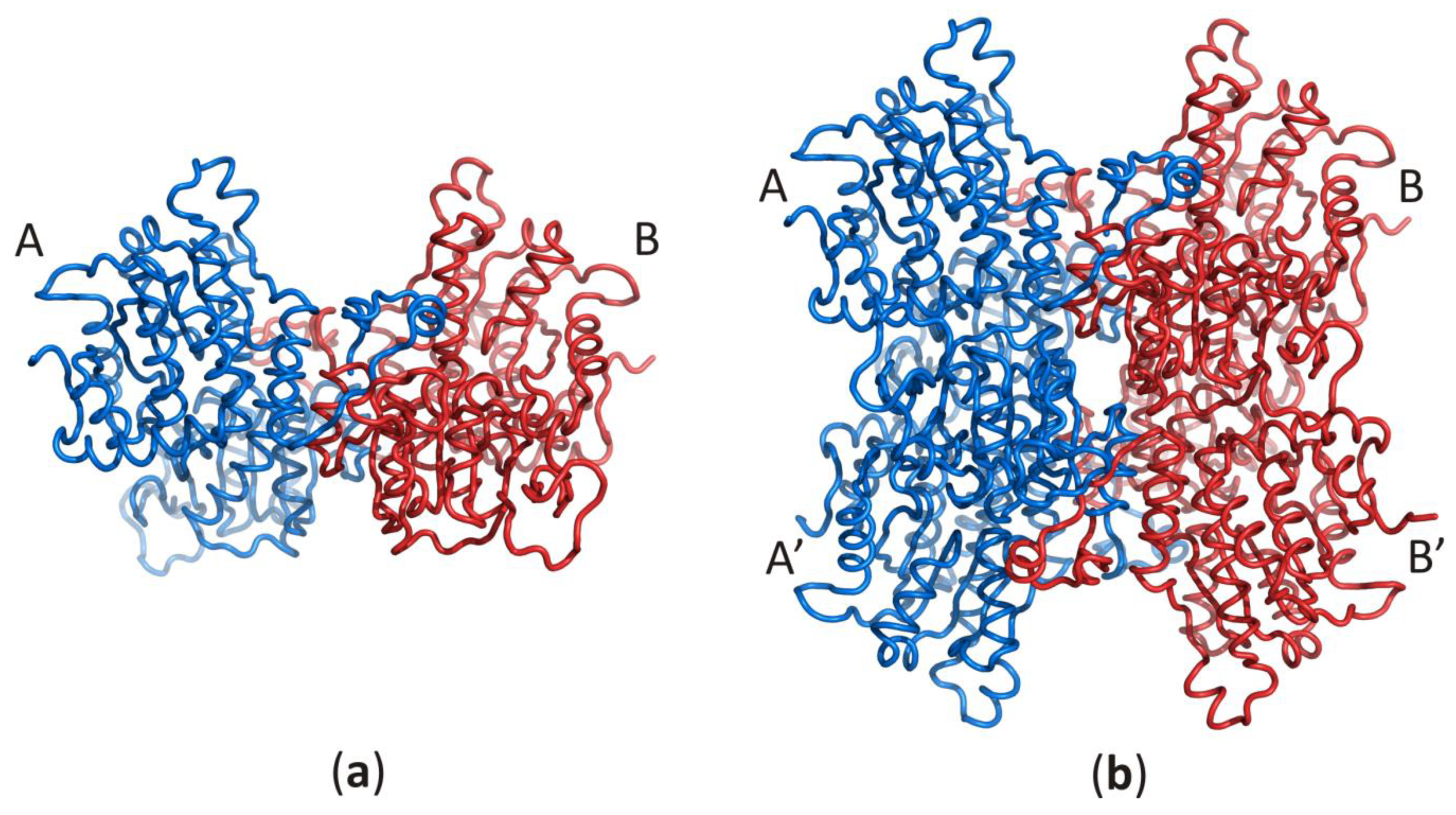
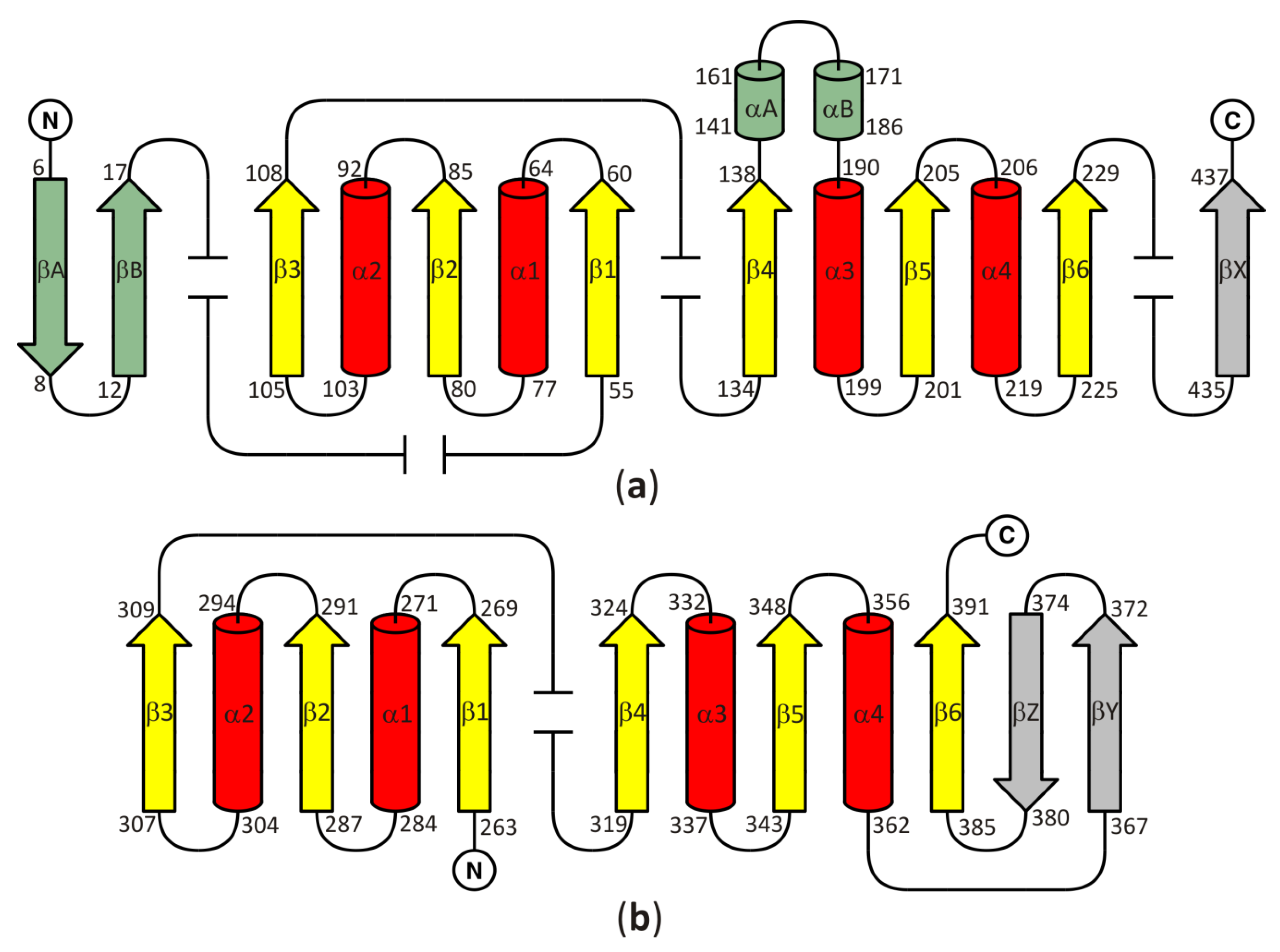
3.1. The Two Principal Domains of SAHase Are Based on the Rossmann Fold
3.2. Ligand Binding Mode in the Substrate- and Cofactor Binding Domains
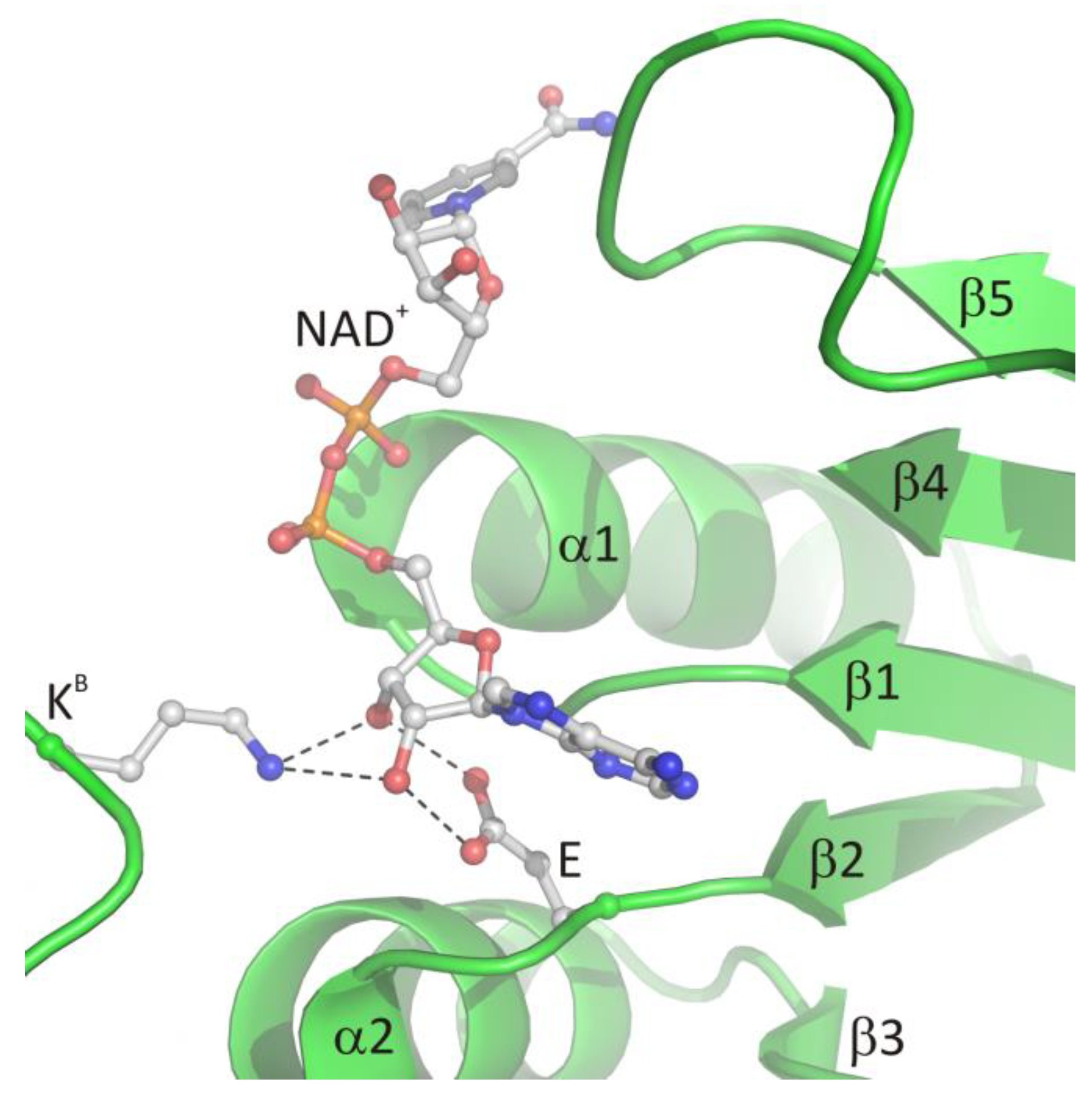
3.2.1. The Role of the Loop β1, Helix α1, and Chain β2 in Ligand Binding
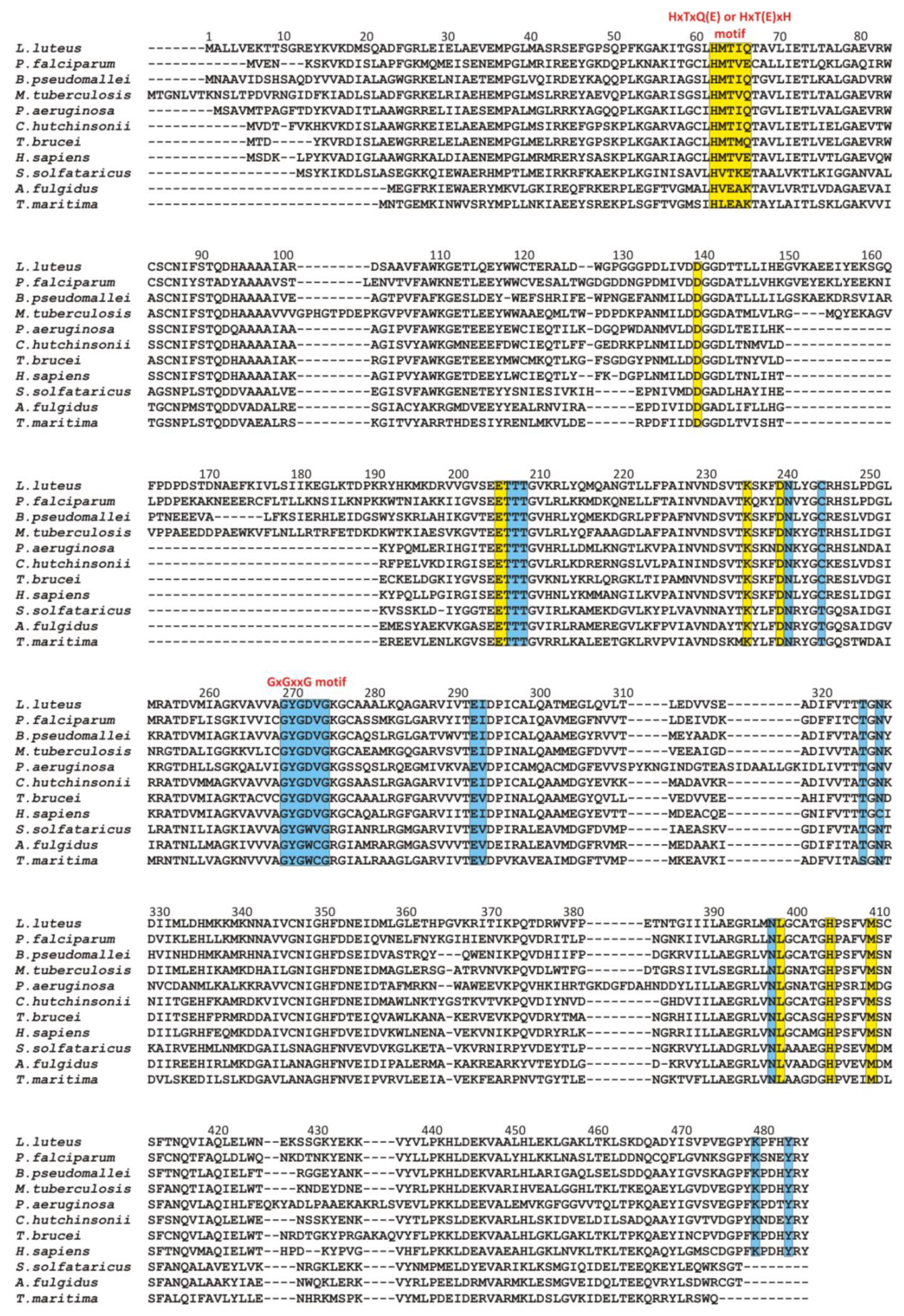
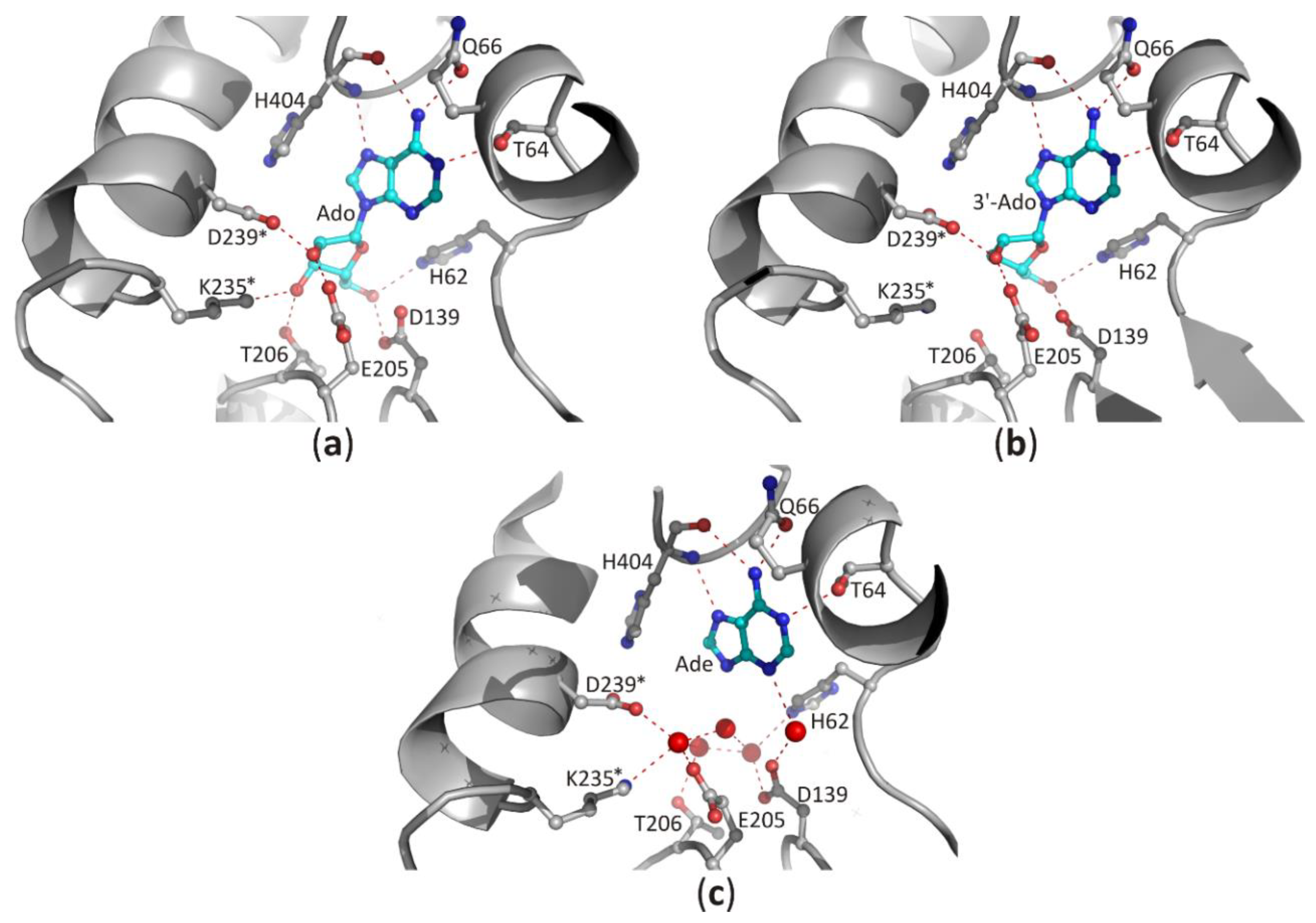
3.2.2. Recognition of the Purine Ring of a Ligand and Its Orientation around N-glycosidic Bond
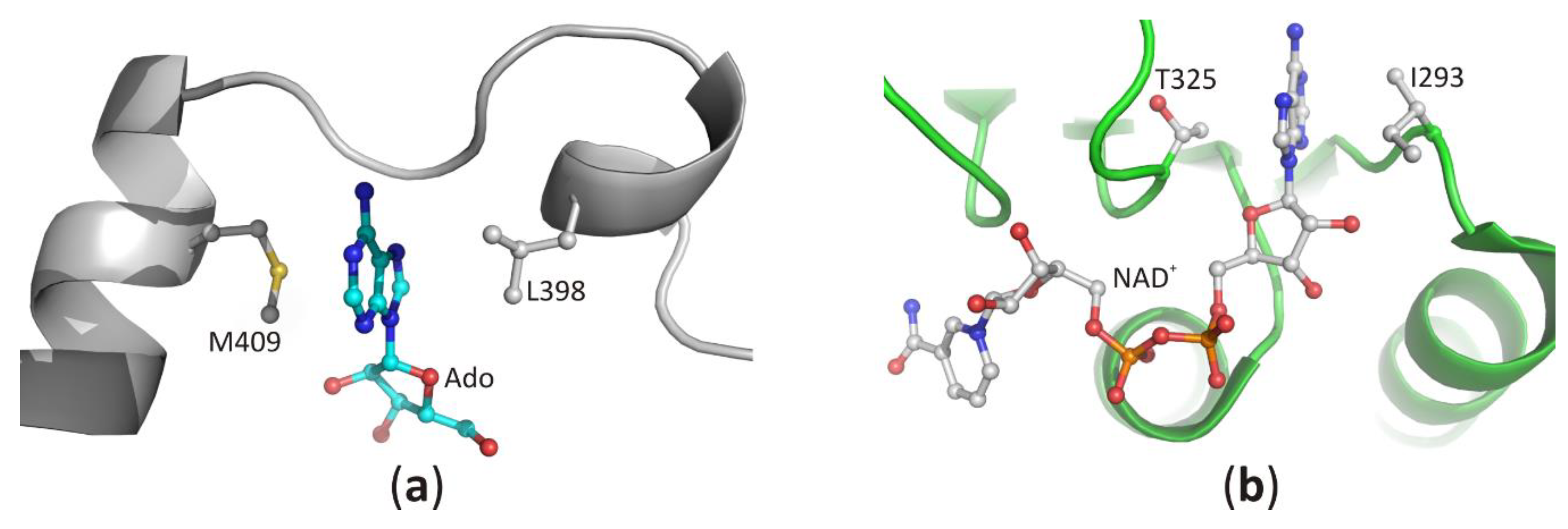
3.2.3. Interaction of the Ligand Sugar Moiety with the Macromolecular Environment
3.2.4. Interaction of Phosphate Groups of Nucleotides with the Macromolecular Environment
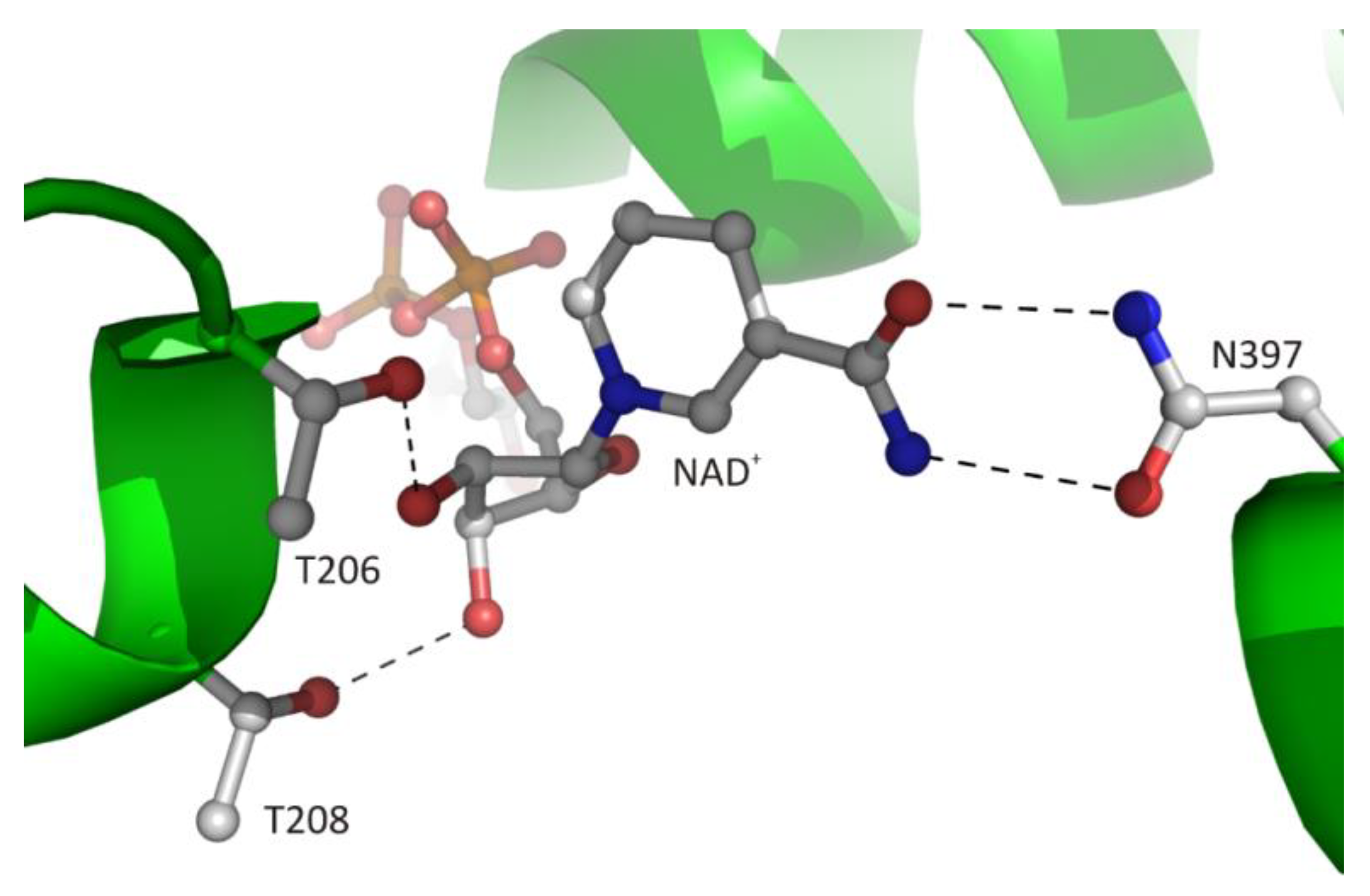
3.2.5. Interaction of the Nicotinamide Moiety with the Macromolecular Environment
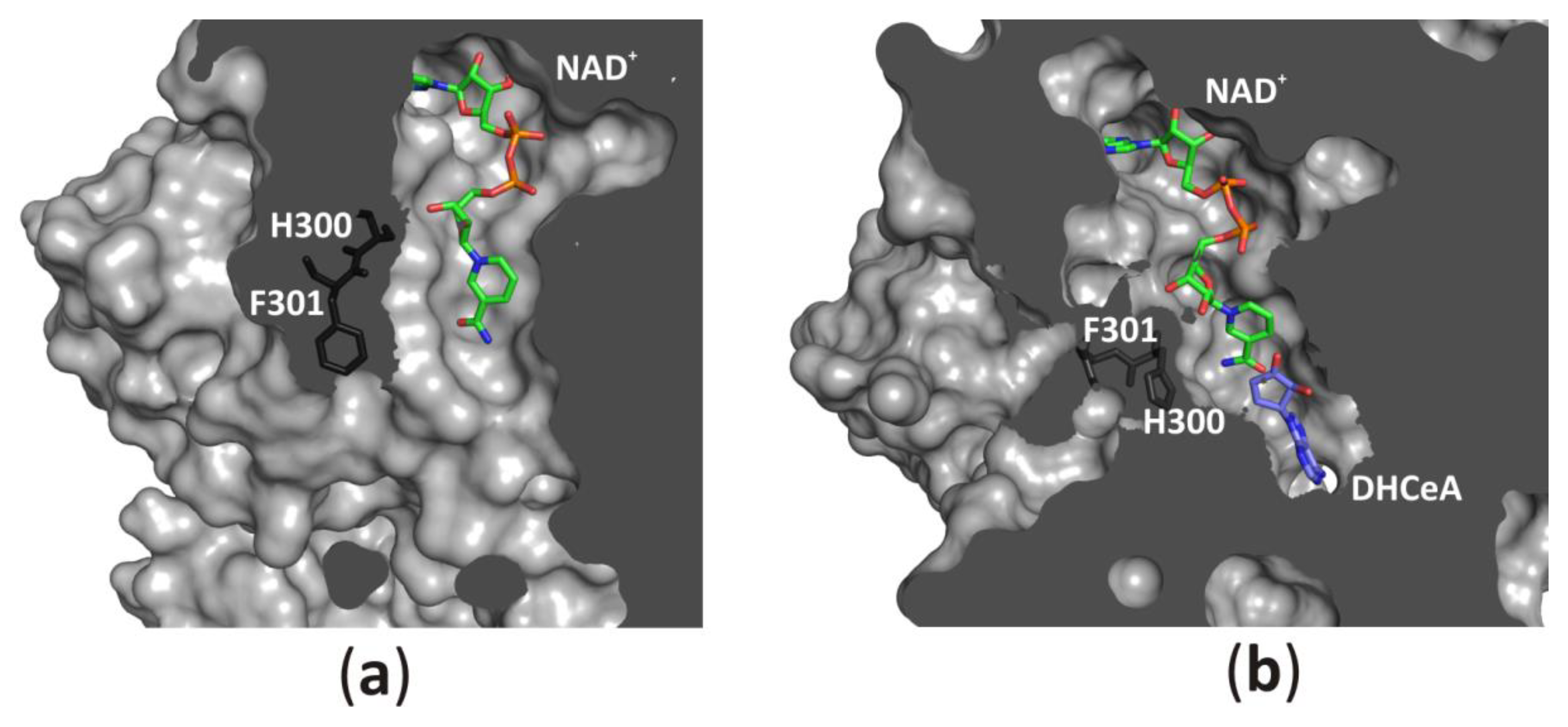
3.2.6. An Access to the Ligand-Binding Pockets
4. Conclusions
Funding
Conflicts of Interest
References
- Rossmann, M.G.; Moras, D.; Olsen, K.W. Chemical and biological evolution of nucleotide-binding protein. Nature 1974, 250, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Mazumder, R.; Vasudevan, S.; Koonin, E.V. Trends in protein evolution inferred from sequence and structure analysis. Curr. Opin. Struct. Biol. 2002, 12, 392–399. [Google Scholar] [CrossRef]
- Xie, L.; Bourne, P.E. Detecting evolutionary relationships across existing fold space, using sequence order-independent profile-profile alignments. Proc. Natl. Acad. Sci. USA 2008, 105, 5441–5446. [Google Scholar] [CrossRef] [Green Version]
- Edwards, H.; Abeln, S.; Deane, C.M. Exploring fold space preferences of new-born and ancient protein superfamilies. PLoS Comput. Biol. 2013, 9. [Google Scholar] [CrossRef] [Green Version]
- Bukhari, S.A.; Caetano-Anollés, G. Origin and evolution of protein fold designs inferred from phylogenomic analysis of CATH domain structures in proteomes. PLoS Comput. Biol. 2013, 9, e1003009. [Google Scholar] [CrossRef] [Green Version]
- Laurino, P.; Tóth-Petróczy, Á.; Meana-Pañeda, R.; Lin, W.; Truhlar, D.G.; Tawfik, D.S. An Ancient Fingerprint Indicates the Common Ancestry of Rossmann-Fold Enzymes Utilizing Different Ribose-Based Cofactors. PLoS Biol. 2016, 14, e1002396. [Google Scholar] [CrossRef] [Green Version]
- Chouhan, B.P.S.; Maimaiti, S.; Gade, M.; Laurino, P. Rossmann-Fold Methyltransferases: Taking a “β-Turn” around Their Cofactor, S-Adenosylmethionine. Biochemistry 2019, 58, 166–170. [Google Scholar] [CrossRef]
- Turner, M.A.; Yuan, C.S.; Borchardt, R.T.; Hershfield, M.S.; Smith, G.D.; Howell, P.L. Structure determination of selenomethionyl S-adenosylhomocysteine hydrolase using data at a single wavelength. Nat. Struct. Biol. 1998, 5, 369–376. [Google Scholar] [CrossRef]
- Hu, Y.; Komoto, J.; Huang, Y.; Gomi, T.; Ogawa, H.; Takata, Y.; Fujioka, M.; Takusagawa, F. Crystal structure of S-adenosylhomocysteine hydrolase from rat liver. Biochemistry 1999, 38, 8323–8333. [Google Scholar] [CrossRef] [PubMed]
- Poulton, J.E.; Butt, V.S. Purification and properties of S-adenosyl-l-methionine: Caffeic acid O-methyltransferase from leaves of spinach beet (Beta vulgaris L.). Biochim. Biophys. Acta 1975, 403, 301–314. [Google Scholar] [CrossRef]
- Chiang, P.K.; Cantoni, G.L. Perturbation of biochemical transmethylations by 3-deazaadenosine in vivo. Biochem. Pharmacol. 1979, 28, 1897–1902. [Google Scholar] [CrossRef]
- Chiang, P.K. Biological effects of inhibitors of S-adenosylhomocysteine hydrolase. Pharmacol. Ther. 1998, 77, 115–134. [Google Scholar] [CrossRef]
- Stepkowski, T.; Brzeziński, K.; Legocki, A.B.; Jaskólski, M.; Béna, G. Bayesian phylogenetic analysis reveals two-domain topology of S-adenosylhomocysteine hydrolase protein sequences. Mol. Phylogenet. Evol. 2005, 34, 15–28. [Google Scholar] [CrossRef]
- De La Haba, G.; Cantoni, G.L. The enzymatic synthesis of S-adenosyl-l-homocysteine from adenosine and homocysteine. J. Biol. Chem. 1959, 234, 603–608. [Google Scholar] [PubMed]
- Richards, H.H.; Chiang, P.K.; Cantoni, G.L. Adenosylhomocysteine hydrolase. Crystallization of the purified enzyme and its properties. J. Biol. Chem. 1978, 253, 4476–4480. [Google Scholar]
- Hershfield, M.S.; Krodich, N.M. S-adenosylhomocysteine hydrolase is an adenosine-binding protein: A target for adenosine toxicity. Science 1978, 202, 757–760. [Google Scholar] [CrossRef]
- Kredich, N.M.; Martin, D.V. Role of S-adenosylhomocysteine in adenosinemediated toxicity in cultured mouse T lymphoma cells. Cell 1977, 12, 931–938. [Google Scholar] [CrossRef]
- Nygård, O.; Nordrehaug, J.E.; Refsum, H.; Ueland, P.M.; Farstad, M.; Vollset, S.E. Plasma homocysteine levels and mortality in patients with coronary artery disease. N. Engl. J. Med. 1997, 337, 230–236. [Google Scholar] [CrossRef]
- Kusakabe, Y.; Ishihara, M.; Umeda, T.; Kuroda, D.; Nakanishi, M.; Kitade, Y.; Gouda, H.; Nakamura, K.T.; Tanaka, N. Structural insights into the reaction mechanism of S-adenosyl-l-homocysteine hydrolase. Sci. Rep. 2015, 5, 16641. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Nakanishi, M.; Kusakabe, Y.; Shiraiwa, K.; Yabe, S.; Ito, Y.; Kitade, Y.; Nakamura, K.T. Crystal structure of S-adenosyl-l-homocysteine hydrolase from the human malaria parasite Plasmodium falciparum. J. Mol. Biol. 2004, 343, 1007–1017. [Google Scholar] [CrossRef]
- Brzezinski, K.; Dauter, Z.; Jaskolski, M. High-resolution structures of complexes of plant S-adenosyl-l-homocysteine hydrolase (Lupinus luteus). Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 218–231. [Google Scholar] [CrossRef] [Green Version]
- Reddy, M.C.M.; Kuppan, G.; Shetty, N.D.; Owen, J.L.; Ioerger, T.R.; Sacchettini, J.C. Crystal structures of Mycobacterium tuberculosis S-adenosyl-l-homocysteine hydrolase in ternary complex with substrate and inhibitors. Protein Sci. Publ. Protein Soc. 2008, 17, 2134–2144. [Google Scholar] [CrossRef]
- Manszewski, T.; Singh, K.; Imiolczyk, B.; Jaskolski, M. An enzyme captured in two conformational states: Crystal structure of S-adenosyl-l-homocysteine hydrolase from Bradyrhizobium elkanii. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 2422–2432. [Google Scholar] [CrossRef]
- Manszewski, T.; Szpotkowski, K.; Jaskolski, M. Crystallographic and SAXS studies of S-adenosyl-l-homocysteine hydrolase from Bradyrhizobium elkanii. IUCrJ 2017, 4, 271–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czyrko, J.; Jaskolski, M.; Brzezinski, K. Crystal Structure of S-adenosyl-l-homocysteine Hydrolase from Cytophaga hutchinsonii, a Case of Combination of Crystallographic and Non-crystallographic Symmetry. Croat. Chem. Acta 2018, 91, 153–162. [Google Scholar] [CrossRef]
- Czyrko, J.; Sliwiak, J.; Imiolczyk, B.; Gdaniec, Z.; Jaskolski, M.; Brzezinski, K. Metal-cation regulation of enzyme dynamics is a key factor influencing the activity of S-adenosyl-l-homocysteine hydrolase from Pseudomonas aeruginosa. Sci. Rep. 2018, 8, 11334. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chen, C.-C.; Ko, T.-P.; Xiao, X.; Yang, Y.; Huang, C.-H.; Qian, G.; Shao, W.; Guo, R.-T. Crystal structures of S-adenosylhomocysteine hydrolase from the thermophilic bacterium Thermotoga maritima. J. Struct. Biol. 2015, 190, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Brzezinski, K.; Czyrko, J.; Sliwiak, J.; Nalewajko-Sieliwoniuk, E.; Jaskolski, M.; Nocek, B.; Dauter, Z. S-adenosyl-l-homocysteine hydrolase from a hyperthermophile (Thermotoga maritima) is expressed in Escherichia coli in inactive form-Biochemical and structural studies. Int. J. Biol. Macromol. 2017, 104, 584–596. [Google Scholar] [CrossRef]
- Guranowski, A.; Pawelkiewicz, J. Adenosylhomocysteinase from yellow lupin seeds. Purification and properties. Eur. J. Biochem. 1977, 80, 517–523. [Google Scholar] [CrossRef]
- Brzezinski, K.; Bujacz, G.; Jaskolski, M. Purification, crystallization and preliminary crystallographic studies of plant S-adenosyl-l-homocysteine hydrolase (Lupinus luteus). Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun. 2008, 64, 671–673. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- The PyMOL Molecular Graphics System, Version 1.8. Schrödinger, LLC. Available online: https://pymol.org/2/support.html?#citing (accessed on 16 December 2020).
- Bond, C.S. TopDraw: A sketchpad for protein structure topology cartoons. Bioinformatics 2003, 19, 311–312. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Hu, Y.; Yin, D.H.; Turner, M.A.; Wang, M.; Borchardt, R.T.; Howell, P.L.; Kuczera, K.; Schowen, R.L. Catalytic strategy of S-adenosyl-l-homocysteine hydrolase: Transition-state stabilization and the avoidance of abortive reactions. Biochemistry 2003, 42, 1900–1909. [Google Scholar] [CrossRef]
- Denessiouk, K.A.; Rantanen, V.V.; Johnson, M.S. Adenine recognition: A motif present in ATP-, CoA-, NAD-, NADP-, and FAD-dependent proteins. Proteins 2001, 44, 282–291. [Google Scholar] [CrossRef]
- Vidal, L.S.; Kelly, C.L.; Mordaka, P.M.; Heap, J.T. Review of NAD(P)H-dependent oxidoreductases: Properties, engineering and application. Biochim. Biophys. Acta BBA Proteins Proteom. 2018, 1866, 327–347. [Google Scholar] [CrossRef]
- Grzechowiak, M.; Sliwiak, J.; Jaskolski, M.; Ruszkowski, M. Structural Studies of Glutamate Dehydrogenase (Isoform 1) From Arabidopsis thaliana, an Important Enzyme at the Branch-Point Between Carbon and Nitrogen Metabolism. Front. Plant Sci. 2020, 11, 754. [Google Scholar] [CrossRef]
- Palmer, J.L.; Abeles, R.H. The mechanism of action of S-adenosylhomocysteinase. J. Biol. Chem. 1979, 254, 1217–1226. [Google Scholar] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Nucleoside Ligand Bound | Conformation | PDB Entry |
|---|---|---|---|
| Homo sapiens | neplanocin | closed | 1LI4 [38] |
| Rattus norvegicus | none | open | 1B3R [9] |
| Mus musculus | Ado | closed | 5AXA [19] |
| Plasmodium falciparum | Ado | closed | 1V8B [20] |
| Trypanosoma brucei | adenine | closed | 3H9U |
| Leishmania major | Ado | closed | 3G1U |
| Cryptosporidium parvum | Ado | closed | 5HM8 |
| Acanthamoeba castellanii | Ado | closed | 6UK3 |
| Naegleria fowleri | Ado | closed | 5V96 |
| Lupinus luteus | Ado | closed | 3OND [21] |
| Lupinus luteus | adenine | closed | 3ONE [21] |
| Lupinus luteus | 3′-deoxyadenosine | closed | 3ONF [21] |
| Mycobacterium tuberculosis | Ado | closed | 3CE6 [22] |
| Bradyrhizobium elkanii | Ado | Closed 1 | 4LVC [23] |
| Bradyrhizobium elkanii | none | Open 1 | 4LVC [23] |
| Cytophaga hutchinsonii | Ado | closed | 6GBN [25] |
| Pseudomonas aeruginosa | Ado | closed | 6F3M [26] |
| Pseudomonas aeruginosa | 3′-deoxyadenosine | closed | 6F3P [26] |
| Pseudomonas aeruginosa | adenine | closed | 6F3Q [26] |
| Burkholderia pseudomalei | none | open | 3D64 |
| Burkholderia pseudomalei | 9-β-d-arabinofuranosyl-adenine | closed | 3GLQ |
| Brucella abortus | Ado | closed | 3N58 |
| Elizabethkingia anopheles | Ado | closed | 6APH |
| Thermotoga maritima | none | open | 3X2E [27] |
| Thermotoga maritima | none | Closed 2 | 3X2F [27] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brzezinski, K. S-adenosyl-l-homocysteine Hydrolase: A Structural Perspective on the Enzyme with Two Rossmann-Fold Domains. Biomolecules 2020, 10, 1682. https://doi.org/10.3390/biom10121682
Brzezinski K. S-adenosyl-l-homocysteine Hydrolase: A Structural Perspective on the Enzyme with Two Rossmann-Fold Domains. Biomolecules. 2020; 10(12):1682. https://doi.org/10.3390/biom10121682
Chicago/Turabian StyleBrzezinski, Krzysztof. 2020. "S-adenosyl-l-homocysteine Hydrolase: A Structural Perspective on the Enzyme with Two Rossmann-Fold Domains" Biomolecules 10, no. 12: 1682. https://doi.org/10.3390/biom10121682
APA StyleBrzezinski, K. (2020). S-adenosyl-l-homocysteine Hydrolase: A Structural Perspective on the Enzyme with Two Rossmann-Fold Domains. Biomolecules, 10(12), 1682. https://doi.org/10.3390/biom10121682