Recent Synthetic Approaches towards Small Molecule Reactivators of p53

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
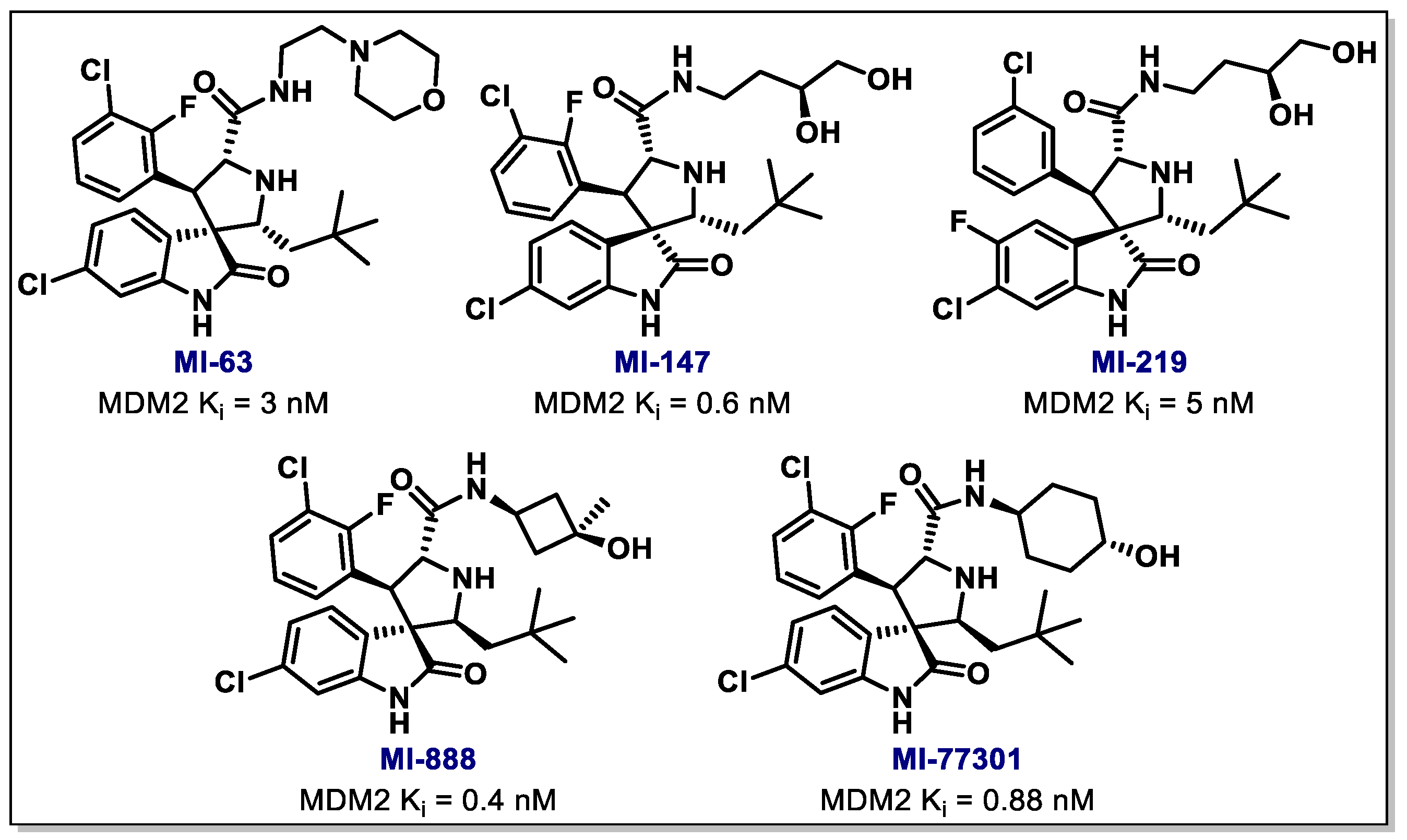{kind=link}
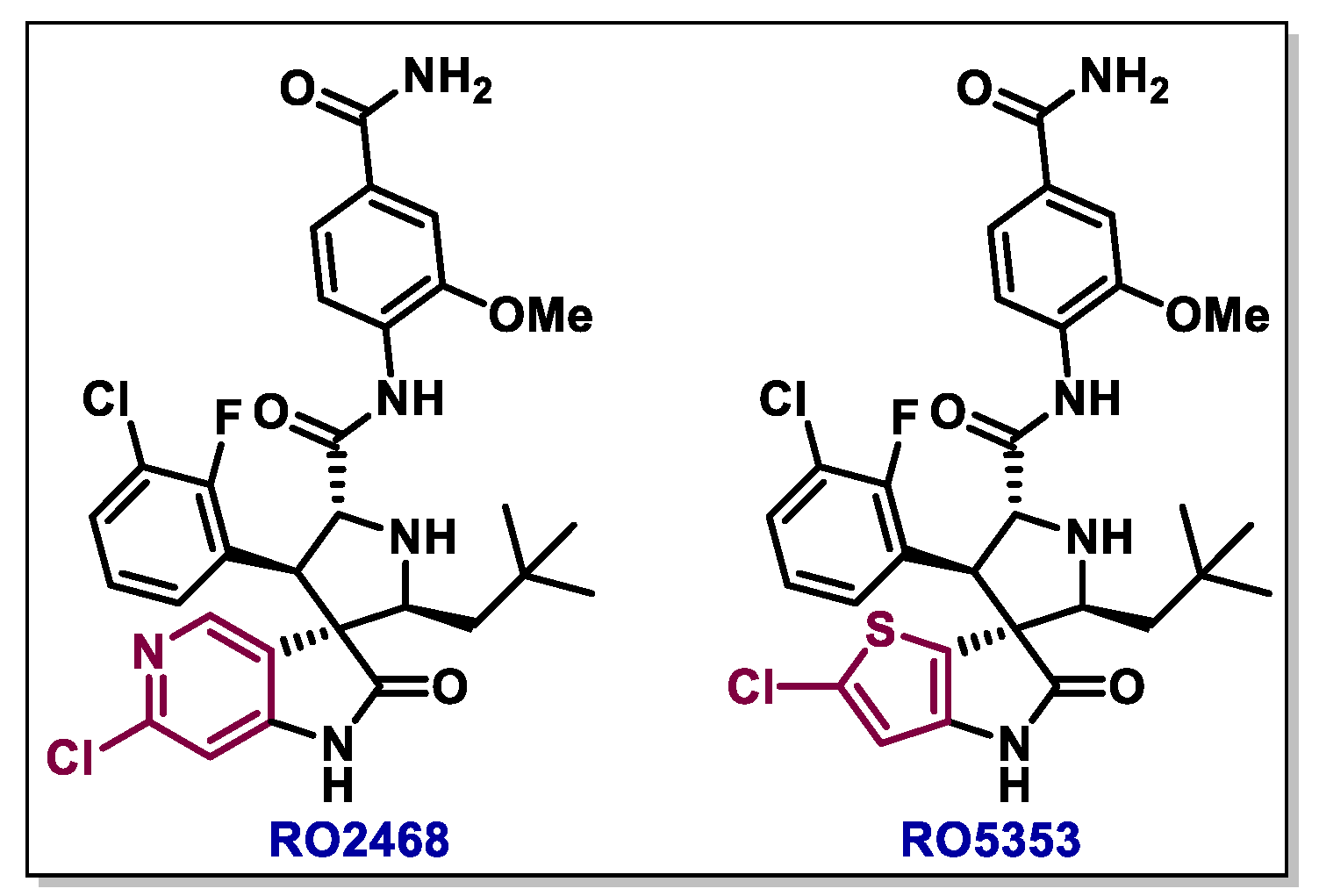{kind=link}
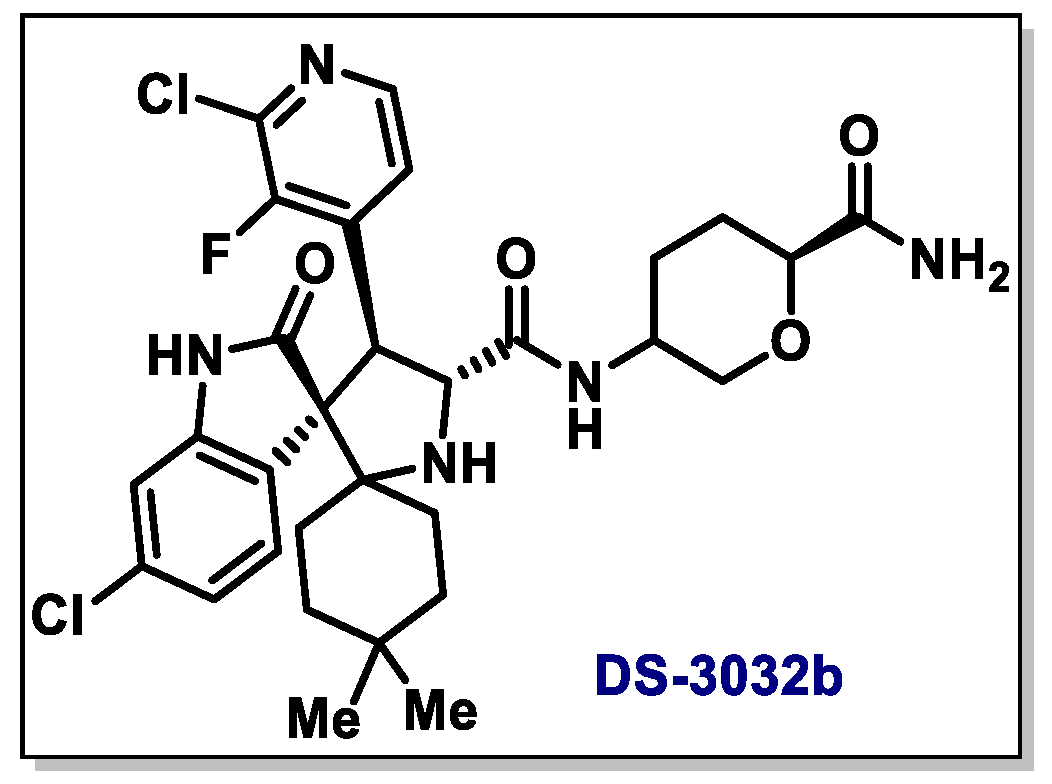{kind=link}
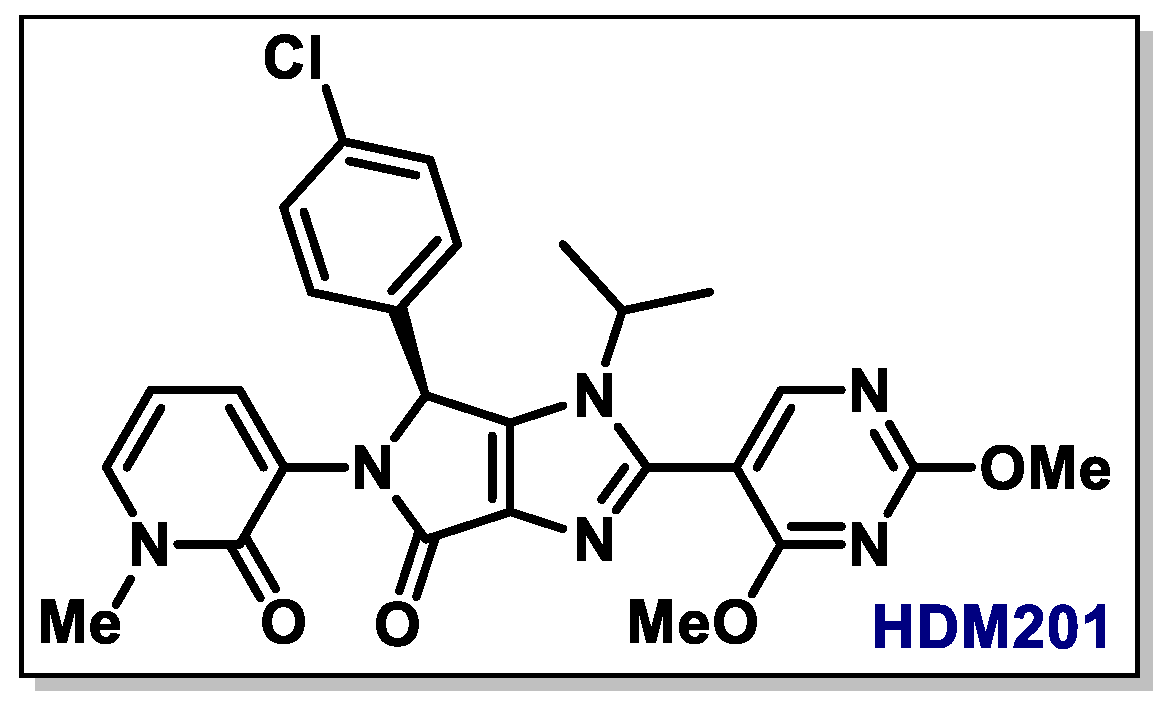{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
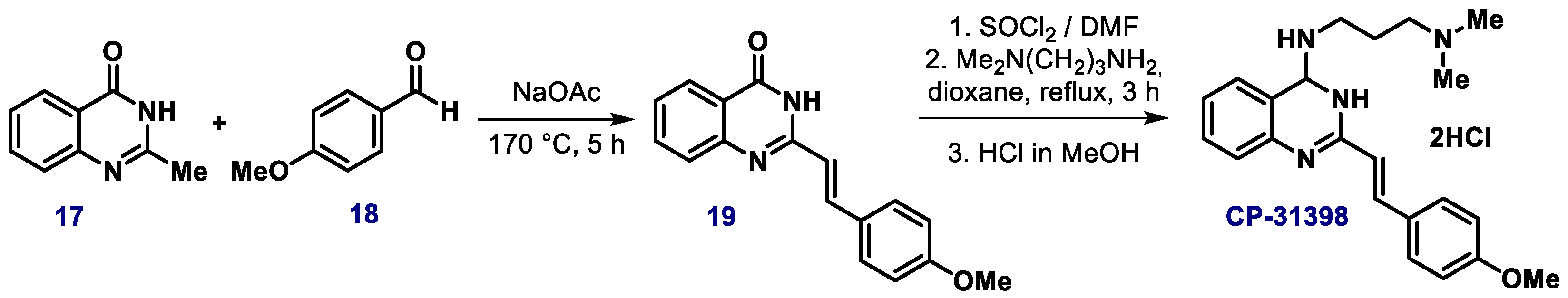{kind=link}
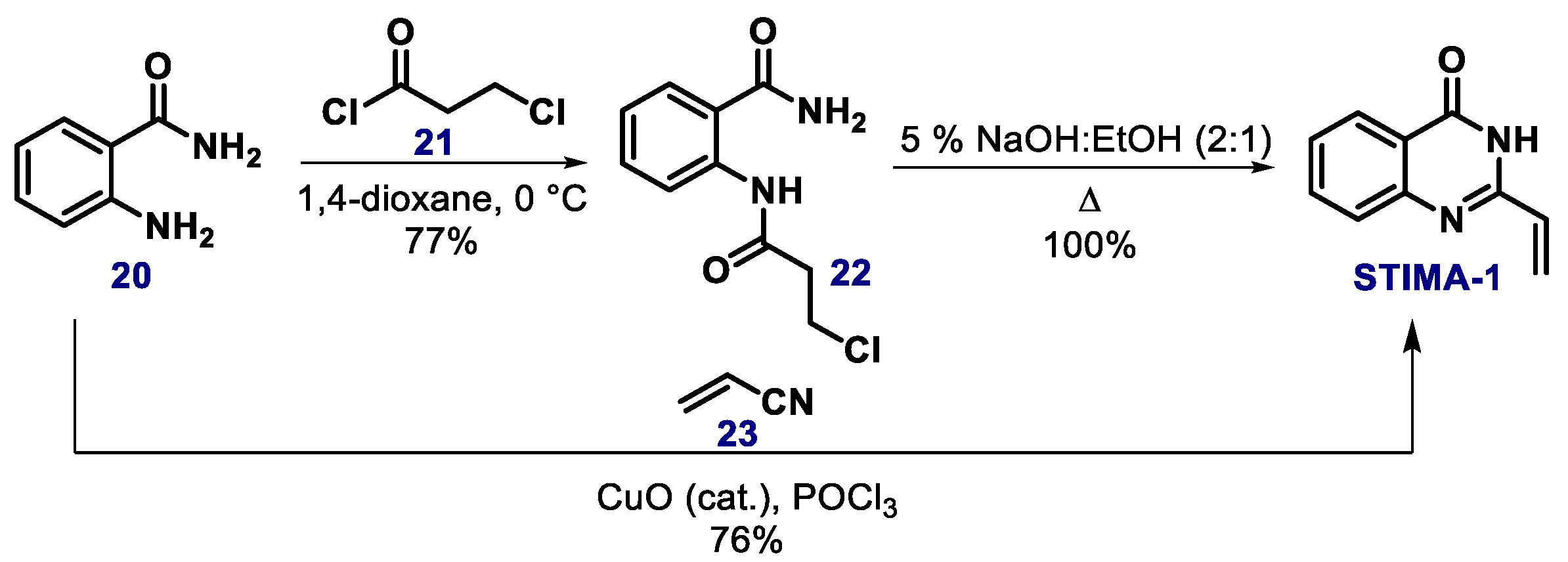{kind=link}
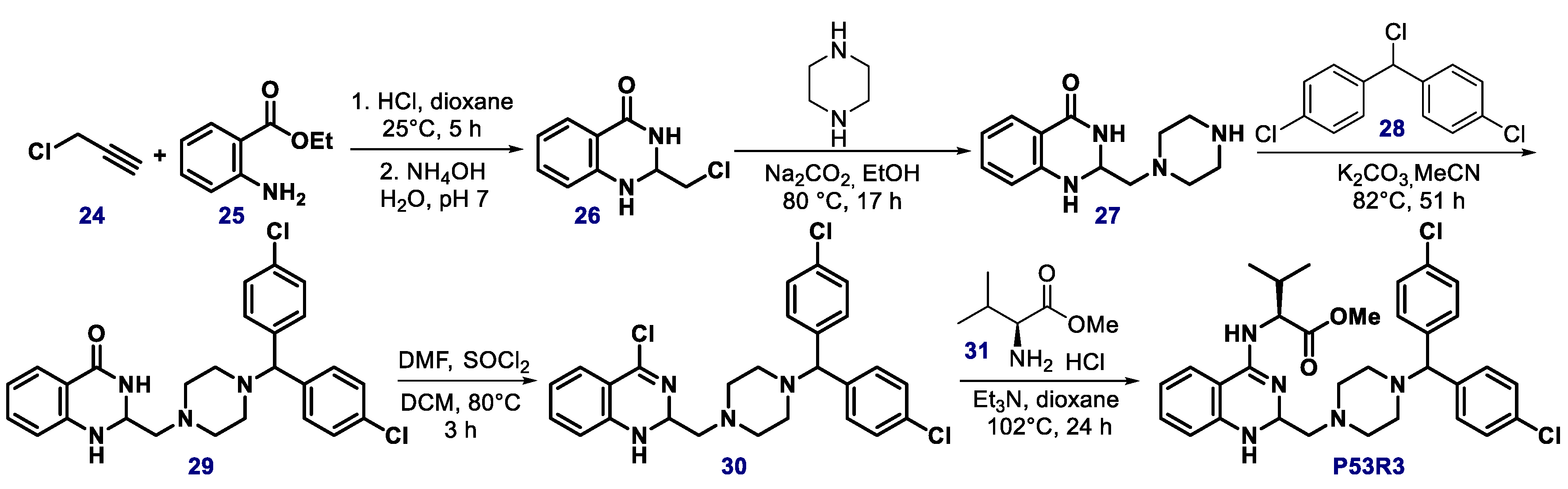{kind=link}
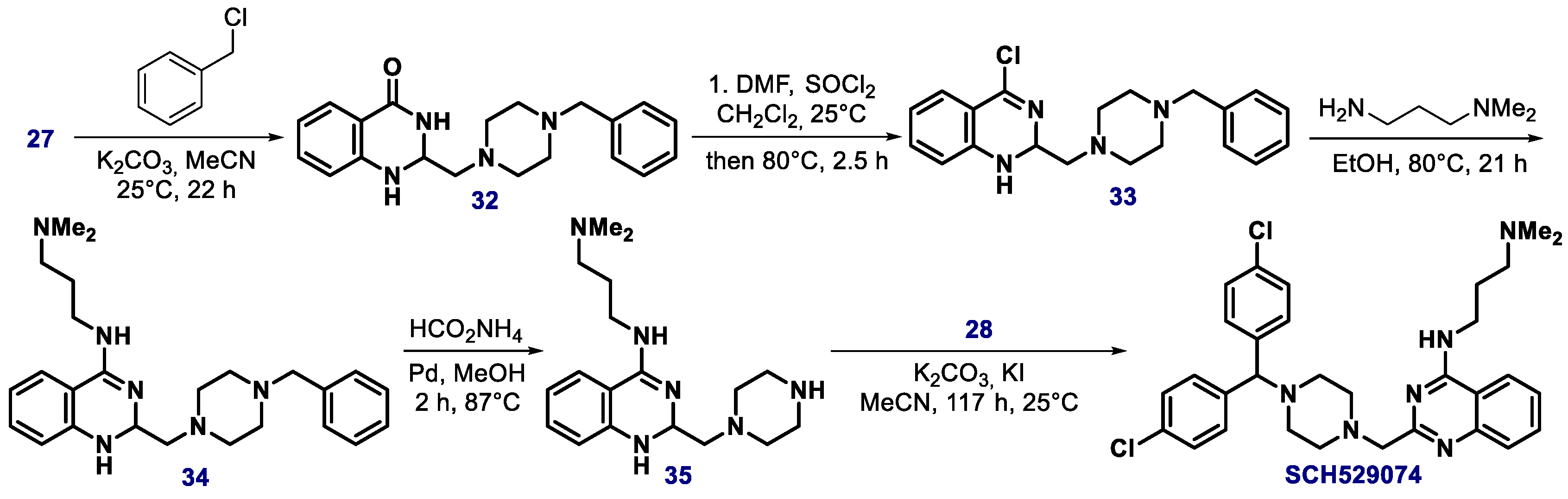{kind=link}
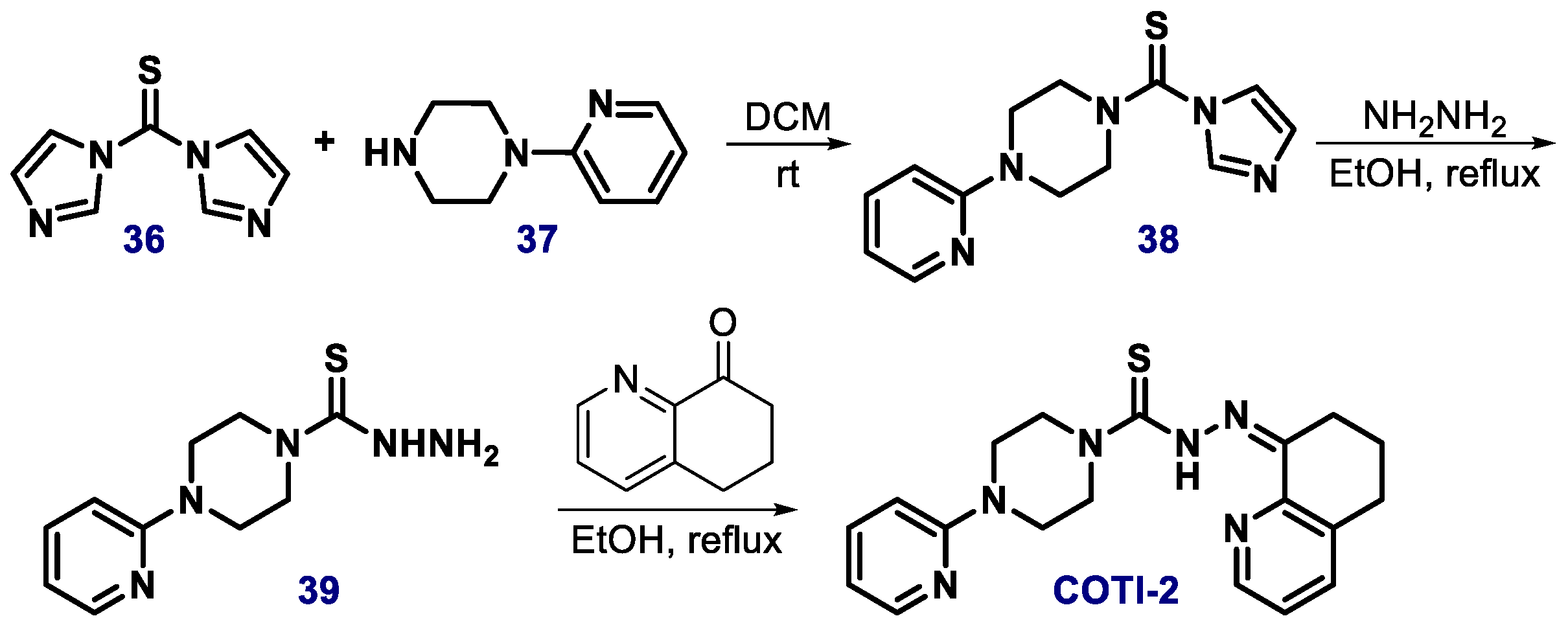{kind=link}
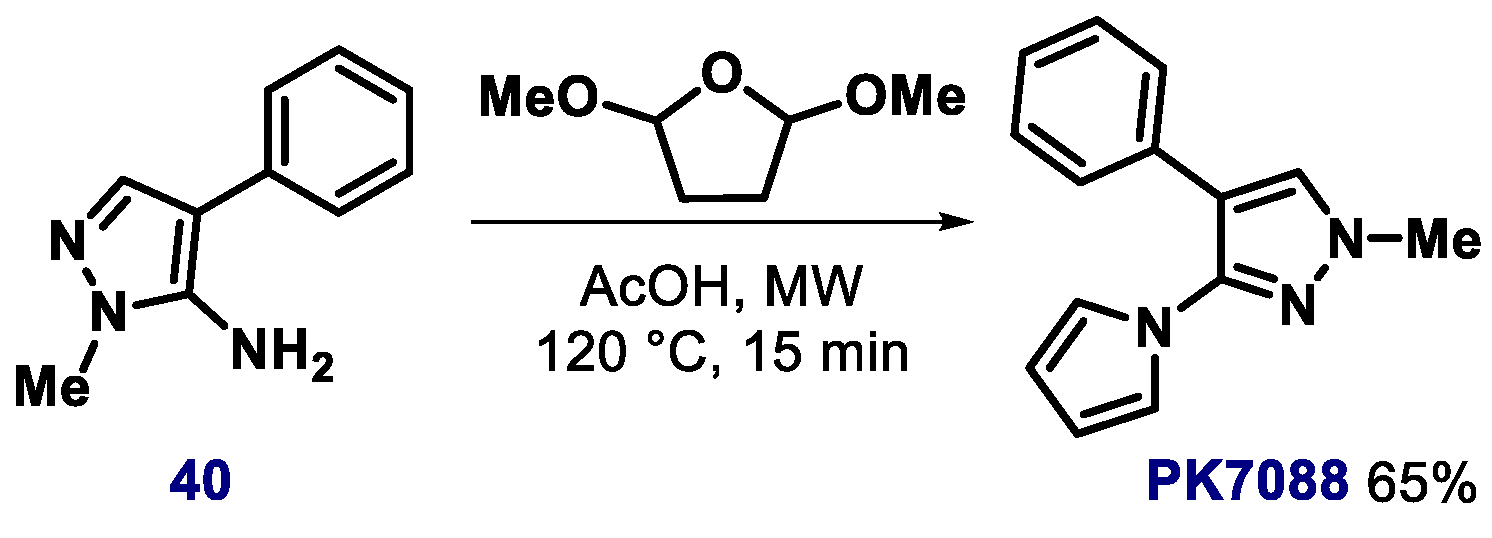{kind=link}
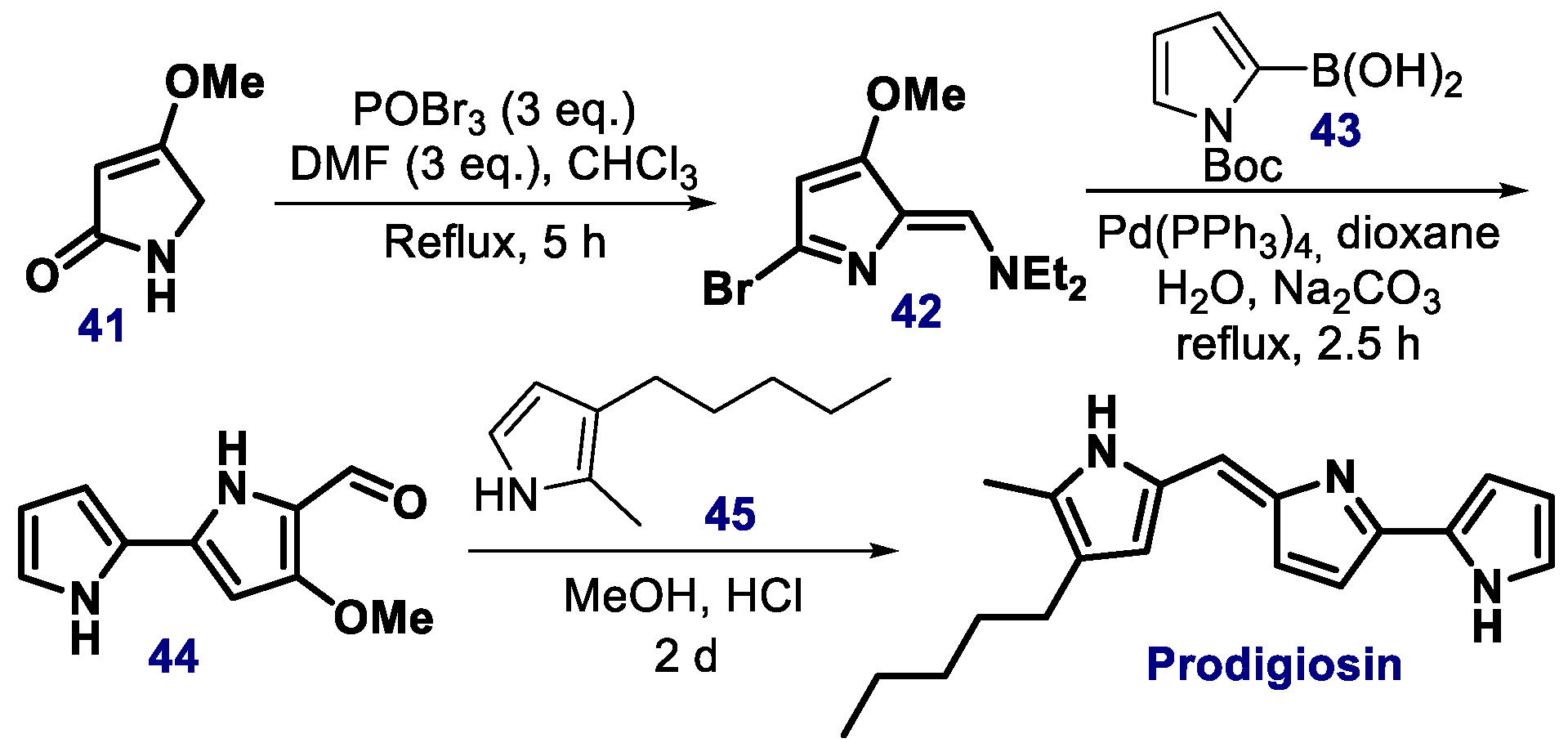{kind=link}
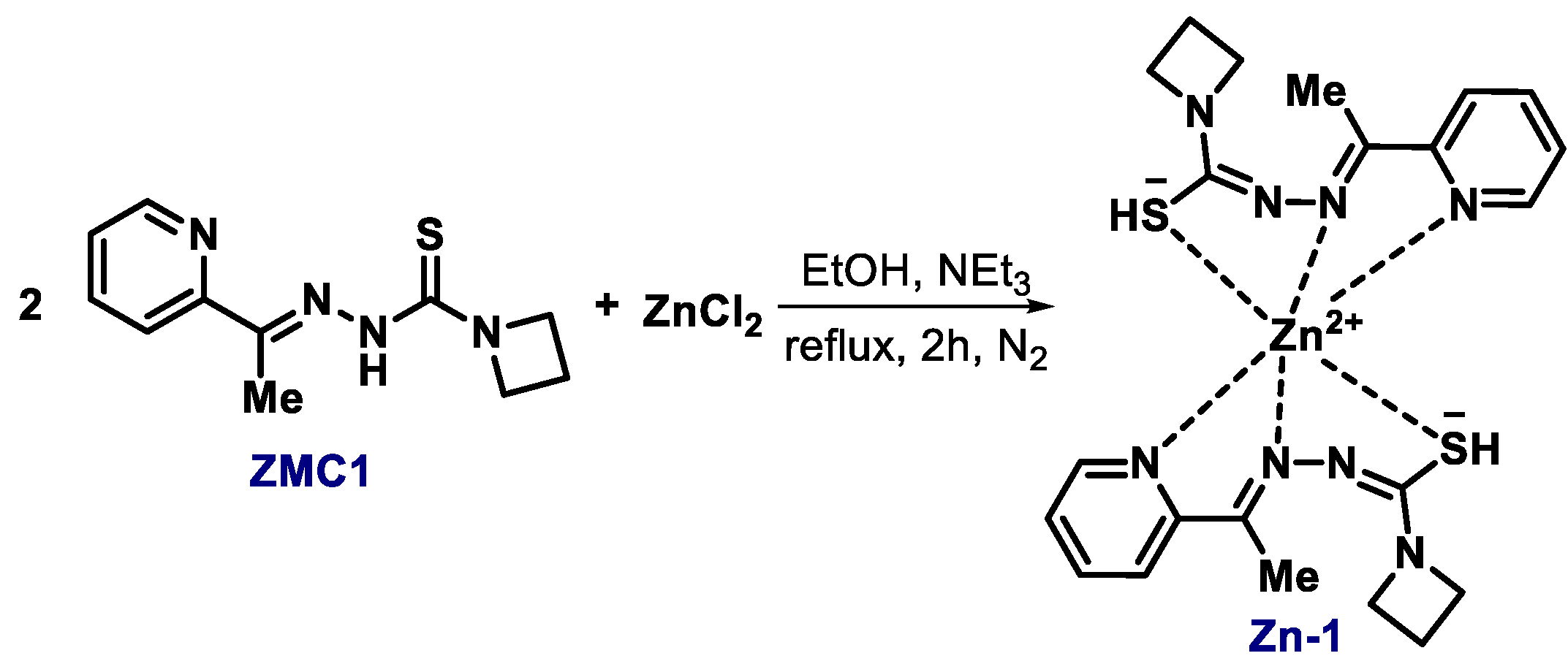{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
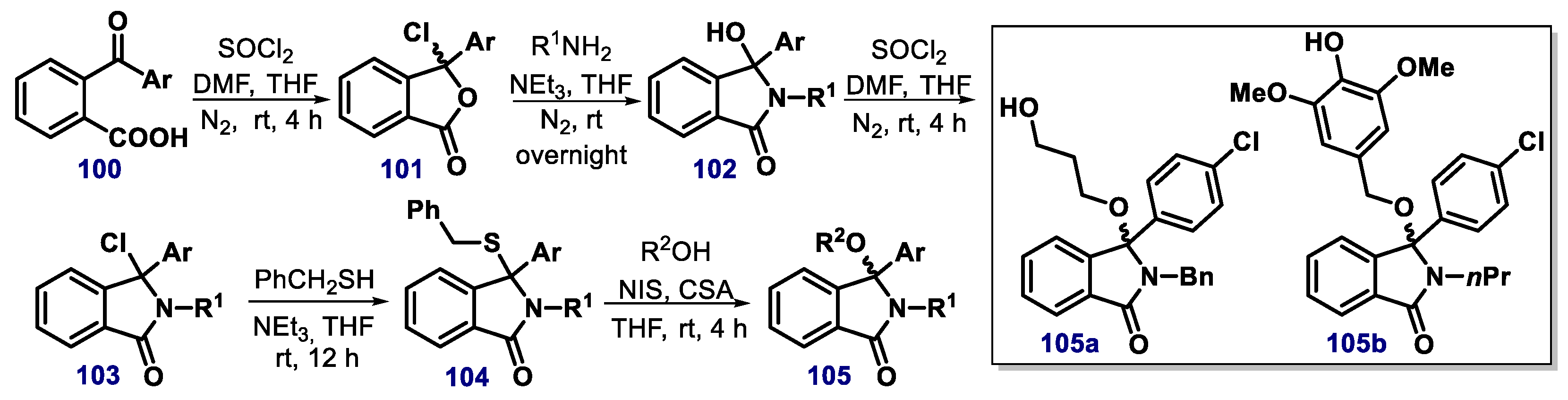{kind=link}
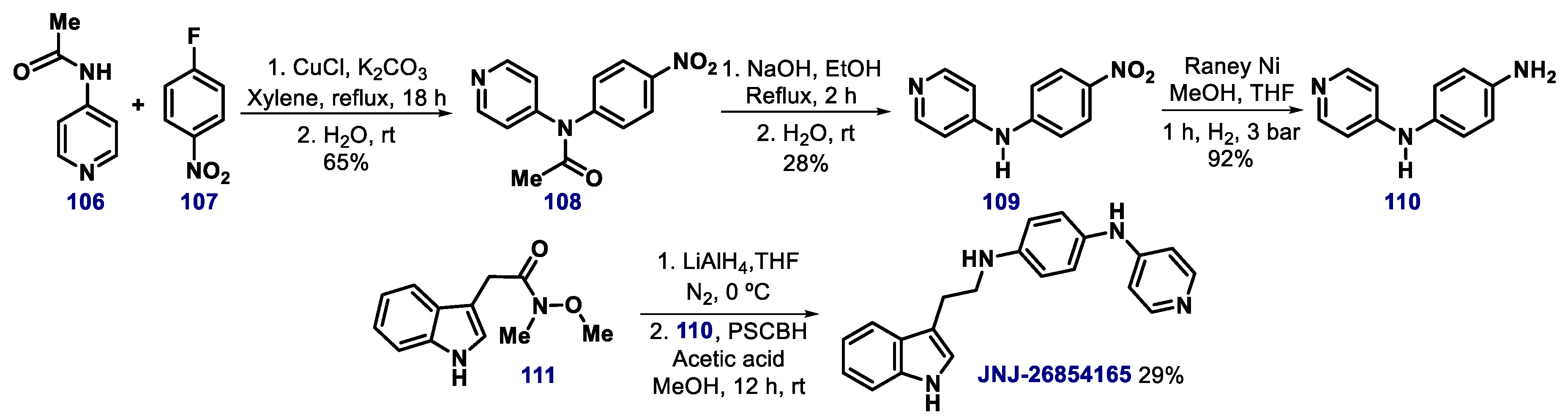{kind=link}
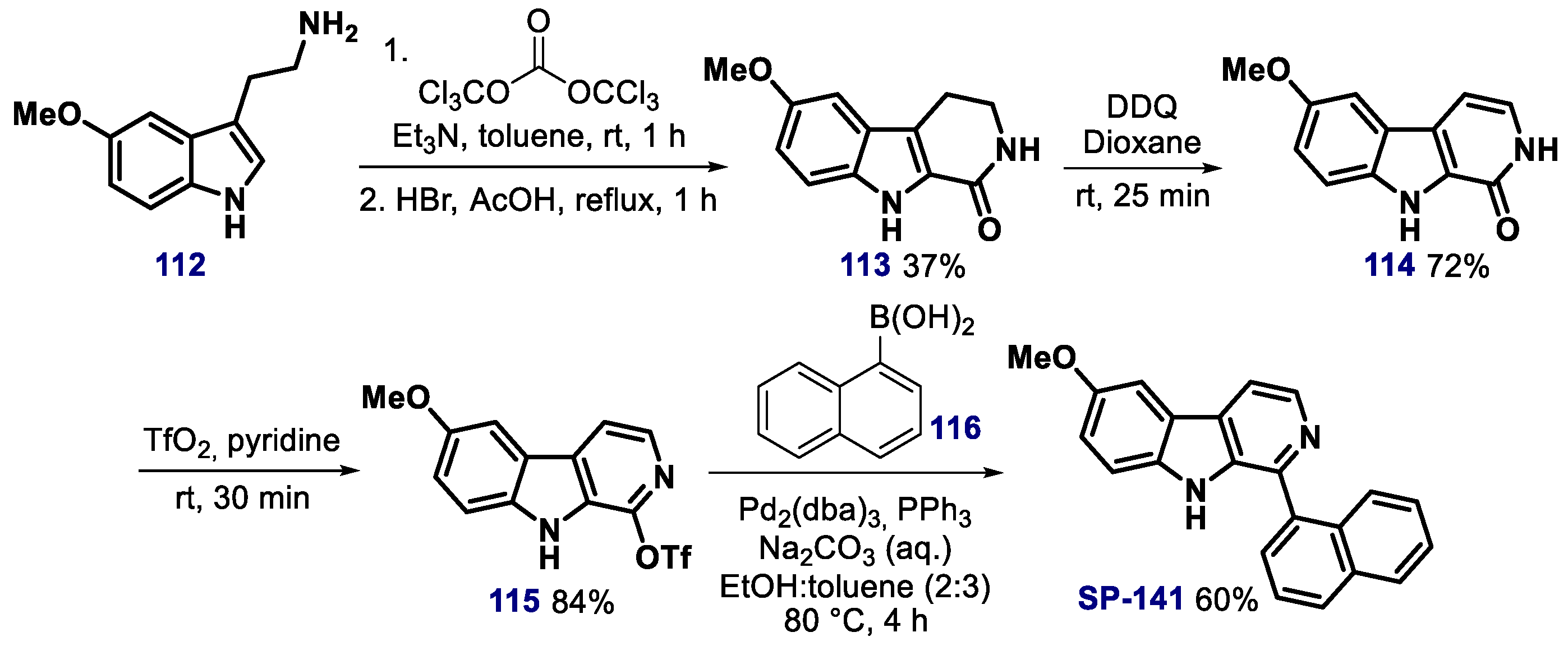{kind=link}
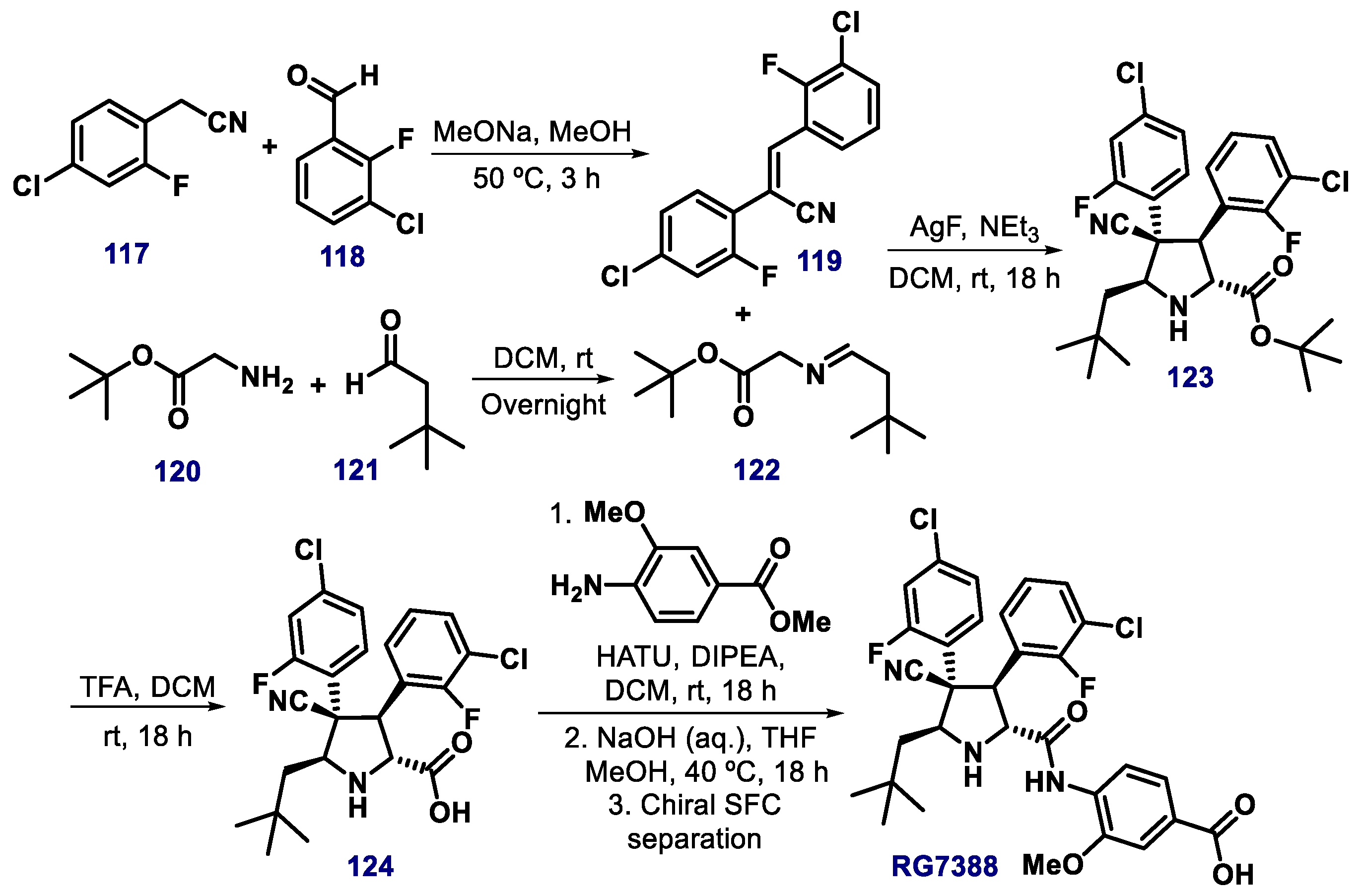{kind=link}
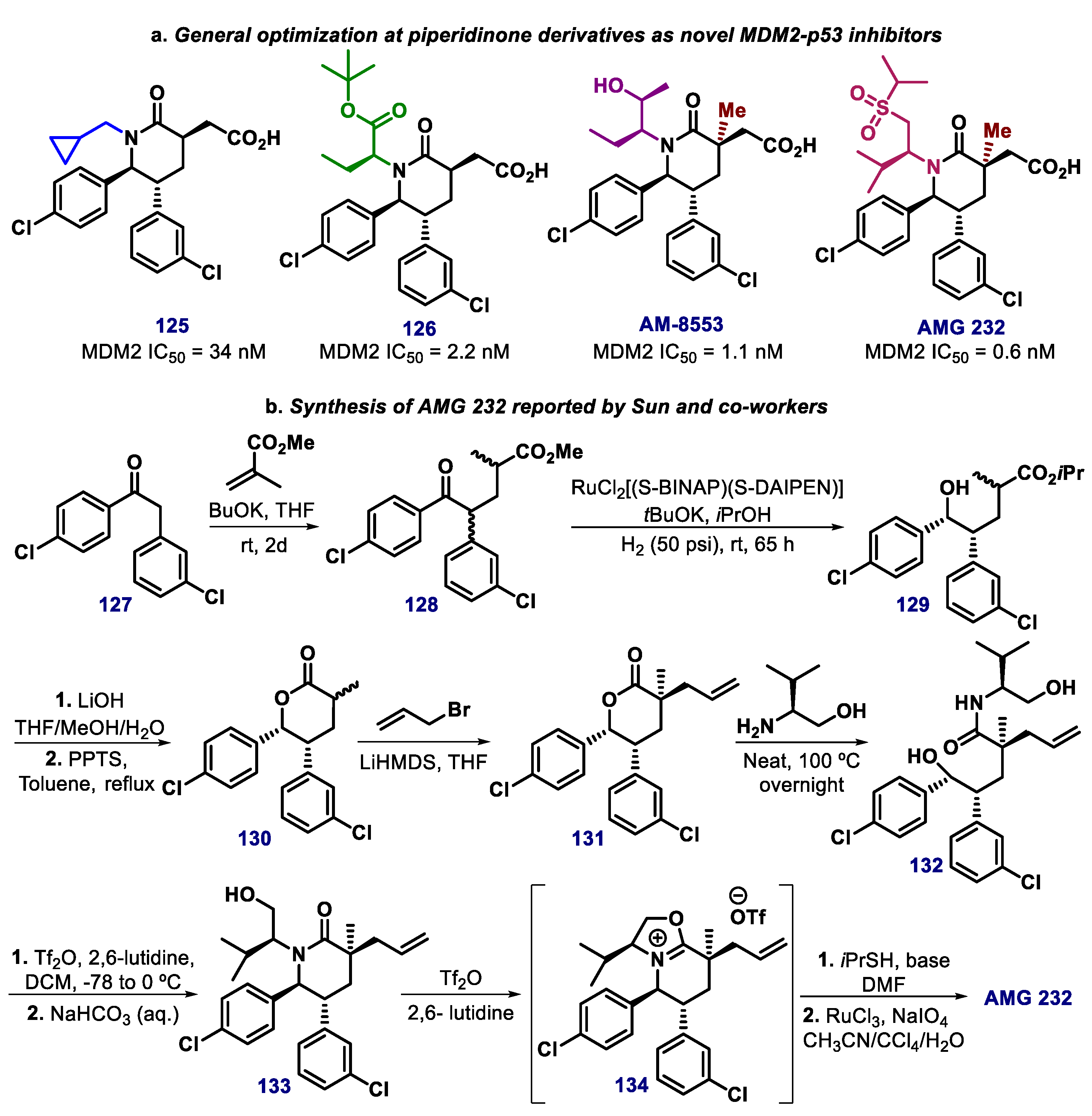{kind=link}
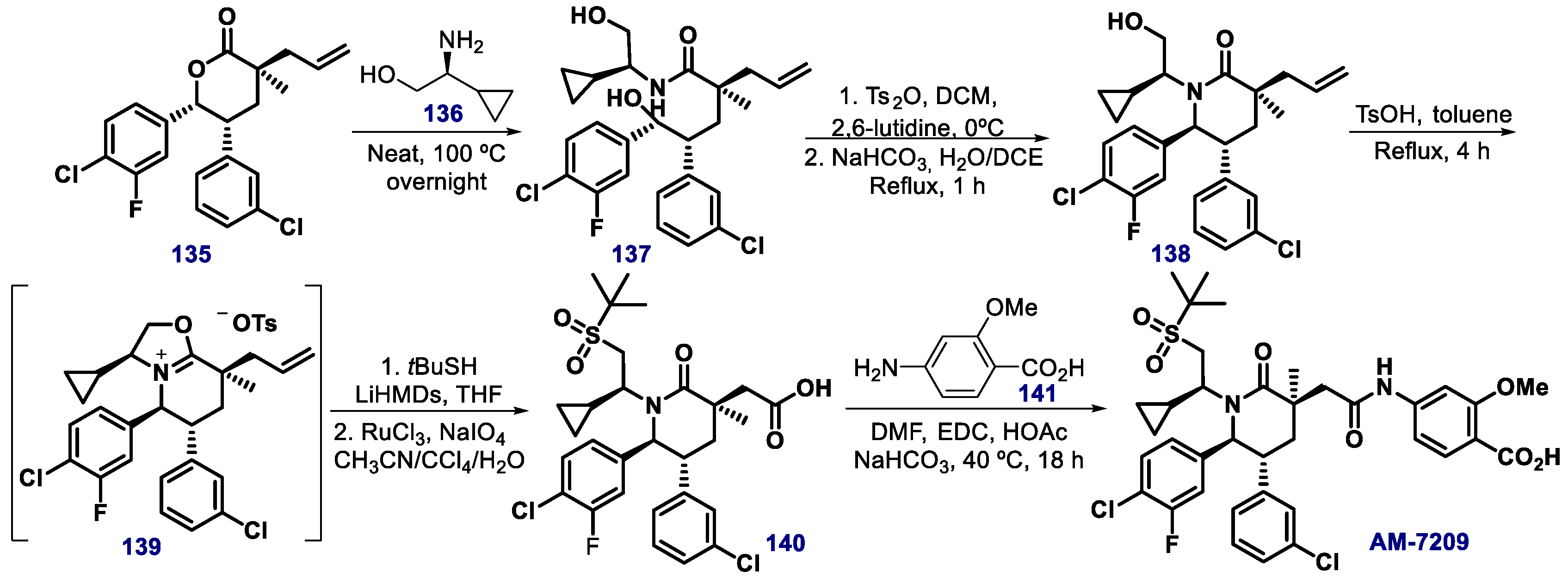{kind=link}
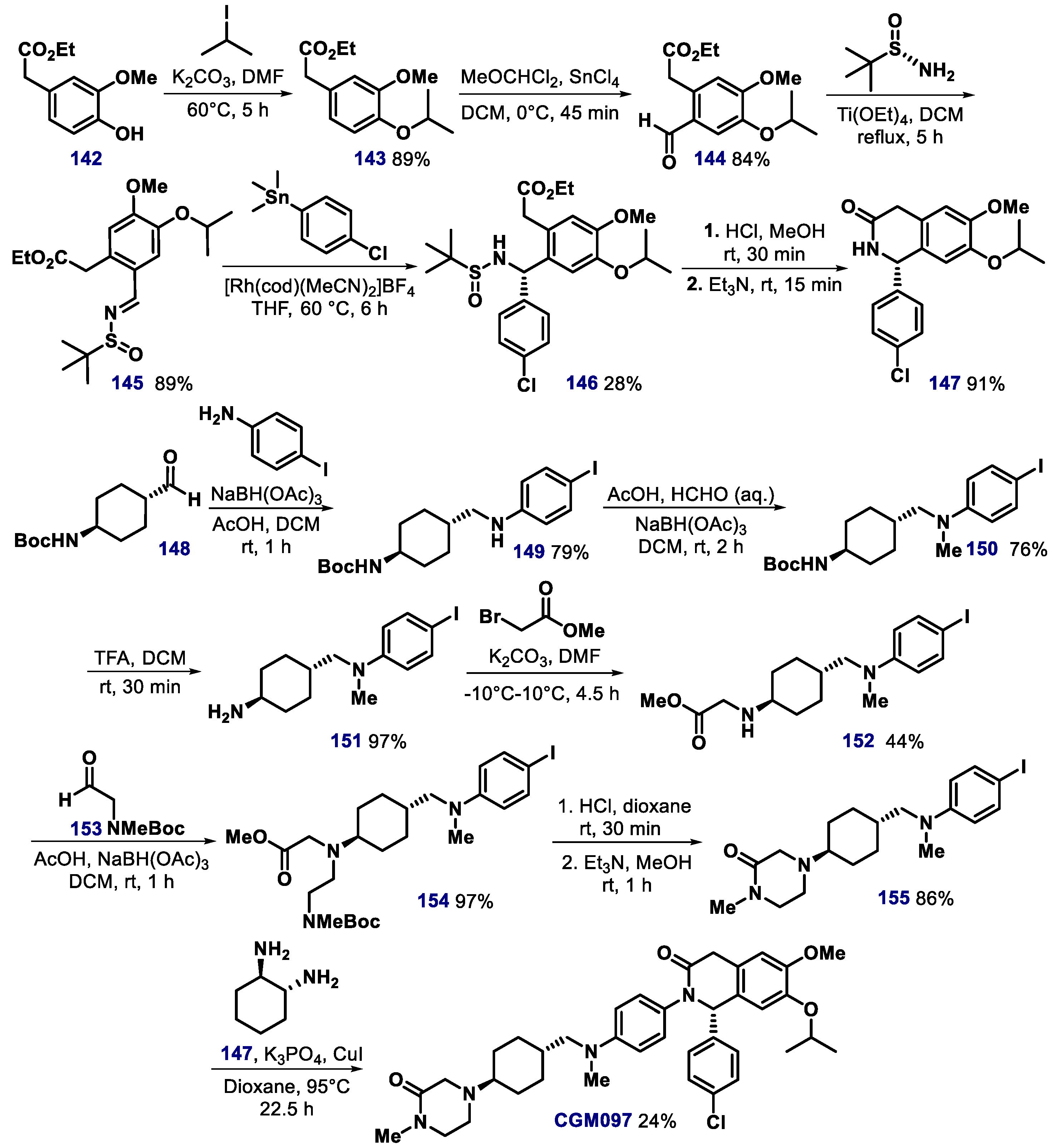{kind=link}
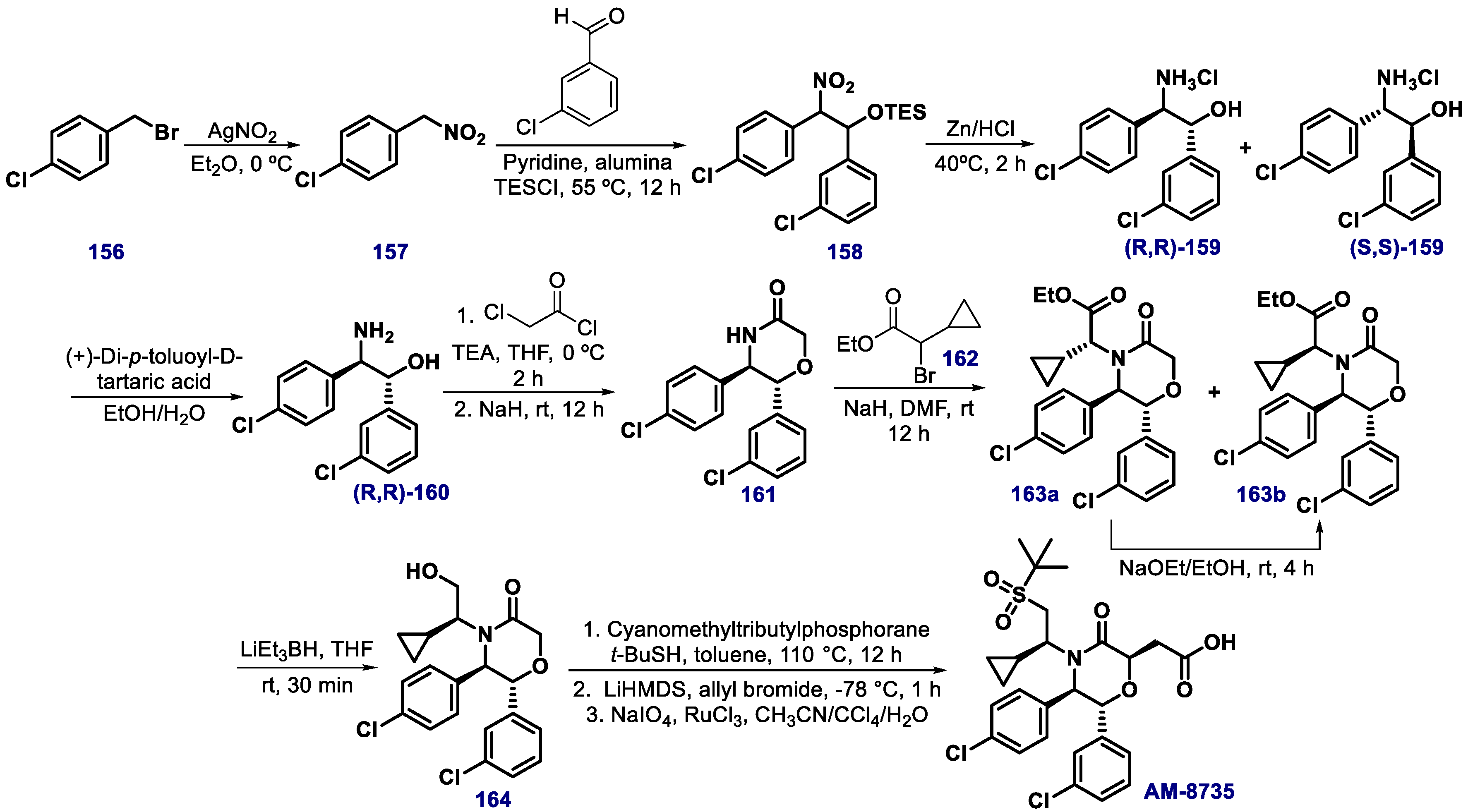{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
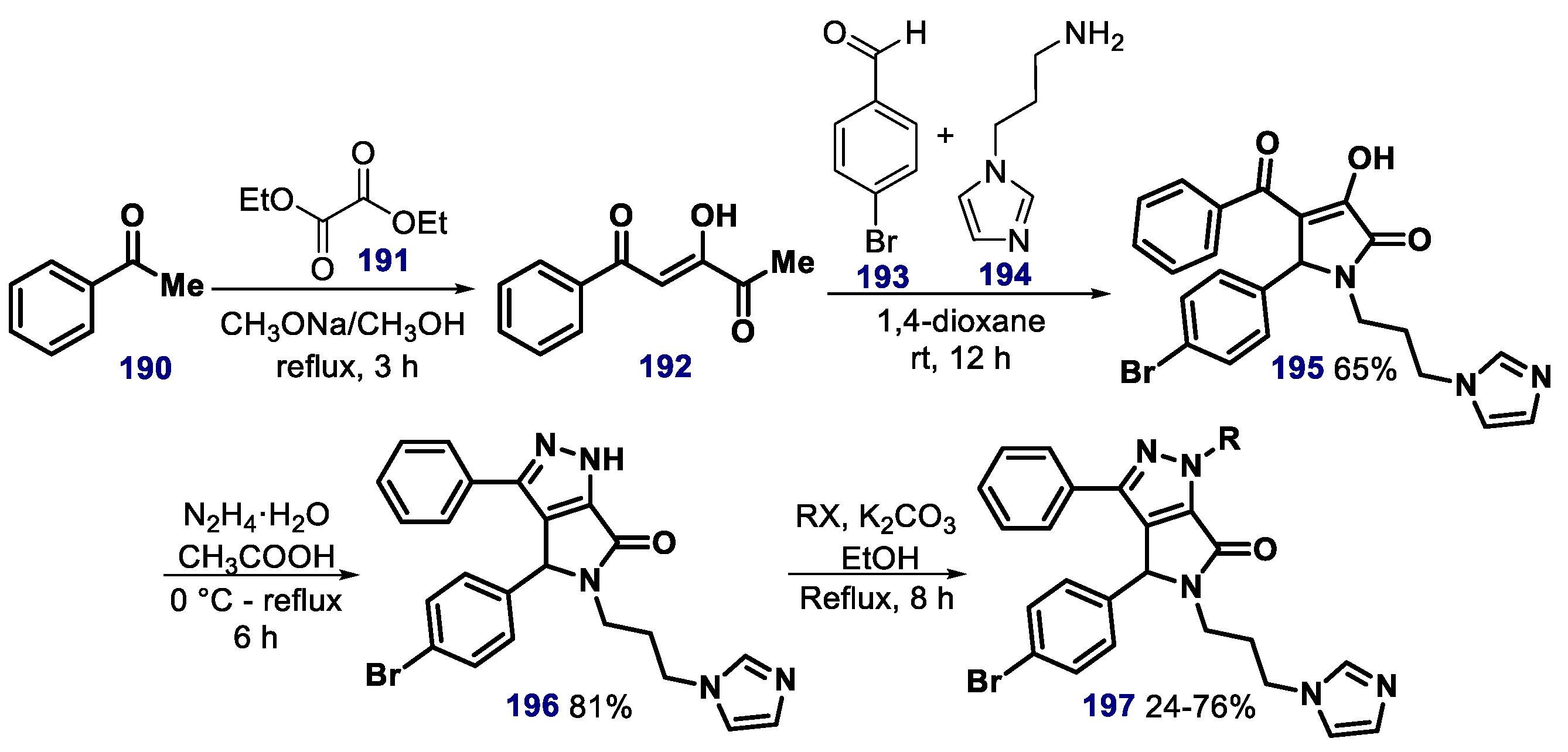{kind=link}
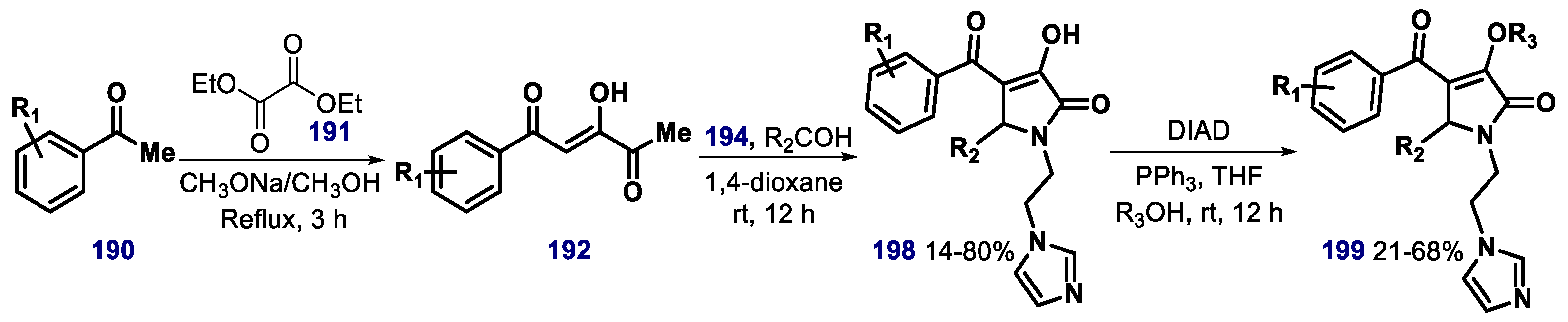{kind=link}
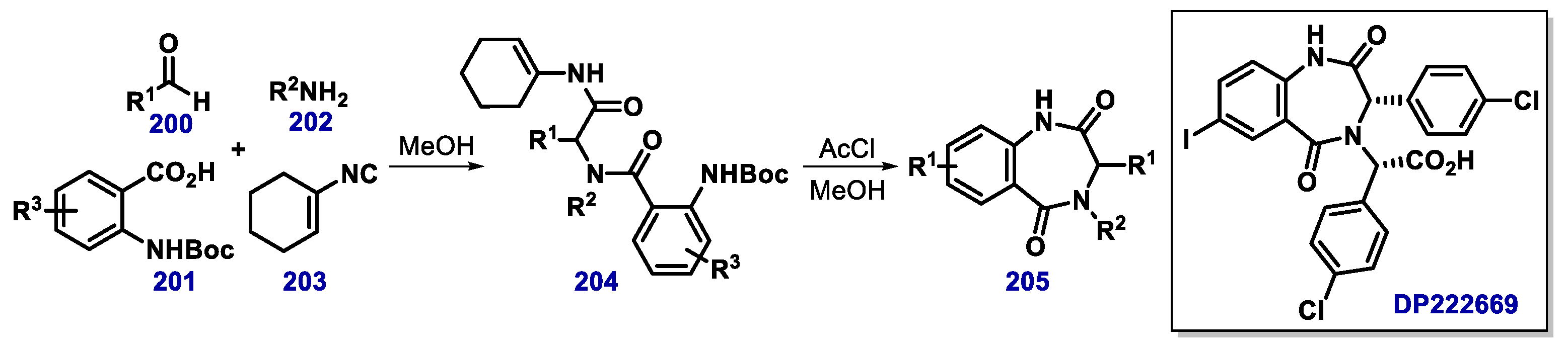{kind=link}
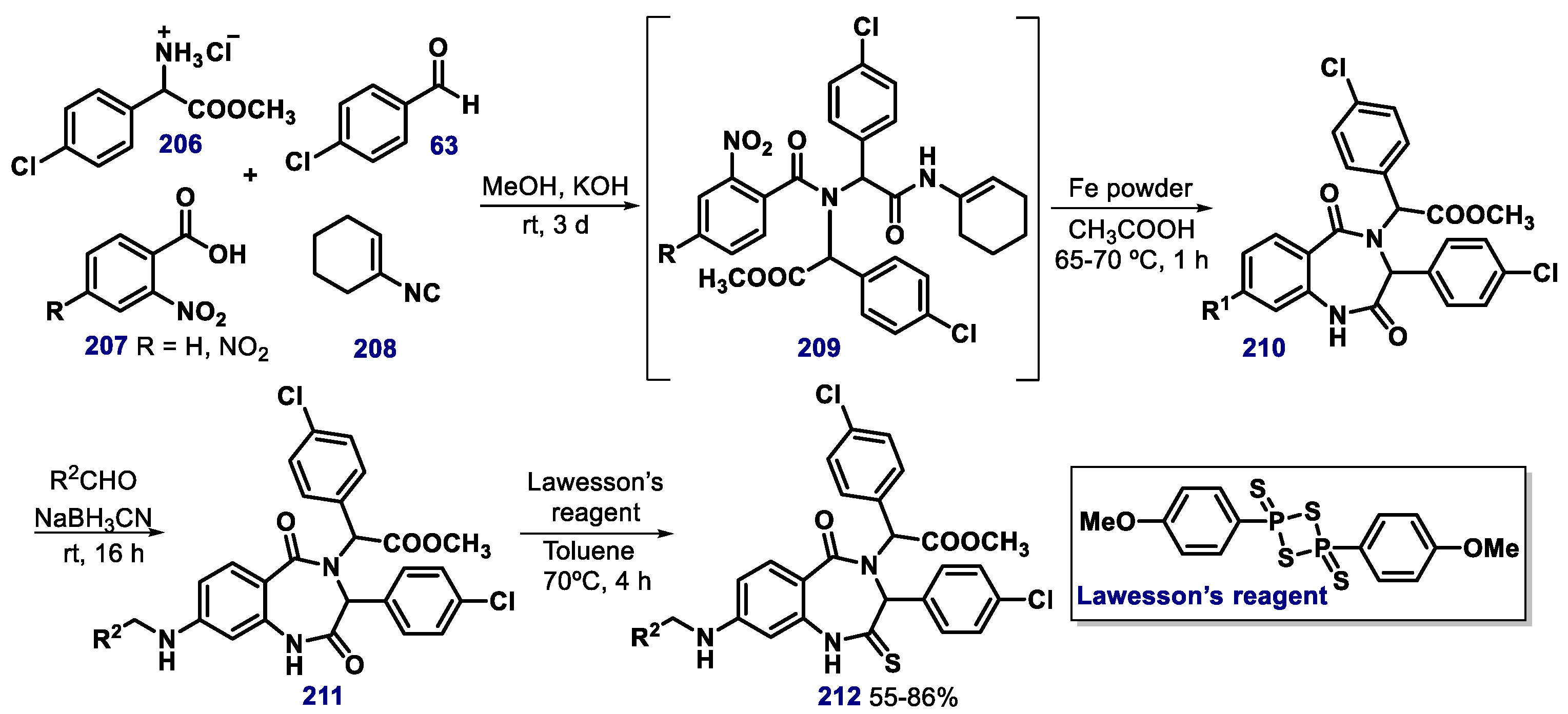{kind=link}
{kind=link}
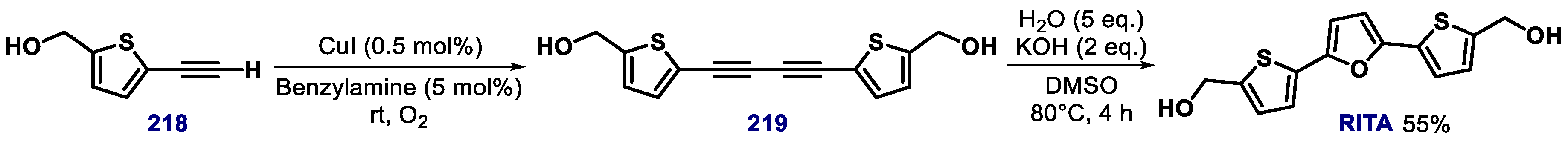{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Targeting Mutant p53: Restoration of the p53 Function
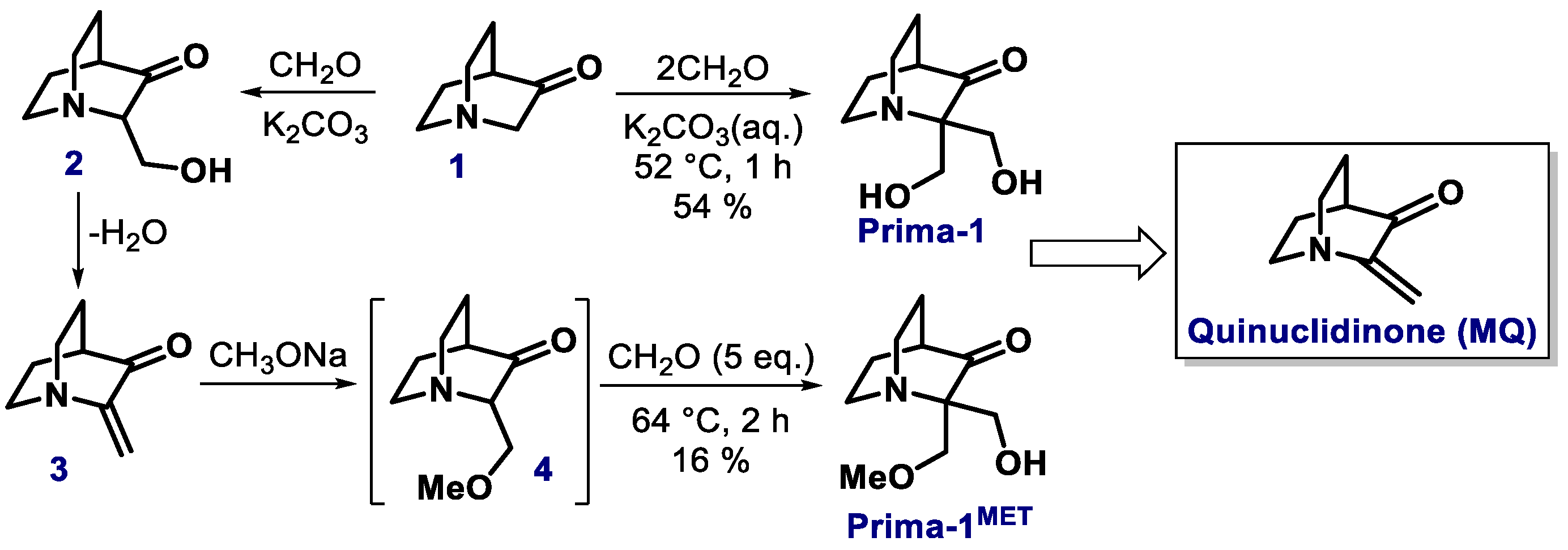
2.1. Quinuclidinone Derivatives
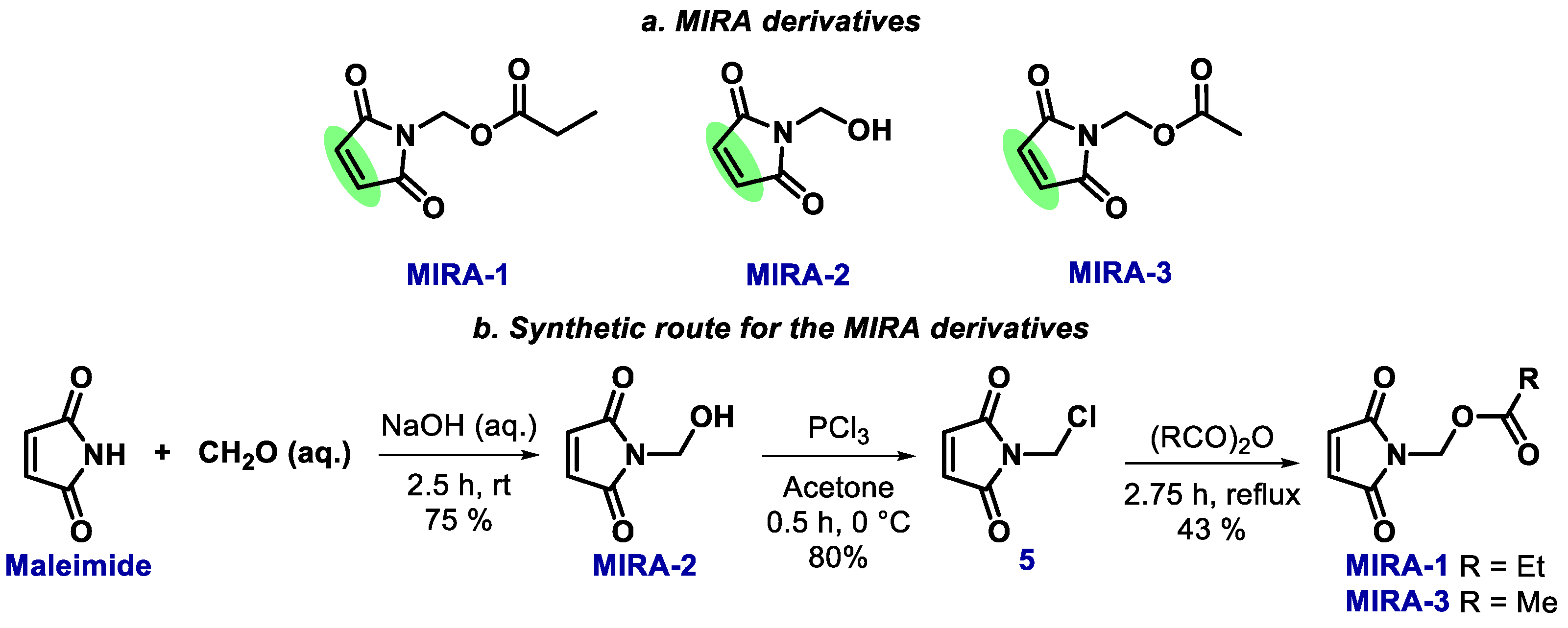
2.2. Maleimide Derivatives
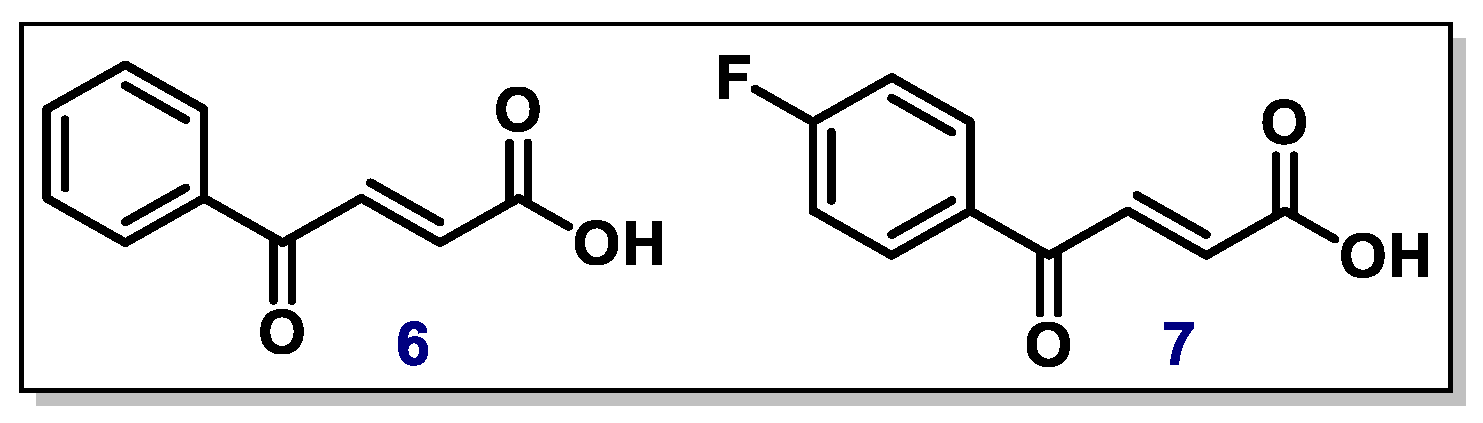
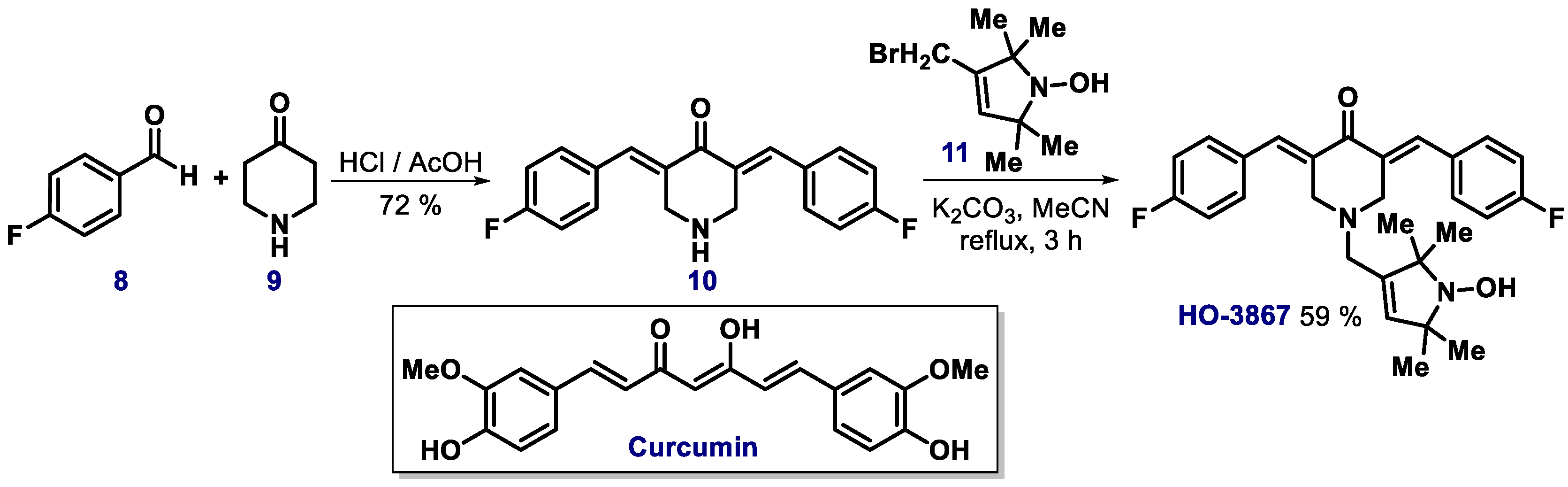
2.3. α,β-Unsaturated Carbonyl Compounds
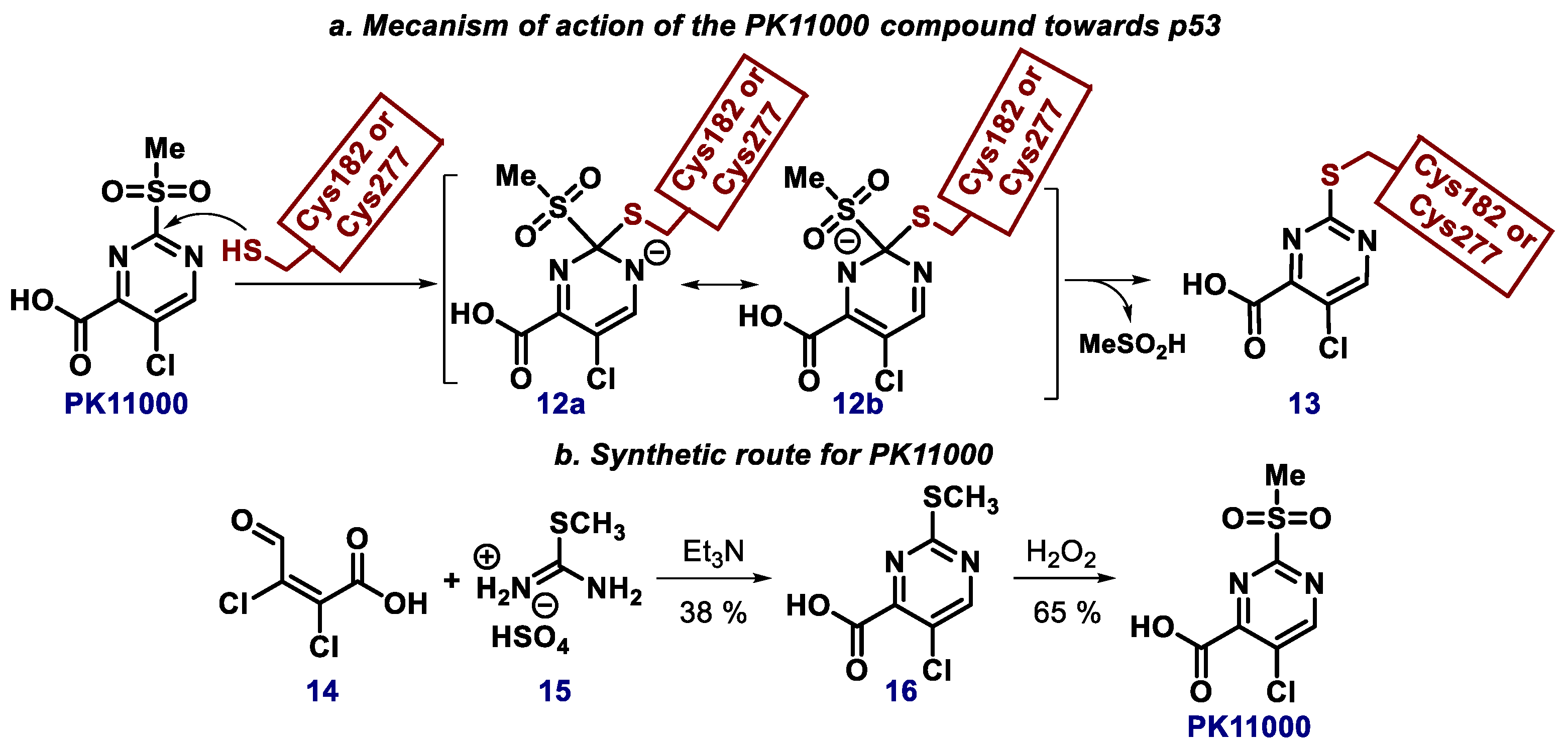
2.4. Pyrimidine, Quinazoline, and Quinoline Derivatives
2.5. Pyrrol and Pyrazole and Derivatives
2.6. Zinc Metallochaperones
2.7. Other Classes of Compounds for the Reactivation of Wild-Type p53
3. When p53 Is Inactivated by Oncogenes: Small Molecules Antagonists of p53-MDM2/MDMX
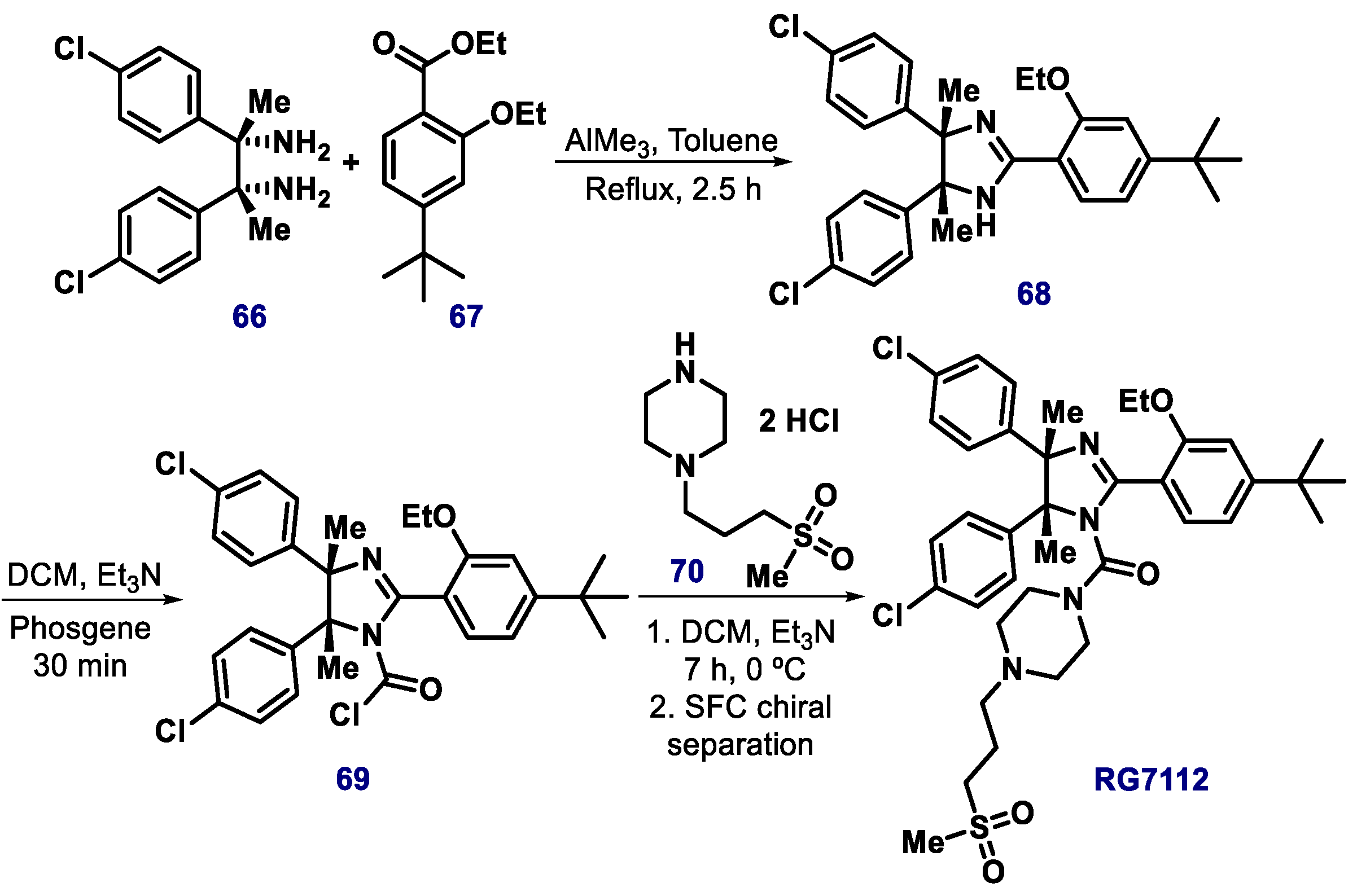
3.1. Cis-Imidazoline Derivatives
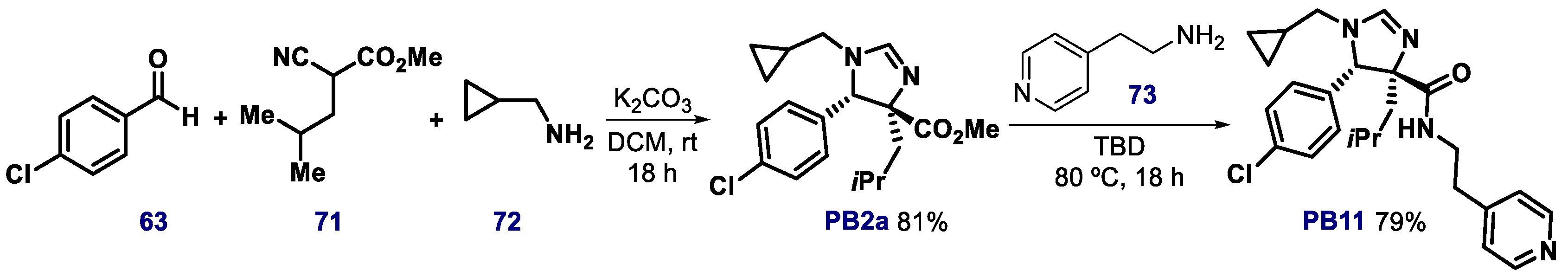
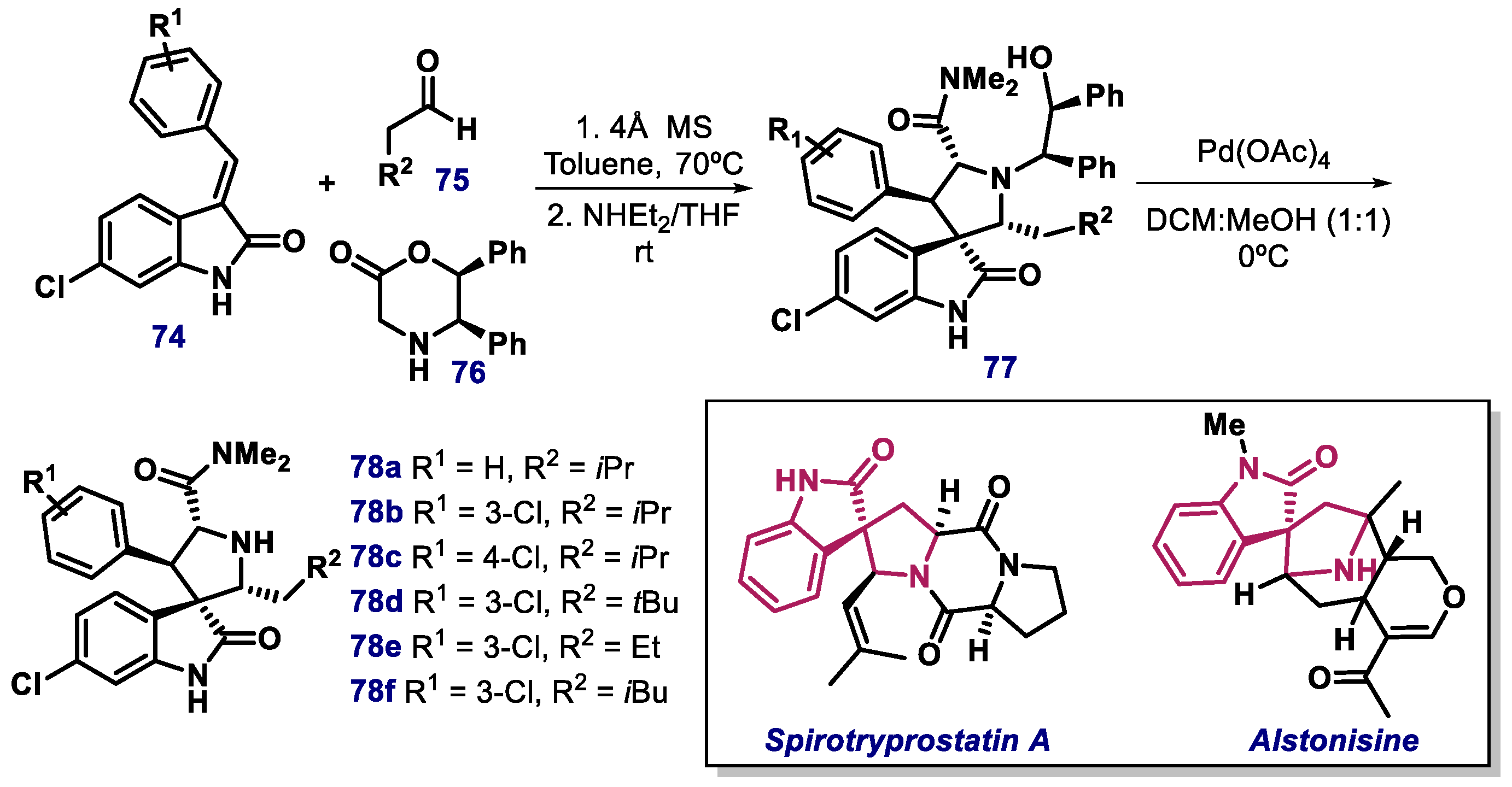
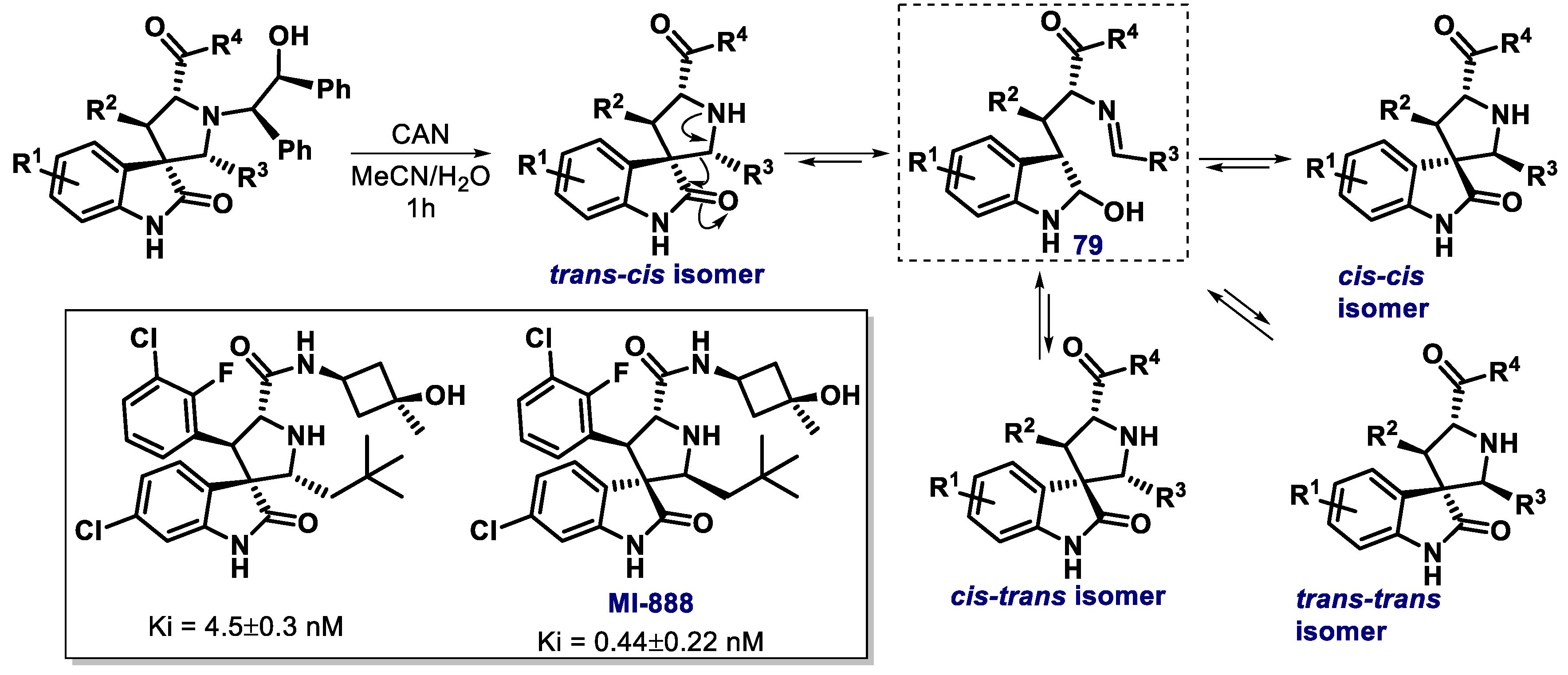
3.2. Spiro-Oxindole, Indole, and Carbazole Derivatives
3.3. Pyrrol/Pyrrolidine, Piperidine, and Morpholine Derivatives
3.4. Pyrimidine and Quinazoline Derivatives
3.5. Pyrazole and Pyrrolone Derivatives
3.6. Benzodiazepinone Derivatives
3.7. Benzofuroxan and Furan Derivatives
4. Small Molecules That Reactivate the p53 Protein through Other Pathways
5. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, D.P.; Cheo, C.F.; Lain, S. p53-based cancer therapy. Cold Spring Harb. Perspect. Biol. 2010, 2, a001222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, D.P. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.; Piwnica-Worms, D.; Piwnica-Worms, H. Contribution of p53 to metastasis. Cancer Discov. 2014, 4, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Mowat, M.R. p53 in tumor progression: Life, death, and everything. Adv. Cancer Res. 1998, 74, 25–48. [Google Scholar]
- Pflaum, J.; Schlosser, S.; Müller, M. p53 family and cellular stress responses in cancer. Front. Oncol. 2014, 4, 285. [Google Scholar] [CrossRef]
- Gupta, A.; Shah, K.; Oza, M.J.; Behl, T. Reactivation of p53 gene by MDM2 inhibitors: A novel therapy for cancer treatment. Biomed. Pharmacother. 2019, 109, 484–492. [Google Scholar] [CrossRef]
- Wassman, C.D.; Baronio, R.; Demir, O.; Wallentine, B.D.; Chen, C.K.; Hall, L.V.; Salehi, F.; Lin, D.W.; Chung, B.P.; Hatfield, G.W.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407. [Google Scholar] [CrossRef]
- Levine, A.J. p53, The cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.J.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Silva, J.L.; Cino, E.A.; Soares, I.N.; Ferreira, V.F.; de Oliveira, G.A.P. Targeting the prion-like aggregation of mutant p53 to combat cancer. Acc. Chem. Res. 2018, 51, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Wiman, K.G. Pharmacological reactivation of mutant p53: From protein structure to the cancer patient. Oncogene 2010, 29, 4245–4252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, B.A.; Coffey, H.A.; Morin, M.J.; Rastinejad, F. Pharmacological rescue of mutant p53 conformation and function. Science 1999, 286, 2507–2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Li, C.; Chen, L.; Sebti, S.; Chen, J. Rescue of mutant p53 transcription function by ellipticine. Oncogene 2003, 22, 4478–4487. [Google Scholar] [CrossRef] [Green Version]
- Demma, M.; Maxwel, E.; Ramos, R.; Liang, L.; Li, C.; Hesk, D.; Rossman, R.; Mallams, A.; Doll, R.; Liu, M.; et al. SCH529074, A small molecule activator of mutant p53, which binds p53 DNA binding domain (DBD), restores growth-suppressive function to mutant p53 and interrupts HDM2-mediated ubiquitination of wild type p53. J. Biol. Chem. 2010, 285, 10198–10212. [Google Scholar] [CrossRef] [Green Version]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef]
- Loh, S.N. Follow the mutations: Toward class-specific, small-molecule reactivation of p53. Biomolecules 2020, 10, 303. [Google Scholar] [CrossRef] [Green Version]
- Lopes, E.A.; Gomes, S.; Saraiva, L.; Santos, M.M.M. Small molecules targeting mutant P53: A promising approach for cancer treatment. Curr. Med. Chem. 2019, 26, 7323–7336. [Google Scholar] [CrossRef]
- Chène, P. Inhibiting the p53-MDM2 interaction: Na importante target for cancer therapy. Nat. Rev. Cancer 2003, 3, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Lemos, A.; Leão, M.; Soares, J.; Palmeira, A.; Pinto, M.; Saraiva, L.; Sousa, M.E. Medicinal chemistry strategies to disrupt the p53–MDM2/MDMX interaction. Med. Res. Rev. 2016, 36, 789–844. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X.; Wang, G.; Yang, Y.; Yuan, Y.; Ouyang, L. The past, present and future of potential small-molecule drugs targeting p53-MDM2/MDMX for cancer therapy. Eur. J. Med. Chem. 2019, 176, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Beloglazkina, A.; Zyk, N.; Majouga, A.; Beloglazkina, E. Recent small-molecule inhibitors of the p53–MDM2 protein–protein interaction. Molecules 2020, 25, 1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espadinha, M.; Barcherini, V.; Lopes, E.A.; Santos, M.M.M. An update on MDMX and dual MDM2/X inhibitors. Curr. Top. Med. Chem. 2018, 18, 647–660. [Google Scholar] [CrossRef]
- Rusiecki, R.; Witkowski, J.; Joanna, J.-A. MDM2-p53 interaction inhibitors: The current state-of-art and updated patent review (2010-Present). Recent Pat. Anticancer Drug Discov. 2019, 14, 324–369. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Kornblau, S.M.; Ruvolo, V.; Dilip, A.; Duvvuri, S.; Davis, R.E.; Zhang, M.; Wang, Z.; Coombes, K.R.; Zhang, N.; et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood 2013, 121, 4166–4174. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Pribluda, A.; Goldstein, R.E.; Morgenstern, Y.; Brachya, G.; Cojocaru, G.; Snir-Alkalay, I.; Burstain, I.; Haffner-Krausz, R.; Jung, S.; et al. CKIa ablation highlights a critical role for p53 in invasiveness control. Nature 2011, 470, 409–413. [Google Scholar] [CrossRef]
- Pribluda, A.; Elyada, E.; Wiener, Z.; Hamza, H.; Goldstein, R.E.; Biton, M.; Burstain, I.; Morgenstern, Y.; Brachya, G.; Billauer, H.; et al. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell 2013, 24, 242–256. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.; Luo, J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim. Biophys. Acta 2010, 1804, 1684–1689. [Google Scholar] [CrossRef] [Green Version]
- Ano Bom, A.P.D.; Rangel, L.P.; Costa, D.C.F.; de Oliveira, G.A.P.; Sanches, D.; Braga, C.A.; Gava, L.M.; Ramos, C.H.I.; Cepeda, A.O.T.; Stumbo, A.C. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: Implications for cancer. J. Biol. Chem. 2012, 287, 28152–28162. [Google Scholar] [CrossRef] [Green Version]
- Levy, C.B.; Stumbo, A.C.; Ano Bom, A.P.D.; Portari, E.; Cordeiro, Y.; Silva, J.L.; De Moura-Gallo, C.V. Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors. Int. J. Biochem. Cell Biol. 2011, 43, 60–64. [Google Scholar] [CrossRef]
- Silva, J.L.; Gallo, C.V.; Costa, D.C.; Rangel, L.P. Prion-like aggregation of mutant p53 in cancer. Trends Biochem. Sci. 2014, 39, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Herrero, A.B.; Rojas, E.A.; Misiewicz-Krzeminska, I.; Krzeminski, P.; Gutiérrez, N.C. Molecular mechanisms of p53 deregulation in cancer: An overview in multiple myeloma. Int. J. Mol. Sci. 2016, 17, 2003. [Google Scholar] [CrossRef] [PubMed]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A designed inhibitor of p53 aggregation rescues p53 tumor suppression in ovarian carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaar, J.L.; Basse, N.; Joerger, A.C.; Stephens, E.; Rutherford, T.J.; Fersht, A.R. Estabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. 2010, 19, 2267–2278. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS Function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Narayanan, S.; Vazquez, A.; Carpizo, D.R. Small molecule compounds targeting the p53 pathway: Are we finally making progress. Apoptosis 2014, 19, 1055–1068. [Google Scholar] [CrossRef] [Green Version]
- Selivanova, G. Wild type p53 reactivation: From lab bench to clinic. FEBS Lett. 2014, 588, 2628–2638. [Google Scholar] [CrossRef] [Green Version]
- Selivanova, G. Therapeutic targeting of p53 by small molecules. Semin. Cancer Biol. 2010, 20, 46–56. [Google Scholar] [CrossRef]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 2772. Cell Death Dis. 2018, 9, 439. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.N.; Issaeva, N.; Shlov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor supressor function to mutante p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Reddy, N.L.; Hill, J.; Ye, L.; Fernandes, P.B.; Stout, D.M. Identification and structure–activity relationship studies of 3-methylene-2-norbornanone as potent anti-proliferative agents presumably working through p53 mediated apoptosis. Bioorg. Med. Chem. Lett. 2004, 14, 5645–5649. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Iwakuma, T. Targeting oncogenic mutant p53 for cancer therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The National Cancer Institute Website. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/prima-1-analog-apr-246?redirect=true (accessed on 12 July 2019).
- Rangel, L.P.; Ferretti, G.D.S.; Costa, C.L.; Andrade, S.M.M.V.; Carvalho, R.S.; Costa, D.C.F.; Silva, J.L. p53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells. J. Biol. Chem. 2019, 294, 3670–3682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, A.T. Systems with bridgehead nitrogen. b-ketols of the 1-azabicyclo[2.2.2]octane series. J. Org. Chem. 1996, 31, 1053–1059. [Google Scholar] [CrossRef]
- Saha, M.N.; Chen, Y.; Chen, G.; Chang, H. Small molecule MIRA-1 induces in vitro and in vivo anti-myeloma activity and synergizes with current anti-myeloma agents. Brit. J. Cancer 2014, 110, 2224–2231. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.N.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, N.; Bergman, J.; Selivanova, G.; Wiman, K.G. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J. Biol. Chem. 2005, 280, 30384–30391. [Google Scholar] [CrossRef] [Green Version]
- Bou-Hanna, C.; Jarry, A.; Lode, L.; Schmitz, I.; Schulze-Osthoff, K.; Kury, S.; Bezieau, S.; Mosnier, J.F.; Laboisse, C.L. Acute cytotoxicity of MIRA-1/NSC19630, a mutant p53-reactivating small molecule, against human normal and cancer cells via a caspase-9-dependent apoptosis. Cancer Lett. 2015, 359, 211–217. [Google Scholar] [CrossRef]
- Ferreira, V.F.; Souza, M.C.B.V.; Cunha, A.C.; Perreira, L.O.R.; Ferreira, M.L.G. Recent advances in the synthesis of pyrroles. Org. Prep. Proced. Int. 2001, 33, 411–454. [Google Scholar] [CrossRef]
- Tawney, P.O.; Snyder, R.H.; Bryan, C.E.; Conger, R.P.; Dovell, F.S.; Kelly, R.J.; Stiteler, C.H. The Chemistry of maleimide and its derivatives. I. N-Carbamylmaleimide. J. Org. Chem. 1960, 25, 56–60. [Google Scholar] [CrossRef]
- Tawney, P.O.; Snyder, R.H.; Conger, R.P.; Leibbrand, K.A.; Stiteler, C.H.; Williams, A.R. The chemistry of maleimide and its derivatives. II. Maleimide and N-methylolmaleimide. J. Org. Chem. 1961, 26, 15–21. [Google Scholar] [CrossRef]
- Makarov, M.V.; Odinets, I.L.; Lyssenko, K.A.; Rybalkina, E.Y.; Kosilkin, I.V.; Antipin, M.Y.; Timofeeva, T.V. N-Alkylated 3,5-bis(arylidene)-4-piperidones. Synthetic approaches, X-ray structure and anticancer activity. J. Heterocyclic Chem. 2008, 45, 729–736. [Google Scholar] [CrossRef]
- Selvendiran, K.; Tong, L.; Bratasz, A.; Kuppusamy, L.M.; Ahmed, S.; Ravi, Y.; Trigg, N.J.; Rivera, B.K.; Kalai, T.; Hideg, K.; et al. Anticancer efficacy of a difluorodiarylidenyl piperidone (HO-3867) in human ovarian cancer cells and tumor xenografts. Mol. Cancer Ther. 2010, 9, 1169–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madan, E.; Parker, T.M.; Bauer, M.R.; Dhiman, A.; Pelham, C.J.; Nagane, M.; Kuppusamy, M.L.; Holmes, M.; Holmes, T.R.; Shaik, K.; et al. The curcumin analog HO-3867 selectively kills cancer cells by converting mutant p53 protein to transcriptionally active wildtype p53. J. Biol. Chem. 2018, 293, 4262–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fersht, A.R.; Bauer, M.R. 2-Sulfonylpyrimidines. International Patent Application No. PCT/GB2016/052544, 23 February 2017. [Google Scholar]
- Bauera, M.R.; Joergera, A.C.; Fersht, A.R. 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl. Acad. Sci. USA 2016, 113, e5271–e5280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Synnott, N.C.; Bauer, M.R.; Madden, S.; Murray, A.; Klinger, R.; O’Donovan, N.; Duffy, M.J. Mutant p53 as a therapeutic target for the treatment of triple-negative breast cancer: Preclinical investigation with the anti-p53 drug, PK11007. Cancer Lett. 2018, 414, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.M.; Luo, S.J.; Zhang, Z.X.; Ma, Y.X. Efficient synthesis of a new pyrimidine derivative. Synth. Commun. 2002, 32, 153–157. [Google Scholar] [CrossRef]
- Jiang, J.B.; Hesson, D.P.; Dusak, B.A.; Dexter, D.L.; Kang, G.J.; Hamel, E. Synthesis and biological evaluation of 2-styrylquinazolin-4(3H)-ones, a new class of antimitotic anticancer agents which inhibit tubulin polymerization. J. Med. Chem. 1990, 33, 1721–1728. [Google Scholar] [CrossRef]
- Madka, V.; Zhang, Y.; Li, Q.; Mohammed, A.; Sindhwani, P.; Lightfoot, S.; Wu, X.R.; Kopelovich, L.; Rao, C.V. p53-stabilizing agent CP-31398 prevents growth and invasion of urothelial cancer of the bladder in transgenic UPII-SV40T Mice. Neoplasia 2013, 15, 966–974. [Google Scholar] [CrossRef] [Green Version]
- Wischhusen, J.; Naumann, U.; Ohgaki, H.; Rastinejad, F.; Weller, M. CP-31398, a novel p53-stabilizing agent, induces p53-dependent and p53-independent glioma cell death. Oncogene 2003, 22, 8233–8245. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Zhu, Y.; Han, L.; Kim, A.L.; Kopelovich, L.; Bickers, D.R.; Athar, M. CP-31398 restores mutant p53 tumor suppressor function and inhibits UVB-induced skin carcinogenesis in mice. J. Clin. Invest. 2007, 117, 3753–3764. [Google Scholar] [CrossRef] [Green Version]
- Rao, C.V.; Patlolla, J.M.; Qian, L.; Zhang, Y.; Brewer, M.; Mohammed, A.; Desai, D.; Amin, S.; Lightfoot, S.; Kopelovich, L. Chemopreventive effects of the p53-modulating agents CP-31398 and PRIMA-1 in tobacco carcinogen–induced lung tumorigenesis in A/J mice. Neoplasia 2013, 15, 1018–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, H.S.; Hwang, I.Y.; Marshall, E.S.; Lindsay, B.S.; Denny, W.A.; Gilchrist, C.; Joseph, W.R.; Greenhalgh, D.; Richardson, E.; Kestell, P.; et al. Therapeutic reactivation of mutant p53 protein by quinazoline derivatives. Invest. New Drug. 2012, 30, 2035. [Google Scholar] [CrossRef] [PubMed]
- Zache, N.; Lambert, J.M.R.; Rökaeus, N.; Shen, J.; Hainaut, P.; Bergman, J.; Wiman, K.G.; Bykov, V.J.N. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol. Oncol. 2008, 2, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witt, A.; Bergman, J. Synthesis and reactions of some 2-vinyl-3H-quinazolin-4-ones. Tetrahedron 2000, 56, 7245–7253. [Google Scholar] [CrossRef]
- Abe, T.; Kida, K.; Yamada, K. A copper-catalyzed Ritter-type cascade via iminoketene for the synthesis of quinazolin-4(3H)-ones and diazocines. Chem. Commun. 2017, 53, 4362. [Google Scholar] [CrossRef]
- Weinmann, L.; Wischhusen, J.; Demma, M.J.; Naumann, U.; Roth, P.; DasMahapatra, B.; Weller, M. A novel p53 rescue compound induces p53-dependent growth arrest and sensitises glioma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 2008, 15, 718–729. [Google Scholar] [CrossRef]
- Mallams, A.; Dasmahapatra, B.; Neustadt, B.R.; Demma, M.; Vaccaro, H. Quinazoline derivatives useful in cancer treatment. International Patent Application No. PCT/US2006/027114, 25 January 2017. [Google Scholar]
- Nenkov, M.; Ma, Y.; Haase, D.; Zhou, Z.; Li, Y.; Petersen, I.; Lu, G.; Chen, Y. Growth inhibitory role of the p53 activator SCH 529074 in non-small cell lung cancer cells expressing mutant p53. Oncol. Rep. 2020, 43, 2073–2082. [Google Scholar] [CrossRef]
- Salim, K.Y.; Vareki, S.M.; Danter, W.R.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef] [Green Version]
- Salim, K.Y.; Vareki, S.M.; Danter, W.R.; Koropatnick, J. COTI-2, a new anticancer drug currently under clinical investigation, targets mutant p53 and negatively modulates the PI3K/AKT/mTOR pathway. Eur. J. Cancer 2016, 69, S19. [Google Scholar] [CrossRef]
- Synnott, N.C.; O’Connell, D.; Crown, J.; Duffy, M.J. COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Res. Treat. 2020, 179, 47–56. [Google Scholar] [CrossRef]
- Danter, W.R.; Brown, M.; Lepifre, F. Compounds and method for treatment of cancer. International Patent Application No. PCT/CA2008/000045, 17 July 2008. [Google Scholar]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef]
- Yip, C.-H.; Yarkoni, O.; Ajioka, J.; Wan, K.-L.; Nathan, S. Recent advancements in high-level synthesis of the promising clinical drug, prodigiosin. Appl. Microbiol. Biot. 2019, 103, 1667–1680. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, H.; Holden, K.G. The synthesis of prodigiosin. J. Am. Chem. Soc. 1962, 84, 635–642. [Google Scholar] [CrossRef]
- Dairi, K.; Tripathy, S.; Attardo, G.; Lavallée, J.-F. Two-step synthesis of the bipyrrole precursor of prodigiosin. Tet. Lett. 2006, 47, 2605–2606. [Google Scholar] [CrossRef]
- Hainaut, P.; Milner, J. A structural role for metal ions in the “wild-type” conformation of the tumor suppressor protein p53. Cancer Res. 1993, 53, 1739–1742. [Google Scholar]
- Puca, R.; Nardinocchi, L.; Porru, M.; Simon, A.J.; Rechavi, G.; Leonetti, C.; Givol, D.; D’Orazi, G. Restoring p53 active conformation with zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle 2011, 10, 1679–1689. [Google Scholar] [CrossRef]
- Blanden, A.R.; Yu, X.; Loh, S.N.; Levine, A.J.; Carpizo, D.R. Reactivating mutant p53 using small molecules as zinc metallochaperones: Awakening a sleeping giant in cancer. Drug Discov. 2015, 20, 1391–1397. [Google Scholar] [CrossRef] [Green Version]
- Na, B.; Yu, X.; Withers, T.; Gilleran, J.; Yao, M.; Foo, T.K.; Chen, C.; Moore, D.; Lin, Y.; Kimball, S.D.; et al. Therapeutic targeting of BRCA1 and TP53 mutant breast cancer through mutant p53 reactivation. NJP Breast Cancer 2019, 5, 14. [Google Scholar] [CrossRef] [Green Version]
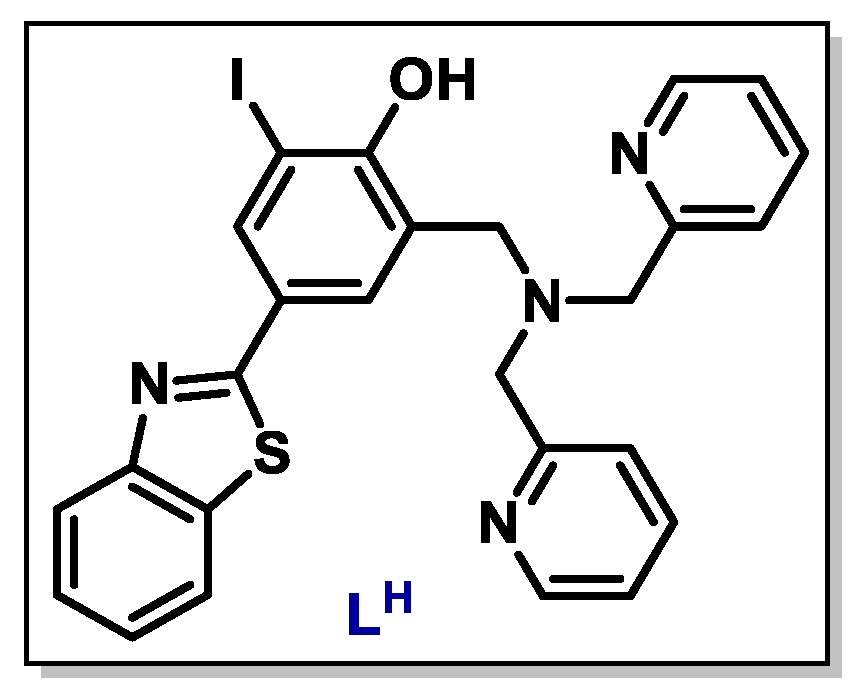
- Miller, J.J.; Blanchet, A.; Orvain, C.; Nouchikian, L.; Reviriot, Y.; Clarke, R.M.; Martelino, D.; Wilson, D.; Gaiddon, C.; Storr, T. Bifunctional ligand design for modulating mutant p53 aggregation in cancer. Chem. Sci. 2019, 10, 10802–10814. [Google Scholar] [CrossRef] [Green Version]
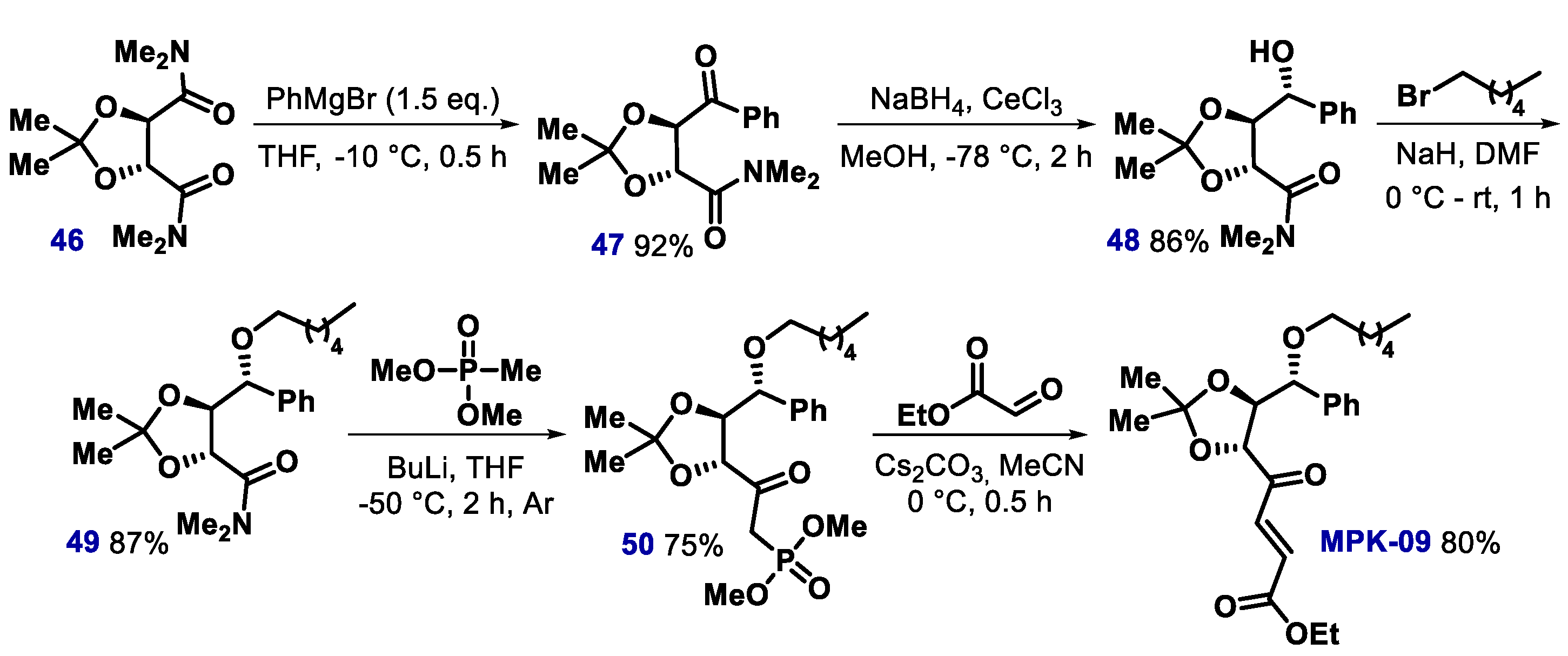
- Metri, P.K.; Naz, S.; Kondaiah, P.; Prasad, K.R. MPK-09, a small molecule inspired from bioactive styryllactone restores the wild-type function of mutant p53. ACS Chem. Biol. 2013, 8, 1429–1434. [Google Scholar] [CrossRef]
- Mereyala, H.B.; Joe, M. Cytotoxic activity of styryl lactones and their derivatives. Curr. Med. Chem. Anticancer Agents 2001, 1, 293–300. [Google Scholar] [CrossRef]
- Wilcken, R.; Wang, G.Z.; Boeckler, F.M.; Fersht, A.R. Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition. Proc. Natl. Acad. Sci. USA 2012, 109, 13584–13589. [Google Scholar] [CrossRef] [Green Version]
- Wilcken, R.; Zimmermann, M.O.; Bauer, M.R.; Rutherford, T.J.; Fersht, A.R.; Joerger, A.C.; Boeckler, F.M. Experimental and theoretical evaluation of the ethynyl moiety as a halogen bioisostere. ACS Chem. Biol. 2015, 10, 2725–2732. [Google Scholar] [CrossRef] [Green Version]
- Mazumder, P.M.; Sasmal, D. Mycotoxins - limits and regulations. Anc. Sci. Life 2001, 20, 1–19. [Google Scholar]
- Hiraki, M.; Hwang, S.Y.; Cao, S.; Ramadhar, T.R.; Byun, S.; Yoon, K.W.; Lee, J.H.; Chu, K.; Gurkar, A.U.; Kolev, V.; et al. Small molecule reactivation of mutant p53 to wt-like p53 through the p53-Hsp40 regulatory axis. Chem. Biol. 2015, 22, 1206–1216. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele specific p53 mutant reactivation. Cancer Cell 2012, 21, 614–625. [Google Scholar] [CrossRef] [Green Version]
- Ferraz da Costa, D.C.; Campos, N.P.C.; Santos, R.A.; Guedes-da-Silva, F.H.; Martins-Dinis, M.M.D.C.; Zanphorlin, L.; Ramos, C.; Rangel, L.P.; Silva, J.L. Resveratrol prevents p53 aggregation in vitro and in breast cancer cells. Oncotarget 2018, 9, 29112–29122. [Google Scholar]
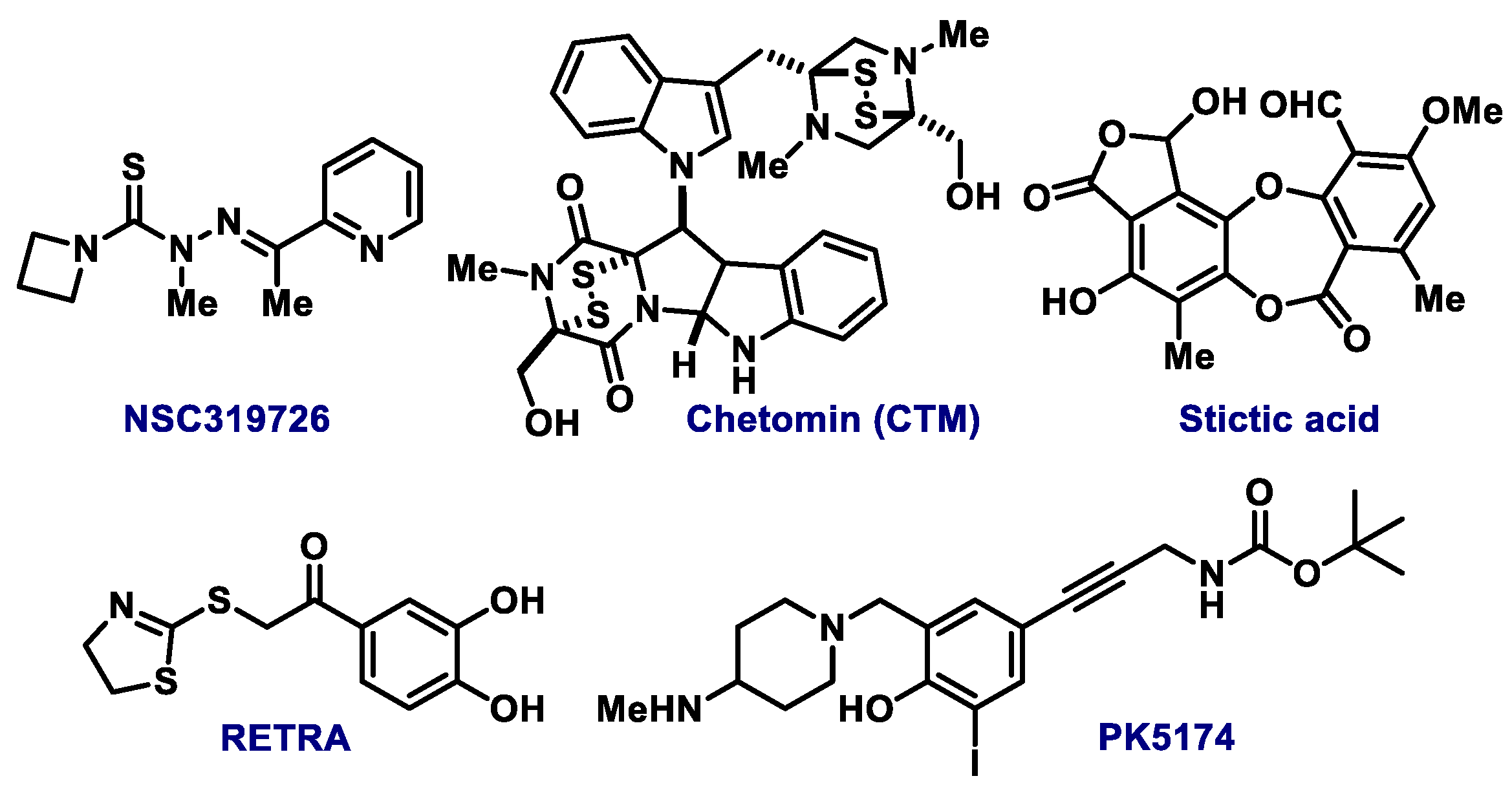
- Kravchenko, J.E.; Ilyinskaya, G.V.; Komarov, P.G.; Agapova, L.S.; Kochetkov, D.V.; Strom, E.; Frolova, E.I.; Kovriga, I.; Gudkov, A.V.; Feinstein, E.; et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. PNAS 2008, 105, 6302–6307. [Google Scholar] [CrossRef] [Green Version]
- Sonnemann, J.; Grauel, D.; Blümel, L.; Hentschel, J.; Marx, C.; Blumrich, A.; Focke, K.; Becker, S.; Wittig, S.; Schinkel, S.; et al. RETRA exerts anticancer activity in Ewing’s sarcoma cells independent of their TP53 status. Eur. J. Cancer 2015, 51, 841–851. [Google Scholar] [CrossRef]
- Deb, S.P.; Deb, S. Mutant p53 and MDM2 in Cancer, 1st ed.; Springer: New York, NY, USA, 2014. [Google Scholar]
- Ribeiro, C.J.; Rodrigues, C.M.; Moreira, R.; Santos, M.M. Chemical variations on the p53 reactivation theme. Pharmaceuticals 2016, 9, 25. [Google Scholar] [CrossRef]
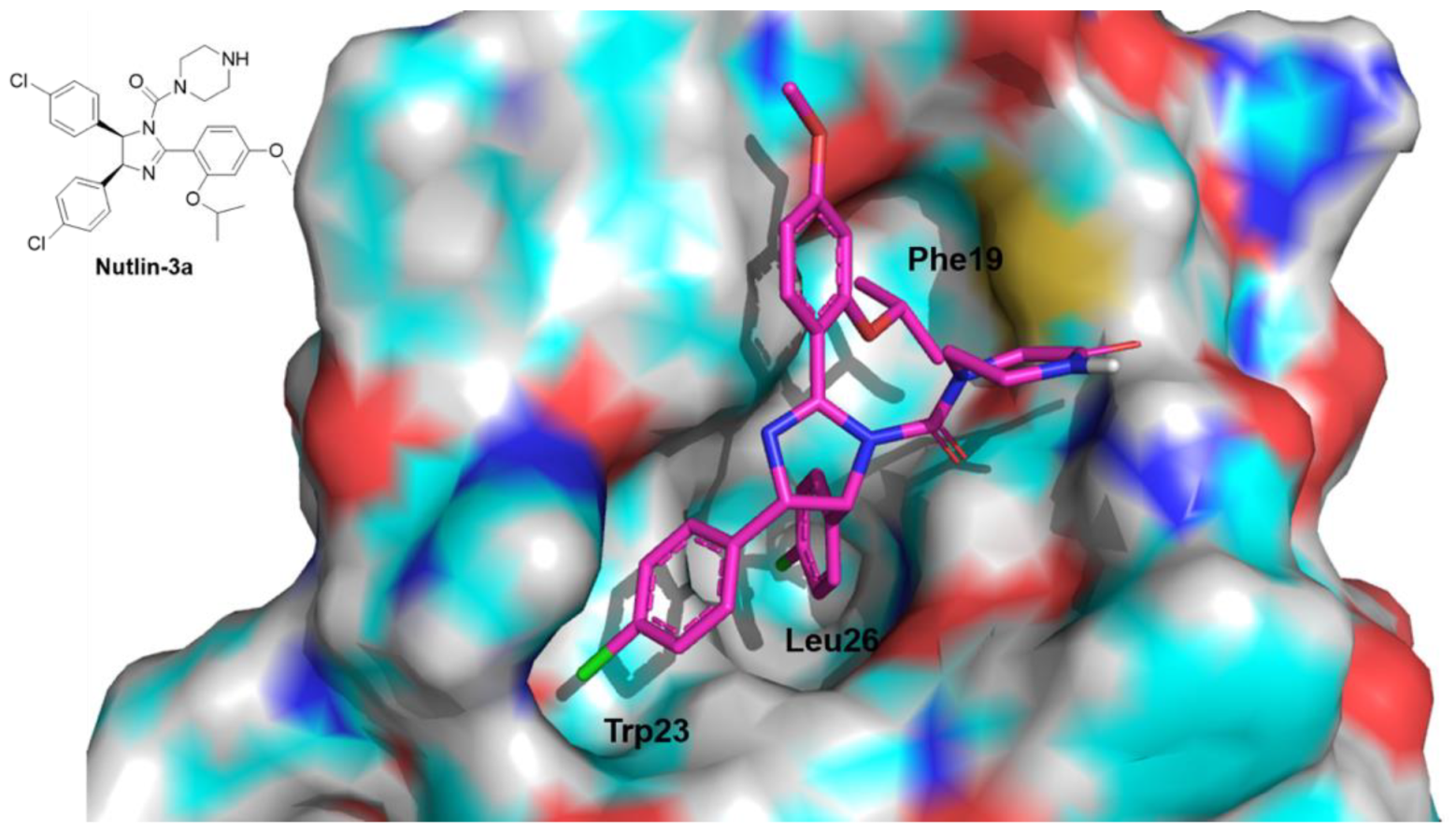
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Neochoritis, C.G.; Wang, K.; Estrada-Ortiz, N.; Herdtweck, E.; Kubica, K.; Twarda, A.; Zak, K.M.; Holak, T.A.; Dömling, A. 2,3´-Bis(1’H-indole) heterocycles: New p53/MDM2/MDMX antagonists. Bioorg. Med. Chem. Lett. 2015, 25, 5661–5666. [Google Scholar] [CrossRef] [Green Version]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Qiu, S.; Ding, Y.; Gao, W.; Stuckey, J.; Krajewski, K.; Roller, P.P.; Tomita, Y.; et al. Structure-based design of potent non-peptide MDM2 inhibitors. J. Am. Chem. Soc. 2005, 127, 10130–10131. [Google Scholar] [CrossRef]
- Kong, N.; Liu, E.A.; Vu, B.T. Cis-imidazolines as MDM2 Inhibitors. International Patent Application No. PCT/EP2002/013905, 26 June 2003. [Google Scholar]
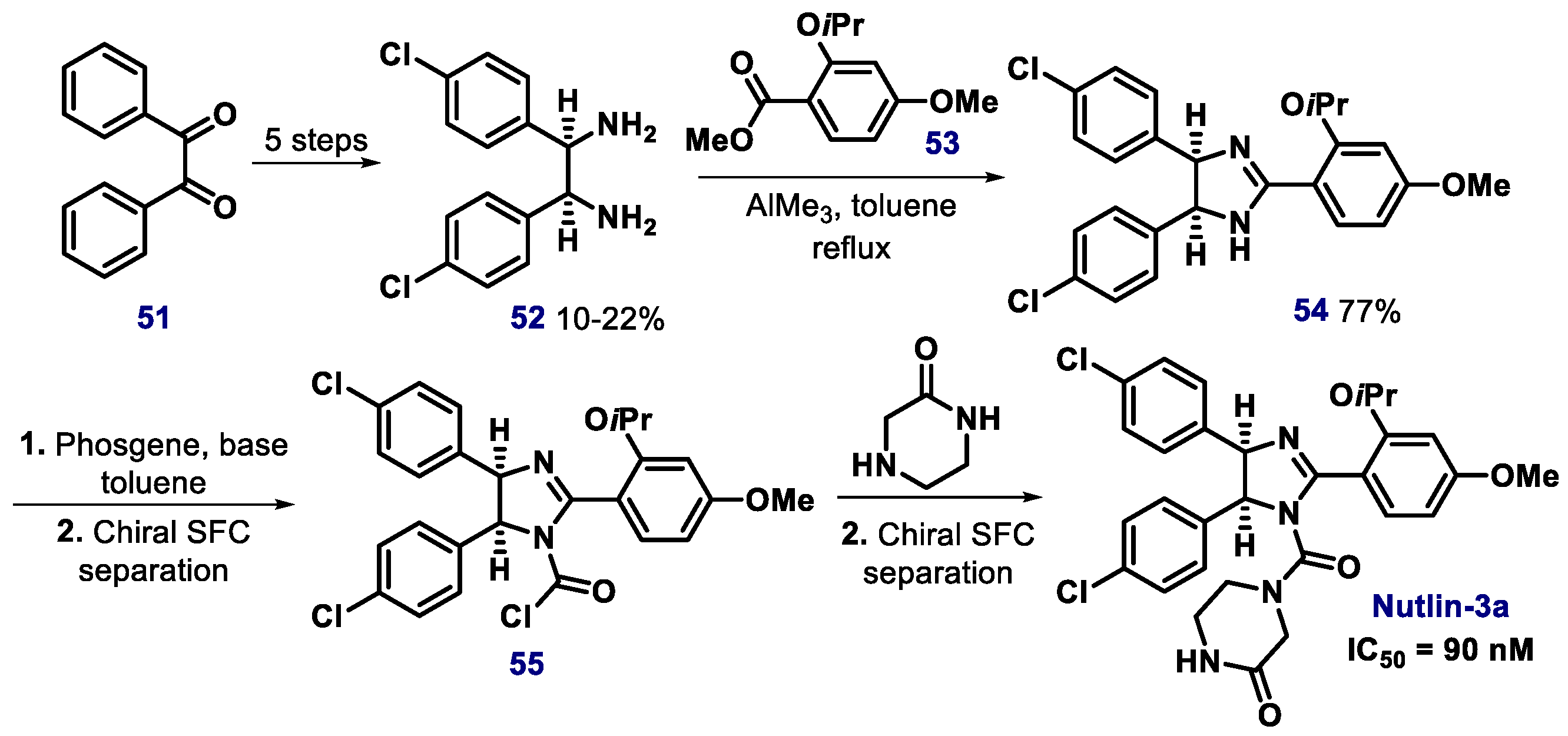
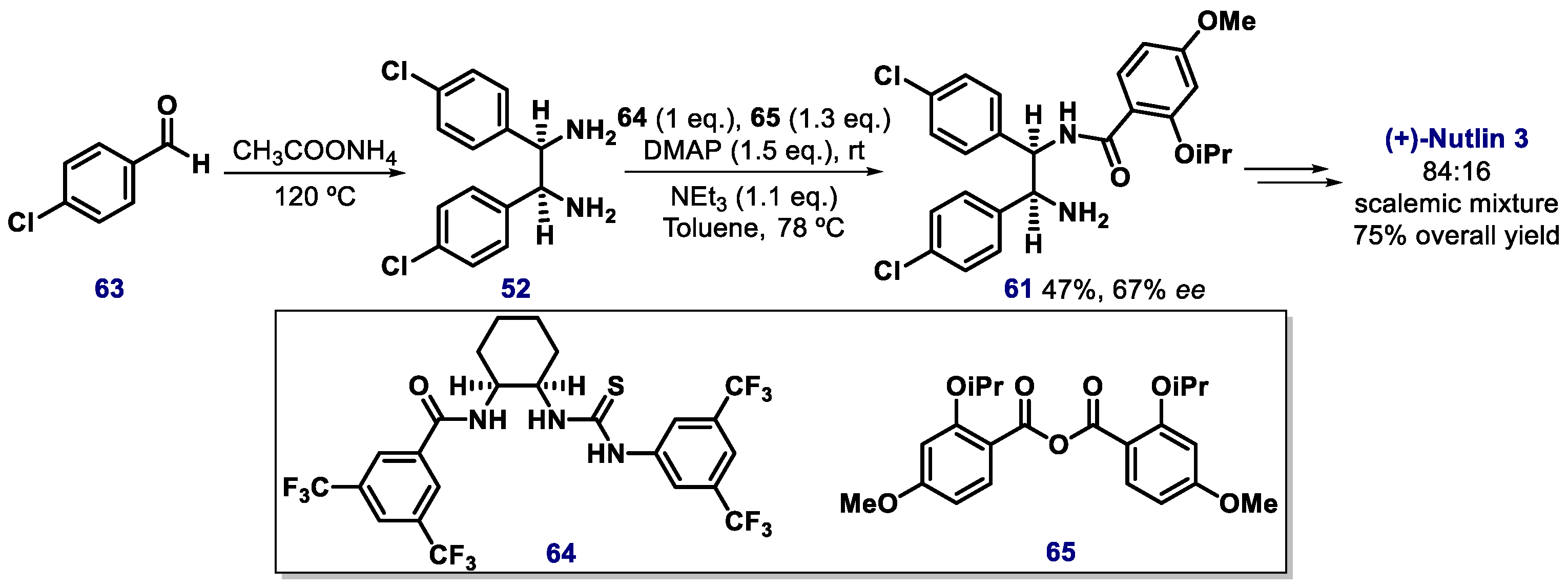
- Fantinati, A.; Bianco, S.; Cristofori, V.; Cavazzini, A.; Catani, M.; Zanirato, V.; Pacifico, S.; Rimondi, E.; Milani, D.; Voltan, R.; et al. Expeditious synthesis and biological characterization of enantio-enriched (-)-Nutlin-3. ChemistrySelect 2017, 2, 8504–8508. [Google Scholar] [CrossRef]
- Bartkovitz, D.J.; Cai, J.; Chu, X.J.; Li, H.; Lovey, A.J.; Vu, B.T.; Zhao, C. Chiral Cis-Imidazolines. International Patent Application No. PCT/EP2008/063053, 16 April 2009. [Google Scholar]
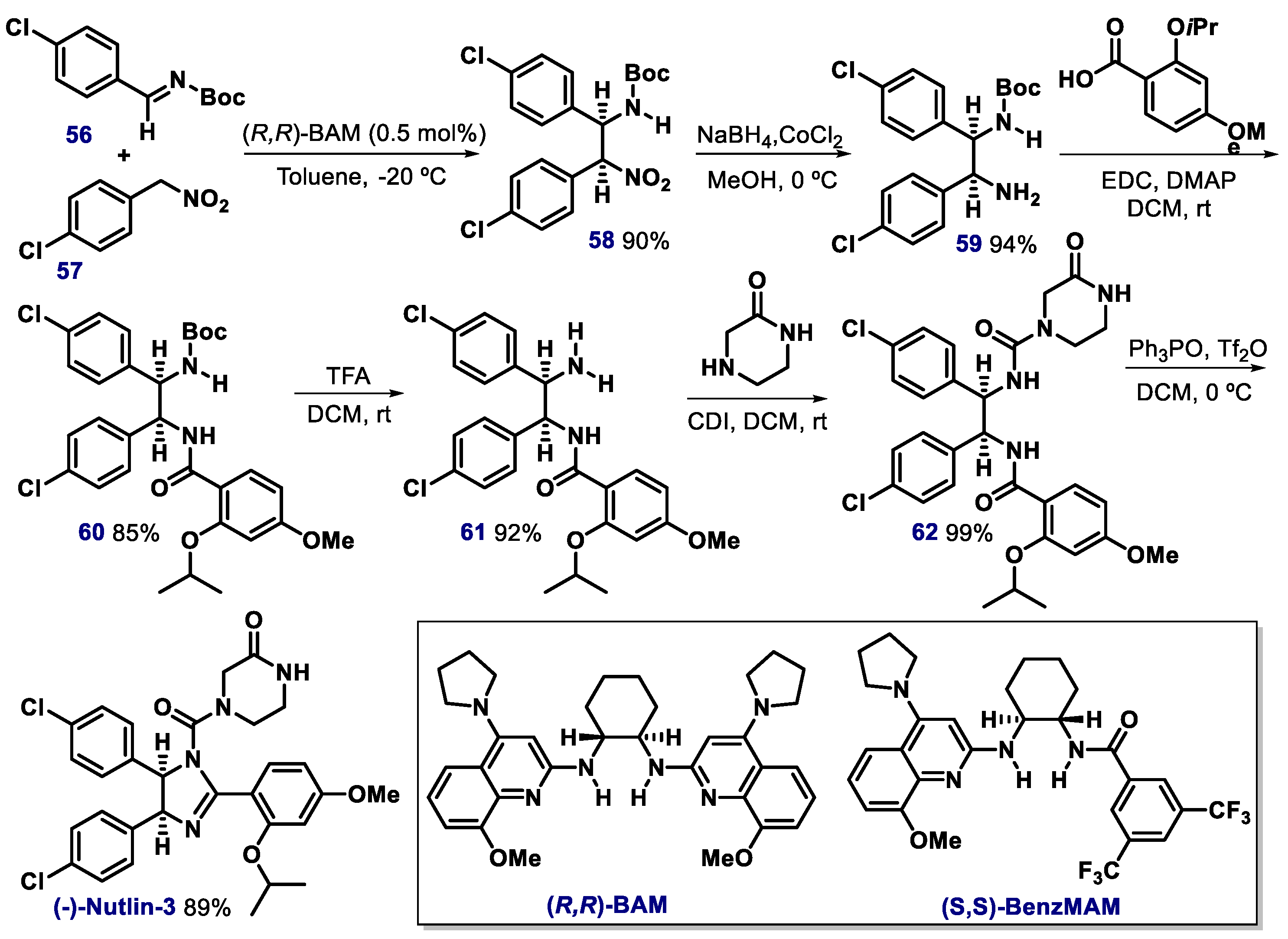
- Davis, T.A.; Johnston, J.N. Catalytic, enantioselective synthesis of stilbene cis-diamines: A concise preparation of (-)-Nutlin-3, a potent p53/MDM2. Chem. Sci. 2011, 2, 1076–1079. [Google Scholar] [CrossRef]
- Davis, T.A.; Vilgelm, A.E.; Richmond, A.; Johnston, J.N. Preparation of (−)-Nutlin-3 using enantioselective organocatalysis at decagram scale. J. Org. Chem. 2013, 78, 10605–10616. [Google Scholar] [CrossRef] [Green Version]
- U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT00623870 (accessed on 12 July 2019).
- Vara, B.A.; Mayasundari, A.; Tellis, J.C.; Danneman, M.W.; Arredondo, V.; Davis, T.A.; Min, J.; Finch, K.; Guy, R.K.; Johnston, J.N. Organocatalytic, diastereo- and enantioselective synthesis of nonsymmetric cis-stilbene diamines: A platform for the preparation of single-enantiomer cis-imidazolines for protein−protein inhibition. J. Org. Chem. 2014, 79, 6913–6938. [Google Scholar] [CrossRef] [Green Version]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [Green Version]
- Tovar, C.; Graves, B.; Packman, K.; Filipovic, Z.; Higgins, B.; Xia, M.; Tardell, C.; Garrido, R.; Lee, E.; Kolinsky, K.; et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013, 73, 2587–2597. [Google Scholar] [CrossRef] [Green Version]
- Neochoritis, C.; Estrada-Ortiz, N.; Khoury, K.; Domling, A. p53-MDM2 and MDMX Antagonists. Annu. Rep. Med. Chem. 2014, 49, 167–184. [Google Scholar]
- Ray-Coquard, I.; Blay, J.Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.; et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef]
- Czarna, A.; Beck, B.; Srivastava, S.; Popowicz, G.M.; Wolf, S.; Huang, Y.; Bista, M.; Holak, T.A.; Dömling, A. Robust generation of lead compounds for protein–protein interactions by computational and MCR chemistry: p53/HDM2 antagonists. Angew. Chem. Int. Ed. Engl. 2010, 49, 5352–5356. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.J.; Lafrance, L.V.; Calvo, R.R.; Milkiewicz, K.L.; Gupta, V.; Lattanze, J.; Ramachandren, K.; Carver, T.E.; Petrella, E.C.; Cummings, M.D.; et al. 1,4-Benzodiazepine-2,5-diones as small molecule antagonists of the HDM2–p53 interaction: Discovery and SAR. Bioorg. Med. Chem. Lett. 2005, 15, 765–770. [Google Scholar] [CrossRef]
- Hussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar]
- Yu, S.; Qin, D.; Shangary, S.; Chen, J.; Wang, G.; Ding, K.; McEachern, D.; Qiu, S.; Nikolovska-Coleska, Z.; Miller, R.; et al. Potent and orally active small-molecule inhibitors of the MDM2−p53 interaction. J. Med. Chem. 2009, 52, 7970–7973. [Google Scholar] [CrossRef] [Green Version]
- Sebahar, P.R.; Williams, R.M. The asymmetric total synthesis of (+)-and (-)-spirotryprostatin B. J. Am. Chem. Soc. 2000, 122, 5666–5667. [Google Scholar] [CrossRef]
- Ding, K.; Wang, G.; Deschamps, J.R.; Parrish, D.A.; Wang, S. Synthesis of spirooxindoles via asymmetric 1,3-dipolar cycloaddition. Tetrahedron Lett. 2005, 46, 5949–5951. [Google Scholar] [CrossRef]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2006, 49, 3432–3435. [Google Scholar] [CrossRef]
- Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R.S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 3393–3398. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yu, S.; Sun, W.; Liu, L.; Lu, J.; McEachern, D.; Shargary, S.; Bernard, D.; Li, X.; Zhao, T.; et al. A potent small-molecule inhibitor of the MDM2−p53 interaction (MI-888) achieved complete and durable tumor regression in mice. J. Med. Chem. 2013, 56, 5553–5561. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, L.; Sun, W.; Lu, J.; McEachern, D.; Li, X.; Yu, S.; Bernard, D.; Ochsenbein, P.; Ferey, V.; et al. Diastereomeric spirooxindoles as highly potent and efficacious MDM2 inhibitors. J. Am. Chem. 2013, 135, 7223–7234. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barrière, C.; Stuckey, J.; Meagher, J.; Bai, L.; Liu, L.; et al. SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-molecule inhibitors of the MDM2–p53 protein–protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef]
- Shangary, S.; Wang, S. Targeting the MDM2-p53 interaction for cancer therapy. Clin. Cancer Res. 2008, 14, 5318–5324. [Google Scholar] [CrossRef] [Green Version]
- Bill, K.L.; Garnett, J.; Meaux, I.; Ma, X.; Creighton, C.J.; Bolshakov, S.; Barriere, C.; Debussche, L.; Lazar, A.J.; Prudner, B.C.; et al. SAR405838: A novel and potent inhibitor of the MDM2:p53 axis for the treatment of dedifferentiated liposarcoma. Clin. Cancer Res. 2016, 22, 1150–1160. [Google Scholar] [CrossRef] [Green Version]
- Jonge, M.; Weger, V.A.; Dickson, M.A.; Langenberg, M.; Le Cesne, A.; Wagner, A.J.; Hsu, K.; Zheng, W.; Macé, S.; Tuffal, G.; et al. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur. J. Cancer. 2017, 76, 144–151. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT01985191 (accessed on 12 July 2019).
- von Richter, O.; Massimini, G.; Scheible, H.; Udvaros, I.; Johne, A. Pimasertib, a selective oral MEK1/2 inhibitor: Absolute bioavailability, mass balance, elimination route, and metabolite profile in cancer patients. Br. J. Clin. Pharmacol. 2016, 82, 1498–1508. [Google Scholar] [CrossRef] [Green Version]
- Bartkovitz, D.J.; Chu, X.J.; Ding, Q.; Graves, B.J.; Jiang, N.; Zhang, J.; Zhang, Z. Spiroindolinone Pyrrolidines. International Patent Application No. PCT/EP2010/068353, 9 June 2011. [Google Scholar]
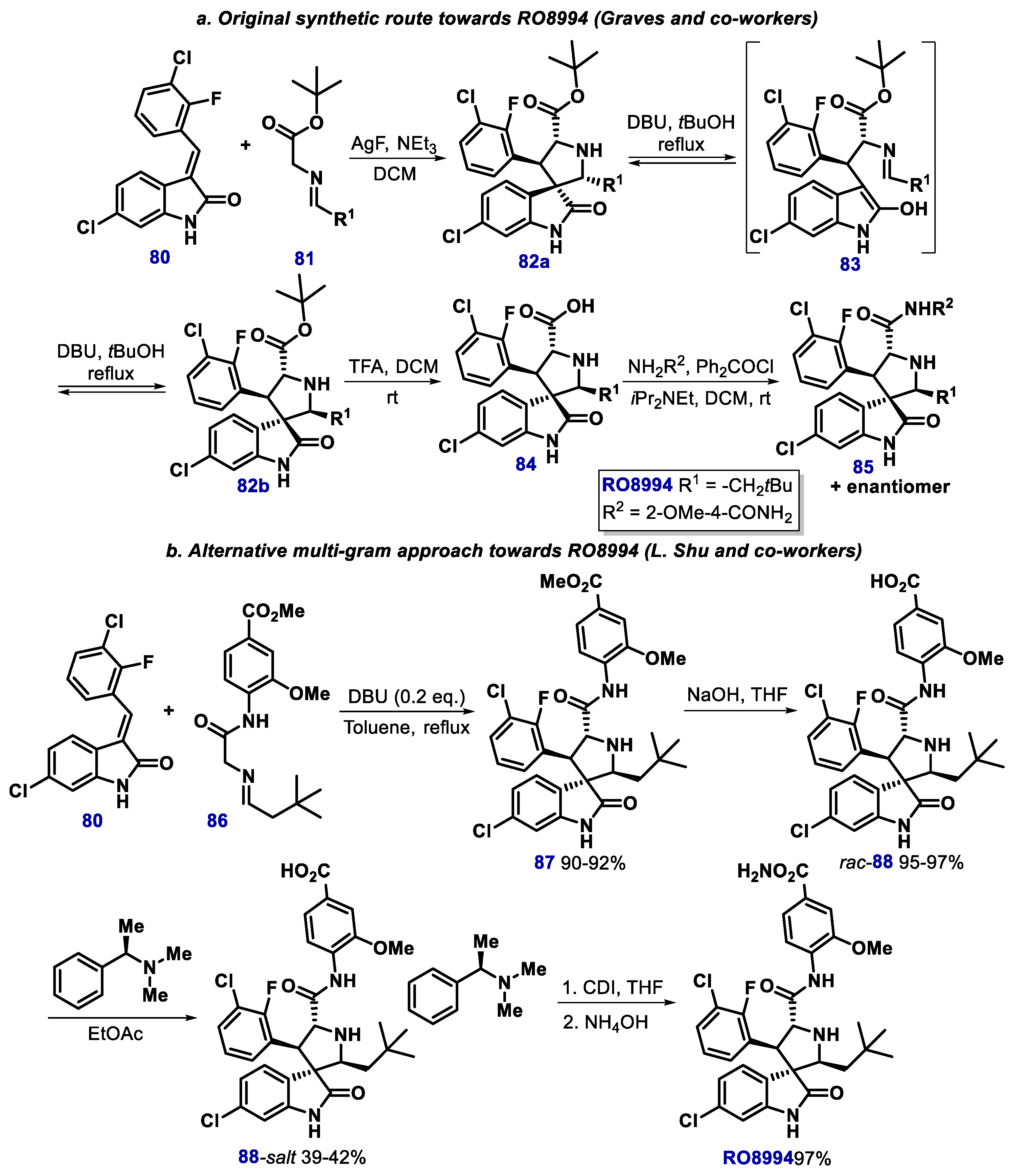
- Zhang, Z.; Ding, Q.; Liu, J.J.; Zhang, J.; Jiang, N.; Chu, X.J.; Bartkovitz, D.; Luk, K.C.; Janson, C.; Tovar, C.; et al. Discovery of potent and selective spiroindolinone MDM2 inhibitor, RO8994, for cancer therapy. Bioorg. Med. Chem. 2014, 22, 4001–4009. [Google Scholar] [CrossRef]
- Shu, L.; Li, Z.; Gu, C.; Fishlock, D. Synthesis of a Spiroindolinone Pyrrolidinecarboxamide MDM2 Antagonist. Org. Process Res. Dev. 2013, 17, 247–256. [Google Scholar] [CrossRef]
- Zhang, Z.; Chu, X.J.; Liu, J.J.; Ding, Q.; Zhang, J.; Bartkovitz, D.; Jiang, N.; Karnachi, P.; So, S.S.; Tovar, C.; et al. Discovery of Potent and Orally Active p53-MDM2 Inhibitors RO5353 and RO2468 for Potential Clinical Development. ACS Med. Chem. Lett. 2014, 5, 124–127. [Google Scholar] [CrossRef] [Green Version]
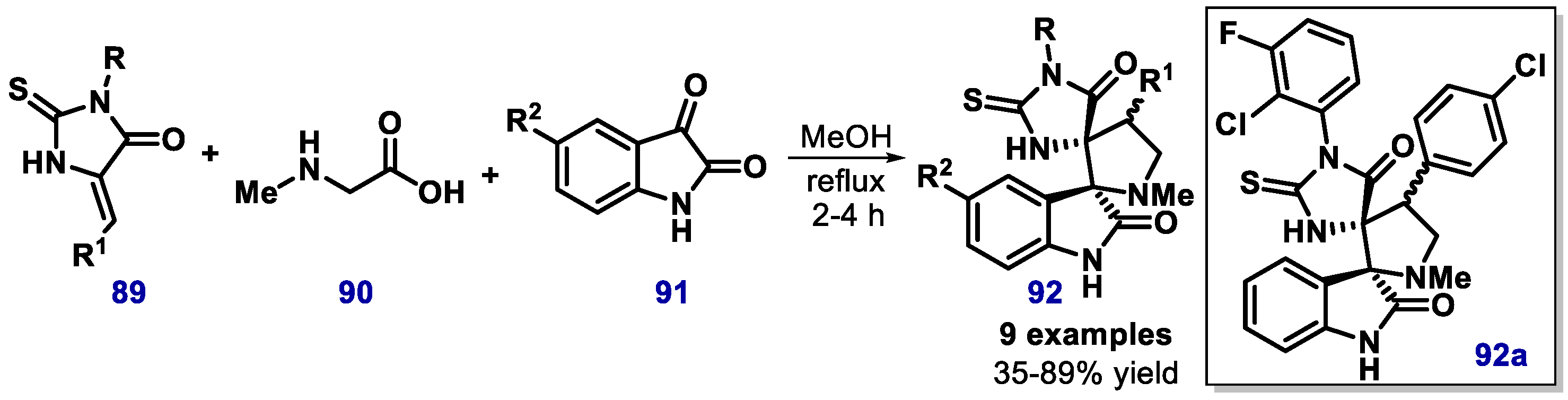
- Ivanenkov, Y.A.; Vasilevski, S.V.; Beloglazkina, E.K.; Kukushkin, M.E.; Machulkin, A.E.; Veselov, M.S.; Chufarova, N.V.; Chernyaginab, E.S.; Vanzcoo, A.S.; Zyk, N.V.; et al. Design, synthesis and biological evaluation of novel potent MDM2/p53 small-molecule inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Amaral, J.D.; Silva, D.; Rodrigues, C.M.P.; Solá, S.; Santos, M.M.M. A novel small molecule p53 stabilizer for brain cell differentiation. Front. Chem. 2019, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT02319369 (accessed on 16 July 2019).
- U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT01877382 (accessed on 16 July 2019).
- U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT01451437 (accessed on 16 July 2019).
- U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/show/NCT01463696 (accessed on 16 July 2019).
- Liao, G.; Yang, D.; Ma, L.; Li, W.; Hu, L.; Zeng, L.; Wu, P.; Duan, L.; Liu, Z. The development of piperidinones as potent MDM2-P53 protein-protein interaction inhibitors for cancer therapy. Eur. J. Med. Chem. 2018, 159, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.J.; Banerji, U.; Mahipal, A.; Somaiah, N.; Hirsch, H.; Fancourt, C.; Johnson-Levonas, A.O.; Lam, R.; Meister, A.K.; Russo, G.; et al. Phase I trial of the human double minute 2 inhibitor MK-8242 in patients with advanced solid tumors. J. Clin. Oncol. 2017, 35, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Gojo, I.; Patnaik, M.M.; Minden, M.D.; Kantarjian, H.; Johnson-Levonas, A.O.; Fancourt, C.; Lam, R.; Jones, M.B.; Knox, C.D.; et al. A phase I trial of the human double minute 2 inhibitor (MK-8242) in patients with refractory/recurrent acute myelogenous leukemia (AML). Leuk. Res. 2016, 48, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zeng, S.X.; Lu, H. Targeting p53-MDM2-MDMX loop for cancer therapy. Subcell. Biochem. 2014, 85, 281–319. [Google Scholar]
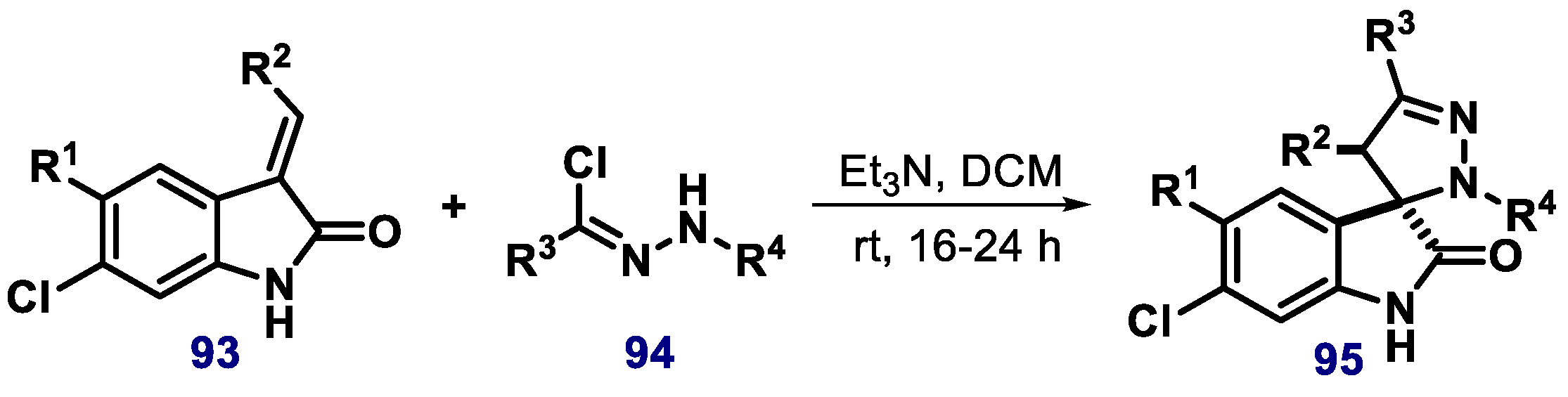
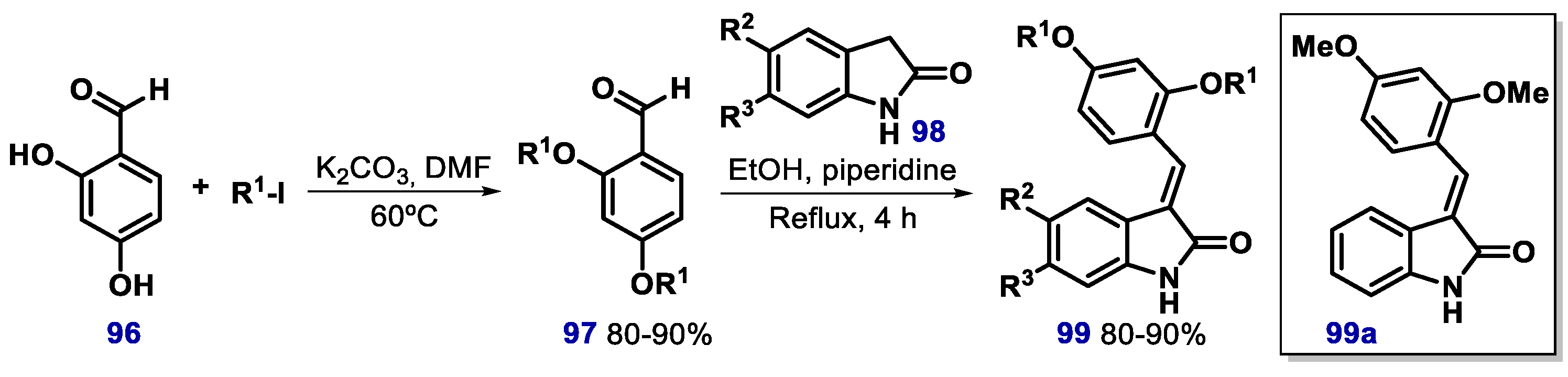
- Zheng, G.H.; Shen, J.J.; Zhan, Y.C.; Yi, H.; Xue, S.T.; Wang, Z.; Ji, X.Y.; Li, Z.R. Design, synthesis and in vitro and in vivo antitumour activity of 3-benzylideneindolin-2-one derivatives, a novel class of small-molecule inhibitors of the MDM2-p53 interaction. Eur. J. Med. Chem. 2014, 81, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Hardcastle, I.R.; Ahmed, S.U.; Atkins, H.; Farnie, G.; Golding, B.T.; Griffin, R.J.; Guyenne, S.; Hutton, C.; Kallblad, P.; Kemp, S.J.; et al. Small-molecule inhibitors of the MDM2-p53 protein–protein interaction based on an isoindolinone scaffold. J. Med. Chem. 2006, 49, 6209–6221. [Google Scholar] [CrossRef]
- Hardcastle, I.R.; Liu, J.; Valeur, E.; Watson, A.; Ahmed, S.U.; Blackburn, T.J.; Bennaceur, K.; Clegg, W.; Drummond, C.; Endicott, J.A.; et al. Isoindolinone inhibitors of the Murine Double Minute 2 (MDM2)-p53 protein–protein interaction: Structure-activity studies leading to improved potency. J. Med. Chem. 2011, 54, 1233–1243. [Google Scholar] [CrossRef]
- Chargari, C.; Leteur, C.; Angevin, E.; Bashir, T.; Schoentjes, B.; Arts, J.; Janicot, M.; Bourhis, J.; Deutsch, E. Preclinical assessment of JNJ-26854165 (Serdemetan1), a novel tryptamine compound with radiosensitizing activity in vitro and in tumor xenografts. Cancer Lett. 2011, 312, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Burks, J.K.; Arts, J.; Andreeff, M. The novel tryptamine derivative JNJ- 26854165 induces wild-type p53- and E2F1-mediated apoptosis in acute myeloid and lymphoid leukemias. Mol. Cancer Ther. 2010, 9, 2545–2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabernero, J.; Dirix, L.; Schoffski, P.; Cervantes, A.; Lopez-Martin, J.A.; Capdevila, J.; van Beijsterveldt, L.; Platero, S.; Hall, B.; Yuan, Z.; et al. A phase I first-in-human pharmacokinetic and pharmacodynamic study of serdemetan in patients with advanced solid tumors. Clin. Cancer Res. 2011, 17, 6313–6321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacrampe, J.F.A.; Meyer, C.; Ligny, Y.A.E.; Csoka, I.C.F.; Van Hijfte, L.; Arts, J.; Schoentjes, B.; Wermuth, C.G.; Giethlen, B.; Contreras, J.-M.; et al. Inhibitors of the interaction between MDM2 and P53. International Patent Application No. PCT/EP2005/054604, 30 March 2006. [Google Scholar]
- Wang, W.; Qin, J.-J.; Voruganti, S.; Srivenugopal, K.S.; Nag, S.; Patil, S.; Sharma, H.; Wang, M.-H.; Wang, H.; Buolamwini, J.K.; et al. The pyrido[b]indole MDM2 inhibitor SP-141 exerts potent therapeutic effects in breast cancer models. Nat. Commun. 2014, 5, 5086. [Google Scholar] [CrossRef] [Green Version]
- Foley, C.A.; Al-Issa, Y.A.; Hiller, K.P.; Mulcahy, S.P. Synthesis and structure-activity relationships of 1-aryl-β-carbolines as affinity probes for the 5-hydroxytryptamine receptor. ACS Omega 2019, 4, 9807–9812. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef]
- Rimmler, G.; Alker, A.; Bosco, M.; Diodone, R.; Fishlock, D.; Hildbrand, S.; Kuhn, B.; Moessner, C.; Peters, C.; Rege, P.D.; et al. Practical synthesis of MDM2 antagonist RG7388. Part 2: Development of the Cu(I) catalyzed [3 + 2] asymmetric cycloaddition process for the manufacture of idasanutlin. Org. Process Res. Dev. 2016, 20, 2050–2056. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/study/NCT01773408 (accessed on 16 July 2019).
- Michelsen, K.; Jordan, J.B.; Long, A.M.; Lewis, J.K.; Yang, E.; Rew, Y.; Zhou, J.; Yakowec, P.; Schnier, P.D.; Huang, X.; et al. Ordering of the N-terminus of human MDM2 by small molecule inhibitors. J. Am. Chem. Soc. 2012, 134, 17059–17067. [Google Scholar] [CrossRef]
- Rew, Y.; Sun, D.; Turiso, F.G.L.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Deignan, J.; Fox, B.M.; et al. Structure-based design of novel inhibitors of the MDM2−p53 interaction. J. Med. Chem. 2012, 55, 4936–4954. [Google Scholar] [CrossRef]
- Yu, M.; Wang, Y.; Zhu, J.; Bartberger, M.D.; Canon, J.; Chen, A.; Chow, D.; Eksterowicz, J.; Fox, B.; Fu, J.; et al. Discovery of potent and simplified piperidinone-based inhibitors of the MDM2–p53 interaction. ACS Med. Chem. Lett. 2014, 5, 894–899. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Li, Z.; Rew, Y.; Gribble, M.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chen, X.; Chow, D.; et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2−p53 inhibitor in clinical development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT02110355 (accessed on 16 July 2019).
- Rew, Y.; Sun, D.; Yan, X.; Beck, H.P.; Canon, J.; Chen, A.; Duquette, J.; Eksterowicz, J.; Fox, B.M.; Fu, J.; et al. Discovery of AM-7209, a Potent and Selective 4.Amidobenzoic Acid Inhibitor of the MDM2.p53 Interaction. J. Med. Chem. 2014, 57, 10499–10511. [Google Scholar] [CrossRef]
- Holzer, P.; Masuya, K.; Furet, P.; Kallen, J.; Valat-Stachyra, T.S.; Ferretti, S.; Berghausen, J.; Bouisset-Leonard, M.; Buschmann, N.; Pissot-Soldermann, C.; et al. Discovery of a dihydroisoquinolinone derivative (NVP-CGM097) – a highly potent and selective MDM2 inhibitor undergoing phase 1 clinical trials I p53wt tumors. J. Med. Chem. 2015, 58, 6348–6358. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT01760525 (accessed on 16 July 2019).
- Townsend, E.C.; De Souza, T.; Murakami, M.A.; Montero, J.; Stevenson, K.; Christie, A.L.; Christodolou, A.N.; Vojinovic, U.; Kopp, N.; Barzaghi-Rinaudo, P.; et al. The MDM2 inhibitor NVP-CGM097 is highly active in a randomized preclinical trial of B-cell acute lymphoblastic leukemia patient derived xenografts. Blood 2015, 126, 797. [Google Scholar] [CrossRef]
- de Turiso, F.G.-L.; Sun, D.; Rew, Y.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Correll, T.L.; Huang, X.; et al. Rational design and binding mode duality of MDM2–p53 inhibitors. Med. Chem. 2013, 56, 4053–4070. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.Z.; Eksterowicz, J.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Duquette, J.; Fox, B.M.; Fu, J.; et al. Selective and potent morpholinone inhibitors of the MDM2-p53 protein−protein interaction. J. Med. Chem. 2014, 57, 2472–2488. [Google Scholar] [CrossRef]
- Yang, Y.; Ludwig, R.L.; Jensen, J.P.; Pierre, S.A.; Medaglia, M.V.; Davydov, I.V.; Safiran, Y.J.; Oberoi, P.; Kenten, J.H.; Phillips, A.C.; et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell 2005, 7, 547–559. [Google Scholar] [CrossRef] [Green Version]
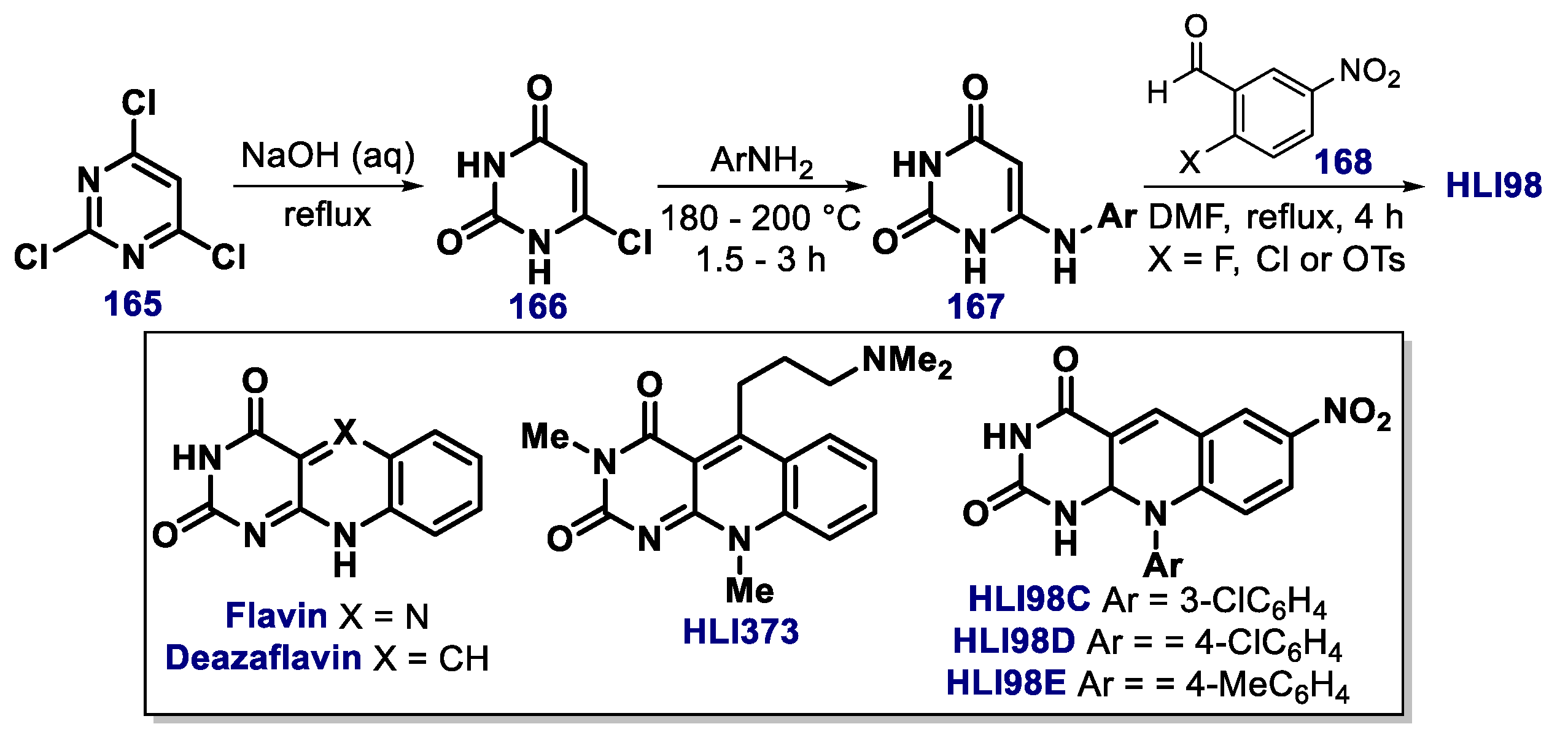
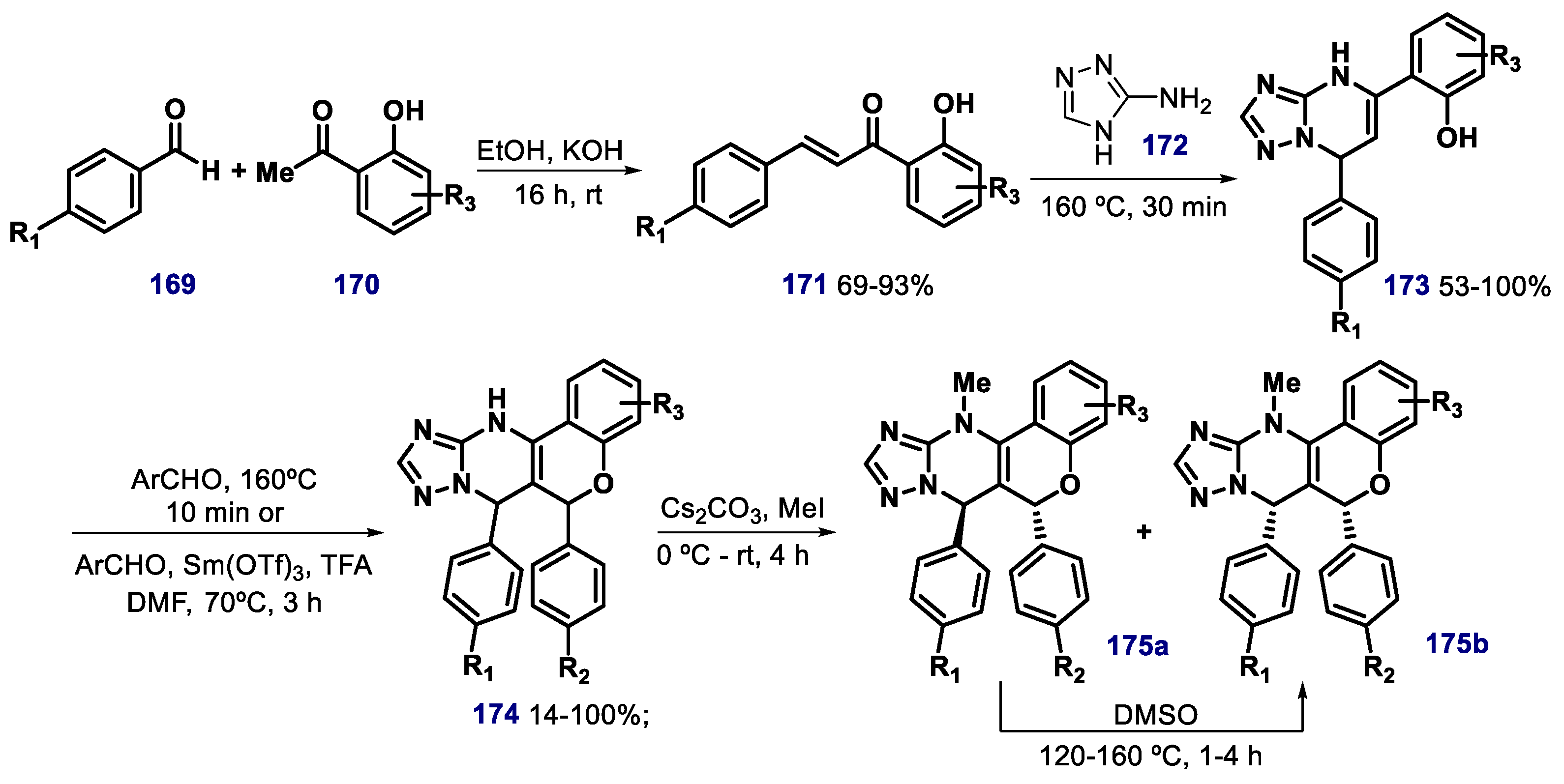
- Dickens, M.P.; Roxburgh, P.; Hock, A.; Mezna, M.; Kellam, B.; Vousden, K.H.; Fischer, P.M. 5-Deazaflavin derivatives as inhibitors of p53 ubiquitination by HDM2. Bioorg. Med. Chem. 2013, 21, 6868–6877. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.G.; Bourbeau, M.P.; Wohlhieter, G.E.; Bartberger, M.D.; Michelsen, K.; Hungate, R.; Gadwood, R.C.; Gaston, R.D.; Evans, B.; Mann, L.W.; et al. Discovery and optimization of chromenotriazolopyrimidines as potent inhibitors of the Mouse Double Minute 2-tumor protein 53 protein–protein interaction. J. Med. Chem. 2009, 52, 7044–7053. [Google Scholar] [CrossRef]
- Furet, P.; Masuya, K.; Kallen, J.; Stachyra-Valat, T.; Ruetz, S.; Guagnano, V.; Holzer, P.; Mah, R.; Stutz, S.; Vaupel, A.; et al. Discovery of a novel class of highly potent inhibitors of the p53-MDM2 interaction by structure-based design starting from a conformational argument. Bioorg. Med. Chem. Lett. 2016, 26, 4837–4841. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT02343172 (accessed on 16 July 2019).
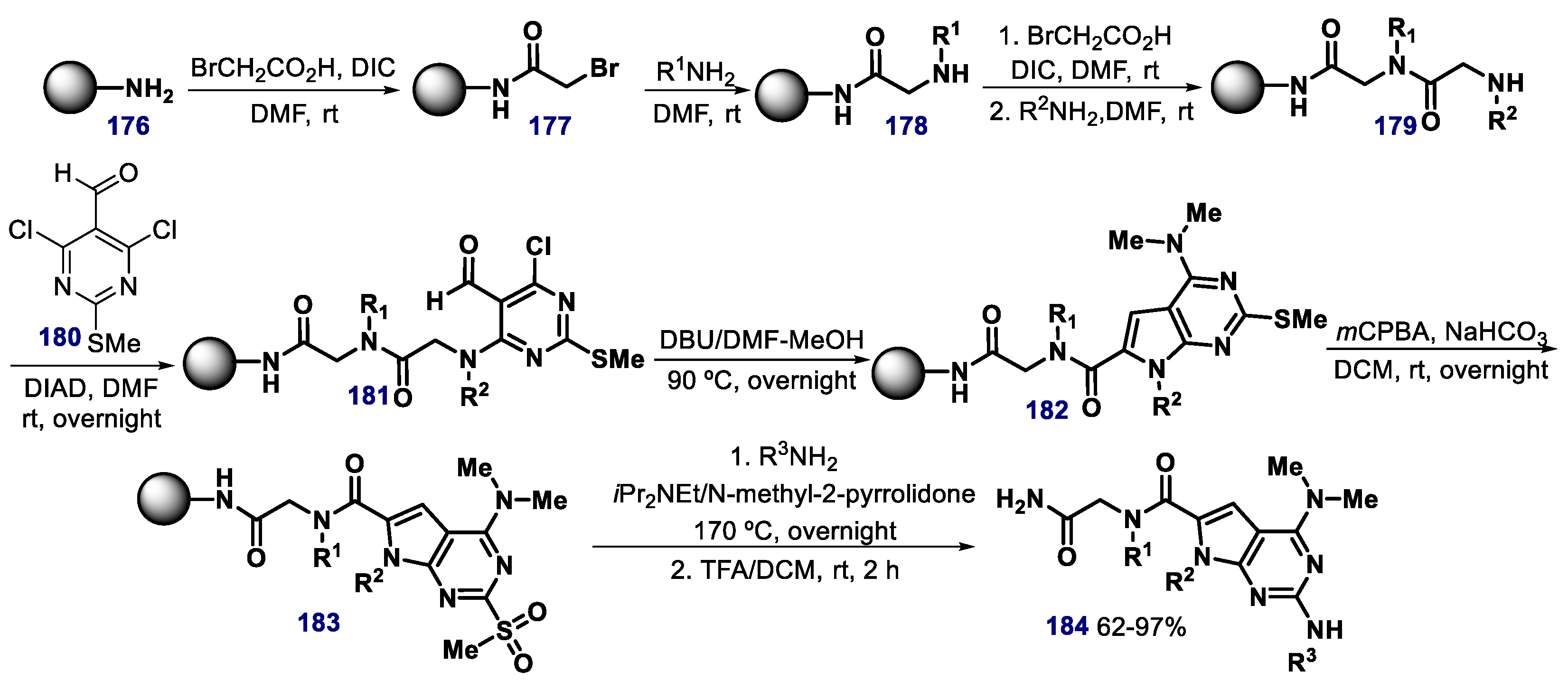
- Lee, J.H.; Zhang, Q.; Jo, S.; Chai, S.C.; Oh, M.; Im, W.; Lu, H.; Lim, H.-S. Novel pyrrolopyrimidine-based R-helix mimetics: Cell-permeable inhibitors of protein–protein interactions. J. Am. Chem. Soc. 2011, 133, 676–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danovi, D.; Meulmeester, E.; Pasini, D.; Migliorini, D.; Capra, M.; Frenk, R.; de Graaf, P.; Francoz, S.; Gasparini, P.; Gobbi, A.; et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol. Cell. Biol. 2004, 24, 5835–5843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, F.; Schulz, J.; Bohnke, A.; Blumke, K.; Kappler, M.; Bache, M.; Schmidt, H.; Wurl, P.; Taubert, H.; Hauptmann, S. Significance of HDMX-S (or MDM4) mRNA splice variant overexpression and HDMX gene amplification on primary soft tissue sarcoma prognosis. Int. J. Cancer 2005, 117, 469–475. [Google Scholar] [CrossRef]
- Reed, D.; Shen, Y.; Shelat, A.A.; Arnold, L.A.; Ferreira, A.M.; Zhu, F.; Mills, N.; Smithson, D.C.; Regni, C.A.; Bashford, D.; et al. Identification and characterization of the first small molecule inhibitor of MDMX. J. Biol. Chem. 2010, 285, 10786–10796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
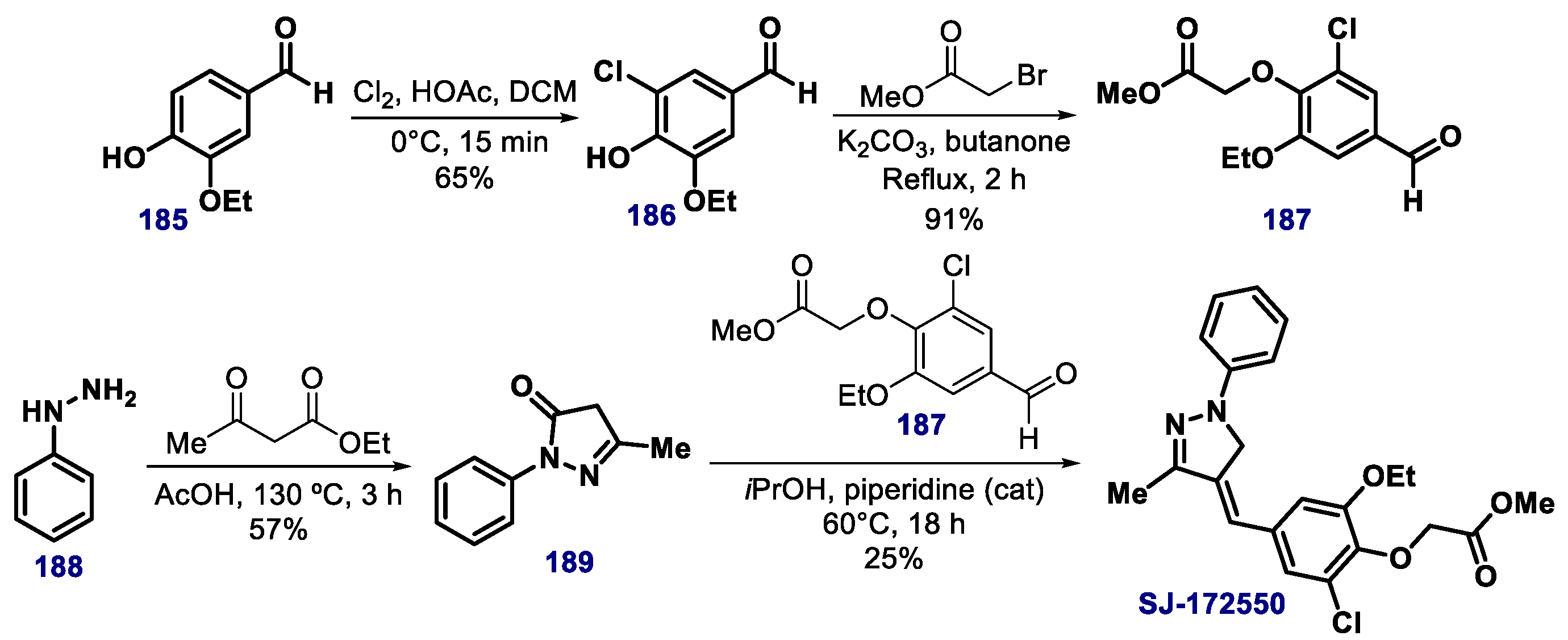
- Stefaniak, J.; Lewis, A.M.; Conole, D.; Galan, S.R.G.; Bataille, C.J.R.; Wynne, G.M.; Castaldi, M.P.; Lundbäck, T.; Russell, A.J.; Huber, K.V.M. chemical instability and promiscuity of arylmethylidenepyrazolinone-based MDMX Inhibitors. ACS Chem. Biol. 2018, 13, 2849–2854. [Google Scholar] [CrossRef] [PubMed]
- Noushini, S.; Alipour, E.; Emami, S.; Safavi, M.; Ardestani, S.K.; Gohari, A.R.; Shafiee, A.; Foroumadi, A. Synthesis and cytotoxic properties of novel (E)-3-benzylidene-7-methoxychroman-4-one derivatives. Daru 2013, 21, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, C.; Miao, Z.; Wu, Y.; Guo, Z.; Li, J.; Yao, J.; Xing, C.; Sheng, C.; Zhang, W. Double-edged swords as cancer therapeutics: Novel, orally active, small molecules simultaneously inhibit p53-MDM2 interaction and the NF-kB pathway. J. Med. Chem. 2014, 57, 567–577. [Google Scholar] [CrossRef]
- Zhuang, C.; Miao, Z.; Zhu, L.; Dong, G.; Guo, Z.; Wang, S.; Zhang, Y.; Wu, Y.; Yao, J.; Sheng, C.; et al. Discovery, Synthesis, and biological evaluation of orally active pyrrolidone derivatives as novel inhibitors of p53-MDM2 protein–protein interaction. J. Med. Chem. 2012, 55, 9630–9642. [Google Scholar] [CrossRef]
- Grasberger, B.L.; Lu, T.; Schubert, C.; Parks, D.J.; Carver, T.E.; Koblish, H.K.; Cummings, M.D.; LaFrance, L.V.; Milkiewicz, K.L.; Calvo, R.R.; et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J. Med. Chem. 2005, 48, 909–912. [Google Scholar] [CrossRef]
- Zhuang, C.; Miao, Z.; Zhu, L.; Zhang, Y.; Guo, Z.; Yao, J.; Dong, G.; Wang, S.; Liu, Y.; Chen, H.; et al. Synthesis and biological evaluation of thio-benzodiazepines as novel small molecule inhibitors of the p53-MDM2 protein–protein interaction. Eur. J. Med. Chem. 2011, 46, 5654–5661. [Google Scholar] [CrossRef]
- Wang, H.; Ma, X.; Ren, S.; Buolamwini, J.K.; Yan, C. A small-molecule inhibitor of MDMX activates p53 and induces apoptosis. Mol. Cancer Ther. 2011, 10, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
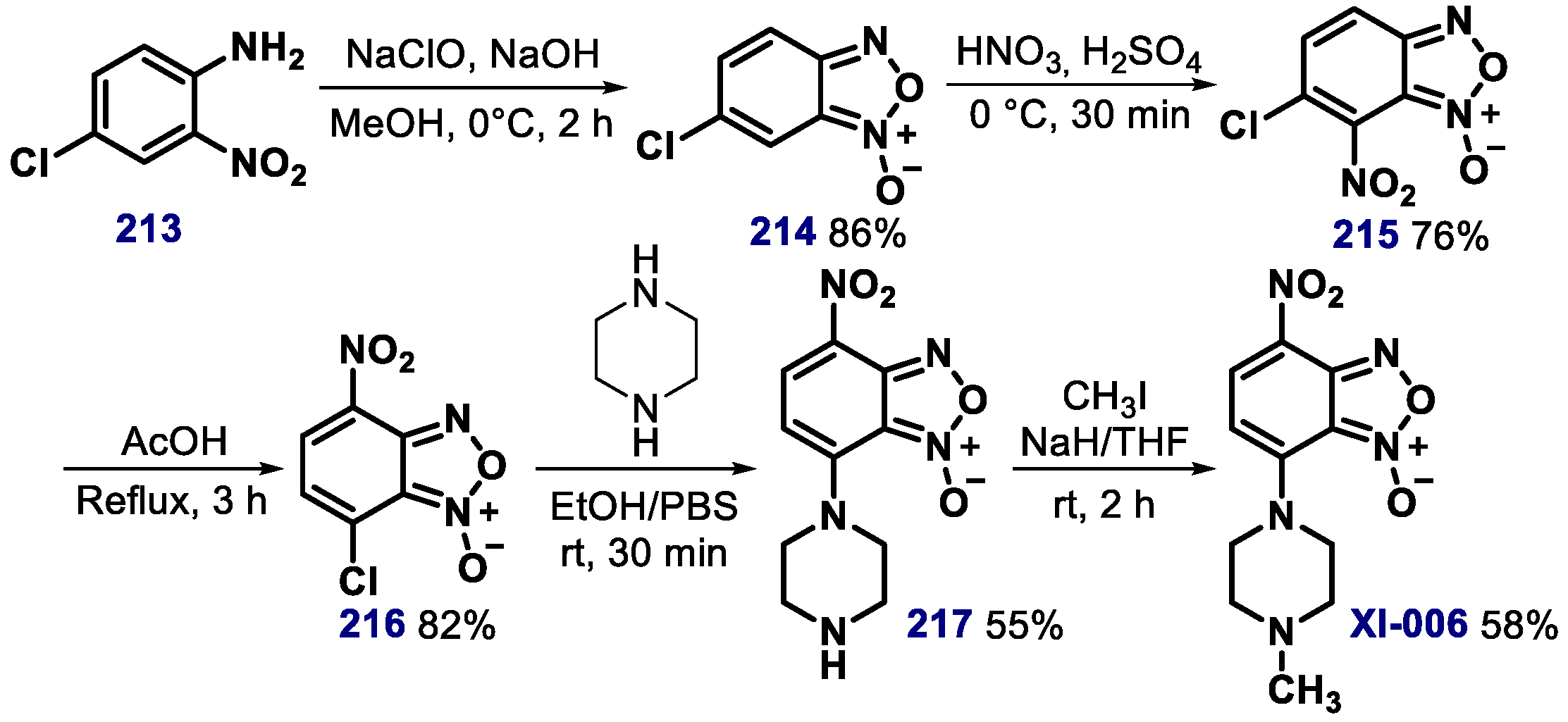
- Pishas, K.I.; Adwal, A.; Neuhaus, S.J.; Clayer, M.T.; Farshid, G.; Staudacher, A.H.; Callen, D.F. XI-006 induces potent p53-independent apoptosis in Ewing sarcoma. Sci. Rep. 2015, 5, 11465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, C.; Yonghao, Y.; Wang, L. Application of benzofuroxan derivative to resisting plant pathogenic fungi. Chinese Patent Application No. 201310003647.5, 3 April 2013. [Google Scholar]
- Issaeva, N.; Bozko, P.; Engel, M.; Protopopova, M.; Verhoef1, L.G.G.C.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53–HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Burmakin, K.; Shi, Y.; Hedström, E.; Kogner, P.; Selivanova, G. Dual targeting of wild-type and mutant p53 by small molecule RITA results in the inhibition of N-Myc and key survival oncogenes and kills neuroblastoma cells in vivo and in vitro. Clin. Cancer Res. 2013, 19, 5092–5103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Ding, C.; Li, L.; Gao, C.; Jiang, Y.; Tan, C.; Hua, R. Synthesis and antiproliferative activity of RITA and its analogs. Tet. Lett. 2014, 55, 6635–6638. [Google Scholar] [CrossRef]
- Lain, S.; Midgley, C.; Sparks, A.; Lane, E.B.; Lane, D.P. An inhibitor of nuclear export activates the p53 response and induces the localization of HDM2 and p53 to U1A-positive nuclear bodies associated with the PODs. Exp. Cell. Res. 1999, 248, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Freedman, D.A.; Levine, A.J. Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol. Cell. Biol. 1998, 18, 7288–7293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
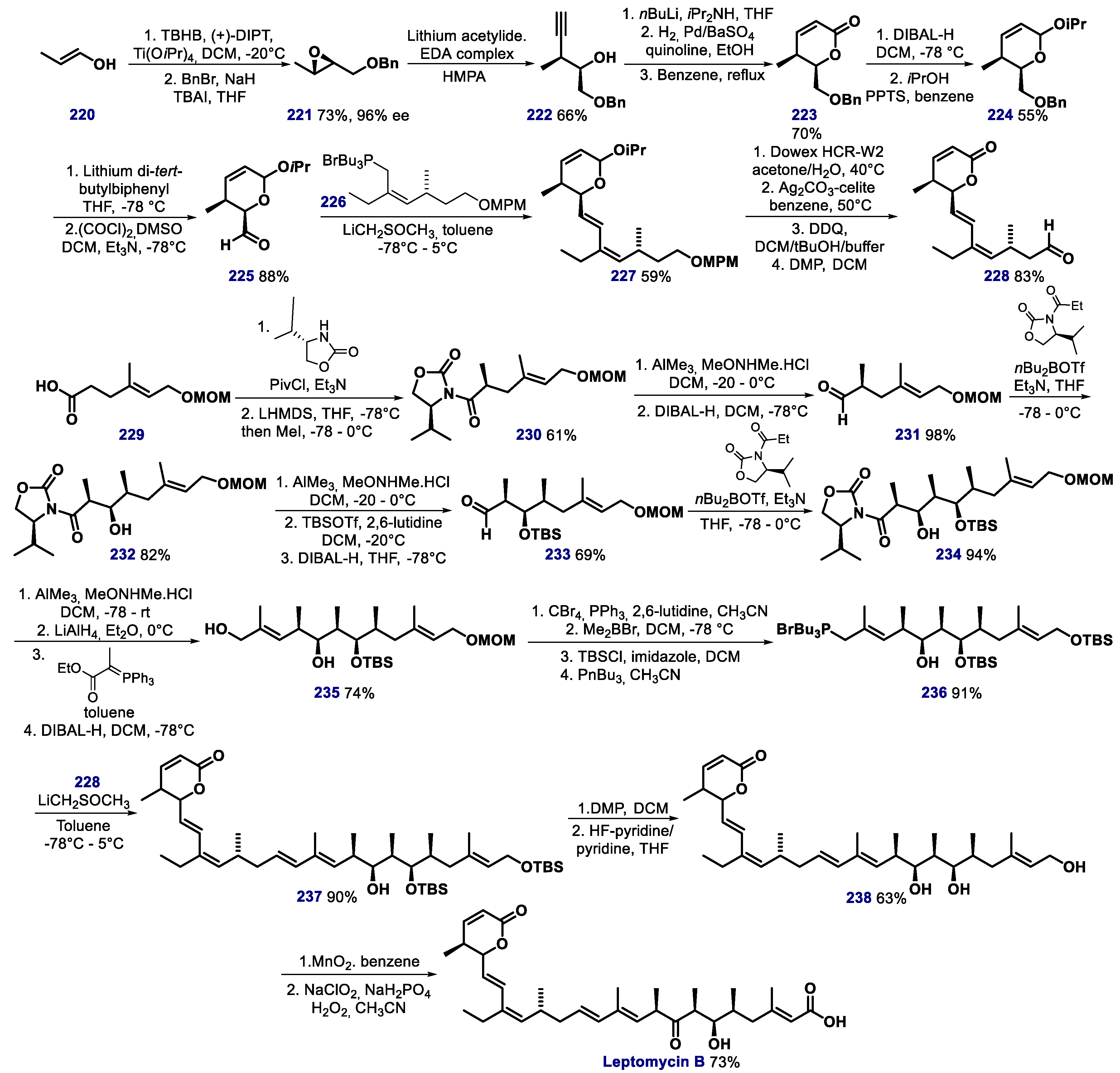
- Kudo, N.; Matsumori, N.; Taoka, H.; Fujiwara, D.; Schreiner, E.P.; Wolff, B.; Yoshida, M.; Horinouchi, S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. USA 1999, 96, 9112–9117. [Google Scholar] [CrossRef] [Green Version]
- Lain, S.; Xirodimas, D.; Lane, D.P. Accumulating active p53 in the nucleus by inhibition of nuclear export: A novel strategy to promote the p53 tumor suppressor function. Exp. Cell. Res. 1999, 253, 315–324. [Google Scholar] [CrossRef]
- Shao, C.; Lu, C.; Chen, L.; Koty, P.P.; Cobos, E.; Gao, W. p53-dependent anticancer effects of leptomycin B on lung adenocarcinoma. Cancer Chemother. Pharmacol. 2011, 67, 1369–1380. [Google Scholar] [CrossRef]
- Mutka, S.C.; Yang, W.Q.; Dong, S.D.; Ward, S.L.; Craig, D.A.; Timmermans, P.B.M.W.M.; Murli, S. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res. 2009, 69, 510–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, M.; Wang, W.; Tsutsui, Y.; Sugimoto, M.; Murakami, N. Absolute stereostructure and total synthesis of leptomycin B. Tetrahedron Lett. 1998, 39, 8291–8294. [Google Scholar] [CrossRef]
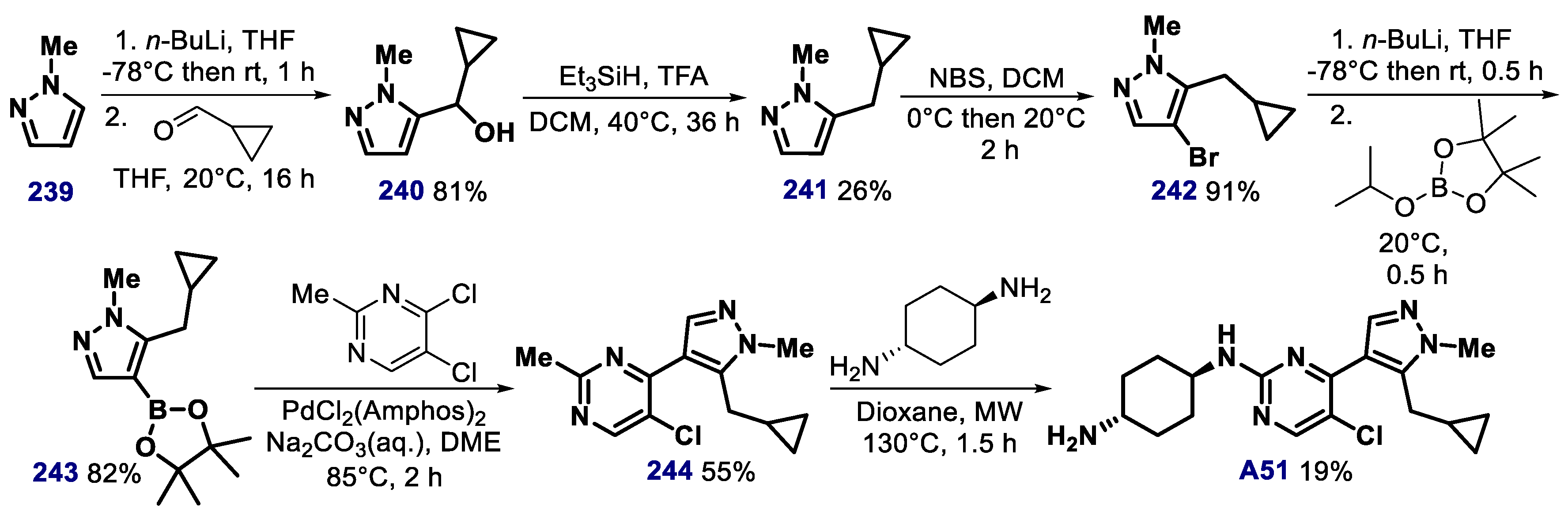
- Jaras, M.; Miller, P.G.; Chu, L.P.; Puram, R.V.; Fink, E.C.; Schneider, R.K.; Al- Shahrour, F.; Peña, P.; Breyfogle, L.J.; Hartwell, K.A.; et al. Csnk1a1 inhibition has p53-dependent therapeutic efficacy in acute myeloid leukemia. J. Exp. Med. 2014, 211, 605–612. [Google Scholar] [CrossRef]
- Minzel, W.; Venkatachalam, A.; Fink, A.; Hung, E.; Brachya, G.; Burstain, I.; Shaham, M.; Rivlin, A.; Omer, I.; Zinger, A.; et al. Small molecules co-targeting CKIa and the transcriptional kinases CDK7/9 control AML in preclinical models. Cell 2018, 175, 171–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [Green Version]
- Van Leeuwen, I.; Lain, S. Sirtuins and p53. Adv. Cancer Res. 2009, 102, 171–195. [Google Scholar]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Langley, E.; Pearson, M.; Faretta, M.; Bauer, U.M.; Frye, R.A.; Minucci, S.; Pelicci, P.G.; Kouzarides, T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002, 21, 2383–2396. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Lain, S.; Hollick, J.J.; Campbell, J.; Staples, O.D.; Higgins, M.; Aoubala, M.; McCarthy, A.R.; Appleyard, V.; Murray, K.E.; Baker, L.; et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 2008, 13, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
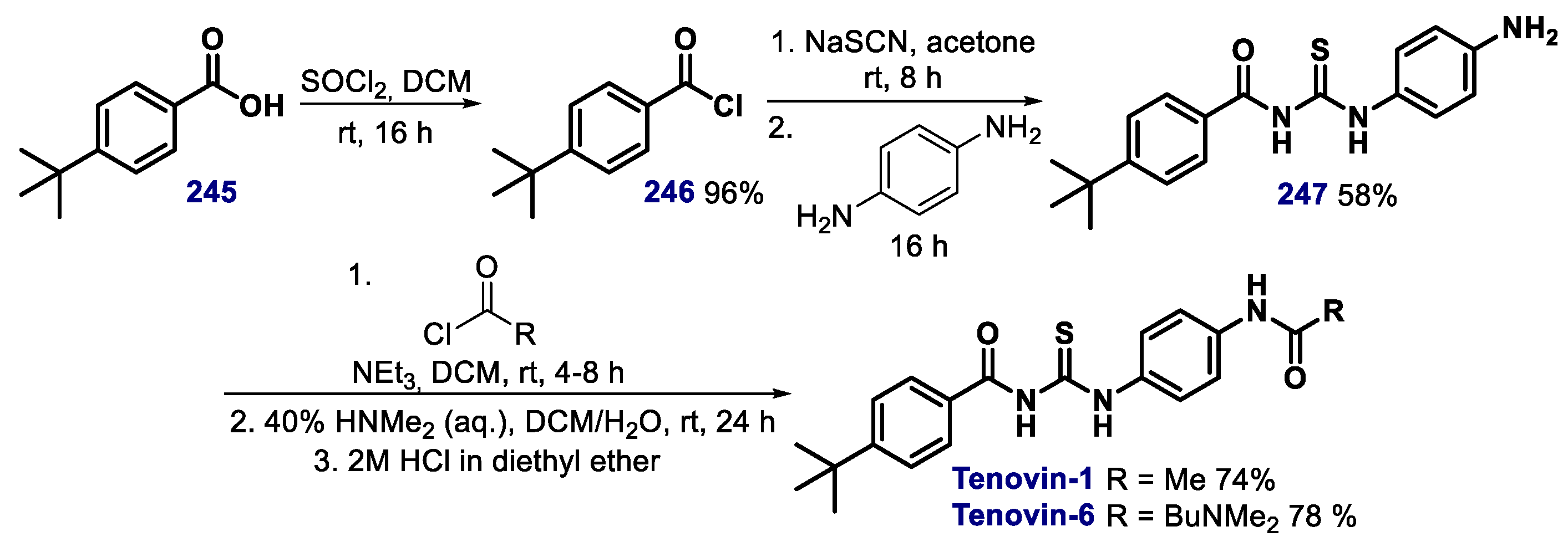
- McCarthy, A.R.; Pirrie, L.; Hollick, J.J.; Ronseaux, S.; Campbell, J.; Higgins, M.; Staples, O.D.; Tran, F.; Slawin, A.M.Z.; Lain, S.; et al. Synthesis and biological characterisation of sirtuin inhibitors based on the tenovins. Bioorg. Med. Chem. 2012, 20, 1779–1793. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zeng, S.X.; Zhang, Y.; Zhang, Y.; Ding, D.; Ye, Q.; Meroueh, S.O.; Lu, H. A small molecule Inauhzin inhibits SIRT1 activity and suppresses tumour growth through activation of p53. EMBO Mol. Med. 2012, 4, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ding, D.; Zeng, S.X.; Ye, Q.-Z.; Lu, H. Structure and activity analysis of inauhzin analogs as novel antitumor compounds that induce p53 and inhibit cell growth. PLoS ONE 2012, 7, e46294. [Google Scholar] [CrossRef]
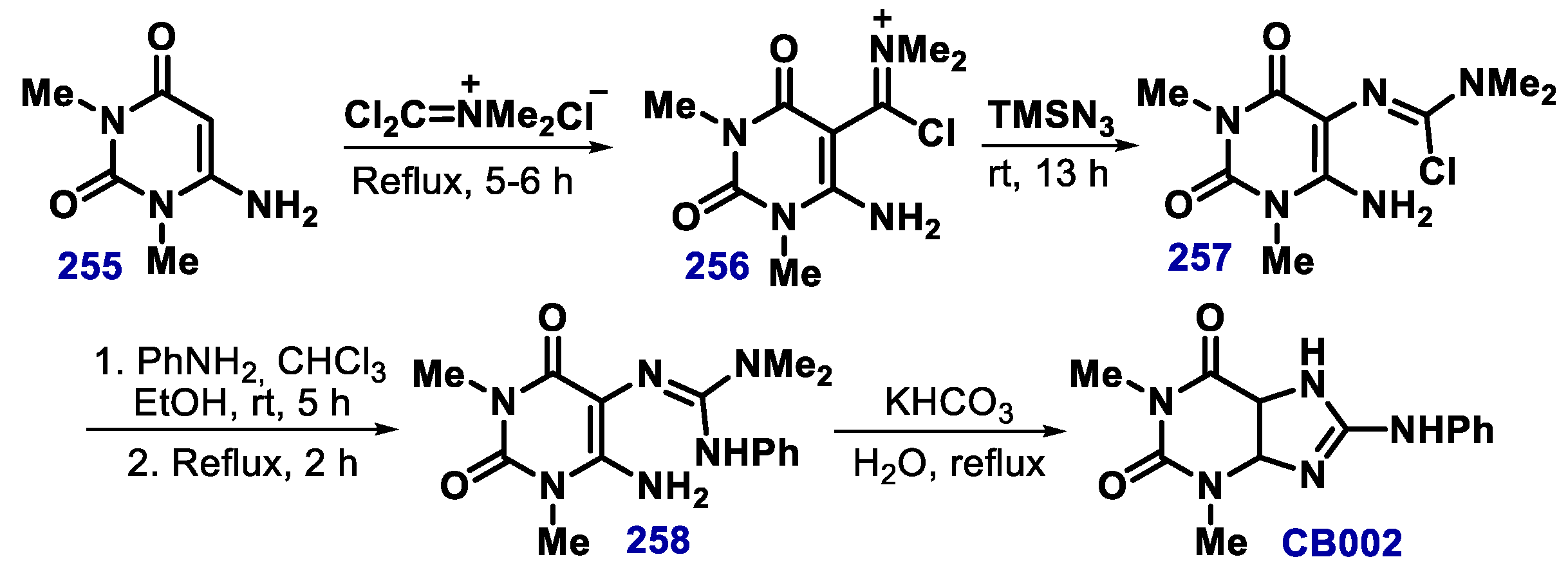
- Kokel, B. A new entry to the imidazo[4,5-d] pyrimidine system. The reaction of 1,3-dimethyl-6-aminouracil with N,N-dimethyldichloromethyleniminium chloride (phosgeniminium chloride) and trimethylsilyl azide, a novel and convenient “one pot” synthesis of 8-N-arylaminotheophyllines (2-N-arylamino-4,6-dimethylimidazo[4,5-d]pyrimidine-(4H,6H)-5,7-diones), starting from 1,3-dimethyl-6-aminouracil. Heterocycl. Chem. 1994, 31, 1185. [Google Scholar]
- Lukman, S.; Lane, D.P.; Verma, C.S. Mapping the structural and dynamical features of multiple p53 DNA binding domains: Insights into loop 1 intrinsic dynamics. PLoS ONE 2013, 8, e80221. [Google Scholar] [CrossRef] [Green Version]
- Cino, E.A.; Soares, I.N.; Pedrote, M.M.; de Oliveira, G.A.; Silva, J.L. Aggregation tendencies in the p53 family are modulated by backbone hydrogen bonds. Sci. Rep. 2016, 6, 32535. [Google Scholar] [CrossRef] [Green Version]
- Lima, I.; Navalkar, A.; Maji, S.K.; Silva, J.L.; de Oliveira, G.A.P.; Cino, E.A. Biophysical characterization of p53 core domain aggregates. Biochem. J. 2020, 477, 111–120. [Google Scholar] [CrossRef]
- de Oliveira, G.A.P.; Petronilho, E.C.; Pedrote, M.M.; Marques, M.A.; Vieira, T.C.R.G.; Cino, E.A.; Silva, J.L. The status of p53 oligomeric and aggregation states in cancer. Biomolecules 2020, 10, 548. [Google Scholar] [CrossRef] [Green Version]























































© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, J.L.; Lima, C.G.S.; Rangel, L.P.; Ferretti, G.D.S.; Pauli, F.P.; Ribeiro, R.C.B.; da Silva, T.d.B.; da Silva, F.C.; Ferreira, V.F. Recent Synthetic Approaches towards Small Molecule Reactivators of p53. Biomolecules 2020, 10, 635. https://doi.org/10.3390/biom10040635
Silva JL, Lima CGS, Rangel LP, Ferretti GDS, Pauli FP, Ribeiro RCB, da Silva TdB, da Silva FC, Ferreira VF. Recent Synthetic Approaches towards Small Molecule Reactivators of p53. Biomolecules. 2020; 10(4):635. https://doi.org/10.3390/biom10040635
Chicago/Turabian StyleSilva, Jerson L., Carolina G. S. Lima, Luciana P. Rangel, Giulia D. S. Ferretti, Fernanda P. Pauli, Ruan C. B. Ribeiro, Thais de B. da Silva, Fernando C. da Silva, and Vitor F. Ferreira. 2020. "Recent Synthetic Approaches towards Small Molecule Reactivators of p53" Biomolecules 10, no. 4: 635. https://doi.org/10.3390/biom10040635
APA StyleSilva, J. L., Lima, C. G. S., Rangel, L. P., Ferretti, G. D. S., Pauli, F. P., Ribeiro, R. C. B., da Silva, T. d. B., da Silva, F. C., & Ferreira, V. F. (2020). Recent Synthetic Approaches towards Small Molecule Reactivators of p53. Biomolecules, 10(4), 635. https://doi.org/10.3390/biom10040635