Cystathionine-β-synthase: Molecular Regulation and Pharmacological Inhibition




Abstract
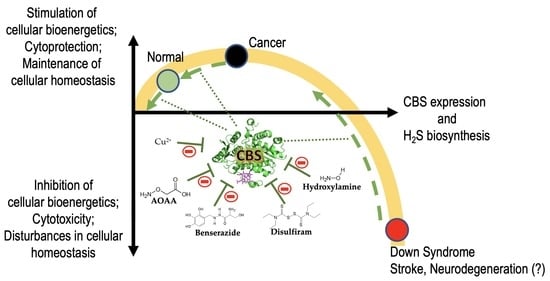
:
1. CBS: Discovery, Regulation, and Physiological Roles
1.1. CBS: Discovery and Early Studies
1.2. The Molecular Organization of Human CBS
1.3. Regulation of CBS Expression
1.3.1. Physiological Factors Regulating CBS
1.3.2. CBS Regulation by Exogenous Factors
1.4. Distribution of CBS in Various Cells and Tissues
1.5. Subcellular Distribution and Translocation of CBS
1.6. Physiological Roles of CBS
1.7. Homocystinuria
2. The Biochemistry of CBS
2.1. Organization of the Active Site of CBS
2.2. H2S Biosynthesis and Other CBS-Catalyzed Biochemical Reactions
3. Physiological Regulation of CBS Enzymatic Activity
3.1. Allosteric Activation of CBS by SAM
3.2. Post-Translational Modifications of CBS Affecting Its Activity or Expression
4. Disease Conditions in Which Inhibition of CBS is Expected to Be Beneficial
4.1. Down Syndrome
4.2. Cancer
5. Pharmacological Inhibitors of CBS
5.1. The “Classical CBS Inhibitor”: Aminooxyacetate
5.1.1. Discovery and Early Studies
5.1.2. The Mode of AOAA’s Inhibitory Effect: the AOAA-PLP Interaction
5.1.3. Effects of AOAA in Mammalian Cells and Tissues In Vitro and In Vivo
5.1.4. AOAA as a “CBS Inhibitor” (or a Broad Inhibitor of H2S Biosynthesis) In Vitro and In Vivo
5.1.5. The Lack of AOAA’s Selectivity as a Pharmacological Inhibitor
5.2. Potentially Repurposable CBS Inhibitors
5.2.1. Benserazide
5.2.2. 2,3,4-Trihydroxybenzylhydrazine, an Active Metabolite of Benserazide
5.2.3. Disulfiram
5.3. Additional Classes of CBS Inhibitors
5.3.1. Hydroxylamine
5.3.2. Copper
5.3.3. NSC67078
5.3.4. Sikokianin C
5.3.5. CH004
5.3.6. 6S and Related Inhibitors
5.3.7. Additional CBS Inhibitors
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- du Vigneaud, V.; Loring, H.S.; Craft, H. The oxidation of the sulfur of homocystine, methionine, and S-methylcysteine in the animal body. J. Biol. Chem. 1934, 105, 481. [Google Scholar]
- Binkley, F.; du Vigneaud, V. The formation of cysteine from homocysteine and serine by liver tissue of rats. J. Biol. Chem. 1942, 144, 507. [Google Scholar]
- Mudd, S.H.; Finkelstein, J.D.; Irreverre, F.; Laster, L. Transsulfuration in mammals. Microassays and tissue distributions of three enzymes of the pathway. J. Boil. Chem. 1965, 240, 4382–4392. [Google Scholar]
- Braunstein, A.; Goryachenkova, E.; Lac, N.D. Reactions catalysed by serine sulfhydrase from chicken liver. Biochim. et Biophys. Acta (BBA) Enzym. 1969, 171, 366–368. [Google Scholar] [CrossRef]
- Porter, P.N.; Grishaver, M.S.; Jones, O.W. Characterization of human cystathionine beta-synthase. Evidence for the identity of human L-serine dehydratase and cystathionine beta-synthase. Biochim. et Biophys. Acta 1974, 364, 128–139. [Google Scholar] [CrossRef]
- Kraus, J.P.; E Rosenberg, L. Cystathionine beta-synthase from human liver: Improved purification scheme and additional characterization of the enzyme in crude and pure form. Arch. Biochem. Biophys. 1983, 222, 44–52. [Google Scholar] [CrossRef]
- Tudball, N.; Reed, M.A. Purification and properties of cystathionine synthase from human liver. Biochem. Biophys. Res. Commun. 1975, 67, 550–555. [Google Scholar] [CrossRef]
- E Braunstein, A.; Goryachenkova, E.V. The pyridoxal-phosphate-dependent enzymes exclusively catalyzing reactions of beta-replacement. Biochimie 1976, 58, 5–17. [Google Scholar] [CrossRef]
- Kraus, J.; Packman, S.; Fowler, B.; E Rosenberg, L. Purification and properties of cystathionine beta-synthase from human liver. Evidence for identical subunits. J. Boil. Chem. 1978, 253, 6523–6528. [Google Scholar]
- Stipanuk, M.H.; Beck, P.W. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem. J. 1982, 206, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Szabo, C.; Papapetropoulos, A. International Union of Basic and Clinical Pharmacology. CII: Pharmacological Modulation of H2S Levels: H2S Donors and H2S Biosynthesis Inhibitors. Pharmacol. Rev. 2017, 69, 497–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münke, M.; Kraus, J.P.; Ohura, T.; Francke, U. The gene for cystathionine β-synthase (CBS) maps to the subtelomeric region on human chromosome 21q and to proximal mouse chromosome. Am. J. Hum. Genet. 1988, 42, 550–559. [Google Scholar]
- Kraus, J.P.; Oliveriusova, J.; Sokolová, J.; Kraus, E.; Vlček, Č.; De Franchis, R.; MacLean, K.N.; Bao, L.; Bukovská, G.; Patterson, D.; et al. The Human Cystathionine β-Synthase (CBS) Gene: Complete Sequence, Alternative Splicing, and Polymorphisms. Genomics 1998, 52, 312–324. [Google Scholar] [CrossRef]
- Miles, E.W.; Kraus, J.P. Cystathionine β-Synthase: Structure, Function, Regulation, and Location of Homocystinuria-causing Mutations. J. Boil. Chem. 2004, 279, 29871–29874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, M.; Janosik, M.; Kery, V.; Kraus, J.P.; Burkhard, P. Structure of human cystathionine beta-synthase: A unique pyridoxal 5’-phosphate-dependent heme protein. EMBO J. 2001, 20, 3910–3916. [Google Scholar] [CrossRef] [Green Version]
- Ereno-Orbea, J.; Majtan, T.; Oyenarte, I.; Kraus, J.P.; Martinez-Cruz, L.A. Structural basis of regulation and oligomerization of human cystathionine beta-synthase, the central enzyme of transsulfuration. Proc. Nat. Acad. Sci. USA 2013, 110, E3790–E3799. [Google Scholar] [CrossRef] [Green Version]
- Taoka, S.; Lepore, B.W.; Kabil, O.; Ojha, S.; Ringe, D.; Banerjee, R. Human cystathionine beta-synthase is a heme sensor protein. Evidence that the redox sensor is heme and not the vicinal cysteines in the CXXC motif seen in the crystal structure of the truncated enzyme. Biochemistry 2002, 41, 10454–10461. [Google Scholar] [CrossRef]
- Kumar, A.; Wißbrock, A.; Goradia, N.; Bellstedt, P.; Ramachandran, R.; Imhof, D.; Ohlenschläger, O. Heme interaction of the intrinsically disordered N-terminal peptide segment of human cystathionine-β-synthase. Sci. Rep. 2018, 8, 2474. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Bellstedt, P.; Wiedemann, C.; Wißbrock, A.; Imhof, D.; Ramachandran, R.; Ohlenschläger, O. NMR experiments on the transient interaction of the intrinsically disordered N-terminal peptide of cystathionine-β-synthase with heme. J. Magn. Reson. 2019, 308, 106561. [Google Scholar] [CrossRef]
- Bublil, E.M.; Majtan, T.; Park, I.; Carrillo, R.S.; Hůlková, H.; Krijt, J.; Kožich, V.; Kraus, J.P. Enzyme replacement with PEGylated cystathionine β-synthase ameliorates homocystinuria in murine model. J. Clin. Investig. 2016, 126, 2372–2384. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Madzelan, P.; Banerjee, R. Properties of an unusual heme cofactor in PLP-dependent cystathionine beta-synthase. Nat. Prod. Rep. 2007, 24, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Ereño-Orbea, J.; Oyenarte, I.; Martínez-Cruz, L.A. CBS domains: Ligand binding sites and conformational variability. Arch. Biochem. Biophys. 2013, 540, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; A Konrad, M.; Matherly, L.H.; Taub, J.W. Transcriptional regulation of the human cystathionine beta-synthase -1b basal promoter: Synergistic transactivation by transcription factors NF-Y and Sp1/Sp. Biochem. J. 2001, 357, 97–105. [Google Scholar]
- Bouwman, P.; Philipsen, S. Regulation of the activity of Sp1-related transcription factors. Mol. Cell. Endocrinol. 2002, 195, 27–38. [Google Scholar] [CrossRef]
- Hourihan, J.M.; Kenna, G.; Hayes, J. The Gasotransmitter Hydrogen Sulfide Induces Nrf2-Target Genes by Inactivating the Keap1 Ubiquitin Ligase Substrate Adaptor Through Formation of a Disulfide Bond Between Cys-226 and Cys-613. Antioxidants Redox Signal. 2013, 19, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Tocmo, R.; Parkin, K. S-1-propenylmercaptocysteine protects murine hepatocytes against oxidative stress via persulfidation of Keap1 and activation of Nrf2. Free Radic. Boil. Med. 2019, 143, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Sperandeo, M.P.; De Franchis, R.; Andria, G.; Sebastio, G. A 68-bp insertion found in a homocystinuric patient is a common variant and is skipped by alternative splicing of the cystathionine beta-synthase mRNA. Am. J. Hum. Genet. 1996, 59, 1391–1393. [Google Scholar] [PubMed]
- Kriebitzsch, C.; Verlinden, L.; Eelen, G.; Van Schoor, N.M.; Swart, K.; Lips, P.; Meyer, M.; Pike, J.W.; Boonen, S.; Carlberg, C.; et al. 1,25-dihydroxyvitamin D3 influences cellular homocysteine levels in murine preosteoblastic MC3T3-E1 cells by direct regulation of cystathionine β-synthase. J. Bone Miner. Res. 2011, 26, 2991–3000. [Google Scholar] [CrossRef] [Green Version]
- Lechuga, T.J.; Qi, Q.-R.; Kim, T.; Magness, R.; Chen, N.-B. E2β stimulates ovine uterine artery endothelial cell H2S production in vitro by estrogen receptor-dependent upregulation of cystathionine β-synthase and cystathionine γ-lyase expression†. Boil. Reprod. 2018, 100, 514–522. [Google Scholar] [CrossRef]
- Sheibani, L.; Lechuga, T.J.; Zhang, H.; Hameed, A.; Wing, D.A.; Kumar, S.; Rosenfeld, C.R.; Chen, N.-B. Augmented H2S production via cystathionine-beta-synthase upregulation plays a role in pregnancy-associated uterine vasodilation†. Boil. Reprod. 2017, 96, 664–672. [Google Scholar] [CrossRef]
- Vitvitsky, V.; Prudova, A.; Stabler, S.; Dayal, S.; Lentz, S.R.; Banerjee, R. Testosterone regulation of renal cystathionine beta-synthase: Implications for sex-dependent differences in plasma homocysteine levels. Am. J. Physiol. Renal Physiol. 2007, 293, F594–F600. [Google Scholar] [CrossRef]
- Ratnam, S.; Maclean, K.N.; Jacobs, R.L.; Brosnan, M.E.; Kraus, J.P.; Brosnan, J.T. Hormonal regulation of cystathionine beta-synthase expression in liver. J. Biol. Chem. 2002, 277, 42912–42918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Wu, S.; Gao, X.; Zhang, Z.; Gong, J.; Zhan, R.; Wang, X.; Wang, W.; Qian, L.-J. Inhibition of cystathionine β-synthase is associated with glucocorticoids over-secretion in psychological stress-induced hyperhomocystinemia rat liver. Cell Stress Chaperon. 2013, 18, 631–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, N.; Peng, Y.-J.; Kumar, G.K.; Luo, W.; Hu, H.; Shimoda, L.A.; Suematsu, M.; Prabhakar, N.R.; Semenza, G.L. Hypoxia-inducible factors regulate human and rat cystathionine β-synthase gene expression. Biochem. J. 2014, 458, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Siow, Y.L.; O, K. Ischemia/reperfusion reduces transcription factor Sp1-mediated cystathionine beta-synthase expression in the kidney. J. Biol. Chem. 2010, 285, 18225–18233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talaei, F.; Bouma, H.; Van Der Graaf, A.C.; Strijkstra, A.M.; Schmidt, M.; Henning, R.H. Serotonin and Dopamine Protect from Hypothermia/Rewarming Damage through the CBS/ H2S Pathway. PLoS ONE 2011, 6, e22568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maclean, K.N.; Janosík, M.; Kraus, E.; Kozich, V.; Allen, R.H.; Raab, B.K.; Kraus, J.P. Cystathionine beta-synthase is coordinately regulated with proliferation through a redox-sensitive mechanism in cultured human cells and Saccharomyces cerevisiae. J. Cell. Physiol. 2002, 192, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Wang, E.A.; Gould, S.; Stein, E.; Kaur, S.; Lim, L.; Amarnath, S.; Fowler, D.H.; Roberts, D.D. Hydrogen Sulfide Is an Endogenous Potentiator of T Cell Activation*. J. Boil. Chem. 2011, 287, 4211–4221. [Google Scholar] [CrossRef] [Green Version]
- Enokido, Y.; Suzuki, E.; Iwasawa, K.; Namekata, K.; Okazawa, H.; Kimura, H. Cystathionine β-synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J. 2005, 19, 1854–1856. [Google Scholar] [CrossRef]
- Sarkar, S.A.; Lee, C.E.; Tipney, H.R.; Karimpour-Fard, A.; Dinella, J.D.; Juhl, K.; Walters, J.A.; Hutton, J.C.; Hunter, L. Synergizing genomic analysis with biological knowledge to identify and validate novel genes in pancreatic development. Pancreas 2012, 41, 962–969. [Google Scholar] [CrossRef] [Green Version]
- Bruintjes, J.; Henning, R.H.; Douwenga, W.; Van Der Zee, E. Hippocampal cystathionine beta synthase in young and aged mice. Neurosci. Lett. 2014, 563, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Predmore, B.L.; Alendy, M.J.; Ahmed, K.I.; Leeuwenburgh, C.; Julian, D. The hydrogen sulfide signaling system: Changes during aging and the benefits of caloric restriction. AGE 2010, 32, 467–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hine, C.; Harputlugil, E.; Zhang, Y.; Ruckenstuhl, C.; Lee, B.C.; Brace, L.; Longchamp, A.; Treviño-Villarreal, J.H.; Mejia, P.; Ozaki, C.K.; et al. Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 2014, 160, 132–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Kim, H.-J.; Lee, M.; Jin, S.H.; Hong, S.H.; Ahn, S.; Kim, S.O.; Shin, N.W.; Lee, S.-T.; Noh, M. Cystathionine metabolic enzymes play a role in the inflammation resolution of human keratinocytes in response to sub-cytotoxic formaldehyde exposure. Toxicol. Appl. Pharmacol. 2016, 310, 185–194. [Google Scholar] [CrossRef]
- Du, C.; Jin, M.; Hong, Y.; Li, Q.; Wang, X.-H.; Xu, J.-M.; Wang, F.; Zhang, Y.; Jia, J.; Liu, C.-F.; et al. Downregulation of cystathionine β-synthase/hydrogen sulfide contributes to rotenone-induced microglia polarization toward M1 type. Biochem. Biophys. Res. Commun. 2014, 451, 239–245. [Google Scholar] [CrossRef]
- Yuan, Y.; Zheng, J.; Zhao, T.; Tang, X.; Hu, N. Uranium-induced rat kidney cell cytotoxicity is mediated by decreased endogenous hydrogen sulfide (H2S) generation involved in reduced Nrf2 levels. Toxicol. Res. 2016, 5, 660–673. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-Y.; Yang, H.-W. Upregulation of CBS/H2S system contributes to asymmetric dimethylarginine-triggered protection against the neurotoxicity of glutamate to PC12 cells by inhibiting NOS/NO pathway. Exp. Cell Res. 2016, 346, 111–118. [Google Scholar] [CrossRef]
- Tang, X.-Q.; Fan, L.-L.; Li, Y.-J.; Shen, X.-T.; Zhuan, Y.-Y.; He, J.-Q.; Xu, J.-H.; Hu, B.; Li, Y.-J. Inhibition of Hydrogen Sulfide Generation Contributes to 1-Methy-4-Phenylpyridinium Ion-Induced Neurotoxicity. Neurotox. Res. 2010, 19, 403–411. [Google Scholar] [CrossRef]
- Zhang, J.; Xie, Y.; Xu, Y.; Pan, Y.; Shao, C. Hydrogen sulfide contributes to hypoxia-induced radioresistance on hepatoma cells. J. Radiat. Res. 2011, 52, 622–628. [Google Scholar] [CrossRef] [Green Version]
- Sarna, L.K.; Sid, V.; Wang, P.; Siow, Y.; House, J.D.; O, K. Tyrosol Attenuates High Fat Diet-Induced Hepatic Oxidative Stress: Potential Involvement of Cystathionine β-Synthase and Cystathionine γ-Lyase. Lipids 2015, 51, 583–590. [Google Scholar] [CrossRef]
- Lu, D.-Z.; Feng, X.-J.; Hu, K.; Jiang, S.; Li, L.; Ma, X.-W.; Fan, H. Inductive effect of Zoletil on cystathionine β-synthase expression in the rat brain. Int. J. Boil. Macromol. 2018, 117, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Sommer, O.; Aug, R.L.; Schmidt, A.J.; Heiser, P.; Schulz, E.; Vedder, H.; Clement, H.-W. Hydrogen Sulfide Affects Radical Formation in the Hippocampus of LPS Treated Rats and the Effect of Antipsychotics on Hydrogen Sulfide Forming Enzymes in Human Cell Lines. Front. Psychol. 2018, 9, 501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Vlcek, C.; Paces, V.; Kraus, J.P. Identification and tissue distribution of human cystathionine beta-synthase mRNA isoforms. Arch. Biochem. Biophys. 1998, 350, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Kabil, O.; Vitvitsky, V.; Xie, P.; Banerjee, R. The Quantitative Significance of the Transsulfuration Enzymes for H2S Production in Murine Tissues. Antioxidants Redox Signal. 2011, 15, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dicker-Brown, A.; A Fonseca, V.; Fink, L.M.; A Kern, P. The effect of glucose and insulin on the activity of methylene tetrahydrofolate reductase and cystathionine-beta-synthase: Studies in hepatocytes. Atheroscler. 2001, 158, 297–301. [Google Scholar] [CrossRef]
- Damba, T.; Zhang, M.; Buist-Homan, M.; Van Goor, H.; Faber, K.N.; Moshage, H. Hydrogen sulfide stimulates activation of hepatic stellate cells through increased cellular bio-energetics. Nitric Oxide 2019, 92, 26–33. [Google Scholar] [CrossRef]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Shan, H.; Wang, Y.; Wang, T.; Liu, W.; Wang, L.; Zhang, L.; Chang, P.; Dong, W.; Chen, X.-P.; et al. The Expression Changes of Cystathionine-β-synthase in Brain Cortex After Traumatic Brain Injury. J. Mol. Neurosci. 2013, 51, 57–67. [Google Scholar] [CrossRef]
- Robert, K.; Vialard, F.; Thiery, E.; Toyama, K.; Sinet, P.-M.; Janel, N.; London, J. Expression of the cystathionine beta synthase (CBS) gene during mouse development and immunolocalization in adult brain. J. Histochem. Cytochem. 2003, 51, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Liu, D.X.; Wang, F.W.; Zhang, Q.; Du, Z.X.; Zhan, J.M.; Yuan, Q.H.; Ling, E.A.; Hao, A.J. L-Cysteine promotes the proliferation and differentiation of neural stem cells via the CBS/H₂S pathway. Neuroscience. 2013, 237, 106–117. [Google Scholar] [CrossRef]
- Huang, P.; Chen, S.; Wang, Y.; Liu, J.; Yao, Q.; Huang, Y.; Li, H.; Zhu, M.; Wang, S.; Li, L.; et al. Down-regulated CBS/H2S pathway is involved in high-salt-induced hypertension in Dahl rats. Nitric Oxide 2015, 46, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhang, J.; Xie, F.; Tan, W.; Wang, S.; Huang, L.; Tao, L.; Xing, Q.; Yuan, Q. Loss of the Protein Cystathionine β-Synthase During Kidney Injury Promotes Renal Tubulointerstitial Fibrosis. Kidney Blood Press. Res. 2017, 42, 428–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, Y.; Kimura, T.; Taniguchi, S.; Souma, M.; Kojima, Y.; Kimura, Y.; Kimura, H.; Niki, I. Glucose-induced production of hydrogen sulfide may protect the pancreatic beta-cells from apoptotic cell death by high glucose. FEBS Lett. 2008, 583, 377–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamizhselvi, R.; Moore, P.K.; Bhatia, M. Hydrogen sulfide acts as a mediator of inflammation in acute pancreatitis: In vitro studies using isolated mouse pancreatic acinar cells. J. Cell. Mol. Med. 2007, 11, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Donovan, J.; Wong, P.S.; Garle, M.J.; Alexander, S.P.H.; Dunn, W.; Ralevic, V. Coronary artery hypoxic vasorelaxation is augmented by perivascular adipose tissue through a mechanism involving hydrogen sulphide and cystathionine-β-synthase. Acta Physiol. 2018, 224, e13126. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Wang, M.-J.; Jin, S.; Bai, Y.-D.; Hou, C.-L.; Ma, F.-F.; Li, X.-H.; Zhu, Y.-C. The H2S Donor NaHS Changes the Expression Pattern of H2S-Producing Enzymes after Myocardial Infarction. Oxidative Med. Cell. Longev. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Dong, Z.; Dimitropoulou, C.; Su, Y. Hydrogen Sulfide Ameliorates Tobacco Smoke-Induced Oxidative Stress and Emphysema in Mice. Antioxidants Redox Signal. 2011, 15, 2121–2134. [Google Scholar] [CrossRef] [Green Version]
- Talaei, F.; Bouma, H.R.; Hylkema, M.N.; Strijkstra, A.M.; Boerema, A.; Schmidt, M.; Henning, R.H. The role of endogenous H2S formation in reversible remodeling of lung tissue during hibernation in the Syrian hamster. J. Exp. Boil. 2012, 215, 2912–2919. [Google Scholar] [CrossRef] [Green Version]
- Rashid, S.; Heer, J.; Garle, M.; Alexander, S.P.H.; Roberts, R. Hydrogen sulphide-induced relaxation of porcine peripheral bronchioles. Br. J. Pharmacol. 2013, 168, 1902–1910. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Liu, D.; Tang, C.; Du, J.-B.; Liu, A.D.; Holmberg, L.; Jin, H. Sulfur dioxide upregulates the inhibited endogenous hydrogen sulfide pathway in rats with pulmonary hypertension induced by high pulmonary blood flow. Biochem. Biophys. Res. Commun. 2013, 433, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-N.; Liu, Y.; Duan, G.-L.; Zhao, W.; Li, X.-H.; Zhu, X.; Ni, X. CBS and CSE Are Critical for Maintenance of Mitochondrial Function and Glucocorticoid Production in Adrenal Cortex. Antioxidants Redox Signal. 2014, 21, 2192–2207. [Google Scholar] [CrossRef] [PubMed]
- Turbat-Herrera, E.A.; Kilpatrick, M.J.; Chen, J.; Meram, A.T.; Cotelingam, J.; Ghali, G.; Kevil, C.G.; Coppola, D.; Shackelford, R.E. Cystathione β-Synthase Is Increased in Thyroid Malignancies. Anticancer. Res. 2018, 38, 6085–6090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magierowski, M.; Magierowska, K.; Surmiak, M.; Hubalewska-Mazgaj, M.; Kwiecien, S.; Wallace, J.L.; Brzozowski, T. The effect of hydrogen sulfide-releasing naproxen (ATB-346) versus naproxen on formation of stress-induced gastric lesions, the regulation of systemic inflammation, hypoxia and alterations in gastric microcirculation. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2017, 68, 749–756. [Google Scholar]
- Tomuschat, C.; O’Donnell, A.M.; Coyle, D.; Puri, P. Reduction of hydrogen sulfide synthesis enzymes cystathionine-β-synthase and cystathionine-γ-lyase in the colon of patients with Hirschsprungs disease. J. Pediatr. Surg. 2018, 53, 525–530. [Google Scholar] [CrossRef]
- Wu, C.; Xu, Z.; Huang, K. Effects of Dietary Selenium on Inflammation and Hydrogen Sulfide in the Gastrointestinal Tract in Chickens. Boil. Trace Element Res. 2016, 174, 428–435. [Google Scholar] [CrossRef]
- Saghazadeh-Dezfuli, M.; Fanaei, H.; Gharib-Naseri, M.K.; Nasri, S.; Mard, S.A. Antidiarrheal effect of sodium hydrosulfide in diabetic rats: In vitro and in vivo studies. Neurogastroenterol. Motil. 2017, 30, e13273. [Google Scholar] [CrossRef]
- Ahmad, A.; Gero, D.; Olah, G.; Szabo, C. Effect of endotoxemia in mice genetically deficient in cystathionine-γ-lyase, cystathionine-β-synthase or 3-mercaptopyruvate sulfurtransferase. Int. J. Mol. Med. 2016, 38, 1683–1692. [Google Scholar] [CrossRef] [Green Version]
- Liang, R.; Yu, W.-D.; Du, J.-B.; Yang, L.-J.; Shang, M.; Guo, J.-Z. Localization of cystathionine beta synthase in mice ovaries and its expression profile during follicular development. Chin. Med. J. 2006, 119, 1877–1883. [Google Scholar] [CrossRef]
- You, X.-J.; Xu, C.; Lu, J.-Q.; Zhu, X.-Y.; Gao, L.; Cui, X.-R.; Li, Y.; Gu, H.; Ni, X. Expression of Cystathionine β-synthase and Cystathionine γ-lyase in Human Pregnant Myometrium and Their Roles in the Control of Uterine Contractility. PLoS ONE 2011, 6, 23788. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.; Vatish, M.; Heptinstall, J.; Wang, R.; Carson, R.J. The endogenous production of hydrogen sulphide in intrauterine tissues. Reprod. Boil. Endocrinol. 2009, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Sen, S.; Kawahara, B.; Mahata, S.K.; Tsai, R.; Yoon, A.; Hwang, L.; Hu-Moore, K.; Villanueva, C.; Vajihuddin, A.; Parameshwar, P.; et al. Cystathionine: A novel oncometabolite in human breast cancer. Arch. Biochem. Biophys. 2016, 604, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Gai, J.-W.; Wang, Y.; Jin, H.; Du, J.-B.; Jin, J. Characterization of Hydrogen Sulfide and Its Synthases, Cystathionine β-Synthase and Cystathionine γ-Lyase, in Human Prostatic Tissue and Cells. Urology 2012, 79, 483.e1–483.e5. [Google Scholar] [CrossRef] [PubMed]
- Gai, J.-W.; Wahafu, W.; Guo, H.; Liu, M.; Wang, X.-C.; Xiao, Y.-X.; Zhang, L.; Xin, Z.-C.; Jin, J. Further evidence of endogenous hydrogen sulphide as a mediator of relaxation in human and rat bladder. Asian J. Androl. 2013, 15, 692–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Xie, Z.-Z.; Chua, J.M.; Wong, P.; Bian, J.-S. Hydrogen sulfide protects testicular germ cells against heat-induced injury. Nitric Oxide 2015, 46, 165–171. [Google Scholar] [CrossRef]
- Teng, H.; Wu, B.; Zhao, K.; Yang, G.; Wu, L.; Wang, R. Oxygen-sensitive mitochondrial accumulation of cystathionine β-synthase mediated by Lon protease. Proc. Natl. Acad. Sci. USA 2013, 110, 12679–12684. [Google Scholar] [CrossRef] [Green Version]
- Szabo, C.; Ransy, C.; Módis, K.; Andriamihaja, M.; Murghes, B.; Coletta, C.; Olah, G.; Yanagi, K.; Bouillaud, F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 2014, 171, 2099–2122. [Google Scholar]
- Szabo, C.; Coletta, C.; Chao, C.; Módis, K.; Szczesny, B.; Papapetropoulos, A.; Hellmich, M.R. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12474–12479. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Saha, S.; Giri, K.; Lanza, I.R.; Nair, K.S.; Jennings, N.B.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Basal, E.; Weaver, A.L.; et al. Cystathionine Beta-Synthase (CBS) Contributes to Advanced Ovarian Cancer Progression and Drug Resistance. PLoS ONE 2013, 8, e79167. [Google Scholar] [CrossRef]
- Panagaki, T.; Randi, E.B.; Augsburger, F.; Szabo, C. Overproduction of H2S, generated by CBS, inhibits mitochondrial Complex IV and suppresses oxidative phosphorylation in Down syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 18769–18771. [Google Scholar] [CrossRef] [Green Version]
- Casique, L.; Kabil, O.; Banerjee, R.; Martinez, J.; De Lucca, M. Characterization of two pathogenic mutations in cystathionine beta-synthase: Different intracellular locations for wild-type and mutant proteins. Gene 2013, 531, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabil, O.; Zhou, Y.; Banerjee, R. Human Cystathionine β-Synthase Is a Target for Sumoylation. Biochem. 2006, 45, 13528–13536. [Google Scholar] [CrossRef] [PubMed]
- Markovic, J.; Borrás, C.; Ortega, Á.; Sastre, J.; Viña, J.; Pallardó, F.V. Glutathione Is Recruited into the Nucleus in Early Phases of Cell Proliferation. J. Boil. Chem. 2007, 282, 20416–20424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, N.; Banerjee, R. Human polycomb 2 protein is a SUMO E3 ligase and alleviates substrate-induced inhibition of cystathionine beta-synthase sumoylation. PLoS ONE 2008, 3, e4032. [Google Scholar] [CrossRef]
- Shen, W.; Gao, C.; Cueto, R.; Liu, L.; Fu, H.; Shao, Y.; Yang, W.Y.; Fang, P.; Choi, E.T.; Wu, Q.; et al. Homocysteine-methionine cycle is a metabolic sensor system controlling methylation-regulated pathological signaling. Redox Boil. 2020, 28, 101322. [Google Scholar] [CrossRef]
- Krijt, J.; Kopecká, J.; Hnizda, A.; Moat, S.; Kluijtmans, L.A.J.; Mayne, P.; Kožich, V. Determination of cystathionine beta-synthase activity in human plasma by LC-MS/MS: Potential use in diagnosis of CBS deficiency. J. Inherit. Metab. Dis. 2010, 34, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Alcaide, P.; Krijt, J.; Ruiz-Sala, P.; Jesina, P.; Ugarte, M.; Kožich, V.; Merinero, B. Enzymatic diagnosis of homocystinuria by determination of cystathionine-ß-synthase activity in plasma using LC-MS/MS. Clin. Chim. Acta 2015, 438, 261–265. [Google Scholar] [CrossRef]
- Watanabè, M.; Osada, J.; Aratani, Y.; Kluckman, K.; Reddick, R.; Malinow, M.R.; Maeda, N. Mice deficient in cystathionine beta-synthase: Animal models for mild and severe homocyst(e)inemia. Proc. Natl. Acad. Sci. USA 1995, 92, 1585–1589. [Google Scholar] [CrossRef] [Green Version]
- Robert, K.; Maurin, N.; Vayssettes, C.; Siauve, N.; Janel, N. Cystathionine beta synthase deficiency affects mouse endochondral ossification. Anat. Rec. A. Discov. Mol. Cell. Evol. Biol. 2005, 282, 1–7. [Google Scholar]
- Robert, K.; Nehmé, J.; Bourdon, E.; Pivert, G.; Friguet, B.; Delcayre, C.; Delabar, J.-M.; Janel, N. Cystathionine beta synthase deficiency promotes oxidative stress, fibrosis, and steatosis in mice liver. Gastroenterol. 2005, 128, 1405–1415. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chen, X.; Tang, B.; Hua, X.; Klein-Szanto, A.; Kruger, W.D. Expression of mutant human cystathionine beta-synthase rescues neonatal lethality but not homocystinuria in a mouse model. Hum. Mol. Genet. 2005, 14, 2201–2208. [Google Scholar] [CrossRef] [PubMed]
- Akahoshi, N.; Kobayashi, C.; Ishizaki, Y.; Izumi, T.; Himi, T.; Suematsu, M.; Ishii, I. Genetic background conversion ameliorates semi-lethality and permits behavioral analyses in cystathionine β-synthase-deficient mice, an animal model for hyperhomocysteinemia. Hum. Mol. Genet. 2008, 17, 1994–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Kühnisch, J.; Mustafa, A.; Lhotak, S.; Schlachterman, A.; Slifker, M.J.; Klein-Szanto, A.; High, K.A.; Austin, R.C.; Kruger, W.D. Mouse models of cystathionine β-synthase deficiency reveal significant threshold effects of hyperhomocysteinemia. FASEB J. 2008, 23, 883–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLean, K.N.; Sikora, J.; Kožich, V.; Jiang, H.; Greiner, L.S.; Kraus, E.; Krijt, J.; Overdier, K.H.; Collard, R.; Brodsky, G.L.; et al. A novel transgenic mouse model of CBS-deficient homocystinuria does not incur hepatic steatosis or fibrosis and exhibits a hypercoagulative phenotype that is ameliorated by betaine treatment. Mol. Genet. Metab. 2010, 101, 153–162. [Google Scholar] [CrossRef] [PubMed]
- MacLean, K.N.; Sikora, J.; Kožich, V.; Jiang, H.; Greiner, L.S.; Kraus, E.; Krijt, J.; Crnic, L.S.; Allen, R.H.; Stabler, S.P.; et al. Cystathionine beta-synthase null homocystinuric mice fail to exhibit altered hemostasis or lowering of plasma homocysteine in response to betaine treatment. Mol. Genet. Metab. 2010, 101, 163–171. [Google Scholar] [CrossRef]
- Magner, M.; Krupková, L.; Honzik, T.; Zeman, J.; Hyánek, J.; Kožich, V. Vascular presentation of cystathionine beta-synthase deficiency in adulthood. J. Inherit. Metab. Dis. 2010, 34, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kruger, W.D. Cystathionine Beta-Synthase Deficiency Causes Fat Loss in Mice. PLoS ONE 2011, 6, e27598. [Google Scholar] [CrossRef]
- Maclean, K.N.; Jiang, H.; Aivazidis, S.; Kim, E.; Shearn, C.T.; Harris, P.S.; Petersen, D.R.; Allen, R.H.; Stabler, S.P.; Roede, J.R. Taurine treatment prevents derangement of the hepatic gamma-glutamyl cycle and methylglyoxal metabolism in a mouse model of classical homocystinuria: Regulatory crosstalk between thiol and sulfinic acid metabolism. FASEB J. 2017, 32, 1265–1280. [Google Scholar] [CrossRef] [Green Version]
- Majtan, T.; Park, I.; Cox, A.; Branchford, B.R.; Di Paola, J.; Bublil, E.M.; Kraus, J.P. Behavior, body composition, and vascular phenotype of homocystinuric mice on methionine-restricted diet or enzyme replacement therapy. FASEB J. 2019, 33, 12477–12486. [Google Scholar] [CrossRef] [Green Version]
- Namekata, K.; Enokido, Y.; Ishii, I.; Nagai, Y.; Harada, T.; Kimura, H. Abnormal Lipid Metabolism in Cystathionine β-Synthase-deficient Mice, an Animal Model for Hyperhomocysteinemia. J. Boil. Chem. 2004, 279, 52961–52969. [Google Scholar] [CrossRef] [Green Version]
- Hamelet, J.; DeMuth, K.; Paul, J.-L.; Delabar, J.-M.; Janel, N. Hyperhomocysteinemia due to cystathionine beta synthase deficiency induces dysregulation of genes involved in hepatic lipid homeostasis in mice. J. Hepatol. 2007, 46, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.; Umanskaya, N.; Wildemann, B.; Colaianni, G.; Widmann, T.; Zallone, A.; Herrmann, W. Stimulation of osteoblast activity by homocysteine. J. Cell. Mol. Med. 2008, 12, 1205–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Hua, Y. Effects of hydrogen sulfide on the expression of alkaline phosphatase, osteocalcin and collagen type I in human periodontal ligament cells induced by tension force stimulation. Mol. Med. Rep. 2016, 14, 3871–3877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, H.; Hua, Y. Hydrogen sulfide regulates bone remodeling and promotes orthodontic tooth movement. Mol. Med. Rep. 2017, 16, 9415–9422. [Google Scholar] [CrossRef]
- Tsai, C.-Y.; Peh, M.T.; Feng, W.; Dymock, B.W.; Moore, P.K. Hydrogen Sulfide Promotes Adipogenesis in 3T3L1 Cells. PLoS ONE 2015, 10, e0119511. [Google Scholar] [CrossRef]
- Eberhardt, R.; Forgione, M.A.; Cap, A.; Leopold, J.A.; Rudd, M.A.; Trolliet, M.; Heydrick, S.; Stark, R.; Klings, E.S.; Moldovan, N.I.; et al. Endothelial dysfunction in a murine model of mild hyperhomocyst(e)inemia. J. Clin. Investig. 2000, 106, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Dayal, S.; Bottiglieri, T.; Arning, E.; Maeda, N.; Malinow, M.R.; Sigmund, C.D.; Heistad, D.D.; Faraci, F.M.; Lentz, S.R. Endothelial dysfunction and elevation of S-adenosylhomocysteine in cystathionine beta-synthase-deficient mice. Circ. Res. 2001, 88, 1203–1209. [Google Scholar] [CrossRef] [Green Version]
- Weiss, N.; Heydrick, S.; Zhang, Y.-Y.; Bierl, C.; Cap, A.; Loscalzo, J. Cellular redox state and endothelial dysfunction in mildly hyperhomocysteinemic cystathionine beta-synthase-deficient mice. Arter. Thromb. Vasc. Boil. 2002, 22, 34–41. [Google Scholar] [CrossRef]
- Baumbach, G.L.; Sigmund, C.D.; Bottiglieri, T.; Lentz, S.R. Structure of cerebral arterioles in cystathionine beta-synthase-deficient mice. Circ. Res. 2002, 91, 931–937. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Jiang, X.; Yang, F.; Gaubatz, J.W.; Ma, L.; Magera, M.J.; Yang, X.; Berger, P.B.; Durante, W.; Pownall, H.J.; et al. Hyperhomocysteinemia accelerates atherosclerosis in cystathionine beta-synthase and apolipoprotein E double knock-out mice with and without dietary perturbation. Blood 2003, 101, 3901–3907. [Google Scholar] [CrossRef]
- Vitvitsky, V.; Dayal, S.; Stabler, S.; Zhou, Y.; Wang, H.; Lentz, S.R.; Banerjee, R. Perturbations in homocysteine-linked redox homeostasis in a murine model for hyperhomocysteinemia. Am. J. Physiol. Integr. Comp. Physiol. 2004, 287, R39–R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayal, S.; Arning, E.; Bottiglieri, T.; BōgerR, H.; Sigmund, C.D.; Faraci, F.M.; Lentz, S.R. Cerebral Vascular Dysfunction Mediated by Superoxide in Hyperhomocysteinemic Mice. Stroke 2004, 35, 1957–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Fang, P.; Jiang, X.; Nelson, J.; Moore, J.K.; Kruger, W.D.; Berretta, R.M.; Houser, S.R.; Yang, X.; Wang, H. Severe hyperhomocysteinemia promotes bone marrow-derived and resident inflammatory monocyte differentiation and atherosclerosis in LDLr/CBS-deficient mice. Circ. Res. 2012, 111, 37–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch-Marcé, M.; Pola, R.; Wecker, A.B.; Silver, M.; Weber, A.; Luedemann, C.; Curry, C.; Murayama, T.; Kearney, M.; Yoon, Y.; et al. Hyperhomocyst(e)inemia impairs angiogenesis in a murine model of limb ischemia. Vasc. Med. 2005, 10, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, H.; Jiang, X.; Yang, F.; Li, Z.; Liao, D.; Trial, J.; Magera, M.J.; Durante, W.; Yang, X.; Wang, H. Hyperhomocysteinemia inhibits post-injury reendothelialization in mice. Cardiovasc. Res. 2005, 69, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Beard, R.; Bearden, S.E. Vascular complications of cystathionine β-synthase deficiency: Future directions for homocysteine-to-hydrogen sulfide research. Am. J. Physiol. Circ. Physiol. 2011, 300, H13–H26. [Google Scholar] [CrossRef]
- Steed, M.M.; Tyagi, S.C. Mechanisms of Cardiovascular Remodeling in Hyperhomocysteinemia. Antioxidants Redox Signal. 2011, 15, 1927–1943. [Google Scholar] [CrossRef] [Green Version]
- Sen, U.; Sathnur, P.B.; Kundu, S.; Givvimani, S.; Coley, D.M.; Mishra, P.K.; Qipshidze, N.; Tyagi, N.; Metreveli, N.; Tyagi, S.C. Increased endogenous H2S generation by CBS, CSE, and 3MST gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia. Am. J. Physiol. Physiol. 2012, 303, C41–C51. [Google Scholar] [CrossRef] [Green Version]
- Kar, S.; Shahshahan, H.R.; Kambis, T.N.; Yadav, S.K.; Li, Z.; Lefer, D.J.; Mishra, P.K. Hydrogen Sulfide Ameliorates Homocysteine-Induced Cardiac Remodeling and Dysfunction. Front. Physiol. 2019, 10, 598. [Google Scholar] [CrossRef]
- Kamat, P.K.; Kalani, A.; Givvimani, S.; Sathnur, P.; Tyagi, S.C.; Tyagi, N. Hydrogen sulfide attenuates neurodegeneration and neurovascular dysfunction induced by intracerebral-administered homocysteine in mice. Neurosci. 2013, 252, 302–319. [Google Scholar] [CrossRef] [Green Version]
- Pushpakumar, S.; Kundu, S.; Sen, U. Endothelial dysfunction: The link between homocysteine and hydrogen sulfide. Curr. Med. Chem. 2014, 21, 3662–3672. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, X.; Shi, S.; Li, H.; Gao, F.; Zhong, X.; Wang, Y. Exogenous hydrogen sulfide protects from endothelial cell damage, platelet activation, and neutrophils extracellular traps formation in hyperhomocysteinemia rats. Exp. Cell Res. 2018, 370, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Ren, L.; Pushpakumar, S.; Sen, U. Hydrogen sulphide mitigates homocysteine-induced apoptosis and matrix remodelling in mesangial cells through Akt/FOXO1 signalling cascade. Cell. Signal. 2019, 61, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Sandhir, R. Hydrogen sulfide suppresses homocysteine-induced glial activation and inflammatory response. Nitric Oxide 2019, 90, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; He, G.-W. Imbalance of Homocysteine and H2S: Significance, Mechanisms, and Therapeutic Promise in Vascular Injury. Oxidative Med. Cell. Longev. 2019, 2019, 7629673. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Sandhir, R. Hydrogen sulfide attenuates hyperhomocysteinemia-induced mitochondrial dysfunctions in brain. Mitochondrion 2019, 50, 158–169. [Google Scholar] [CrossRef]
- Kanagy, N.L.; Szabo, C.; Papapetropoulos, A. Vascular biology of hydrogen sulfide. Am. J. Physiol. Physiol. 2017, 312, C537–C549. [Google Scholar] [CrossRef]
- Szabo, C. Hydrogen sulfide, an enhancer of vascular nitric oxide signaling: Mechanisms and implications. Am. J. Physiol. Physiol. 2016, 312, C3–C15. [Google Scholar] [CrossRef]
- Zhao, W.; Ndisang, J.F.; Wang, R. Modulation of endogenous production of H2S in rat tissues. Can. J. Physiol. Pharmacol. 2003, 81, 848–853. [Google Scholar] [CrossRef]
- Majtan, T.; Krijt, J.; Sokolová, J.; Křížková, M.; Ralat, M.; Kent, J.; Gregory, J.; Kožich, V.; Kraus, J.P.; Iii, J.F.G. Biogenesis of Hydrogen Sulfide and Thioethers by Cystathionine Beta-Synthase. Antioxidants Redox Signal. 2018, 28, 311–323. [Google Scholar] [CrossRef]
- Jensen, K.K.; Geoghagen, N.S.; Jin, L.; Holt, T.G.; Luo, Q.; Malkowitz, L.; Ni, W.; Quan, S.; Waters, M.G.; Zhang, A.; et al. Pharmacological activation and genetic manipulation of cystathionine beta-synthase alter circulating levels of homocysteine and hydrogen sulfide in mice. Eur. J. Pharmacol. 2011, 650, 86–93. [Google Scholar] [CrossRef]
- Basu, P.; Qipshidze, N.; Sen, U.; Givvimani, S.; Munjal, C.; Mishra, P.K.; Tyagi, S.C. Chronic hyperhomocysteinemia causes vascular remodelling by instigating vein phenotype in artery. Arch. Physiol. Biochem. 2011, 117, 270–282. [Google Scholar] [CrossRef]
- Dawe, G.; Han, S.; Bian, J.-S.; Moore, P. Hydrogen sulphide in the hypothalamus causes an ATP-sensitive K+ channel-dependent decrease in blood pressure in freely moving rats. Neuroscience 2008, 152, 169–177. [Google Scholar] [CrossRef]
- Roy, A.; Khan, A.H.; Islam, M.T.; Prieto, M.C.; Majid, D.S. Interdependency of Cystathione -Lyase and Cystathione -Synthase in Hydrogen Sulfide-Induced Blood Pressure Regulation in Rats. Am. J. Hypertens. 2011, 25, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Ding, R.; Lin, S.; Chen, D. The association of cystathionine β synthase (CBS) T833C polymorphism and the risk of stroke: A meta-analysis. J. Neurol. Sci. 2012, 312, 26–30. [Google Scholar] [CrossRef]
- Ufnal, M.; Sikora, M.; Dudek, M. Exogenous hydrogen sulfide produces hemodynamic effects by triggering central neuroregulatory mechanisms. Acta Neurobiol. Exp. 2008, 68, 382–388. [Google Scholar]
- Liu, W.-Q.; Chai, C.; Li, X.-Y.; Yuan, W.-J.; Wang, W.-Z.; Lu, Y. The cardiovascular effects of central hydrogen sulfide are related to K(ATP) channels activation. Physiol. Res. 2011, 60, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.C.; Liu, S.Y.; Guo, R.; Xiao, L.; Xue, H.M.; Guo, Q.; Jin, S.; Wu, Y.M. Cystathionine-β-synthase gene transfer into rostral ventrolateral medulla exacerbates hypertension via nitric oxide in spontaneously hypertensive rats. Am. J. Hypertens. 2015, 28, 1106–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabino, J.P.J.; Soriano, R.N.; Santos, B.M.; Donatti, A.F.; Fernandez, R.R.; Da Silva, G.S.F.; Branco, L.G. Central administration of aminooxyacetate, an inhibitor of H2S production, affects thermoregulatory but not cardiovascular and ventilatory responses to hypercapnia in spontaneously hypertensive rats. Respir. Physiol. Neurobiol. 2019, 263, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Chakraborty, P.K.; Xiong, X.; Dwivedi, S.K.D.; Mustafi, S.B.; Leigh, N.R.; Ramchandran, R.; Mukherjee, P.; Bhattacharya, R. Cystathionine β-synthase regulates endothelial function via protein S -sulfhydration. FASEB J. 2015, 30, 441–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawfik, A.; Markand, S.; Al-Shabrawey, M.; Mayo, J.N.; Reynolds, J.; Bearden, S.E.; Ganapathy, V.; Smith, S.B. Alterations of retinal vasculature in cystathionine-β-synthase heterozygous mice: A model of mild to moderate hyperhomocysteinemia. Am. J. Pathol. 2014, 184, 2573–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.-B.; Feng, L.; Hodges, J.K.; Lechuga, T.J.; Zhang, H. Human trophoblast-derived hydrogen sulfide stimulates placental artery endothelial cell angiogenesis†. Boil. Reprod. 2017, 97, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.; Singh, M.; George, A.K.; Behera, J.; Tyagi, N.; Tyagi, S.C. Hydrogen sulfide improves postischemic neoangiogenesis in the hind limb of cystathionine-β-synthase mutant mice via PPAR-γ/VEGF axis. Physiol. Rep. 2018, 6, e13858. [Google Scholar] [CrossRef] [PubMed]
- Eto, K.; Ogasawara, M.; Umemura, K.; Nagai, Y.; Kimura, H. Hydrogen Sulfide Is Produced in Response to Neuronal Excitation. J. Neurosci. 2002, 22, 3386–3391. [Google Scholar] [CrossRef]
- Loewen, N.L. Health assessment handbook No authors listed. Developmental editor: Regina Daley Ford. Springhouse, PA: Springhouse Corporation, 1985. 454 pages. $18.95, hardcover. J. Nurse-Midwifery 1987, 32, 54. [Google Scholar] [CrossRef]
- Rong, W.; Kimura, H.; Grundy, D. The neurophysiology of hydrogen sulfide. Inflamm. Allergy Drug Targets 2011, 10, 109–117. [Google Scholar] [CrossRef]
- Wang, J.-F.; Li, Y.; Song, J.-N.; Pang, H.-G. Role of hydrogen sulfide in secondary neuronal injury. Neurochem. Int. 2014, 64, 37–47. [Google Scholar] [CrossRef]
- Shefa, U.; Kim, M.-S.; Jeong, N.Y.; Jung, J. Antioxidant and Cell-Signaling Functions of Hydrogen Sulfide in the Central Nervous System. Oxidative Med. Cell. Longev. 2018, 2018, 1–17. [Google Scholar] [CrossRef]
- Hu, H.; Shi, Y.; Chen, Q.; Yang, W.; Zhou, H.; Chen, L.; Tang, Y.; Zheng, Y. Endogenous hydrogen sulfide is involved in regulation of respiration in medullary slice of neonatal rats. Neuroscience 2008, 156, 1074–1082. [Google Scholar] [CrossRef]
- Lee, M.; Schwab, C.; Yu, S.; McGeer, E.; McGeer, P.L. Astrocytes produce the antiinflammatory and neuroprotective agent hydrogen sulfide. Neurobiol. Aging 2009, 30, 1523–1534. [Google Scholar] [CrossRef]
- Austgen, J.R.; Hermann, G.E.; Dantzler, H.A.; Rogers, R.C.; Kline, D.D. Hydrogen sulfide augments synaptic neurotransmission in the nucleus of the solitary tract. J. Neurophysiol. 2011, 106, 1822–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.-B.; Wu, W.-N.; Wang, W.; Gu, X.-H.; Yu, B.; Wei, B.; Yang, Y.-J. Cystathionine-β-synthase-derived hydrogen sulfide is required for amygdalar long-term potentiation and cued fear memory in rats. Pharmacol. Biochem. Behav. 2017, 155, 16–23. [Google Scholar] [CrossRef]
- Ide, M.; Ohnishi, T.; Toyoshima, M.; Balan, S.; Maekawa, M.; Shimamoto-Mitsuyama, C.; Iwayama, Y.; Ohba, H.; Watanabe, A.; Ishii, T.; et al. Excess hydrogen sulfide and polysulfides production underlies a schizophrenia pathophysiology. EMBO Mol. Med. 2019, 11, e10695. [Google Scholar] [CrossRef] [PubMed]
- Eto, K.; Asada, T.; Arima, K.; Makifuchi, T.; Kimura, H. Brain hydrogen sulfide is severely decreased in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2002, 293, 1485–1488. [Google Scholar] [CrossRef]
- Mani, S.; Cao, W.; Wu, L.; Wang, R. Hydrogen sulfide and the liver. Nitric Oxide 2014, 41, 62–71. [Google Scholar] [CrossRef]
- Faverzani, J.L.; Hammerschmidt, T.G.; Sitta, A.; Deon, M.; Wajner, M.; Vargas, C.R. Oxidative Stress in Homocystinuria Due to Cystathionine ß-Synthase Deficiency: Findings in Patients and in Animal Models. Cell. Mol. Neurobiol. 2017, 358, 273–1485. [Google Scholar] [CrossRef]
- Guillén, N.; Navarro, M.A.; Arnal, C.; Noone, E.; Arbones-Mainar, J.M.; Acín, S.; Surra, J.C.; Muniesa, P.; Roche, H.M.; Osada, J. Microarray analysis of hepatic gene expression identifies new genes involved in steatotic liver. Physiol. Genom. 2009, 37, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Esse, R.; Imbard, A.; Florindo, C.; Gupta, S.; Quinlivan, E.P.; Davids, M.; Teerlink, T.; De Almeida, I.T.; Kruger, W.D.; Blom, H.J.; et al. Protein arginine hypomethylation in a mouse model of cystathionine β-synthase deficiency. FASEB J. 2014, 28, 2686–2695. [Google Scholar] [CrossRef] [Green Version]
- Hamelet, J.; Noll, C.; Ripoll, C.; Paul, J.-L.; Janel, N.; Delabar, J.-M. Effect of hyperhomocysteinemia on the protein kinase DYRK1A in liver of mice. Biochem. Biophys. Res. Commun. 2009, 378, 673–677. [Google Scholar] [CrossRef]
- Sarna, L.K.; Siow, Y.L.; O, K. The CBS/CSE system: A potential therapeutic target in NAFLD? Can. J. Physiol. Pharmacol. 2015, 93, 1–11. [Google Scholar] [CrossRef]
- Veeranki, S.; Winchester, L.J.; Tyagi, S.C. Hyperhomocysteinemia associated skeletal muscle weakness involves mitochondrial dysfunction and epigenetic modifications. Biochim. et Biophys. Acta (BBA) Bioenerg. 2015, 1852, 732–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.-Q.; Jiang, L.; Lan, F.; Wei, H.-J.; Xie, M.; Zou, W.; Zhang, P.; Wang, C.-Y.; Xie, Y.-R.; Tang, X.-Q. Inhibited Endogenous H2S Generation and Excessive Autophagy in Hippocampus Contribute to Sleep Deprivation-Induced Cognitive Impairment. Front. Psychol. 2019, 10, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbaux, S.; Plomin, R.; Whitehead, A.S. Polymorphisms of genes controlling homocysteine/folate metabolism and cognitive function. NeuroReport 2000, 11, 1133–1136. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Han, J.; Huang, R.; Sun, J.; Cai, R.; Shen, Y.; Wang, S. Increased Plasma Homocysteine Level is Associated with Executive Dysfunction in Type 2 Diabetic Patients with Mild Cognitive Impairment. J. Alzheimer’s Dis. 2017, 58, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Szabo, C. Both the H 2 S biosynthesis inhibitor aminooxyacetic acid and the mitochondrially targeted H 2 S donor AP39 exert protective effects in a mouse model of burn injury. Pharmacol. Res. 2016, 113, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Behera, J.; Kelly, K.E.; Voor, M.J.; Metreveli, N.; Tyagi, S.C.; Tyagi, N. Hydrogen Sulfide Promotes Bone Homeostasis by Balancing Inflammatory Cytokine Signaling in CBS-Deficient Mice through an Epigenetic Mechanism. Sci. Rep. 2018, 8, 15226. [Google Scholar] [CrossRef]
- Garg, S.; Vitvitsky, V.; Gendelman, H.E.; Banerjee, R. Monocyte differentiation, activation, and mycobacterial killing are linked to transsulfuration-dependent redox metabolism. J. Biol. Chem. 2006, 281, 38712–38720. [Google Scholar]
- Yang, R.; Qu, C.; Zhou, Y.; Konkel, J.E.; Shi, S.; Liu, Y.; Chen, C.; Liu, S.; Liu, D.; Chen, Y.; et al. Hydrogen Sulfide Promotes Tet1- and Tet2-Mediated Foxp3 Demethylation to Drive Regulatory T Cell Differentiation and Maintain Immune Homeostasis. Immunity 2015, 43, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Saini, V.; Chinta, K.C.; Reddy, V.P.; Glasgow, J.N.; Stein, A.; Lamprecht, D.A.; Rahman, A.; MacKenzie, J.S.; Truebody, B.E.; Adamson, J.H.; et al. Hydrogen sulfide stimulates Mycobacterium tuberculosis respiration, growth and pathogenesis. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Zheng, F.; Han, J.; Lu, H.; Cui, C.; Yang, J.; Cui, Q.; Cai, J.; Zhou, Y.; Tang, C.; Xu, G.; et al. Cystathionine beta synthase-hydrogen sulfide system in paraventricular nucleus reduced high fatty diet induced obesity and insulin resistance by brain-adipose axis. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 3281–3291. [Google Scholar] [CrossRef]
- Beltowski, J.; Wójcicka, G.; Jamroz-Wiśniewska, A. Hydrogen sulfide in the regulation of insulin secretion and insulin sensitivity: Implications for the pathogenesis and treatment of diabetes mellitus. Biochem. Pharmacol. 2018, 149, 60–76. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.; Saha, M.N.; Mok, A.; Champsi, O.; Wang, R.; Lobb, I.; Sener, A. Hydrogen Sulfide Induced Erythropoietin Synthesis is Regulated by HIF Proteins. J. Urol. 2016, 196, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, X.; Zhou, G.; Mu, M.-D.; Zhang, F.-L.; Li, F.-M.; Qian, C.; Du, F.; Yung, W.-H.; Qian, Z.-M.; et al. Cystathionine β-synthase is required for body iron homeostasis. Hepatology 2017, 67, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, J.; Juriasingani, S.; Akbari, M.; Shao, P.; Saha, M.N.; Lobb, I.; Bachtler, M.; O Fernandez, B.; Qian, Z.; Van Goor, H.; et al. Endogenous H2S production deficiencies lead to impaired renal erythropoietin production. Can. Urol. Assoc. J. 2018, 13, E210–E219. [Google Scholar] [CrossRef]
- Zhao, P.; Qian, C.; Chen, Y.-J.; Sheng, Y.; Ke, Y.; Qian, Z.-M. Cystathionine β-synthase (CBS) deficiency suppresses erythropoiesis by disrupting expression of heme biosynthetic enzymes and transporter. Cell Death Dis. 2019, 10, 708–711. [Google Scholar] [CrossRef]
- Jhee, K.-H.; Kruger, W.D. The Role of Cystathionine β-Synthase in Homocysteine Metabolism. Antioxidants Redox Signal. 2005, 7, 813–822. [Google Scholar] [CrossRef]
- Kruger, W.D.; Gupta, S. The effect of dietary modulation of sulfur amino acids on cystathionine β synthase-deficient mice. Ann. New York Acad. Sci. 2015, 1363, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Majtan, T.; Pey, A.L.; Ereño-Orbea, J.; Martínez-Cruz, L.; Kraus, J. Targeting Cystathionine Beta-Synthase Misfolding in Homocystinuria by Small Ligands: State of the Art and Future Directions. Curr. Drug Targets 2016, 17, 1. [Google Scholar] [CrossRef]
- Morris, A.A.M.; Kožich, V.; Santra, S.; Andria, G.; Ben-Omran, T.I.M.; Chakrapani, A.B.; Crushell, E.; Henderson, M.J.; Hochuli, M.; Huemer, M.; et al. Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J. Inherit. Metab. Dis. 2016, 40, 49–74. [Google Scholar] [CrossRef] [Green Version]
- Kruger, W.D. Cystathionine β-synthase deficiency: Of mice and men. Mol. Genet. Metab. 2017, 121, 199–205. [Google Scholar] [CrossRef]
- Majtan, T.; Mascarell, P.G.; Martínez-Cruz, L.A.; Kožich, V.; Pey, A.L.; Szabo, C.; Kraus, J.P. Potential Pharmacological Chaperones for Cystathionine Beta-Synthase-Deficient Homocystinuria. Handbook of Experimental Pharmacology 2017, 245, 345–383. [Google Scholar] [CrossRef]
- Bublil, E.M.; Majtan, T. Classical homocystinuria: From cystathionine beta-synthase deficiency to novel enzyme therapies. Biochimie 2019. [Google Scholar] [CrossRef]
- Zaric, B.L.; Obradovic, M.M.; Bajić, V.; A Haidara, M.; Jovanović, M.; Isenovic, E.R. Homocysteine and Hyperhomocysteinaemia. Curr. Med. Chem. 2019, 26, 2948–2961. [Google Scholar] [CrossRef] [PubMed]
- Koutmos, M.; Kabil, O.; Smith, J.L.; Banerjee, R. Structural basis for substrate activation and regulation by cystathionine beta-synthase (cbs) domains in cystathionine {beta}-synthase. Proc. Nat. Acad. Sci. USA 2010, 107, 20958–20963. [Google Scholar]
- Ereno-Orbea, J.; Majtan, T.; Oyenarte, I.; Kraus, J.P.; Martinez-Cruz, L.A. Structural insight into the molecular mechanism of allosteric activation of human cystathionine beta-synthase by s-adenosylmethionine. Proc. Nat. Acad. Sci. USA 2014, 111, E3845–E3852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCorvie, T.J.; Kopec, J.; Hyung, S.J.; Fitzpatrick, F.; Feng, X.; Termine, D.; Strain-Damerell, C.; Vollmar, M.; Fleming, J.; Janz, J.M.; et al. Inter-domain communication of human cystathionine beta-synthase: Structural basis of s-adenosyl-l-methionine activation. J. Biol. Chem. 2014, 289, 36018–36030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhee, K.-H.; McPHIE, P.; Miles, E.W. Yeast cystathionine beta-synthase is a pyridoxal phosphate enzyme but, unlike the human enzyme, is not a heme protein. J. Boil. Chem. 2000, 275, 11541–11544. [Google Scholar] [CrossRef] [Green Version]
- Taoka, S.; Banerjee, R. Stopped-flow Kinetic Analysis of the Reaction Catalyzed by the Full-length Yeast Cystathionine beta -Synthase. J. Boil. Chem. 2002, 277, 22421–22425. [Google Scholar] [CrossRef] [Green Version]
- Aitken, S.M.; Brenner, S.E. Role of Active-Site Residues Thr81, Ser82, Thr85, Gln157, and Tyr158 inYeast Cystathionine β-Synthase Catalysis and Reaction Specificity†. Biochemistry 2004, 43, 1963–1971. [Google Scholar] [CrossRef]
- Lodha, P.H.; Hopwood, E.M.; Manders, A.L.; Aitken, S.M. Residue N84 of Yeast Cystathionine β-Synthase is a Determinant of Reaction Specificity. Biochim. et Biophys. Acta (BBA)Proteins Proteom. 2010, 1804, 1424–1431. [Google Scholar] [CrossRef]
- Aitken, S.M.; Kirsch, J.F. The enzymology of cystathionine biosynthesis: Strategies for the control of substrate and reaction specificity. Arch. Biochem. Biophys. 2005, 433, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.K.; Banerjee, R. Detection of reaction intermediates during human cystathionine beta-synthase-monitored turnover and H2S production. J. Biol. Chem. 2012, 287, 43464–43471. [Google Scholar] [PubMed] [Green Version]
- Lipson, M.H.; Kraus, J.; Rosenberg, L.E. Affinity of Cystathionine β-Synthase for Pyridoxal 5′-Phosphate in Cultured Cells. J. Clin. Investig. 1980, 66, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Mudd, S.H.; Levy, H.L.; Kraus, J.P. Disorders of transsulfuration. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Childs, B., Kinzler, K., Vogelstein, B., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 2007–2056. [Google Scholar]
- Christen, P.; Mehta, P. From cofactor to enzymes. The molecular evolution of pyridoxal-5’-phosphate-dependent enzymes. Chem. Rec. 2001, 1, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Banerjee, R. PLP-dependent H2S biogenesis. Biochim. et Biophys. Acta (BBA) Bioenerg. 2011, 1814, 1518–1527. [Google Scholar] [CrossRef] [Green Version]
- Kabil, O.; Banerjee, R. Enzymology of H2S Biogenesis, Decay and Signaling. Antioxidants Redox Signal. 2014, 20, 770–782. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, R. Catalytic promiscuity and heme-dependent redox regulation of H2S synthesis. Curr. Opin. Chem. Boil. 2017, 37, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Jhee, K.-H.; Kruger, W.D. Production of the Neuromodulator H2S by Cystathionine β-Synthase via the Condensation of Cysteine and Homocysteine. J. Boil. Chem. 2004, 279, 52082–52086. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Padovani, D.; Leslie, R.A.; Chiku, T.; Banerjee, R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J. Biol. Chem. 2009, 284, 22457–22466. [Google Scholar]
- Brosnan, J.T.; Brosnan, M.E.; Bertolo, R.F.; Brunton, J. Methionine: A metabolically unique amino acid. Livest. Sci. 2007, 112, 2–7. [Google Scholar] [CrossRef]
- Pey, A.L.; Majtan, T.; Sanchez-Ruiz, J.M.; Kraus, J.P. Human cystathionine beta-synthase (cbs) contains two classes of binding sites for s-adenosylmethionine (sam): Complex regulation of cbs activity and stability by SAM. Biochem. J. 2013, 449, 109–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prudova, A.; Bauman, Z.; Braun, A.; Vitvitsky, V.; Lu, S.C.; Banerjee, R. S-adenosylmethionine stabilizes cystathionine beta-synthase and modulates redox capacity. Proc. Nat. Acad. Sci. USA 2006, 103, 6489–6494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majtan, T.; Pey, A.L.; Fernández, R.A.; Fernández, J.A.; Martínez-Cruz, L.A.; Kraus, J.P. Domain Organization, Catalysis and Regulation of Eukaryotic Cystathionine Beta-Synthases. PLoS ONE 2014, 9, e105290. [Google Scholar] [CrossRef] [Green Version]
- Pey, A.L.; Martínez-Cruz, L.A.; Kraus, J.P.; Majtan, T. Oligomeric status of human cystathionine beta-synthase modulates AdoMet binding. FEBS Lett. 2016, 590, 4461–4471. [Google Scholar] [CrossRef] [PubMed]
- Portillo, F.; Vázquez, J.; Pajares, M.A. Protein-protein interactions involving enzymes of the mammalian methionine and homocysteine metabolism. Biochime 2020. [Google Scholar] [CrossRef]
- Boutell, J.M.; Wood, J.D.; Harper, P.S.; Jones, A.L. Huntingtin interacts with cystathionine beta-synthase. Hum. Mol. Genet. 1998, 7, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Vicente, J.B.; Malagrinò, F.; Arese, M.; Forte, E.; Sarti, P.; Giuffrè, A. Bioenergetic relevance of hydrogen sulfide and the interplay between gasotransmitters at human cystathionine β-synthase. Biochim. et Biophys. Acta (BBA) Bioenerg. 2016, 1857, 1127–1138. [Google Scholar] [CrossRef]
- Gherasim, C.; Yadav, P.K.; Kabil, O.; Niu, W.N.; Banerjee, R. Nitrite reductase activity and inhibition of H₂S biogenesis by human cystathionine ß-synthase. PLoS ONE 2014, 9, e85544. [Google Scholar] [CrossRef] [Green Version]
- Carballal, S.; Cuevasanta, E.; Yadav, P.K.; Gherasim, C.; Ballou, D.P.; Alvarez, B.; Banerjee, R. Kinetics of Nitrite Reduction and Peroxynitrite Formation by Ferrous Heme in Human Cystathionine β-Synthase. J. Boil. Chem. 2016, 291, 8004–8013. [Google Scholar] [CrossRef] [Green Version]
- Ragunathan, P.; Kumarevel, T.; Agari, Y.; Shinkai, A.; Kuramitsu, S.; Yokoyama, S.; Ponnuraj, K. Crystal structure of ST2348, a CBS domain protein, from hyperthermophilic archaeon Sulfolobus tokodaii. Biochem. Biophys. Res. Commun. 2008, 375, 124–128. [Google Scholar] [CrossRef]
- She, Y.-M.; Xu, X.; Yakunin, A.F.; Dhe-Paganon, S.; Donald, L.J.; Standing, K.G.; Lee, D.C.; Jia, Z.; Cyr, T.D. Mass Spectrometry Following Mild Enzymatic Digestion Reveals Phosphorylation of Recombinant Proteins inEscherichia coliThrough Mechanisms Involving Direct Nucleotide Binding. J. Proteome Res. 2010, 9, 3311–3318. [Google Scholar] [CrossRef] [PubMed]
- Bianca, R.D.D.V.; Mitidieri, E.; Fusco, F.; Russo, A.; Pagliara, V.; Tramontano, T.; Donnarumma, E.; Mirone, V.; Cirino, G.; Russo, G.; et al. Urothelium muscarinic activation phosphorylates CBSSer227 via cGMP/PKG pathway causing human bladder relaxation through H2S production. Sci. Rep. 2016, 6, 31491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolland, T.; Taşan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R.; et al. A proteome-scale map of the human interactome network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef]
- Gauci, S.; Helbig, A.O.; Slijper, M.; Krijgsveld, J.; Heck, A.J.R.; Mohammed, S. Lys-N and Trypsin Cover Complementary Parts of the Phosphoproteome in a Refined SCX-Based Approach. Anal. Chem. 2009, 81, 4493–4501. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative Phosphoproteomics Reveals Widespread Full Phosphorylation Site Occupancy During Mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar] [CrossRef]
- Yu, G.; Xiao, C.; Lu, C.-H.; Jia, H.-T.; Ge, F.; Wang, W.; Yin, X.-F.; Jia, H.-L.; He, J.; He, Q.-Y. Phosphoproteome profile of human lung cancer cell line A549. Mol. BioSyst. 2011, 7, 472–479. [Google Scholar] [CrossRef]
- Santamaria, A.; Wang, B.; Elowe, S.; Malik, R.; Zhang, F.; Bauer, M.; Schmidt, A.; Silljé, H.H.W.; Körner, R.; Nigg, E.A. The Plk1-dependent phosphoproteome of the early mitotic spindle. Mol. Cell. Proteom. 2010, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Rigbolt, K.T.G.; Prokhorova, T.; Akimov, V.; Henningsen, J.; Johansen, P.T.; Kratchmarova, I.; Kassem, M.; Mann, M.; Olsen, J.V.; Blagoev, B. System-Wide Temporal Characterization of the Proteome and Phosphoproteome of Human Embryonic Stem Cell Differentiation. Sci. Signal. 2011, 4, rs3. [Google Scholar] [CrossRef]
- Klammer, M.; Kaminski, M.; Zedler, A.; Oppermann, F.; Blencke, S.; Marx, S.; Müller, S.; Tebbe, A.; Godl, K.; Schaab, C. Phosphosignature predicts dasatinib response in non-small cell lung cancer. Mol. Cell. Proteom. 2012, 11, 651–668. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Di Palma, S.; Preisinger, C.; Peng, M.; Polat, A.N.; Heck, A.J.R.; Mohammed, S. Toward a Comprehensive Characterization of a Human Cancer Cell Phosphoproteome. J. Proteome Res. 2012, 12, 260–271. [Google Scholar] [CrossRef]
- Sharma, K.; D’Souza, R.C.J.; Tyanova, S.; Schaab, C.; Wisniewski, J.R.; Cox, J.; Mann1, M. Ultradeep Human Phosphoproteome Reveals a Distinct Regulatory Nature of Tyr and Ser/Thr-Based Signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, Y.; Song, C.; Cheng, K.; Dong, M.; Wang, F.; Huang, J.; Sun, D.; Wang, L.; Ye, M.; Zou, H. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J. Proteom. 2014, 96, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Boeing, S.; Williamson, L.; Encheva, V.; Gori, I.; Saunders, R.E.; Instrell, R.; Aygün, O.; Rodriguez-Martinez, M.; Weems, J.C.; Kelly, G.; et al. Multiomic Analysis of the UV-Induced DNA Damage Response. Cell Rep. 2016, 15, 1597–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchin, C.; Cesaro, L.; Salvi, M.; Millioni, R.; Iori, E.; Cifani, P.; James, P.; Arrigoni, G.; Pinna, L.A. Quantitative analysis of a phosphoproteome readily altered by the protein kinase CK2 inhibitor quinalizarin in HEK-293T cells. Biochim. et Biophys. Acta (BBA)Proteins Proteom. 2015, 1854, 609–623. [Google Scholar] [CrossRef]
- Stuart, S.A.; Houel, S.; Lee, T.; Wang, N.; Old, W.M.; Ahn, N.G. A phosphoproteomic comparison of B-RAFV600E and MKK1/2 inhibitors in melanoma cells. Mol. Cell. Proteomics. 2015, 14, 1599–1615. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Cptac, N.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Niu, W.-N.; Yadav, P.K.; Adamec, J.; Banerjee, R. S-glutathionylation enhances human cystathionine β-synthase activity under oxidative stress conditions. Antioxidants Redox Signal. 2014, 22, 350–361. [Google Scholar] [CrossRef] [Green Version]
- Niu, W.; Wang, J.; Qian, J.; Wang, M.; Wu, P.; Chen, F.; Yan, S. Allosteric control of human cystathionine β-synthase activity by a redox active disulfide bond. J. Boil. Chem. 2018, 293, 2523–2533. [Google Scholar] [CrossRef] [Green Version]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.F.; Hallenborg, P.; Pedersen, A.-K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.; Vanselow, J.T.; Nielsen, M.M.; et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Boil. 2018, 25, 631–640. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Smith, D. A Practical Review of Proteasome Pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Wang, L.; Anderl, J.; Slifker, M.J.; Kirk, C.; Kruger, W.D. Correction of cystathionine β-synthase deficiency in mice by treatment with proteasome inhibitors. Hum. Mutat. 2013, 34, 1085–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skovby, F.; Kraus, J.P.; E Rosenberg, L. Biosynthesis and proteolytic activation of cystathionine beta-synthase in rat liver. J. Boil. Chem. 1984, 259, 588–593. [Google Scholar]
- Zou, C.G.; Banerjee, R. Tumor necrosis factor-alpha-induced targeted proteolysis of cystathionine beta-synthase modulates redox homeostasis. J. Biol. Chem. 2003, 278, 16802–16808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hnizda, A.; Spiwok, V.; Jurga, V.; Kožich, V.; Kodíček, M.; Kraus, J.P. Cross-Talk between the Catalytic Core and the Regulatory Domain in Cystathionine β-Synthase: Study by Differential Covalent Labeling and Computational Modeling. Biochemistry 2010, 49, 10526–10534. [Google Scholar] [CrossRef]
- Bruno, S.; Schiaretti, F.; Burkhard, P.; Kraus, J.P.; Janosik, M.; Mozzarelli, A. Functional Properties of the Active Core of Human Cystathionine β-Synthase Crystals. J. Boil. Chem. 2000, 276, 16–19. [Google Scholar] [CrossRef] [Green Version]
- Kéry, V.; Poneleit, L.; Kraus, J.P. Trypsin Cleavage of Human Cystathionine β-Synthase into an Evolutionarily Conserved Active Core: Structural and Functional Consequences. Arch. Biochem. Biophys. 1998, 355, 222–232. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox. Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [Green Version]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. (Lond). 2018, 38, 12. [Google Scholar] [CrossRef] [Green Version]
- Santos, I.; Ramos, C.; Mendes, C.; Sequeira, C.; Tomé, C.S.; Fernandes, D.; Mota, P.; Pires, R.F.; Urso, D.; Hipólito, A.; et al. Targeting Glutathione and Cystathionine β-Synthase in Ovarian Cancer Treatment by Selenium-Chrysin Polyurea Dendrimer Nanoformulation. Nutrients 2019, 11, 2523. [Google Scholar] [CrossRef] [Green Version]
- Lejeune, J. Réflexions sur la débilité de l’intelligence des enfants trisomiques. Pont. Acad. Sci. 1975, 3, 1–12. [Google Scholar]
- Chadefaux, B.; Rethore, M.; Raoul, O.; Ceballos, I.; Poissonnier, M.; Gilgenkranz, S.; Allard, D. Cystathionine beta synthase: Gene dosage effect in trisomy. Biochem. Biophys. Res. Commun. 1985, 128, 40–44. [Google Scholar] [CrossRef]
- Lejeune, J. Pathogenesis of mental deficiency in trisomy. Am. J. Med. Genet. Suppl. 1990, 7, 20–30. [Google Scholar]
- Taub, J.W.; Huang, X.; Matherly, L.H.; Stout, M.L.; A Buck, S.; Massey, G.V.; Becton, D.L.; Chang, M.N.; Weinstein, H.J.; Ravindranath, Y. Expression of chromosome 21-localized genes in acute myeloid leukemia: Differences between Down syndrome and non-Down syndrome blast cells and relationship to in vitro sensitivity to cytosine arabinoside and daunorubicin. Blood 1999, 94, 1393–1400. [Google Scholar] [PubMed]
- Ge, Y.; Jensen, T.L.; Matherly, L.H.; Taub, J.W. Transcriptional regulation of the cystathionine-beta-synthase gene in Down syndrome and non-Down syndrome megakaryocytic leukemia cell lines. Blood 2003, 101, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, A.; Kanaumi, T.; Takashima, S.; Enokido, Y.; Nagai, Y.; Kimura, H. Cystathionine beta-synthase is enriched in the brains of Down’s patients. Biochem. Biophys. Res. Commun. 2005, 338, 1547–1550. [Google Scholar]
- Liu, Y.; Borel, C.; Li, L.; Müller, T.; Williams, E.G.; Germain, P.-L.; Buljan, M.; Sajic, T.; Boersema, P.J.; Shao, W.; et al. Systematic proteome and proteostasis profiling in human Trisomy 21 fibroblast cells. Nat. Commun. 2017, 8, 1212. [Google Scholar] [CrossRef]
- London, J.; Ndiaye, F.K.; Bui, L.C.; Souchet, B.; Daubigney, F.; Magnan, C.; Luquet, S.; Dairou, J.; Janel, N.; Rouch, C. Alterations in the Serotonin and Dopamine Pathways by Cystathionine Beta Synthase Overexpression in Murine Brain. Mol. Neurobiol. 2018, 56, 3958–3971. [Google Scholar] [CrossRef]
- Marechal, D.; Brault, V.; Leon, A.; Martin, D.; Lopes Pereira, P.; Loaëc, N.; Birling, M.C.; Friocourt, G.; Blondel, M.; Herault, Y. CBS overdosage is necessary and sufficient to induce cognitive phenotypes in mouse models of Down syndrome and interacts genetically with Dyrk1a. Hum. Mol. Genet. 2019, 28, 1561–1577. [Google Scholar]
- Herault, Y.; Delabar, J.M.; Fisher, E.; Tybulewicz, V.L.; Yu, Y.E.; Brault, V. Rodent models in Down syndrome research: Impact and future opportunities. Dis. Model. Mech. 2017, 10, 1165–1186. [Google Scholar] [CrossRef] [Green Version]
- Pogribna, M.; Melnyk, S.; Pogribny, I.; Chango, A.; Yi, P.; James, S.J. Homocysteine Metabolism in Children with Down Syndrome: In Vitro Modulation. Am. J. Hum. Genet. 2001, 69, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Caracausi, M.; Ghini, V.; Locatelli, C.; Mericio, M.; Piovesan, A.; Antonaros, F.; Pelleri, M.C.; Vitale, L.; Vacca, R.A.; Bedetti, F.; et al. Plasma and urinary metabolomic profiles of Down syndrome correlate with alteration of mitochondrial metabolism. Sci. Rep. 2018, 8, 2977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, K.; Lewis, H.C.; A Hill, A.; Pandey, A.; Jackson, L.P.; Cabral, J.; Smith, K.P.; Liggett, L.A.; Gomez, E.B.; Galbraith, M.; et al. Trisomy 21 consistently activates the interferon response. eLife 2016, 5, 16220. [Google Scholar] [CrossRef] [PubMed]
- Guedj, F.; Pennings, J.L.; Massingham, L.; Wick, H.C.; Siegel, A.E.; Tantravahi, U.; Bianchi, D.W. An Integrated Human/Murine Transcriptome and Pathway Approach To Identify Prenatal Treatments For Down Syndrome. Sci. Rep. 2016, 6, 32353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelleri, M.C.; Cattani, C.; Vitale, L.; Antonaros, F.; Strippoli, P.; Locatelli, C.; Cocchi, G.; Piovesan, A.; Caracausi, M. Integrated Quantitative Transcriptome Maps of Human Trisomy 21 Tissues and Cells. Front. Genet. 2018, 9, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriroopreddy, R.; Sajeed, R.; Raghuraman, P.; Sudandiradoss, C. Differentially expressed gene (DEG) based protein-protein interaction (PPI) network identifies a spectrum of gene interactome, transcriptome and correlated miRNA in nondisjunction Down syndrome. Int. J. Boil. Macromol. 2019, 122, 1080–1089. [Google Scholar] [CrossRef]
- Sobol, M.; Klar, J.; Laan, L.; Shahsavani, M.; Schuster, J.; Annerén, G.; Konzer, A.; Mi, J.; Bergquist, J.; Nordlund, J.; et al. Transcriptome and Proteome Profiling of Neural Induced Pluripotent Stem Cells from Individuals with Down Syndrome Disclose Dynamic Dysregulations of Key Pathways and Cellular Functions. Mol. Neurobiol. 2019, 56, 7113–7127. [Google Scholar] [CrossRef] [Green Version]
- Moreira-Filho, C.A.; Bando, S.Y.; Bertonha, F.B.; Silva, F.N.; Costa, L.D.F.; Ferreira, L.R.; Furlanetto, G.; Chacur, P.; Zerbini, M.C.N.; Carneiro-Sampaio, M. Modular transcriptional repertoire and MicroRNA target analyses characterize genomic dysregulation in the thymus of Down syndrome infants. Oncotarget 2016, 7, 7497–7533. [Google Scholar] [CrossRef]
- Belardinelli, M.-C.; Chabli, A.; Chadefaux-Vekemans, B.; Kamoun, P. Urinary Sulfur Compounds in Down Syndrome. Clin. Chem. 2001, 47, 1500–1501. [Google Scholar] [CrossRef]
- Kamoun, P.; Belardinelli, M.-C.; Chabli, A.; Lallouchi, K.; Chadefaux-Vekemans, B. Endogenous hydrogen sulfide overproduction in Down syndrome. Am. J. Med. Genet. 2002, 116, 310–311. [Google Scholar] [CrossRef]
- Kamoun, P. Mental retardation in Down syndrome: A hydrogen sulfide hpothesis. Med. Hypotheses 2001, 57, 389–392. [Google Scholar] [CrossRef] [Green Version]
- Chance, B.; Schoener, B. High and low energy states of cytochromes. I. In mitochondria. J. Biol. Chem. 1965, 241, 4567–4573. [Google Scholar]
- Nicholls, P.; Marshall, D.C.; Cooper, C.; Wilson, M.T. Sulfide inhibition of and metabolism by cytochrome c oxidase. Biochem. Soc. Trans. 2013, 41, 1312–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, S.H.; Skrajny, B.; Reiffenstein, R.J. ALTERATION OF THE MORPHOLOGY AND NEUROCHEMISTRY OF THE DEVELOPING MAMMALIAN NERVOUS SYSTEM BY HYDROGEN SULPHIDE. Clin. Exp. Pharmacol. Physiol. 1995, 22, 379–380. [Google Scholar] [CrossRef] [PubMed]
- Skrajny, B.; Hannah, R.S.; Roth, S.H. Low concentrations of hydrogen sulphide alter monoamine levels in the developing rat central nervous system. Can. J. Physiol. Pharmacol. 1992, 70, 1515–1518. [Google Scholar] [CrossRef]
- Partlo, L.; Sainsbury, R.S.; Roth, S.H. Effects of repeated hydrogen sulphide (H2S) exposure on learning and memory in the adult rat. NeuroToxicology 2001, 22, 177–189. [Google Scholar] [CrossRef]
- Hannah, R.; Roth, S. Chronic exposure to low concentrations of hydrogen sulfide produces abnormal growth in developing cerebellar Purkinje cells. Neurosci. Lett. 1991, 122, 225–228. [Google Scholar] [CrossRef]
- Vacca, R.A.; Bawari, S.; Valenti, D.; Tewari, D.; Nabavi, S.F.; Shirooie, S.; Sah, A.N.; Volpicella, M.; Braidy, N.; Nabavi, S.M. Down syndrome: Neurobiological alterations and therapeutic targets. Neurosci. Biobehav. Rev. 2019, 98, 234–255. [Google Scholar] [CrossRef]
- Valenti, D.; De Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217. [Google Scholar] [CrossRef]
- Izzo, A.; Mollo, N.; Nitti, M.; Paladino, S.; Cali, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; Barbato, M.; Sarnataro, V.; et al. Mitochondrial dysfunction in down syndrome: Molecular mechanisms and therapeutic targets. Mol. Med. 2018, 24, 2. [Google Scholar] [CrossRef] [Green Version]
- Valenti, D.; Braidy, N.; De Rasmo, D.; Signorile, A.; Rossi, L.; Atanasov, A.G.; Volpicella, M.; Henrion-Caude, A.; Nabavi, S.; Vacca, R.A. Mitochondria as pharmacological targets in Down syndrome. Free. Radic. Boil. Med. 2018, 114, 69–83. [Google Scholar] [CrossRef]
- Abdel-Salam, E.; Abdel-Meguid, I.; Korraa, S. Assessment of immune function in Down syndrome patients. Egypt. J. Med Hum. Genet. 2013, 14, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Kamoun, P.P. Mental retardation in Down syndrome: Two ways to treat. Med. Hypotheses 2019, 131, 109289. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. The re-emerging pathophysiological role of the cystathionine-β-synthase - hydrogen sulfide system in Down syndrome. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Outschoorn, U.; Peiris-Pagès, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2016, 14, 11–31. [Google Scholar] [CrossRef] [PubMed]
- De Vos, J.; Thykjær, T.; Tarte, K.; Ensslen, M.; Raynaud, P.; Requirand, G.; Pellet, F.; Pantesco, V.; Rème, T.; Jourdan, M.; et al. Comparison of gene expression profiling between malignant and normal plasma cells with oligonucleotide arrays. Oncogene 2002, 21, 6848–6857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansel, N.E.; Rahman, A.; Hidalgo, M.; Thuluvath, P.J.; Lillemoe, K.D.; Shulick, R.; Ku, J.-L.; Park, J.-G.; Miyazaki, K.; Ashfaq, R.; et al. Identification of Novel Cellular Targets in Biliary Tract Cancers Using Global Gene Expression Technology. Am. J. Pathol. 2003, 163, 217–229. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W. Expression Profiling of Homocysteine Junction Enzymes in the NCI60 Panel of Human Cancer Cell Lines. Cancer Res. 2005, 65, 1554–1560. [Google Scholar] [CrossRef] [Green Version]
- Ryu, C.S.; Kwak, H.C.; Lee, K.S.; Kang, K.W.; Oh, S.J.; Lee, K.H.; Kim, H.M.; Ma, J.Y.; Kim, S.-K. Sulfur amino acid metabolism in doxorubicin-resistant breast cancer cells. Toxicol. Appl. Pharmacol. 2011, 255, 94–102. [Google Scholar] [CrossRef]
- Módis, K.; Coletta, C.; Asimakopoulou, A.; Szczesny, B.; Chao, C.; Papapetropoulos, A.; Hellmich, M.R.; Szabo, C. Effect of S-adenosyl-L-methionine (SAM), an allosteric activator of cystathionine-β-synthase (CBS) on colorectal cancer cell proliferation and bioenergetics in vitro. Nitric Oxide 2014, 41, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Sanokawa-Akakura, R.; Ostrakhovitch, E.A.; Akakura, S.; Goodwin, S.; Tabibzadeh, S. A H2S-Nampt Dependent Energetic Circuit Is Critical to Survival and Cytoprotection from Damage in Cancer Cells. PLoS ONE 2014, 9, e108537. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, P.K.; Xiong, X.; Mustafi, S.B.; Saha, S.; Dhanasekaran, D.; Mandal, N.A.; McMeekin, S.; Bhattacharya, R.; Mukherjee, P. Role of cystathionine beta synthase in lipid metabolism in ovarian cancer. Oncotarget 2015, 6, 37367–37384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrakhovitch, E.A.; Akakura, S.; Sanokawa-Akakura, R.; Goodwin, S.; Tabibzadeh, S. Dedifferentiation of cancer cells following recovery from a potentially lethal damage is mediated by H2S–Nampt. Exp. Cell Res. 2015, 330, 135–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, S.; Kawahara, B.; Gupta, D.; Tsai, R.; Khachatryan, M.; Chowdhuri, S.R.; Bose, S.; Yoon, A.; Faull, K.; Farias-Eisner, R.; et al. Role of cystathionine β-synthase in human breast Cancer. Free. Radic. Boil. Med. 2015, 86, 228–238. [Google Scholar] [CrossRef]
- Ye, F.; Chen, C.; Qin, J.; Liu, J.; Zheng, C. Genetic profiling reveals an alarming rate of cross-contamination among human cell lines used in China. FASEB J. 2015, 29, 4268–4272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, C.; Zatarain, J.R.; Ding, Y.; Coletta, C.; Mrazek, A.A.; Druzhyna, N.; Johnson, P.; Chen, H.; Hellmich, J.L.; Asimakopoulou, A.; et al. Cystathionine-beta-synthase inhibition for colon cancer: Enhancement of the efficacy of aminooxyacetic acid via the prodrug approach. Mol. Med. 2016, 22, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Ye, J.; Kang, W.; Wang, T.; You, J.; Shi, X. Role of the cystathionine β-synthase/H2S system in liver cancer cells and the inhibitory effect of quinolone-indolone conjugate QIC2 on the system. Oncol. Rep. 2017, 37, 3001–3009. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.; Saide, A.; Cagliani, R.; Cantile, M.; Botti, G.; Russo, G. rpL3 promotes the apoptosis of p53 mutated lung cancer cells by down-regulating CBS and NFκB upon 5-FU treatment. Sci. Rep. 2016, 6, 38369. [Google Scholar] [CrossRef] [Green Version]
- Szczesny, B.; Marcatti, M.; Zatarain, J.R.; Druzhyna, N.; Wiktorowicz, J.E.; Nagy, P.; Hellmich, M.R.; Szabo, C. Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA repair and suppressing cellular bioenergetics. Sci. Rep. 2016, 6, 36125. [Google Scholar] [CrossRef] [Green Version]
- Alix-Panabières, C.; Cayrefourcq, L.; Mazard, T.; Maudelonde, T.; Assenat, E.; Assou, S. Molecular Portrait of Metastasis-Competent Circulating Tumor Cells in Colon Cancer Reveals the Crucial Role of Genes Regulating Energy Metabolism and DNA Repair. Clin. Chem. 2017, 63, 700–713. [Google Scholar] [CrossRef]
- Wróbel, M.; Bronowicka-Adamska, P.; Bentke, A. Hydrogen sulfide generation from L-cysteine in the human glioblastoma-astrocytoma U-87 MG and neuroblastoma SHSY5Y cell lines. Acta Biochim. Pol. 2017, 64, 171–176. [Google Scholar] [CrossRef]
- Niu, W.; Chen, F.; Wang, J.; Qian, J.; Yan, S. Antitumor effect of sikokianin C, a selective cystathionine β-synthase inhibitor, against human colon cancer in vitro and in vivo. MedChemComm 2017, 9, 113–120. [Google Scholar] [CrossRef]
- Phillips, C.M.; Zatarain, J.R.; Nicholls, M.E.; Porter, C.; Widen, S.G.; Thanki, K.; Johnson, P.; Jawad, M.U.; Moyer, M.P.; Randall, J.W.; et al. Upregulation of Cystathionine-β-Synthase in Colonic Epithelia Reprograms Metabolism and Promotes Carcinogenesis. Cancer Res. 2017, 77, 5741–5754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelford, R.E.; Abdulsattar, J.; Wei, E.X.; Cotelingam, J.; Coppola, D.; Herrera, G.A. Increased Nicotinamide Phosphoribosyltransferase and Cystathionine-β-Synthase in Renal Oncocytomas, Renal Urothelial Carcinoma, and Renal Clear Cell Carcinoma. Anticancer. Res. 2017, 37, 3423–3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breza, J.; Soltysova, A.; Hudecova, S.; Penesova, A.; Szadvari, I.; Babula, P.; Chovancova, B.; Lencesova, L.; Pös, O.; Ondrias, K.; et al. Endogenous H2S producing enzymes are involved in apoptosis induction in clear cell renal cell carcinoma. BMC Cancer 2018, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.K.; Murphy, B.; Mustafi, S.B.; Dey, A.; Xiong, X.; Rao, G.; Naz, S.; Zhang, M.; Yang, D.; Dhanasekaran, D.N.; et al. Cystathionine β-synthase regulates mitochondrial morphogenesis in ovarian cancer. FASEB J. 2018, 32, 4145–4157. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, B.; Ramadoss, S.; Chaudhuri, G.; Janzen, C.; Sen, S.; Mascharak, P.K. Carbon monoxide sensitizes cisplatin-resistant ovarian cancer cell lines toward cisplatin via attenuation of levels of glutathione and nuclear metallothionein. J. Inorg. Biochem. 2019, 191, 29–39. [Google Scholar] [CrossRef]
- Oláh, G.; Módis, K.; Törö, G.; Hellmich, M.R.; Szczesny, B.; Szabo, C. Role of endogenous and exogenous nitric oxide, carbon monoxide and hydrogen sulfide in HCT116 colon cancer cell proliferation. Biochem. Pharmacol. 2018, 149, 186–204. [Google Scholar] [CrossRef]
- Untereiner, A.A.; Pavlidou, A.; Druzhyna, N.; Papapetropoulos, A.; Hellmich, M.R.; Szabo, C. Drug resistance induces the upregulation of H2S-producing enzymes in HCT116 colon cancer cells. Biochem. Pharmacol. 2018, 149, 174–185. [Google Scholar] [CrossRef]
- Untereiner, A.A.; Oláh, G.; Módis, K.; Hellmich, M.R.; Szabo, C. H2S-induced S-sulfhydration of lactate dehydrogenase a (LDHA) stimulates cellular bioenergetics in HCT116 colon cancer cells. Biochem. Pharmacol. 2017, 136, 86–98. [Google Scholar] [CrossRef]
- Wahafu, W.; Gai, J.; Song, L.; Ping, H.; Wang, M.; Yang, F.; Niu, Y.; Xing, N. Increased H2S and its synthases in urothelial cell carcinoma of the bladder, and enhanced cisplatin-induced apoptosis following H2S inhibition in EJ cells. Oncol. Lett. 2018, 15, 8484–8490. [Google Scholar] [CrossRef]
- Wang, L.; Hannawi, H.; Liu, Y.; Zhang, X.; Shi, X.; Wang, T. Cystathionine β-synthase Induces Multidrug Resistance and Metastasis in Hepatocellular Carcinoma. Curr. Mol. Med. 2019, 18, 496–506. [Google Scholar] [CrossRef]
- Wang, L.; Cai, H.; Hu, Y.; Liu, F.; Huang, S.; Zhou, Y.; Yu, J.; Xu, J.; Wu, F. A pharmacological probe identifies cystathionine β-synthase as a new negative regulator for ferroptosis. Cell Death Dis. 2018, 9, 1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Song, Y.; Zhou, C.; Bai, Y.; Yuan, D.; Pan, Y.; Shao, C. Blocking Endogenous H2S Signaling Attenuated Radiation-Induced Long-Term Metastasis of Residual HepG2 Cells through Inhibition of EMT. Radiat. Res. 2018, 190, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yue, T.; Huang, Z.; Zhu, J.; Bu, D.; Wang, X.; Pan, Y.; Liu, Y.; Wang, P. Inhibition of hydrogen sulfide synthesis reverses acquired resistance to 5-FU through miR-215-5p-EREG/TYMS axis in colon cancer cells. Cancer Lett. 2019, 466, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yang, Z.; Liu, Z.; Zou, Q.; Yuan, Y. Clinical Significance of CBS and CCL21 in Gallbladder Adenocarcinomas and Squamous Cell/Adenosquamous Carcinomas. Appl. Immunohistochem. Mol. Morphol. 2020, 28, 103–110. [Google Scholar] [CrossRef]
- Liu, N.; Lin, X.; Huang, C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br. J. Cancer. 2020, 122, 279–292. [Google Scholar] [CrossRef]
- Yue, T.; Zuo, S.; Bu, D.; Zhu, J.; Chen, S.; Ma, Y.; Ma, J.; Guo, S.; Wen, L.; Zhang, X.; et al. Aminooxyacetic acid (AOAA) sensitizes colon cancer cells to oxaliplatin via exaggerating apoptosis induced by ROS. J. Cancer 2020, 11, 1828–1838. [Google Scholar] [CrossRef]
- Wang, M.; Yan, J.; Cao, X.; Hua, P.; Li, Z. Hydrogen sulfide modulates epithelial-mesenchymal transition and angiogenesis in non-small cell lung cancer via HIF-1α activation. Biochem. Pharmacol. 2019, 172, 113775. [Google Scholar] [CrossRef]
- Hellmich, M.R.; Coletta, C.; Chao, C.; Szabo, C. The therapeutic potential of cystathionine beta-synthetase/hydrogen sulfide inhibition in cancer. Antioxid. Redox. Signal. 2015, 22, 424–448. [Google Scholar] [CrossRef] [Green Version]
- Szabo, C. Gasotransmitters in cancer: From pathophysiology to experimental therapy. Nat. Rev. Drug Discov. 2015, 15, 185–203. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Blake, S.; Chan, K.T.; Pearson, R.B.; Kang, J. Cystathionine β-Synthase in Physiology and Cancer. BioMed. Res. Int. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Augsburger, F.; Szabo, C. Potential role of the 3-mercaptopyruvate sulfurtransferase (3-MST)—hydrogen sulfide (H2S) pathway in cancer cells. Pharmacol. Res. 2020, 154, 104083. [Google Scholar] [CrossRef] [PubMed]
- Augsburger, F.; Randi, E.; Jendly, M.; Ascencao, K.; Dilek, N.; Szabo, C. Role of 3-Mercaptopyruvate Sulfurtransferase in the Regulation of Proliferation, Migration, and Bioenergetics in Murine Colon Cancer Cells. Biomolecules 2020, 10, 447. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Shi, H.; Liu, Y.; Zhang, W.; Duan, X.; Li, M.; Shi, X.; Wang, T. Cystathionine-γ-lyase promotes the metastasis of breast cancer via the VEGF signaling pathway. Int. J. Oncol. 2019, 55, 73–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Mao, C.; Ouyang, L.; Liu, Y.; Lai, W.; Liu, N.; Shi, Y.; Chen, L.; Xiao, D.; Yu, F.; et al. Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 2019, 26, 2329–2343. [Google Scholar] [CrossRef] [PubMed]
- Thorson, M.K.; Majtan, T.; Kraus, J.P.; Barrios, A.M. Identification of cystathionine beta-synthase inhibitors using a hydrogen sulfide selective probe. Angew. Chem. Int. Ed. Engl. 2013, 52, 4641–4644. [Google Scholar] [CrossRef]
- Zhou, Y.; Yu, J.; Lei, X.; Wu, J.; Niu, Q.; Zhang, Y.; Liu, H.; Christen, P.; Gehring, H.; Wu, F. High-throughput tandem-microwell assay identifies inhibitors of the hydrogen sulfide signaling pathway. Chem. Commun. 2013, 49, 11782. [Google Scholar] [CrossRef]
- Druzhyna, N.; Szczesny, B.; Olah, G.; Modis, K.; Asimakopoulou, A.; Pavlidou, A.; Szoleczky, P.; Gero, D.; Yanagi, K.; Toro, G.; et al. Screening of a composite library of clinically used drugs and well-characterized pharmacological compounds for cystathionine beta-synthase inhibition identifies benserazide as a drug potentially suitable for repurposing for the experimental therapy of colon cancer. Pharmacol. Res. 2016, 113, 18–37. [Google Scholar]
- Zuhra, K.; Sousa, P.M.F.; Paulini, G.; Lemos, A.R.; Kalme, Z.; Bisenieks, I.; Bisenieks, E.; Vigante, B.; Duburs, G.; Bandeiras, T.M.; et al. Screening Pyridine Derivatives against Human Hydrogen Sulfide-synthesizing Enzymes by Orthogonal Methods. Sci. Rep. 2019, 9, 684. [Google Scholar] [CrossRef]
- Werner. Ueber Hydroxylsminessigsaure und Derivate derselben. Berichte Deutsch. Chem. Gesellschaft. 1893, 26, 1567–1571. [Google Scholar] [CrossRef] [Green Version]
- Werner, A.; Sonnenfeld, E. Ueber Hydroxylaminessigsäure und α-Hydroxylaminpropionsäure. Eur. J. Inorg. Chem. 1894, 27, 3350–3354. [Google Scholar] [CrossRef] [Green Version]
- Borek, E.; Clarke, H.T. Carboxymethoxylamine. J. Am. Chem. Soc. 1936, 58, 2020–2021. [Google Scholar] [CrossRef]
- Anchel, M.; Schoenheimer, R. Reagents for the isolation of carbonyl compounds from unsaponifiable material. J. Biol. Chem. 1936, 114, 539–546. [Google Scholar]
- El Mangouri, H.A. The separation of carbonyl compounds from waxes. Biochem. J. 1937, 31, 1978–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huffman, M.N.; MacCorquodale, D.W.; Thayer, S.A.; Doisy, E.A.; Smith, G.V.; Smith, O.W. The isolation of α-dihydrotheelin from human pregnancy urine. J. Biol. Chem. 1940, 134, 591–604. [Google Scholar]
- Anchel, M.; Waelsch, H. The higher fatty aldehydes I. Isolation from small amounts of tissue with acidic carbonyl reagents. J. Biol. Chem. 1942, 145, 605–613. [Google Scholar]
- Richardson, A. Aminooxyacetic Acid Derivatives. J. Med. Chem. 1964, 7, 824–826. [Google Scholar] [CrossRef]
- Van Dijk, J.; Zwagemakers, J.M.A. Oxime ether derivatives, a new class of nonsteroidal antiinflammatory compounds. J. Med. Chem. 1977, 20, 1199–1206. [Google Scholar] [CrossRef]
- Mayer, R.L.; Oechslin, C. Antistreptococciques. Compt. Rend. Acad. Sci. 1937, 205, 181–182. [Google Scholar]
- Burton, H.; McLeod, J.W.; McLeod, T.S.; Mayr-Harting, A. On the Relationships between the Respiratory Activities of Bacteria and their Sensitiveness to Sulphanilamide, p-Hydroxylamino- and p-Nitrobenzenesulphonamide. Br. J. Exp. Pathol. 1940, 21, 288–302. [Google Scholar]
- Clarke, H.T. Bacteriostatic and antibiotic compound and method of preventing bacterial growths. U.S. Patent US2464197A, 15 March 1949. [Google Scholar]
- Dienes, L.; Weinberger, H.J.; Madoff, S. The Transformation Of Typhoid Bacilli Into L Forms Under Various Conditions. J. Bacteriol. 1950, 59, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Favour, C.B. Bacteriological Study of Carboxylmethoxylamine Hemihydrochloride. J. Bacteriol. 1948, 55, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Weikum, J.; Ritzmann, N.; Jelden, N.; Klöckner, A.; Herkersdorf, S.; Josten, M.; Sahl, H.-G.; Grein, F. Sulfide protects staphylococcus aureus from aminoglycoside antibiotics but cannot be regarded as a general defense mechanism against antibiotics. Antimicrob. Agents Chemother. 2018, 62, e00602-18. [Google Scholar]
- Shatalin, K.; Shatalina, E.; Mironov, A.; Nudler, E. H2S: A Universal Defense Against Antibiotics in Bacteria. Science 2011, 334, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Tolliver-Kinsky, T.; Cui, W.; Törö, G.; Lee, S.-J.; Shatalin, K.; Nudler, E.; Szabo, C. H2S, a Bacterial Defense Mechanism against the Host Immune Response. Infect. Immun. 2018, 87, 00272-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, D.B.; E Snell, E. Pyridoxal phosphokinases. II. Effects of inhibitors. J. Boil. Chem. 1961, 236, 2085–2088. [Google Scholar]
- Snell, E.E. Chemical Structure in Relation to Biological Activities of Vitamin B6. Vitam. Horm. 1958, 16, 77–125. [Google Scholar]
- Phillips, R.S. Chemistry and diversity of pyridoxal-5′-phosphate dependent enzymes. Biochim. et Biophys. Acta (BBA)Proteins Proteom. 2015, 1854, 1167–1174. [Google Scholar] [CrossRef]
- Liang, J.; Han, Q.; Tan, Y.; Ding, H.; Li, J. Current Advances on Structure-Function Relationships of Pyridoxal 5’-Phosphate-Dependent Enzymes. Front. Mol. Biosci. 2019, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Gale, E.F. Studies on bacterial amino-acid decarboxylases. Biochem. J. 1945, 39, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Wallach, D.P. Studies on the GABA pathway. I. The inhibition of gamma-aminobutyric acid-alpha-ketoglutaric acid transaminase in vitro and in vivo by U-7524 (amino-oxyacetic acid). Biochem. Pharmacol. 1961, 5, 323–331. [Google Scholar] [CrossRef]
- Segal, H.L.; Rosso, R.G.; Hopper, S.; Weber, M.M. Direct evidence for an increase in enzyme level as the basis for the glucocorticoid-induced increase in glutamic-alanine transaminase activity in rat liver. J. Boil. Chem. 1962, 237, 3189–3195. [Google Scholar]
- Roberts, E.; Simonsen, D.G. Some properties of L-glutamic decarboxylase in mouse brain. Biochem. Pharmacol. 1963, 12, 113–134. [Google Scholar] [CrossRef]
- Free, C.A.; Julius, M.; Arnow, P.; Barry, G.T. Inhibition of alanine racemase by aminoxyacetic acid. Biochim. et Biophys. Acta (BBA) Enzym. 1967, 146, 608–610. [Google Scholar] [CrossRef]
- Leinweber, F.J. Mechanism of histidine decarboxylase inhibition by NSD-1055 and related hydroxylamines. Mol. Pharmacol. 1968, 4, 337–348. [Google Scholar]
- Yonaha, K.; Misono, H.; Yamamoto, T.; Soda, K. D-amino acid aminotransferase of Bacillus sphaericus. Enzymologic and spectrometric properties. J. Boil. Chem. 1975, 250, 6983–6989. [Google Scholar]
- Williamson, J.R.; Jakob, A.; Refino, C. Control of the removal of reducing equivalents from the cytosol in perfused rat liver. J. Boil. Chem. 1971, 246, 7632–7641. [Google Scholar]
- Longshaw, I.D.; Bowen, N.L.; Pogson, C.I. The Pathway of Gluconeogenesis in the Cortex of Guinea-Pig Kidney. Use of Aminooxyacetate as a Transaminase Inhibitor. JBIC J. Boil. Inorg. Chem. 1972, 25, 366–371. [Google Scholar] [CrossRef]
- Rognstad, R.; Clark, D.G. Effects of aminooxyacetate on the metabolism of isolated liver cells. Arch. Biochem. Biophys. 1974, 161, 638–646. [Google Scholar] [CrossRef]
- A John, R.; Charteris, A. The reaction of amino-oxyacetate with pyridoxal phosphate-dependent enzymes. Biochem. J. 1978, 171, 771–779. [Google Scholar] [CrossRef] [Green Version]
- Braunstein, A.; Goryachenkova, E.; Tolosa, E.; Willhardt, I.; Yefremova, L. Specificity and some other properties of liver serine sulphhydrase: Evidence for its identity with cystathionine β-synthase. Biochim. et Biophys. Acta (BBA) Enzym. 1971, 242, 247–260. [Google Scholar] [CrossRef]
- Beeler, T.; E Churchich, J. Reactivity of the phosphopyridoxal groups of cystathionase. J. Boil. Chem. 1976, 251, 5267–5271. [Google Scholar]
- Eliot, A.C.; Kirsch, J.F. Pyridoxal phosphate enzymes: Mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 2004, 73, 383–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kéry, V.; Poneleit, L.; Meyer, J.D.; Manning, M.C.; Kraus, J.P. Binding of Pyridoxal 5‘-Phosphate to the Heme Protein Human Cystathionine β-Synthase. Biochem. 1999, 38, 2716–2724. [Google Scholar] [CrossRef]
- Banerjee, R.; Evande, R.; Kabil, O.; Ojha, S.; Taoka, S. Reaction mechanism and regulation of cystathionine beta-synthase. Biochim. et Biophys. Acta 2003, 1647, 30–35. [Google Scholar] [CrossRef]
- Nadvi, N.A.; Salam, N.K.; Park, J.; Akladios, F.N.; Kapoor, V.; Collyer, C.A.; Gorrell, M.D.; Church, W.B. High resolution crystal structures of human kynurenine aminotransferase-I bound to PLP cofactor, and in complex with aminooxyacetate. Protein Sci. 2017, 26, 727–736. [Google Scholar] [CrossRef]
- Amadasi, A.; Bertoldi, M.; Contestabile, R.; Bettati, S.; Cellini, B.; Di Salvo, M.L.; Voltattorni, C.; Bossa, F.; Mozzarelli, A. Pyridoxal 5’-phosphate enzymes as targets for therapeutic agents. Curr. Med. Chem. 2007, 14, 1291–1324. [Google Scholar] [CrossRef]
- Tu, Y.; Kreinbring, C.A.; Hill, M.; Liu, C.; Petsko, G.A.; McCune, C.D.; Berkowitz, D.B.; Liu, D.; Ringe, D. Crystal Structures of Cystathionine β-Synthase from Saccharomyces cerevisiae: One Enzymatic Step at a Time. Biochem. 2018, 57, 3134–3145. [Google Scholar] [CrossRef]
- Baxter, C.F.; Roberts, E. Elevation of gamma-aminobutyric acid in brain: Selective inhibition of gamma-aminobutyric-alpha-ketoglutaric acid transaminase. J. Boil. Chem. 1961, 236, 3287–3294. [Google Scholar]
- Van Gelder, N.M. The effect of aminooxyacetic acid on the metabolism of gamma-aminobutyric acid in brain. Biochem. Pharmacol. 1966, 15, 533–539. [Google Scholar] [CrossRef]
- Tibbles, J.A.R.; McGreal, D.A. Trial of Amino-oxyacetic Acid, an Anticonvulsant. Can. Med. Assoc. J. 1963, 88, 881–886. [Google Scholar] [PubMed]
- LaVeck, G.D.; De La Cruz, F.; Thomas, D.B. Anticonvulsant Properties of Amino-Oxyacetic Acid: Clinical Studies. J. New Drugs 1962, 2, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Merlis, S. Clinical and Electroencephalographic Correlates of Gaba-Transaminase Inhibition in Man. Pharmacol. 1961, 5, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W. Therapeutic Trends. Am. J. Heal. Pharm. 1963, 20, 367–368. [Google Scholar] [CrossRef]
- Perry, T.L.; Hansen, S. Biochemical Effects in Man and Rat of Three Drugs Which Can Increase Brain Gaba Content. J. Neurochem. 1978, 30, 679–684. [Google Scholar] [CrossRef]
- Perry, T.L.; Wright, J.M.; Hansen, S.; Allan, B.M.; Baird, P.A.; MacLeod, P.M. Failure of aminooxyacetic acid therapy in Huntington disease. Neurology 1980, 30, 772. [Google Scholar] [CrossRef]
- Reed, H.T.; Meltzer, J.; Crews, P.; Norris, C.H.; Quine, D.B.; Guth, P.S. Amino-oxyacetic Acid as a Palliative in Tinnitus. Arch. Otolaryngol.Head Neck Surg. 1985, 111, 803–805. [Google Scholar] [CrossRef]
- Guth, P.S.; Blair, P.; Norris, C.; Risey, J.; Reed, H.T.; Housley, G.D.; Briner, W.; Bryant, G.; Miller, R. Evaluation of Amino-Oxyacetic Acid as a Palliative in Tinnitus. Ann. Otol. Rhinol. Laryngol. 1990, 99, 74–79. [Google Scholar] [CrossRef]
- McHale, D.; Green, J.; Mamalis, P. Amino-oxy-derivatives. Part I. Some α-amino-oxy-acids and α amino-oxy-hydrazides. J. Chem. Soc. (Resumed). 1960, 44, 225–229. [Google Scholar] [CrossRef]
- Price, S.A.; Mamalis, P.; McHale, D.; Green, J. The antimicrobal properties of some alpha-amino-oxy-acids, alpha-amino-oxy-hydrazides, alkoxyamines, alkoxydiguanides and their derivatives. Brit. J. Pharm. Chemother. 1960, 15, 243–246. [Google Scholar] [CrossRef]
- Venos, E.S.; Knodel, M.H.; Radford, C.L.; Berger, B.J. Branched-chain amino acid aminotransferase and methionine formation in Mycobacterium tuberculosis. BMC Microbiol. 2004, 4, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markovic-Housley, Z.; Schirmer, T.; Hohenester, E.; Khomutov, A.R.; Khomutov, R.M.; Karpeisky, M.Y.; Sandmeier, E.; Christen, P.; Jansonius, J.N. Crystal Structures and Solution Studies of Oxime Adducts of Mitochondrial Aspartate Aminotransferase. JBIC J. Boil. Inorg. Chem. 1996, 236, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Berger, L.C.; Wilson, J.; Wood, P.; Berger, B.J. Methionine Regeneration and Aspartate Aminotransferase in Parasitic Protozoa. J. Bacteriol. 2001, 183, 4421–4434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrenger, C.; Müller, I.B.; Schifferdecker, A.J.; Jain, R.; Jordanova, R.; Groves, M.R. Specific Inhibition of the Aspartate Aminotransferase of Plasmodium falciparum. J. Mol. Boil. 2011, 405, 956–971. [Google Scholar] [CrossRef]
- Li, J.; Guo, H.; Galon, E.M.; Gao, Y.; Lee, S.-H.; Liu, M.; Li, Y.; Ji, S.; Jia, H.; Xuan, X. Hydroxylamine and Carboxymethoxylamine Can Inhibit Toxoplasma gondii Growth through an Aspartate Aminotransferase-Independent Pathway. Antimicrob. Agents Chemother. 2020, 64, 64. [Google Scholar] [CrossRef]
- Fitzpatrick, S.M.; Cooper, A.J.; E Duffy, T. Use of beta-methylene-D,L-aspartate to assess the role of aspartate aminotransferase in cerebral oxidative metabolism. J. Neurochem. 1983, 41, 1370–1383. [Google Scholar] [CrossRef]
- Kauppinen, R.A.; Sihra, T.S.; Nicholls, D.G. Aminooxyacetic acid inhibits the malate-aspartate shuttle in isolated nerve terminals and prevents the mitochondria from utilizing glycolytic substrates. Biochim. et Biophys. Acta (BBA) Bioenerg. 1987, 930, 173–178. [Google Scholar] [CrossRef]
- Rubi, B.; Del Arco, A.; Bartley, C.; Satrustegui, J.; Maechler, P. The Malate-Aspartate NADH Shuttle Member Aralar1 Determines Glucose Metabolic Fate, Mitochondrial Activity, and Insulin Secretion in Beta Cells. J. Boil. Chem. 2004, 279, 55659–55666. [Google Scholar] [CrossRef] [Green Version]
- McKenna, M.C.; Waagepetersen, H.S.; Schousboe, A.; Sonnewald, U. Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: Current evidence and pharmacological tools. Biochem. Pharmacol. 2006, 71, 399–407. [Google Scholar] [CrossRef]
- Pardo, B.; Contreras, L.; Satrústegui, J. De novo Synthesis of Glial Glutamate and Glutamine in Young Mice Requires Aspartate Provided by the Neuronal Mitochondrial Aspartate-Glutamate Carrier Aralar/AGC1. Front. Endocrinol. 2013, 4, 149. [Google Scholar] [CrossRef] [Green Version]
- Llorente-Folch, I.; Rueda, C.; Amigo, I.; Del Arco, A.; Saheki, T.; Pardo, B.; Satrustegui, J. Calcium-Regulation of Mitochondrial Respiration Maintains ATP Homeostasis and Requires ARALAR/AGC1-Malate Aspartate Shuttle in Intact Cortical Neurons. J. Neurosci. 2013, 33, 13957–13971. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, J.M.; Nelson, K.K.; Clem, B.F.; Lane, A.N.; Arumugam, S.; Simmons, A.; Eaton, J.W.; Telang, S.; Chesney, J. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 2008, 10, R84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Korangath, P.; Teo, W.W.; Sadik, H.; Han, L.; Mori, N.; Huijts, C.M.; Wildes, F.; Bharti, S.; Zhang, Z.; Santa-Maria, C.A.; et al. Targeting Glutamine Metabolism in Breast Cancer with Aminooxyacetate. Clin. Cancer Res. 2015, 21, 3263–3273. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Chen, H.; Zhang, M.; Zhang, J.; Wei, X.; Ying, W. Malate-aspartate shuttle inhibitor aminooxyacetic acid leads to decreased intracellular ATP levels and altered cell cycle of C6 glioma cells by inhibiting glycolysis. Cancer Lett. 2016, 378, 1–7. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Asimakopoulou, A.; Panopoulos, P.; Chasapis, C.T.; Coletta, C.; Zhou, Z.; Cirino, G.; Giannis, A.; Szabo, C.; Spyroulias, G.A.; Papapetropoulos, A. Selectivity of commonly used pharmacological inhibitors for cystathionine beta synthase (CBS) and cystathionine gamma lyase (CSE). Br. J. Pharmacol. 2013, 169, 922–932. [Google Scholar] [CrossRef] [Green Version]
- Sekiya, J.; Schmidt, A.; Wilson, L.G.; Filner, P. Emission of Hydrogen Sulfide by Leaf Tissue in Response to l-Cysteine. Plant Physiol. 1982, 70, 430–436. [Google Scholar] [CrossRef] [Green Version]
- Julian, D.; Statile, J.L.; E Wohlgemuth, S.; Arp, A.J. Enzymatic hydrogen sulfide production in marine invertebrate tissues. Comp. Biochem. Physiol. Part A: Mol. Integr. Physiol. 2002, 133, 105–115. [Google Scholar] [CrossRef]
- Teague, B.; Asiedu, S.; Moore, P.K. The smooth muscle relaxant effect of hydrogen sulphidein vitro: Evidence for a physiological role to control intestinal contractility. Br. J. Pharmacol. 2002, 137, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, M.; Njie-Mbye, Y.F.; Okpobiri, I.; Zhao, M.; Opere, C.A.; Ohia, S.E. Endogenous Production of Hydrogen Sulfide in Isolated Bovine Eye. Neurochem. Res. 2011, 36, 1540–1545. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Pan, H.; Li, C.; Lan, X.; Liu, B.; Yang, G. Effects of hydrogen sulfide on a rat model of sepsis-associated encephalopathy. Acta Acad. Med. Wuhan 2011, 31, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, R.; Otsuguro, K.-I.; Yamaguchi, S.; Ito, S. Contribution of cysteine aminotransferase and mercaptopyruvate sulfurtransferase to hydrogen sulfide production in peripheral neurons. J. Neurochem. 2014, 130, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Tomasiak, M. The importance of aspartate aminotransferase for platelet aggregation. Haematologia 1986, 19, 101–112. [Google Scholar]
- Bianca, R.D.D.V.; Mitidieri, E.; Di Minno, M.N.D.; Kirkby, N.S.; Warner, T.D.; Di Minno, M.N.D.; Cirino, G.; Sorrentino, R. Hydrogen sulphide pathway contributes to the enhanced human platelet aggregation in hyperhomocysteinemia. Proc. Natl. Acad. Sci. USA 2013, 110, 15812–15817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Vanzo, J.P.; Greig, M.E.; Cronin, M.A. Anticonvulsant properties of amino-oxyacetic acid. Am. J. Physiol. Content 1961, 201, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Bobbin, R.; Gondra, M. Effect of intravenous aminooxyacetic acid on guinea pig cochlear potentials. Neuropharmacology 1973, 12, 1005–1007. [Google Scholar] [CrossRef]
- Wood, J.D.; Peesker, S.J. The role of GABA metabolism in the convulsant and anticonvulsant actions of aminooxyacetic acid. J. Neurochem. 1973, 20, 379–387. [Google Scholar] [CrossRef]
- Wood, J.D.; Peesker, S.J. A dual mechanism for the anticonvulsant action of aminooxyacetic acid. Can. J. Physiol. Pharmacol. 1976, 54, 534–540. [Google Scholar] [CrossRef]
- Buchar, E.; Mašek, K.; Jank, I.; Seifert, J.; Ostrovskaya, R. Effect of aminooxyacetic acid on the metabolism of pentobarbital in mice. J. Neurochem. 1974, 23, 447–449. [Google Scholar] [CrossRef]
- Emson, P.C. Effects of Chronic Treatment with Amino-Oxyacetic Acid or Sodium N-Dipropylacetate on Brain Gaba Levels and The Development and Regression of Cobalt Epileptic Foci in Rats. J. Neurochem. 1976, 27, 1489–1494. [Google Scholar] [CrossRef]
- Katz, R.J.; Liebler, L. GABA involvement in memory consolidation: Evidence from posttrial amino-oxyacetic acid. Psychopharmacology 1978, 56, 191–193. [Google Scholar] [CrossRef] [Green Version]
- Davison, A.J. Aminooxyacetic acid provides transient protection against seizures induced by hyperbaric oxygen. Brain Res. 1983, 276, 384–387. [Google Scholar] [CrossRef]
- Beuter, W.; Cojocel, C.; Müller, W.; Donaubauer, H.H.; Mayer, D. Peroxidative Damage and Nephrotoxicity of Dichlorovinylcysteine in Mice. J. Appl. Toxicol. 1989, 9, 181–186. [Google Scholar] [CrossRef]
- Gomez, R.; Asnis, N.; Tannhauser, S.L.; Barros, H.M. GABA Agonists Differentially Modify Blood Glucose Levels of Diabetic Rats. Jpn. J. Pharmacol. 1999, 80, 327–331. [Google Scholar] [CrossRef] [Green Version]
- Hadadha, M.; Vakili, A.; Bandegi, A.R. Effect of the Inhibition of Hydrogen Sulfide Synthesis on Ischemic Injury and Oxidative Stress Biomarkers in a Transient Model of Focal Cerebral Ischemia in Rats. J. Stroke Cerebrovasc. Dis. 2015, 24, 2676–2684. [Google Scholar] [CrossRef]
- Donovan, L.M.; Moore, M.W.; Gillombardo, C.B.; Chai, S.; Strohl, K. Effects of hydrogen sulfide synthesis inhibitors on posthypoxic ventilatory behavior in the C57BL/6J mouse. Respiration 2011, 82, 522–529. [Google Scholar] [CrossRef] [Green Version]
- Townsend, D.M.; Hanigan, M.H. Inhibition of gamma-glutamyl transpeptidase or cysteine S-conjugate beta-lyase activity blocks the nephrotoxicity of cisplatin in mice. J. Pharmacol. Exp. Ther. 2002, 300, 142–148. [Google Scholar] [CrossRef]
- Katayama, R.; Nagata, S.; Iida, H.; Yamagishi, N.; Yamashita, T.; Furuhama, K. Possible role of cysteine-S-conjugate β-lyase in species differences in cisplatin nephrotoxicity. Food Chem. Toxicol. 2011, 49, 2053–2059. [Google Scholar] [CrossRef] [Green Version]
- Qi, F.; Zhou, Y.; Xiao, Y.; Tao, J.; Gu, J.; Jiang, X.; Xu, G.-Y. Promoter demethylation of cystathionine-β-synthetase gene contributes to inflammatory pain in rats. Pain 2013, 154, 34–45. [Google Scholar] [CrossRef]
- Hao, Y.; Samuels, Y.; Li, Q.; Krokowski, D.; Guan, B.-J.; Wang, C.; Jin, Z.; Dong, B.; Cao, B.; Feng, X.; et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun. 2016, 7, 11971. [Google Scholar] [CrossRef]
- Li, T.; Wang, L.; Hu, Q.; Liu, S.; Bai, X.; Xie, Y.; Zhang, T.; Bo, S.; Gao, X.; Wu, S.; et al. Neuroprotective Roles of l-Cysteine in Attenuating Early Brain Injury and Improving Synaptic Density via the CBS/H2S Pathway Following Subarachnoid Hemorrhage in Rats. Front. Neurol. 2017, 8, 994. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.-M.; Du, A.-L.; Qin, H.-Z.; Jiang, H.-B.; Fu, P.-Y.; Lou, K. Aminooxyacetic acid improves learning and memory in a rat model of chronic alcoholism. Neural Regen. Res. 2018, 13, 1568–1574. [Google Scholar] [CrossRef]
- Du, A.; Dai, X.; Dong, J.; Liu, J.; Zhang, Y.; Fu, P.; Qin, H.; Li, R.; Zhang, R. Effects of aminooxyacetic acid on hippocampal mitochondria in rats with chronic alcoholism: The analysis of learning and memory-related genes. J. Integr. Neurosci. 2019, 18, 451–462. [Google Scholar] [CrossRef] [Green Version]
- He, J.-T.; Li, H.; Yang, L.; Cheng, K.-L. Involvement of Endothelin-1, H2S and Nrf2 in Beneficial Effects of Remote Ischemic Preconditioning in Global Cerebral Ischemia-Induced Vascular Dementia in Mice. Cell. Mol. Neurobiol. 2019, 39, 671–686. [Google Scholar] [CrossRef]
- Spalloni, A.; Greco, V.; Ciriminna, G.; Carregari, V.C.; Marini, F.; Pieroni, L.; Mercuri, N.B.; Urbani, A.; Longone, P. Impact of Pharmacological Inhibition of Hydrogen Sulphide Production in the SOD1G93A-ALS Mouse Model. Int. J. Mol. Sci. 2019, 20, 2550. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, M.K.; Roberts, E. Return of gamma-aminobutyrate transaminase activity in mouse brain after inhibition by aminooxyacetic acid: Chemical and histochemical observations. Biochem. Pharmacol. 1967, 16, 1138–1140. [Google Scholar] [CrossRef]
- Roberts, E.; Wein, J.; Simonsen, D.G. γ-Aminobutyric Acid (γABA), Vitamin B6, and Neuronal Function—A Speculative Synthesis. Vitam. Horm. 1964, 22, 503–559. [Google Scholar] [CrossRef]
- Hopper, S.; Segal, H.L. Kinetic studies of rat liver glutamicalanine transaminase. J. Boil. Chem. 1962, 237, 3189–3195. [Google Scholar]
- Rofe, A.M.; Edwards, J.B. Oxalate synthesis in isolated rat hepatocytes: The effects of hydroxypyruvate and amino-oxyacetate. Biochem. Med. 1978, 20, 323–335. [Google Scholar] [CrossRef]
- Martinez-Carrion, M.; Jenkins, W.T. D-Alanine-D-glutamate transaminase. II. Inhibitors and the mechanism of transamination of D-amino acids. J. Boil. Chem. 1965, 240, 3547–3552. [Google Scholar]
- DaVanzo, J.; Kang, L.; Ruckart, R.; Daugherty, M. Inhibition of pyridoxal phosphokinase by aminooxyacetic acid. Biochem. Pharmacol. 1966, 15, 124–126. [Google Scholar] [CrossRef]
- Free, C.A.; Majchrowicz, E.; Hess, S.M. Mechanism of inhibition of histidine decarboxylase by rhodanines. Biochem. Pharmacol. 1971, 20, 1421–1428. [Google Scholar] [CrossRef]
- Wilson, O.H.; Holden, J.T. Arginine transport and metabolism in osmotically shocked and unshocked cells of Escherichia coli W. J. Boil. Chem. 1969, 244, 2737–2742. [Google Scholar]
- Braunstein, A.E. 10 Amino Group Transfer. Peptidomics of Cancer-Derived Enzyme Products 1973, 9, 379–481. [Google Scholar] [CrossRef]
- Raunio, R.P.; Lindberg, R.K.; Jenkins, W. Effects of ligands and pH on the reactions of aspartate aminotransferase with aminooxyacetate and hydroxylamine. Arch. Biochem. Biophys. 1984, 233, 43–49. [Google Scholar] [CrossRef]
- Teraoka, T.; Ohta, J.; Abe, T.; Inoue, H.; Ubuka, T. Inhibition of sulfate-forming activity in rat liver mitochondria by (aminooxy)acetate. Amino Acids 1993, 5, 245–251. [Google Scholar] [CrossRef]
- Willetts, A.; Turner, J. L-Threonine acetaldehyde-lyase in a strain of Bacillus subtilis. Biochim. et Biophys. Acta (BBA)Gen. Subj. 1971, 252, 105–110. [Google Scholar] [CrossRef]
- Davies, L.; Johnston, G. Serine hydroxymethyltransferase in the central nervous system: Regional and subcellular distribution studies. Brain Res. 1973, 54, 149–156. [Google Scholar] [CrossRef]
- Jenkins, C.; Rogers, L.; Kerr, M. Inhibition of glycollate metabolism by amino-oxyacetate: Consequences for photosynthesis. Phytochemistry 1983, 22, 19–23. [Google Scholar] [CrossRef]
- Kaczorowski, G.; Shaw, L.; F-Entes, M.; Walsh, C. Coupling of alanine racemase and D-alanine dehydrogenase to active transport of amino acids in Escherichia coli B membrane vesicles. J. Boil. Chem. 1975, 250, 2855–2865. [Google Scholar]
- Akopyan, T.N.; Braunstein, A.E.; Goryachenkova, E.V. Beta-cyanoalanine synthase: Purification and characterization. Proc. Natl. Acad. Sci. USA 1975, 72, 1617–1621. [Google Scholar] [CrossRef] [Green Version]
- Amrhein, N.; Gödeke, K.H.; Kefeli, V. The estimation of relative intracellular phenylalanine ammonia-lyase (PAL)-activities and the modulation in vivo and in vitro by competitive inhibitors. Berichte Deutsch. Bot. Gesellschaft. 1976, 89, 247–259. [Google Scholar]
- Turski, W.A.; Gramsbergen, J.B.P.; Traitler, H.; Schwarcz, R. Rat Brain Slices Produce and Liberate Kynurenic Acid upon Exposure to l-Kynurenine. J. Neurochem. 1989, 52, 1629–1636. [Google Scholar] [CrossRef]
- Mazelis, M.; Miflin, B.; Pratt, H.M. A chloroplast-localized diaminopimelate decarboxylase in higher plants. FEBS Lett. 1976, 64, 197–200. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.W.; Goldberg, A.L. Leucine inhibits oxidation of glucose and pyruvate in skeletal muscles during fasting. J. Boil. Chem. 1978, 253, 3696–3701. [Google Scholar]
- Ohisalo, J.J.; Andersson, S.M.; Pispa, J.P.; Hannah, R.; Sahib, M.K.; Snape, B.M.; A Badawy, A.; Evans, M.; Mukhtar, H.; Murti, C.R.K.; et al. Partial purification and properties of frog liver tyrosine aminotransferase. Biochem. J. 1977, 163, 411–417. [Google Scholar] [CrossRef] [Green Version]
- Paris, C.G.; Magasanik, B. Purification and properties of aromatic amino acid aminotransferase from Klebsiella aerogenes. J. Bacteriol. 1981, 145, 266–271. [Google Scholar] [CrossRef] [Green Version]
- Oppici, E.; Montioli, R.; Dindo, M.; Maccari, L.; Porcari, V.; Lorenzetto, A.; Chellini, S.; Voltattorni, C.; Cellini, B. The Chaperoning Activity of Amino-oxyacetic Acid on Folding-Defective Variants of Human Alanine:Glyoxylate Aminotransferase Causing Primary Hyperoxaluria Type I. ACS Chem. Boil. 2015, 10, 2227–2236. [Google Scholar] [CrossRef]
- Yu, Y.-B.; Adams, D.O.; Yang, S.F. 1-Aminocyclopropanecarboxylate synthase, a key enzyme in ethylene biosynthesis. Arch. Biochem. Biophys. 1979, 198, 280–286. [Google Scholar] [CrossRef]
- Tobkin, H.E.; Mazelis, M. Alliin lyase: Preparation and characterization of the homogeneous enzyme from onion bulbs. Arch. Biochem. Biophys. 1979, 193, 150–157. [Google Scholar] [CrossRef]
- Todorović, B.; Glick, B.R. The interconversion of ACC deaminase and d-cysteine desulfhydrase by directed mutagenesis. Planta 2008, 229, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Tomisawa, H.; Ichimoto, N.; Takanohashi, Y.; Ichihara, S.; Fukazawa, H.; Tateishi, M. Purification and Characterization of Cysteine Conjugate Transaminases from Rat Liver. Xenobiotica 1988, 18, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Reichert, P.; Urban, P.-F. Purification and properties of rat brain cysteine sulfinate decarboxylase (EC 4.1.1.29). Neurochem. Int. 1986, 9, 315–321. [Google Scholar] [CrossRef]
- Griffith, O.W. Cysteinesulfinate metabolism. altered partitioning between transamination and decarboxylation following administration of beta-methyleneaspartate. J. Boil. Chem. 1983, 258, 1591–1598. [Google Scholar]
- Borek, S.; Morkunas, I.; Ratajczak, W.; Ratajczak, L. Metabolism of amino acids in germinating yellow lupin seeds III. Breakdown of arginine in sugar-starved organs cultivated in vitro. Acta Physiol. Plant. 2001, 23, 141–148. [Google Scholar] [CrossRef]
- Dumora, C.; Lacoste, A.; Cassaigne, A. Purification and Properties of 2-Aminoethylphosphonate: Pyruvate Aminotransferase from Pseudomonas aeruginusa. JBIC J. Boil. Inorg. Chem. 1983, 133, 119–125. [Google Scholar] [CrossRef]
- Ilag, L.L.; Jahn, D.; Eggertsson, G.; Söll, D. The Escherichia coli hemL gene encodes glutamate 1-semialdehyde aminotransferase. J. Bacteriol. 1991, 173, 3408–3413. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.-H. Rat brain aspartate ?-decarboxylase. A comparative study with the liver enzyme. Neurochem. Int. 1985, 7, 351–355. [Google Scholar] [CrossRef]
- Lash, L.H.; A Elfarra, A.; Anders, M.W. Renal cysteine conjugate beta-lyase. Bioactivation of nephrotoxic cysteine S-conjugates in mitochondrial outer membrane. J. Boil. Chem. 1986, 261, 5930–5935. [Google Scholar]
- Wu, J.-Y.; Roberts, E. Properties Of Brain L-Glutamate Decarboxylase: Inhibition Studies. J. Neurochem. 1974, 23, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Denner, L.A.; Wei, S.C.; Lin, H.S.; Lin, C.T.; Wu, J.Y. Brain L-glutamate decarboxylase: Purification and subunit structure. Proc. Natl. Acad. Sci. USA 1987, 84, 668–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolosker, H.; Sheth, K.N.; Takahashi, M.; Mothet, J.-P.; Brady, R.O.; Ferris, C.D.; Snyder, S.H. Purification of serine racemase: Biosynthesis of the neuromodulator d-serine. Proc. Natl. Acad. Sci. USA 1999, 96, 721–725. [Google Scholar] [CrossRef] [Green Version]
- Meijer, A.; Van Dam, K. The metabolic significance of anion transport in mitochondria. Biochim. et Biophys. Acta (BBA)Rev. Bioenerg. 1974, 346, 213–244. [Google Scholar] [CrossRef]
- Smith, S.B.; Briggs, S.; Triebwasser, K.C.; A Freedland, R. Re-evaluation of amino-oxyacetate as an inhibitor. Biochem. J. 1977, 162, 453–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Minkler, P.; Grove, D.; Wang, R.; Willard, B.; Dweik, R.; Hine, C. Non-enzymatic hydrogen sulfide production from cysteine in blood is catalyzed by iron and vitamin B 6. Commun. Boil. 2019, 2, 194. [Google Scholar] [CrossRef] [Green Version]
- Bünger, R.; Glanert, S.; Sommer, O.; Gerlach, E. Inhibition by (Aminooxy)acetate of the Malate-Aspartate Cycle in the Isolated Working Guinea Pig Heart. Hoppe-Seyler´s Z. Phys. Chem. 1980, 361, 907–914. [Google Scholar] [CrossRef]
- Ochs, R.S.; Harris, R.A. Aminooxyacetate inhibits gluconeogenesis by isolated chicken hepatocytes. Biochim. et Biophys. Acta (BBA) Gen. Subj. 1980, 632, 260–269. [Google Scholar] [CrossRef]
- Sherry, A.D.; Zhao, P.; Wiethoff, A.J.; Jeffrey, F.M.; Malloy, C.R. Effects of aminooxyacetate on glutamate compartmentation and TCA cycle kinetics in rat hearts. Am. J. Physiol. Content 1998, 274, H591–H599. [Google Scholar] [CrossRef]
- Wejksza, K.; Rzeski, W.; Okuno, E.; Kandefer-Szerszeń, M.; Albrecht, J.; Turski, W.A. Demonstration of Kynurenine Aminotransferases I and II and Characterization of Kynurenic Acid Synthesis in Oligodendrocyte Cell Line (OLN-93). Neurochem. Res. 2005, 30, 963–968. [Google Scholar] [CrossRef]
- Walczak, K.; Dabrowski, W.; Langner, E.; Zgrajka, W.; Piłat, J.; Kocki, T.; Rzeski, W.; Turski, W.A. Kynurenic acid synthesis and kynurenine aminotransferases expression in colon derived normal and cancer cells. Scand. J. Gastroenterol. 2011, 46, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S. (Ed.) Targeting the Broadly Pathogenic Kynurenine Pathway; Springer International Publishing: New York, NY, USA, 2015. [Google Scholar]
- Girard, A.; Sandulesco, G. Betaines and process for preparing the same. U.S. Patent US2045132A, 23 June 1936. [Google Scholar]
- Astwood, E.B.; Bissell, A.; Hughes, A.M. Further studies on the chemical nature of compounds which inhibit the function of the thyroid gland. Endocrinology 1945, 37, 456–481. [Google Scholar] [CrossRef]
- Dieke, S.H. Thiosemicarbazide: A New Toxic Derivative of Thiourea. Exp. Boil. Med. 1949, 70, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Pleasure, H. Psychiatric and Neurological Side-Effects of Isoniazid and Iproniazid. Arch. Neurol. Psychiatry 1954, 72, 313. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.L.; Bain, J. Convulsive Effect of Hydrazides: Relationship to Pyridoxine. Int. Rev. Neurobiol. 1961, 3, 319–348. [Google Scholar] [CrossRef]
- Killam, K.F.; A Bain, J. Convulsant hydrazides. I. In vitro and in vivo inhibition of vitamin B6 enzymes by convulsant hydrazides. J. Pharmacol. Exp. Ther. 1957, 119, 255–262. [Google Scholar] [PubMed]
- Killam, K.F. Convulsant hydrazides. II. Comparison of electrical changes and enzyme inhibition induced by the administration of thiosemicarbazide. J. Pharmacol. Exp. Ther. 1957, 119, 263–271. [Google Scholar]
- Prescott, B.; Kauffmann, G.; James, W.D. Means of increasing the tolerated dose of isoniazid in mice. IV. Certain keto acids. Antibiot. Chemother. (Northfield, Ill.) 1958, 8, 349–353. [Google Scholar]
- Blaschko, H. A haft-century of research on catecholamine biosynthesis. J. Appl. Cardiol. 1987, 2, 171–183. [Google Scholar]
- Brodie, B.; Kuntzman, R.; Hirsch, C.; Costa, E. Effects of decarboxylase inhibition on the biosynthesis of brain monoamines. Life Sci. 1962, 1, 81–84. [Google Scholar] [CrossRef]
- Drain, D.J.; Horlington, M.; Lazare, R.; A Poulter, G. The effect of alpha-methyl DOPA and some other decarboxylase inhibitors on brain 5-hydroxtryptamine. Life Sci. 1962, 1, 93–97. [Google Scholar] [CrossRef]
- Lovenberg, W.; Weissbach, H.; Udenfriend, S. Aromatic L-amino acid decarboxylase. J. Biol. Chem. 1962, 237, 89–93. [Google Scholar] [PubMed]
- Porter, C.; Watson, L.; Titus, D.; Totaro, J.; Byer, S. Inhibition of dopa decarboxylase by the hydrazino analog of α-methyldopa. Biochem. Pharmacol. 1962, 11, 1067–1077. [Google Scholar] [CrossRef]
- Sourkes, T.L. Inhibition of dihydroxy-phenylalanine decarboxylase by derivatives of phenylalanine. Arch. Biochem. Biophys. 1954, 51, 444–456. [Google Scholar] [CrossRef]
- Burkard, W.P.; Gey, K.F.; Pletscher, A. A new inhibitor of decarboxylase of aromatic amino acids. Cell. Mol. Life Sci. 1962, 18, 411–412. [Google Scholar] [CrossRef]
- Burkard, W.; Gey, K.; Pletscher, A. Inhibition of decarboxylase of aromatic amino acids by 2,3,4-trihydroxybenzylhydrazine and its seryl derivative. Arch. Biochem. Biophys. 1964, 107, 187–196. [Google Scholar] [CrossRef]
- Bartholini, G.; Burkard, W.P.; Pletscher, A.; Bates, H.M. Increase of Cerebral Catecholamines caused by 3,4-Dihydroxyphenylalanine after Inhibition of Peripheral Decarboxylase. Nature 1967, 215, 852–853. [Google Scholar] [CrossRef]
- Tissot, R.; Bartholini, G.; Pletscher, A. Drug-Induced Changes of Extracerebral Dopa Metabolism in Man. Arch. Neurol. 1969, 20, 187–190. [Google Scholar] [CrossRef]
- Barbeau, A.; Joffroy, L.G.; Mars, H. Treatment of Parkinson’s disease with levodopa and Ro 4-Clin. Pharmacol. Ther. 1971, 12, 353–359. [Google Scholar] [CrossRef]
- Rinne, U.; Birket-Smith, E.; Dupont, E.; Hansen, E.; Hyyppä, M.; Marttila, R.; Mikkelsen, B.; Pakkenberg, H.; Presthus, J. Levodopa alone and in combination with a peripheral decarboxylase inhibitor benserazide (Madopar®) in the treatment of Parkinson’s disease. J. Neurol. 1975, 211, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.; Fischer, P.-A.; Jacobi, P.; Maxion, H. Wirkungsvergleich von l-dopa und der kombination l-dopa+ decarboxylasehemmer beim Parkinson-syndrom. Archiv Psychiatr. Nervenkrankh. 1973, 217, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Kuhn, W. Homocysteine levels after acute levodopa intake in patients with Parkinson’s disease. Mov. Disord. 2009, 24, 1339–1343. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Xu, S.; Cao, L.-D.; Wu, Q.-Y. The effect of levodopa benserazide hydrochloride on homocysteinemia levels in patients with Parkinson’s disease and treatment of hyperhomocysteinemia. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2409–2412. [Google Scholar] [PubMed]
- Schwartz, D.E.; Jordan, J.C.; Ziegler, W.H. Pharmacokinetics of the decarboxylase inhibitor benserazide in man; its tissue distribution in the rat. Eur. J. Clin. Pharmacol. 1974, 7, 39–45. [Google Scholar] [CrossRef]
- E Schwartz, D.; Brandt, R. Pharmacokinetic and metabolic studies of the decarboxylase inhibitor benserazide in animals and man. Arzneimittelforschung 1978, 28, 302–307. [Google Scholar]
- Voltattorni, C.B.; Minelli, A.; Borri, P. The interaction of 2,3,4-trihydroxybenzylhydrazine with dopa decarboxylase from pig kidney. Life Sci. 1981, 28, 103–108. [Google Scholar] [CrossRef]
- Burkhard, P.; Dominici, P.; Borri-Voltattorni, C.; Jansonius, J.N.; Malashkevich, V.N. Structural insight into Parkinson’s disease treatment from drug-inhibited DOPA decarboxylase. Nat. Struct. Biol. 2001, 8, 963–967. [Google Scholar] [CrossRef]
- Treseder, S.A.; Rose, S.; Jenner, P. The central aromatic amino acid DOPA decarboxylase inhibitor, NSD-1015, does not inhibit L-DOPA-induced circling in unilateral 6-OHDA-lesioned-rats. Eur. J. Neurosci. 2001, 13, 162–170. [Google Scholar] [CrossRef]
- Grodzki, M. Ueber äthylirte Sulfoharnstoffe. Berichte Deutsch. Chem. Gesellschaft. 1881, 14, 2754–2758. [Google Scholar] [CrossRef] [Green Version]
- Twiss, D.; Bazier, S.; Thomas, F. The dithiocarbamate accelerators of vulcanization. J. Soc. Chem. Ind. 1922, 41, 81T–88T. [Google Scholar]
- Williams, E. Effects of alcohol on workers with carbon disulfide. JAMA 1937, 109, 1472–1473. [Google Scholar]
- Hald, J.; Jacobsen, E. A drug sensitizing the organism to ethyl alcohol. Lancet 1948, 2, 1001–1004. [Google Scholar] [CrossRef]
- Kragh, H. From disulfiram to antabuse: The invention of a drug. Bull. Hist. Chem. 2008, 33, 82–88. [Google Scholar]
- Koppaka, V.; Thompson, D.C.; Chen, Y.; Ellermann, M.; Nicolaou, K.C.; Juvonen, R.O.; Petersen, D.; Deitrich, R.A.; Hurley, T.D.; Vasiliou, V. Aldehyde dehydrogenase inhibitors: A comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol. Rev. 2012, 64, 520–539. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; McLeod, H.L.; Cassidy, J. Disulfiram-mediated inhibition of NF-κB activity enhances cytotoxicity of 5-fluorouracil in human colorectal cancer cell lines. Int. J. Cancer. 2003, 104, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Cui, Q.C.; Yang, H.; Dou, Q.P. Disulfiram, a Clinically Used Anti-Alcoholism Drug and Copper-Binding Agent, Induces Apoptotic Cell Death in Breast Cancer Cultures and Xenografts via Inhibition of the Proteasome Activity. Cancer Res. 2006, 66, 10425–10433. [Google Scholar] [CrossRef] [Green Version]
- Iljin, K.; Ketola, K.; Vainio, P.; Halonen, P.; Kohonen, P.; Fey, V.; Kallioniemi, O.-P.; Grafström, R.; Perälä, M. High-Throughput Cell-Based Screening of 4910 Known Drugs and Drug-like Small Molecules Identifies Disulfiram as an Inhibitor of Prostate Cancer Cell Growth. Clin. Cancer Res. 2009, 15, 6070–6078. [Google Scholar] [CrossRef] [Green Version]
- Safi, R.; Nelson, E.; Chitneni, S.K.; Franz, K.J.; George, D.J.; Zalutsky, M.R.; McDonnell, D.P. Copper signaling axis as a target for prostate cancer therapeutics. Cancer Res. 2014, 74, 5819–5831. [Google Scholar] [CrossRef] [Green Version]
- Zha, J.; Chen, F.; Dong, H.; Shi, P.; Yao, Y.; Xu, B.; Li, R.; Wang, S.; Li, P.; Wang, W.; et al. Disulfiram targeting lymphoid malignant cell lines via ROS-JNK activation as well as Nrf2 and NF-kB pathway inhibition. J. Transl. Med. 2014, 12, 163. [Google Scholar] [CrossRef] [Green Version]
- Papaioannou, M.; Mylonas, I.; Kast, R.E.; Brüning, A. Disulfiram/copper causes redox-related proteotoxicity and concomitant heat shock response in ovarian cancer cells that is augmented by auranofin-mediated thioredoxin inhibition. Oncoscience 2013, 1, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, J.; Wang, Y.; Zhang, X.; Zhang, N.; Liu, L.; Soukup, K.; Michelakos, T.; Hong, T.; DeLeo, A.; Cai, L.; et al. A novel chemoradiation targeting stem and nonstem pancreatic cancer cells by repurposing disulfiram. Cancer Lett. 2017, 409, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Skrott, Z.; Mistrik, M.; Andersen, K.K.; Friis, S.; Majera, D.; Gursky, J.; Oždian, T.; Bartkova, J.; Turi, Z.; Moudry, P.; et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL. Nature 2017, 552, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Lossen, W. Ueber das Hydroxylamin. J. Prakt. Chem. 1865, 96, 462–465. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, H.E.; Kipping, F.S. III.—Studies of the terpenes and allied compounds. The formation of ketones by the interaction of camphor and agents such as sulphuric acid and zinc chloride. J. Chem. Soc., Trans. 1893, 63, 75–99. [Google Scholar] [CrossRef] [Green Version]
- Kipping, F.S. Action of phosphoric anhydride on fatty acids. Part I. J. Chem. Soc. Transact. 1890, 57, 532–540. [Google Scholar] [CrossRef] [Green Version]
- Gaffron, H. REDUCTION OF CARBON DIOXIDE COUPLED WITH THE OXYHYDROGEN REACTION IN ALGAE. J. Gen. Physiol. 1942, 26, 241–267. [Google Scholar] [CrossRef] [Green Version]
- Sevag, M.; Shelburne, M. The respiration of Streptococcus pyogenes: II. The inhibition of respiration and growth by sulfanilamide; the inhibition of respiration by hydroxylamine and its sulfonamide and other derivatives. J. Bacteriol. 1942, 43, 421. [Google Scholar]
- Blaschko, H. The mechanism of catalase inhibitions. Biochem. J. 1935, 29, 2303–2312. [Google Scholar] [CrossRef] [Green Version]
- Baxter, C.F.; Roberts, E. Elevation of gamma-aminobutyric acid in rat brain with hydroxylamine. Proc. Soc. Exp. Boil. Med. 1959, 101, 811–815. [Google Scholar] [CrossRef]
- Nakamura, M.; Horie, Y. Partial Purification and Properties of Alanine and Aspartate Aminotransferases in the Midgut Tissue of the Silkworm, Bombyx mori (Lepidoptera: Bombycidae). Appl. Èntomol. Zoōl. 1986, 21, 236–243. [Google Scholar] [CrossRef] [Green Version]
- I Nakada, H. Glutamic-Glycine Transaminase from Rat Liver. J. Boil. Chem. 1964, 239, 468–471. [Google Scholar]
- Asada, Y.; Sawa, Y.; Tanizawa, K.; Soda, K. Purification and Characterization of Yeast L-Kynurenine Aminotransferase with Broad Substrate Specificity. J. Biochem. 1986, 99, 1101–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ames, B.N.; Horecker, B.L. The biosynthesis of histidine: Imidazoleacetol phosphate transaminase. J. Boil. Chem. 1956, 220, 113–128. [Google Scholar]
- Kito, K.; Sanada, Y.; Katunuma, N. Mode of Inhibition of Ornithine Aminotransferase by L-Canaline. J. Biochem. 1978, 83, 201–206. [Google Scholar] [CrossRef]
- Iversen, H.; Gustafsson, L.; Leone, A.; Wiklund, N. Smooth muscle relaxing effects of NO, nitrosothiols and a nerve-induced relaxing factor released in guinea-pig colon. Br. J. Pharmacol. 1994, 113, 1088–1092. [Google Scholar] [CrossRef] [Green Version]
- Correia, N.A.; Oliveira, R.B.; Ballejo, G. Pharmacological profile of nitrergic nerve-, nitric oxide-, nitrosoglutathione- and hydroxylamine-induced relaxations of the rat duodenum. Life Sci. 2000, 68, 709–717. [Google Scholar] [CrossRef]
- Izato, Y.-I.; Koshi, M.; Miyake, A. Initial Decomposition Pathways of Aqueous Hydroxylamine Solutions. J. Phys. Chem. B 2017, 121, 4502–4511. [Google Scholar] [CrossRef]
- Han, Y.; Qin, J.; Chang, X.; Yang, Z.; Tang, X.; Du, J. Hydrogen sulfide may improve the hippocampal damage induced by recurrent febrile seizures in rats. Biochem. Biophys. Res. Commun. 2005, 327, 431–436. [Google Scholar] [CrossRef]
- Xu, G.Y.; Winston, J.H.; Shenoy, M.; Zhou, S.; Chen, J.D.; Pasricha, P.J. The endogenous hydrogen sulfide producing enzyme cystathionine-beta synthase contributes to visceral hypersensitivity in a rat model of irritable bowel syndrome. Mol. Pain. 2009, 5, 44. [Google Scholar] [CrossRef] [Green Version]
- Roa-Coria, J.E.; Farias, J.P.; Barragan-Iglesias, P.; Quiñonez-Bastidas, G.N.; Zúñiga-Romero, Á.; Huerta-Cruz, J.C.; Reyes-García, J.; Flores-Murrieta, F.J.; Granados-Soto, V.; Rocha-González, H.I. Possible involvement of peripheral TRP channels in the hydrogen sulfide-induced hyperalgesia in diabetic rats. BMC Neurosci. 2019, 20, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cudkowicz, G. Effect of hydroxylamine and sodium azide on the growth of various transplantable tumors. Tumori J. 1955, 41, 181–185. [Google Scholar] [CrossRef]
- Tarnowski, G.S.; Kreis, W.; A Schmid, F.; Cappuccino, J.G.; Burchenal, J.H. Effect of hydroxylamine (NSC-26250) and related compounds on growth of transplanted animal tumors. Cancer Res. 1966, 26, 1279–1301. [Google Scholar] [PubMed]
- Stenbäck, F.; Weisburger, J.; Williams, G.M. Hydroxylamine effects on cryptogenic neoplasm development in C3H mice. Cancer Lett. 1987, 38, 73–85. [Google Scholar] [CrossRef]
- Underwood, E. Trace Elements in Human and Animal Nutrition; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Liu, J.; Chakraborty, S.; Hosseinzadeh, P.; Yü, Y.; Tian, S.; Petrík, I.; Bhagi, A.; Lu, Y. Metalloproteins Containing Cytochrome, Iron–Sulfur, or Copper Redox Centers. Chem. Rev. 2014, 114, 4366–4469. [Google Scholar] [CrossRef]
- Letelier, M.E.; Lepe, A.M.; Faundez, M.; Salazar, J.; Marín, R.; Aracena, P.; Speisky, H. Possible mechanisms underlying copper-induced damage in biological membranes leading to cellular toxicity. Chem. Interactions 2005, 151, 71–82. [Google Scholar] [CrossRef]
- Krajewska, B. Mono- (Ag, Hg) and di- (Cu, Hg) valent metal ions effects on the activity of jack bean urease. Probing the modes of metal binding to the enzyme. J. Enzym. Inhib. Med. Chem. 2008, 23, 535–542. [Google Scholar] [CrossRef]
- Matsuo, Y.; Greenberg, D.M. A crystalline enzyme that cleaves homoserine and cystathionine. J. Biol. Chem. 1958, 230, 545–560. [Google Scholar]
- Bar-Or, D.; Rael, L.; Thomas, G.; Kraus, J. Inhibitory Effect of Copper on Cystathionine β-Synthase Activity: Protective Effect of an Analog of the Human Albumin N-Terminus. Protein Pept. Lett. 2005, 12, 271–273. [Google Scholar] [CrossRef]
- Szabo, C. A timeline of hydrogen sulfide (H2S) research: From environmental toxin to biological mediator. Biochem. Pharmacol. 2018, 149, 5–19. [Google Scholar] [CrossRef]
- DeNoyer, D.; Masaldan, S.; La Fontaine, S.; Cater, M.A. Targeting copper in cancer therapy: ‘Copper That Cancer’. Metallomics 2015, 7, 1459–1476. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-F.; Orvig, C.; Liang, H. Multi-Target Metal-Based Anticancer Agents. Curr. Top. Med. Chem. 2017, 17, 3131–3145. [Google Scholar] [CrossRef] [PubMed]
- Denoyer, D.; Clatworthy, S.A.S.; Cater, M.A. Copper complexes in cancer therapy. Met. Ions Life Sci. 2018, 18. [Google Scholar]
- Katarzyna, M.; Anna, S.; Zielinska-Blizniewska, H.; Ireneusz, M.; Szczepanska, A.K.; Malinowska, K.; Majsterek, I. An Evaluation of the Antioxidant and Anticancer Properties of Complex Compounds of Copper (II), Platinum (II), Palladium (II) and Ruthenium (III) for Use in Cancer Therapy. Mini-Reviews Med. Chem. 2018, 18, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- McMahon, A.; Chen, W.; Li, F. Old wine in new bottles: Advanced drug delivery systems for disulfiram-based cancer therapy. J. Control. Release 2020, 319, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Leow, P.-C.; Tian, Q.; Ong, Z.Y.; Yang, Z.; Ee, P.L.R. Antitumor activity of natural compounds, curcumin and PKF118-310, as Wnt/β-catenin antagonists against human osteosarcoma cells. Investig. New Drugs 2009, 28, 766–782. [Google Scholar] [CrossRef]
- Antony, L.; Van Der Schoor, F.; Dalrymple, S.L.; Isaacs, J.T. Androgen receptor (AR) suppresses normal human prostate epithelial cell proliferation via AR/β-catenin/TCF-4 complex inhibition ofc-MYCtranscription. Prostate 2014, 74, 1118–1131. [Google Scholar] [CrossRef] [Green Version]
- Paluszczak, J.; Kleszcz, R.; Studzińska-Sroka, E.; Krajka-Kuźniak, V. Lichen-derived caperatic acid and physodic acid inhibit Wnt signaling in colorectal cancer cells. Mol. Cell. Biochem. 2017, 441, 109–124. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Zhong, L.; Hendriks, J.; Post, J.N.; Karperien, M. The Effects of the WNT-Signaling Modulators BIO and PKF118-310 on the Chondrogenic Differentiation of Human Mesenchymal Stem Cells. Int. J. Mol. Sci. 2018, 19, 561. [Google Scholar] [CrossRef] [Green Version]
- Choi, G.; Lee, J.; Ji, J.Y.; Woo, J.; Kang, N.S.; Cho, S.Y.; Kim, H.R.; Ha, J.D.; Han, S.Y. Discovery of a potent small molecule SIRT1/2 inhibitor with anticancer effects. Int. J. Oncol. 2013, 43, 1205–1211. [Google Scholar] [CrossRef] [Green Version]
- Franci, G.; Sarno, F.; Nebbioso, A.; Altucci, L. Identification and characterization of PKF118-310 as a KDM4A inhibitor. Epigenetics 2016, 12, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Van Veen, A.; Mertens, W. On the isolation of a toxic bacterial pigment (provisional communication). Proc. Konink. Akad. Wetensch. (Amsterdam). 1933, 36, 666–670. [Google Scholar]
- Ham, J.H.; Melanson, R.A.; Rush, M.C. Burkholderia glumae: Next major pathogen of rice? Mol. Plant. Pathol. 2011, 12, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, P.A.; Johannes, A.G.; Cox, H.C.; Berends, W. On toxoflavin, the yellow poison of pseudomonas cocovenenans. Recl. des Trav. Chim. des Pays-bas 2010, 79, 255–267. [Google Scholar] [CrossRef]
- A Machlowitz, R.; Fisher, W.P.; McKay, B.S.; A Tytell, A.; Charney, J. Xanthothricin, a new antibiotic. Antibiot. Chemother. (Northfield, Ill.) 1954, 4, 259–261. [Google Scholar]
- Latuasan, H.; Berends, W. On the origin of the toxicity of toxoflavin. Biochim. et Biophys. Acta (BBA) Bioenerg. 1961, 52, 502–508. [Google Scholar] [CrossRef]
- Akimenko, V.K.; Golovchenko, N.; Medentsev, A.G.; Esipov, S. Effect of xanthothricin on the respiratory chain. FEBS Lett. 1974, 46, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Niu, W.; Wu, P.; Chen, F.; Wang, J.; Shang, X.; Xu, C. Discovery of selective cystathionine β-synthase inhibitors by high-throughput screening with a fluorescent thiol probe. MedChemComm 2016, 8, 198–201. [Google Scholar] [CrossRef]
- Nunome, S.; Ishiyama, A.; Kobayashi, M.; Otoguro, K.; Kiyohara, H.; Yamada, H.; Omura, S. In Vitro Antimalarial Activity of Biflavonoids from Wikstroemia indica. Planta Medica 2004, 70, 76–78. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.-Y.; Unehara, T.; Kitanaka, S. Anti-inflammatory activity of new guaiane type sesquiterpene from Wikstroemia indica. Chem. Pharm. Bull. 2005, 53, 137–139. [Google Scholar] [CrossRef] [Green Version]
- Aitken, S.M.; Brenner, S.E. Kinetics of the Yeast Cystathionine β-Synthase Forward and Reverse Reactions: Continuous Assays and the Equilibrium Constant for the Reaction†. Biochemistry 2003, 42, 571–578. [Google Scholar] [CrossRef] [PubMed]
- McCune, C.D.; Chan, S.J.; Beio, M.L.; Shen, W.; Chung, W.J.; Szczesniak, L.M.; Chai, C.; Koh, S.Q.; Wong, P.T.-H.; Berkowitz, D.B. “Zipped Synthesis” by Cross-Metathesis Provides a Cystathionine β-Synthase Inhibitor that Attenuates Cellular H2S Levels and Reduces Neuronal Infarction in a Rat Ischemic Stroke Model. ACS Central Sci. 2016, 2, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Bauvy, C.; Codogno, P. Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. J. Biol. Chem. 2003, 278, 16667–16674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Wei, W.; Chai, H.; Xie, X. Aurintricarboxylic Acid Ameliorates Experimental Autoimmune Encephalomyelitis by Blocking Chemokine-Mediated Pathogenic Cell Migration and Infiltration. J. Immunol. 2012, 190, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laufenberg, L.J.; Kazi, A.A.; Lang, C.H. Salutary effect of aurintricarboxylic acid on endotoxin- and sepsis-induced changes in muscle protein synthesis and inflammation. Shock 2014, 41, 420–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Wang, L.; Han, X.; Zhou, Y.; Zhang, T.; Wang, L.; Hong, T.; Zhang, W.; Guo, X.-X.; Sun, J.; et al. Discovery of a Bioactive Inhibitor with a New Scaffold for Cystathionine γ-Lyase. J. Med. Chem. 2018, 62, 1677–1683. [Google Scholar] [CrossRef]
- Thorson, M.K.; Van Wagoner, R.M.; Harper, M.K.; Ireland, C.M.; Majtan, T.; Kraus, J.P.; Barrios, A.M. Marine natural products as inhibitors of cystathionine beta-synthase activity. Bioorganic Med. Chem. Lett. 2015, 25, 1064–1066. [Google Scholar] [CrossRef]
- Hanaoka, K.; Sasakura, K.; Suwanai, Y.; Toma-Fukai, S.; Shimamoto, K.; Takano, Y.; Shibuya, N.; Terai, T.; Komatsu, T.; Ueno, T.; et al. Discovery and Mechanistic Characterization of Selective Inhibitors of H2S-producing Enzyme: 3-Mercaptopyruvate Sulfurtransferase (3MST) Targeting Active-site Cysteine Persulfide. Sci. Rep. 2017, 7, 40227. [Google Scholar] [CrossRef]
- Kožich, V.; Majtan, T. A key leader in homocystinuria research: Jan P. Kraus (1942–2019). Hum. Mutat. 2019, 40, 1909. [Google Scholar] [CrossRef] [Green Version]
- Fowler, B.; Kožich, V. Jan Kraus PhD., Professor of Pediatrics at the University of Colorado Anschutz Medical Campus, Aurora, CO, USA (b London, UK, June 5, 1942; q 1965 Faculty of Science Prague, d July 4, 2019, CO, USA). J. Inherit. Metab. Dis. 2020, 43, 379–380. [Google Scholar] [CrossRef]













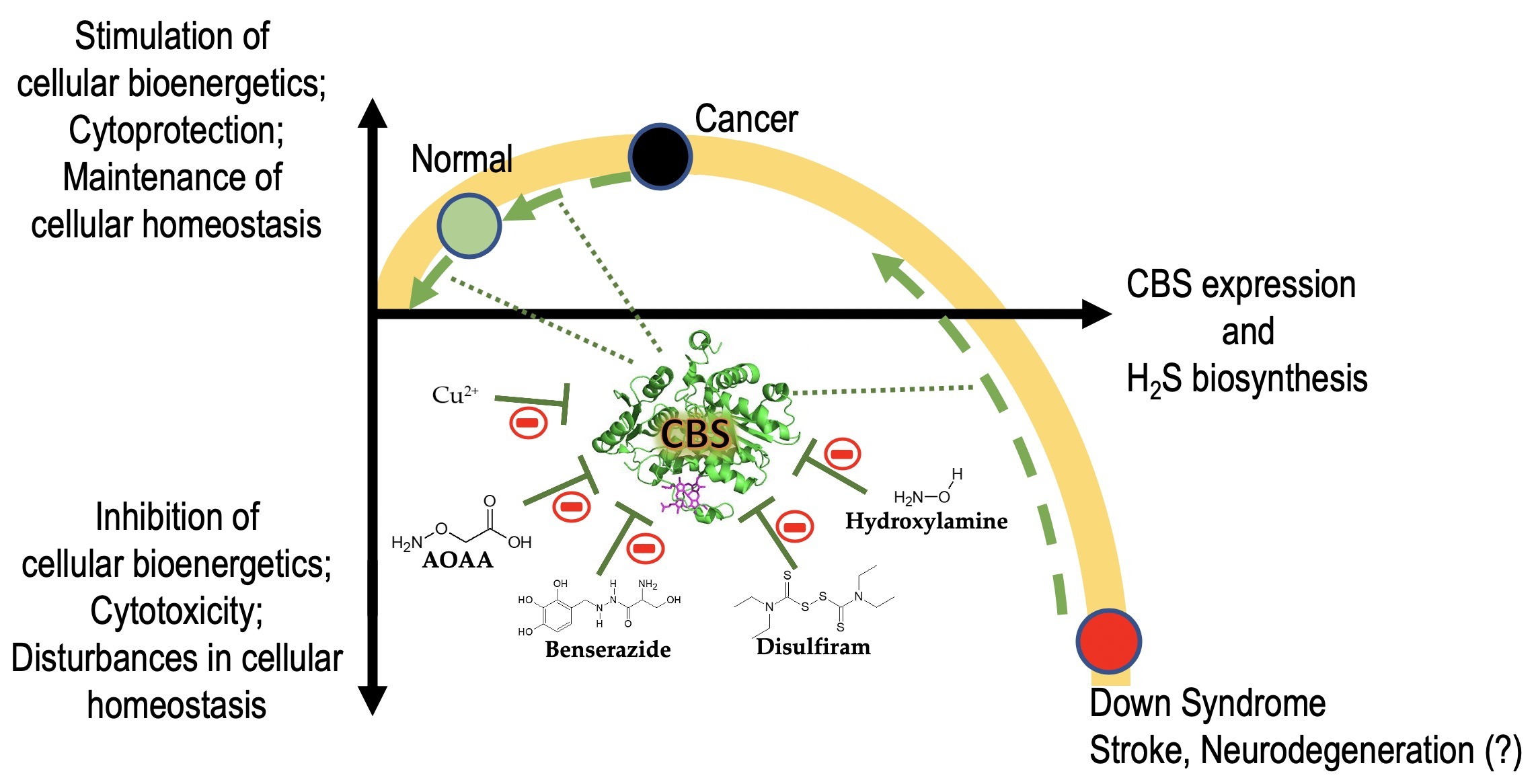{kind=link}
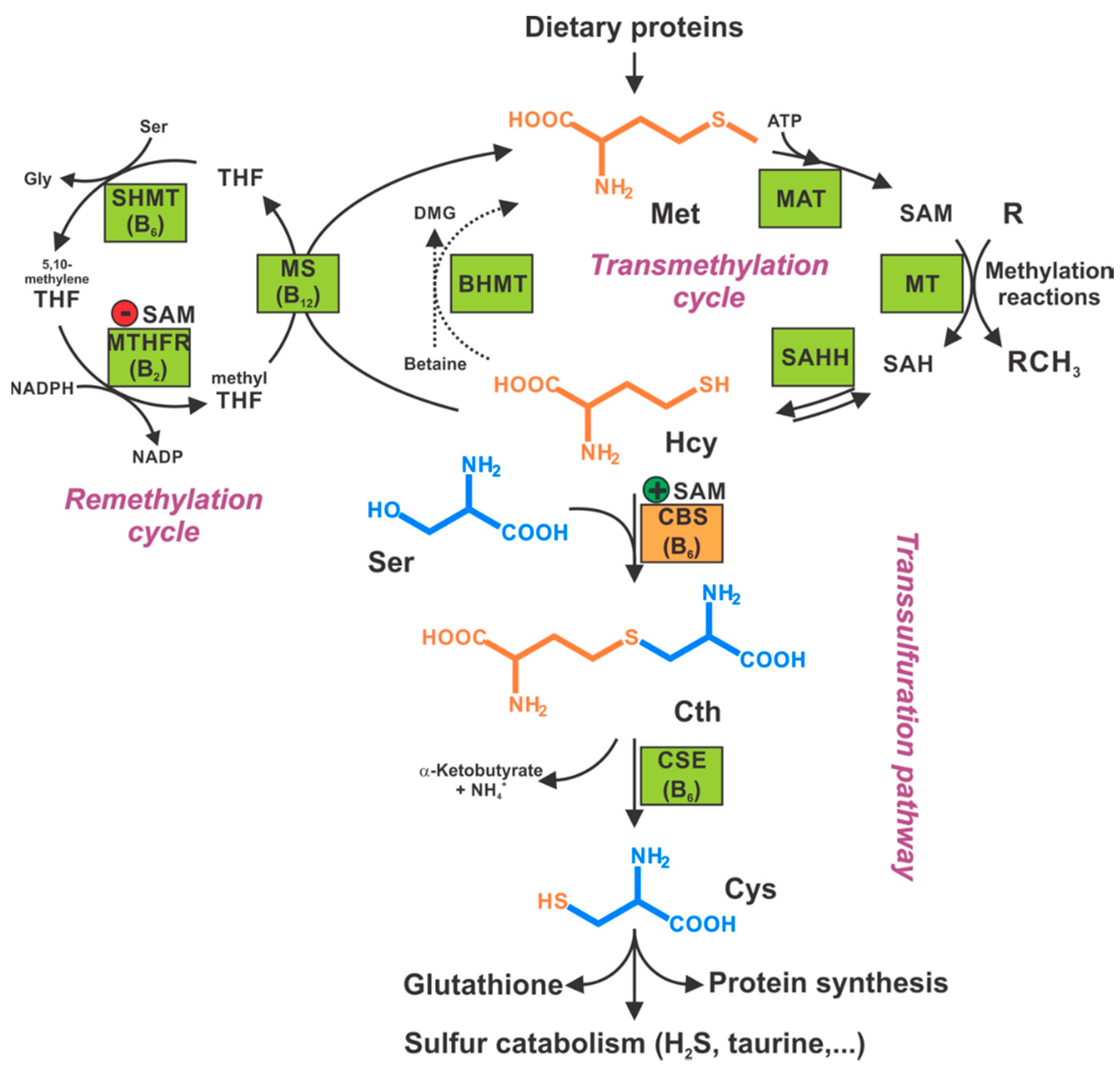{kind=link}
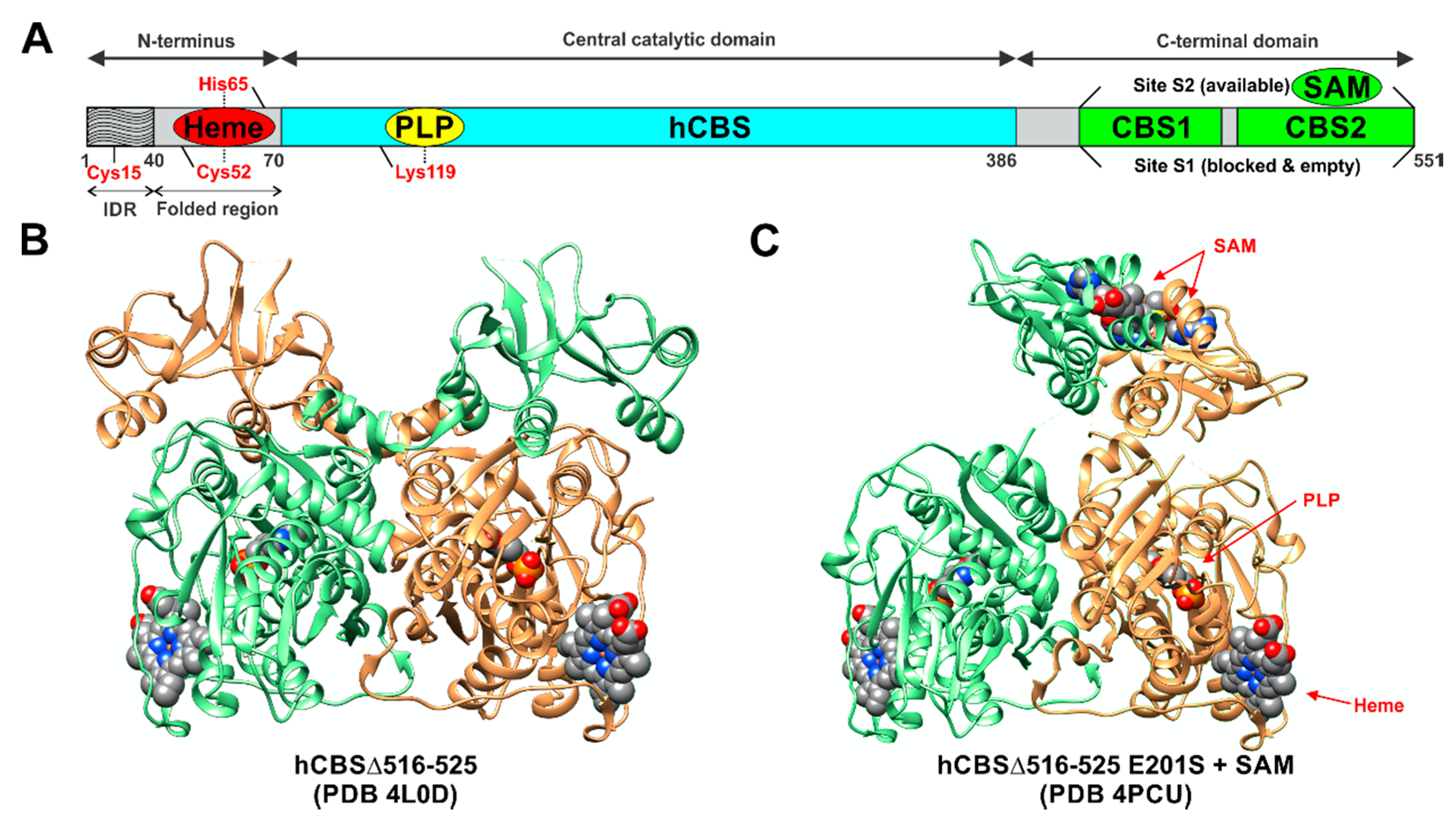{kind=link}
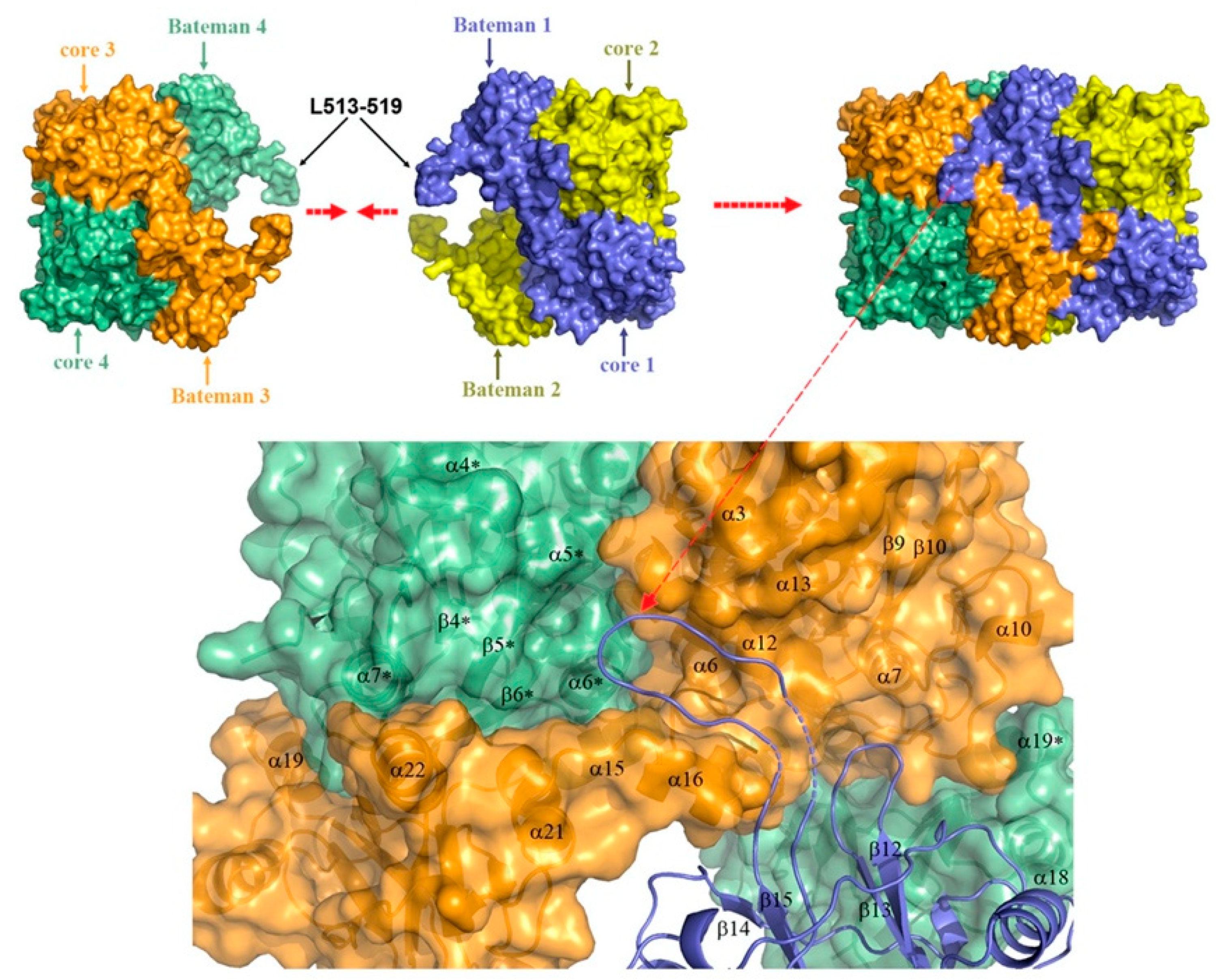{kind=link}
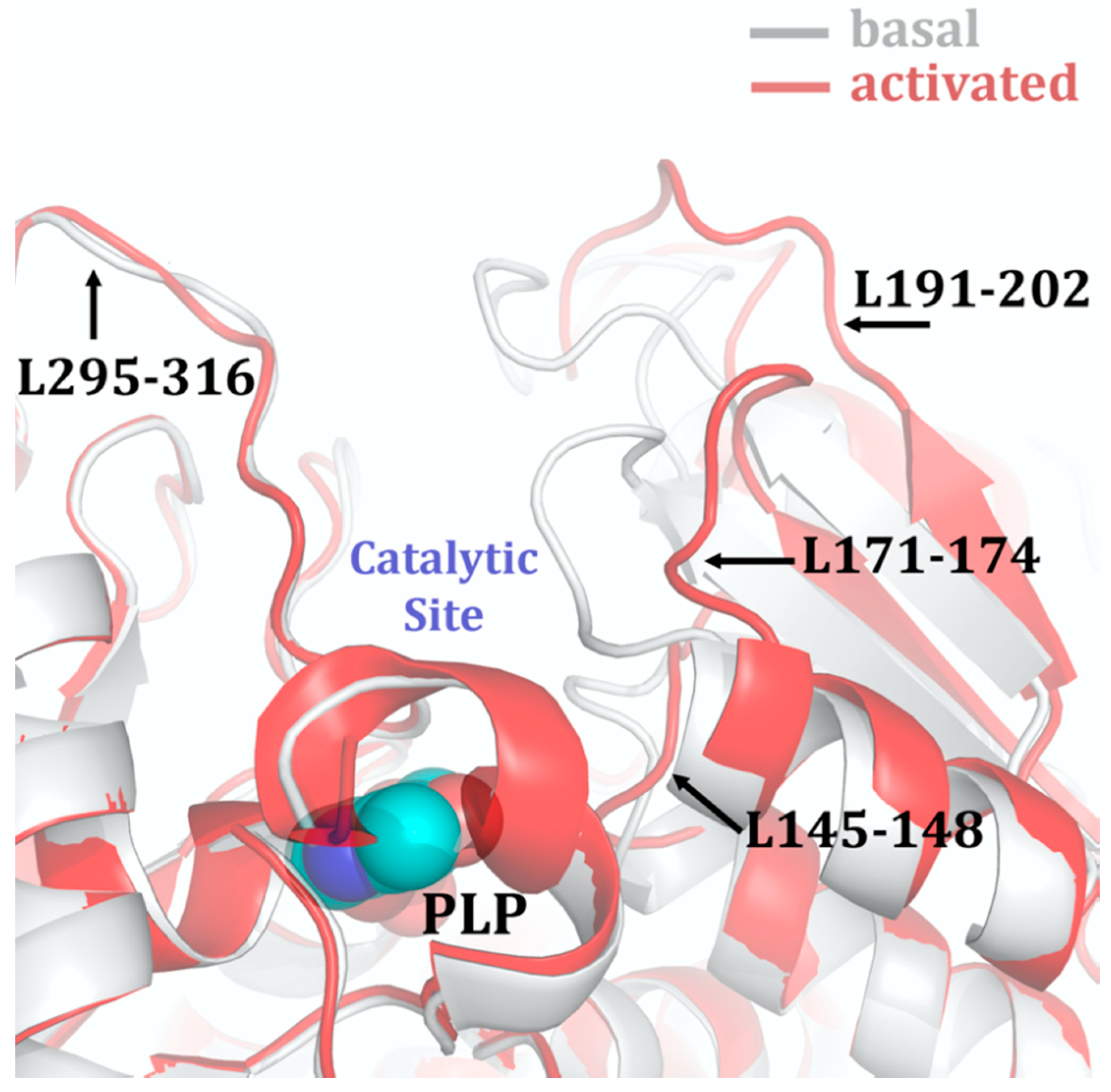{kind=link}
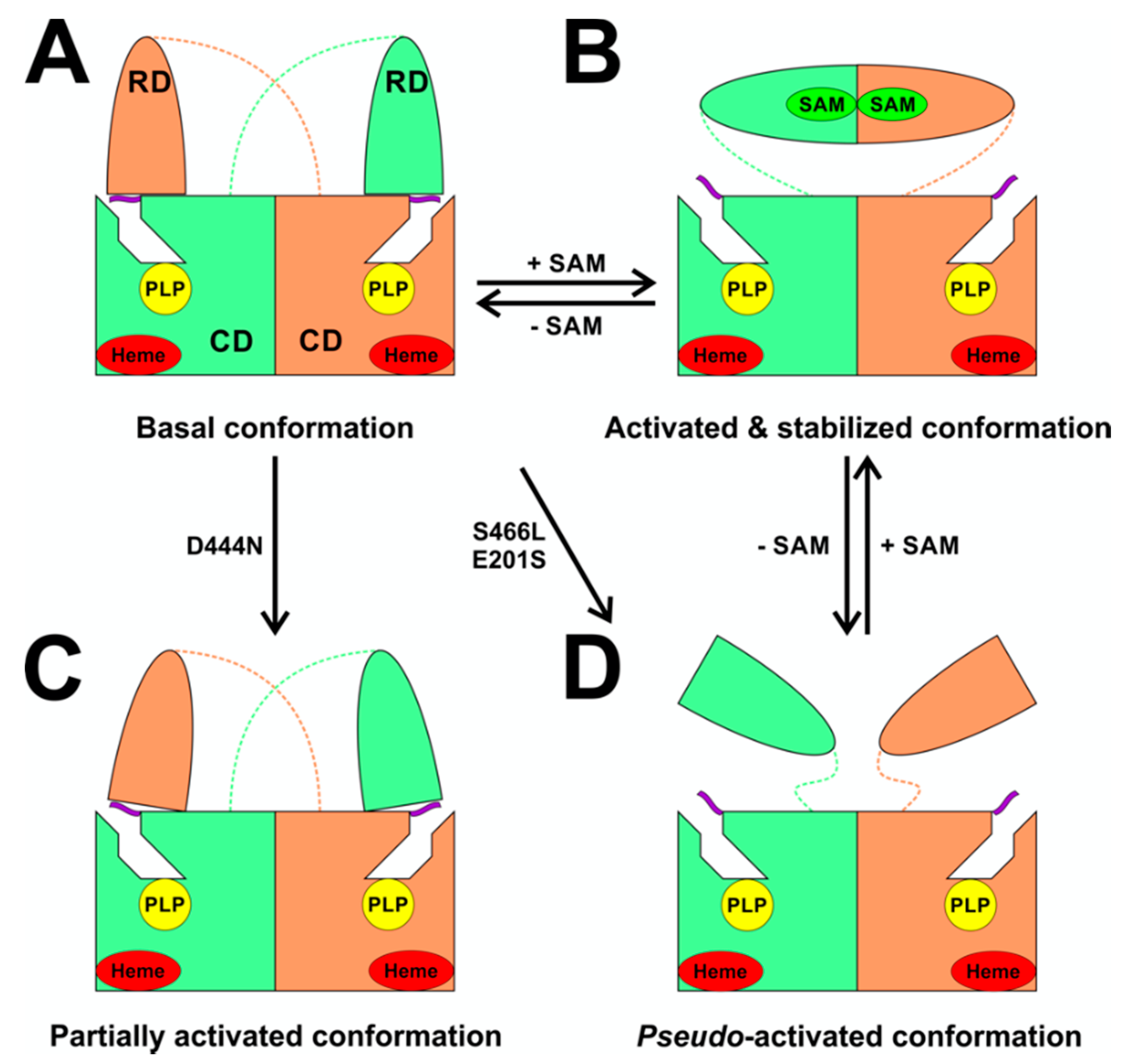{kind=link}
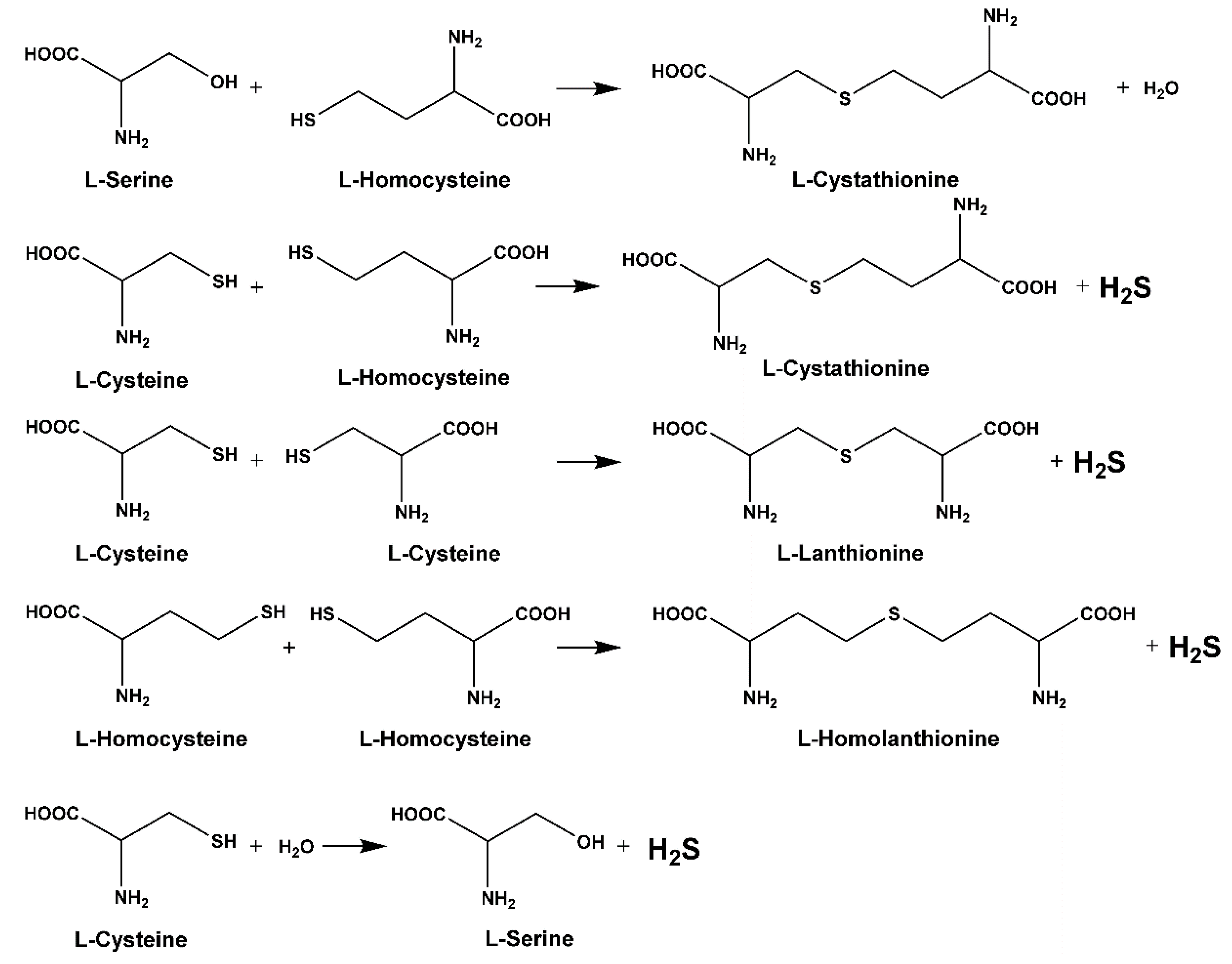{kind=link}
{kind=link}
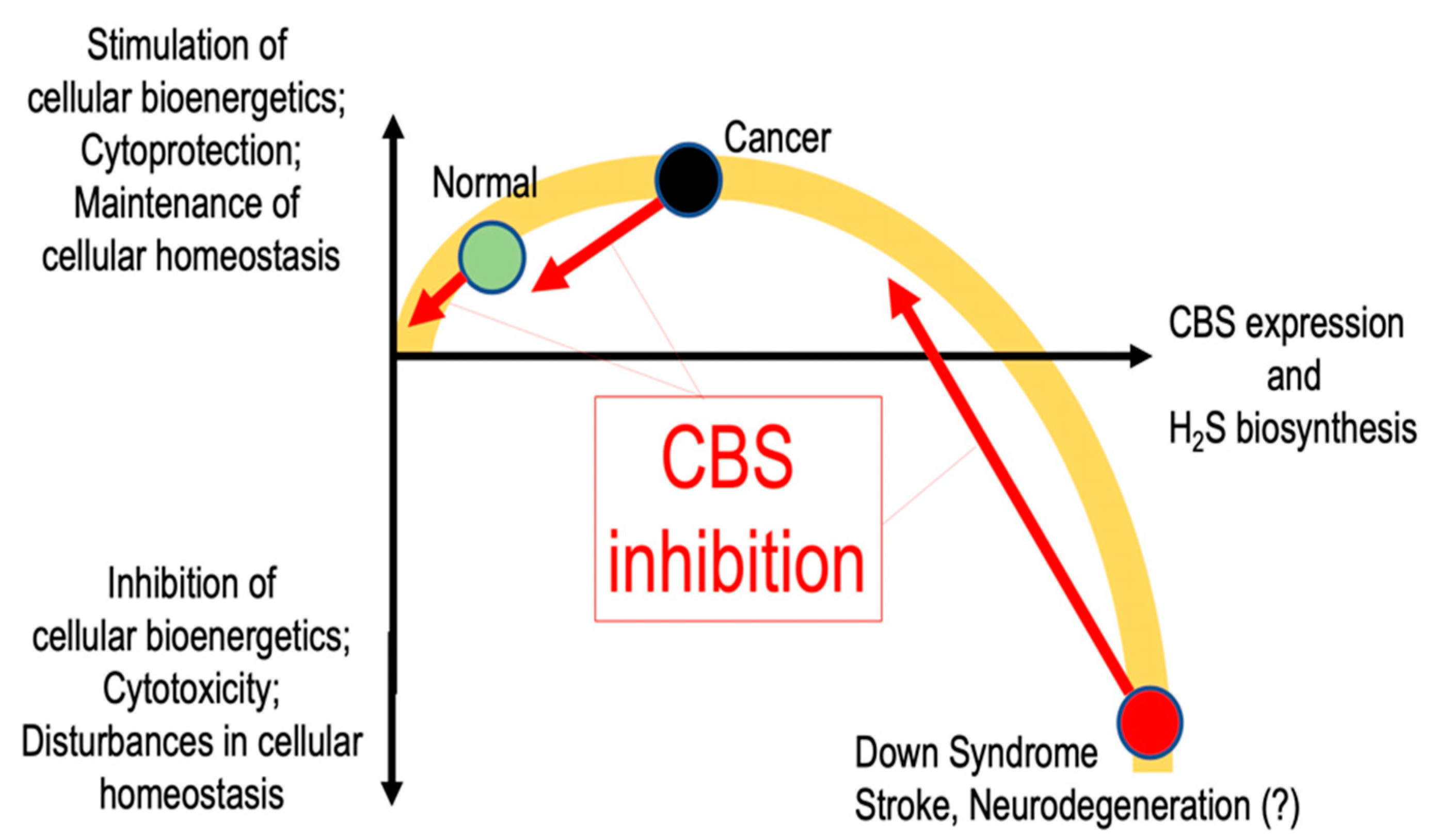{kind=link}
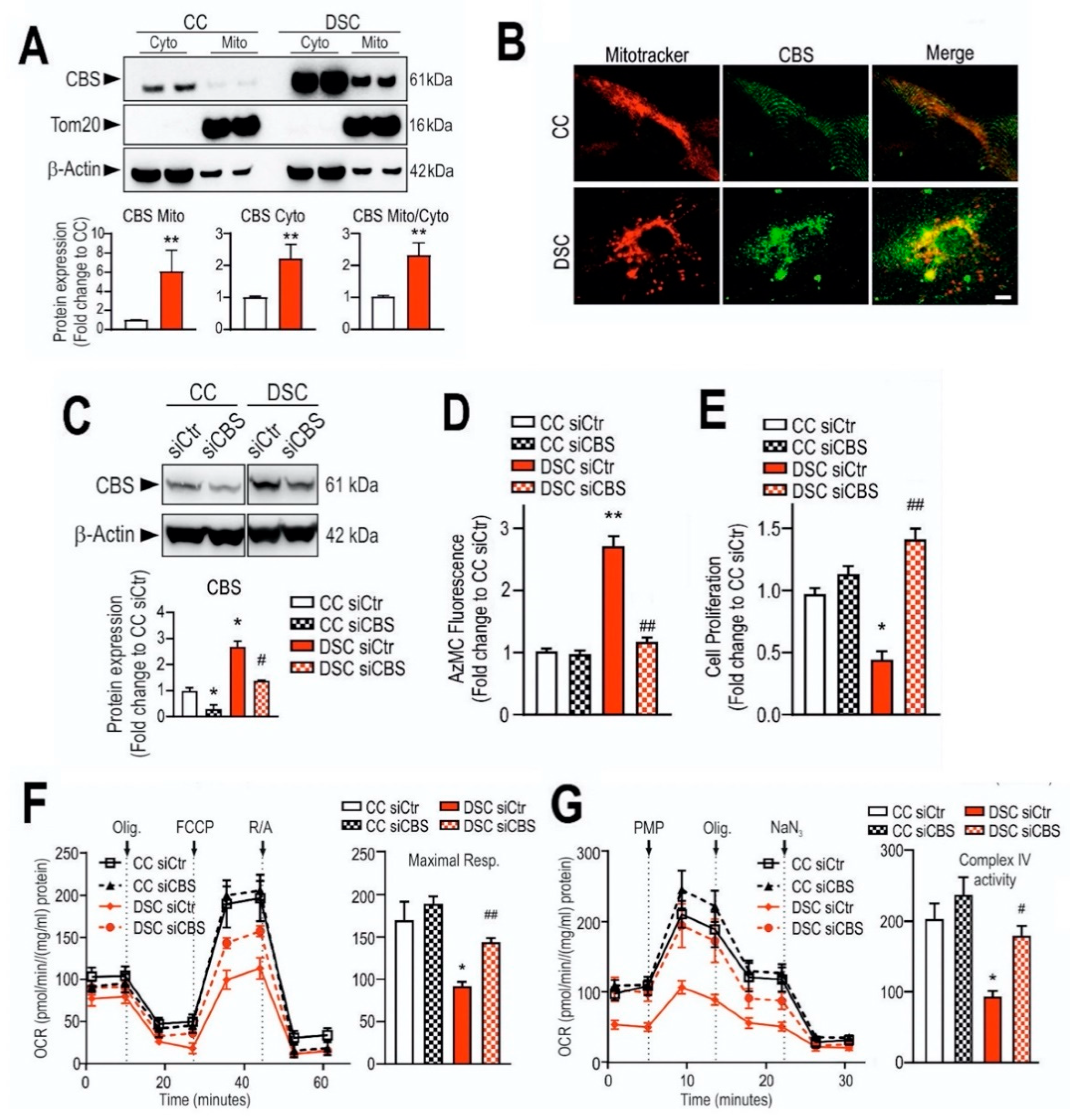{kind=link}
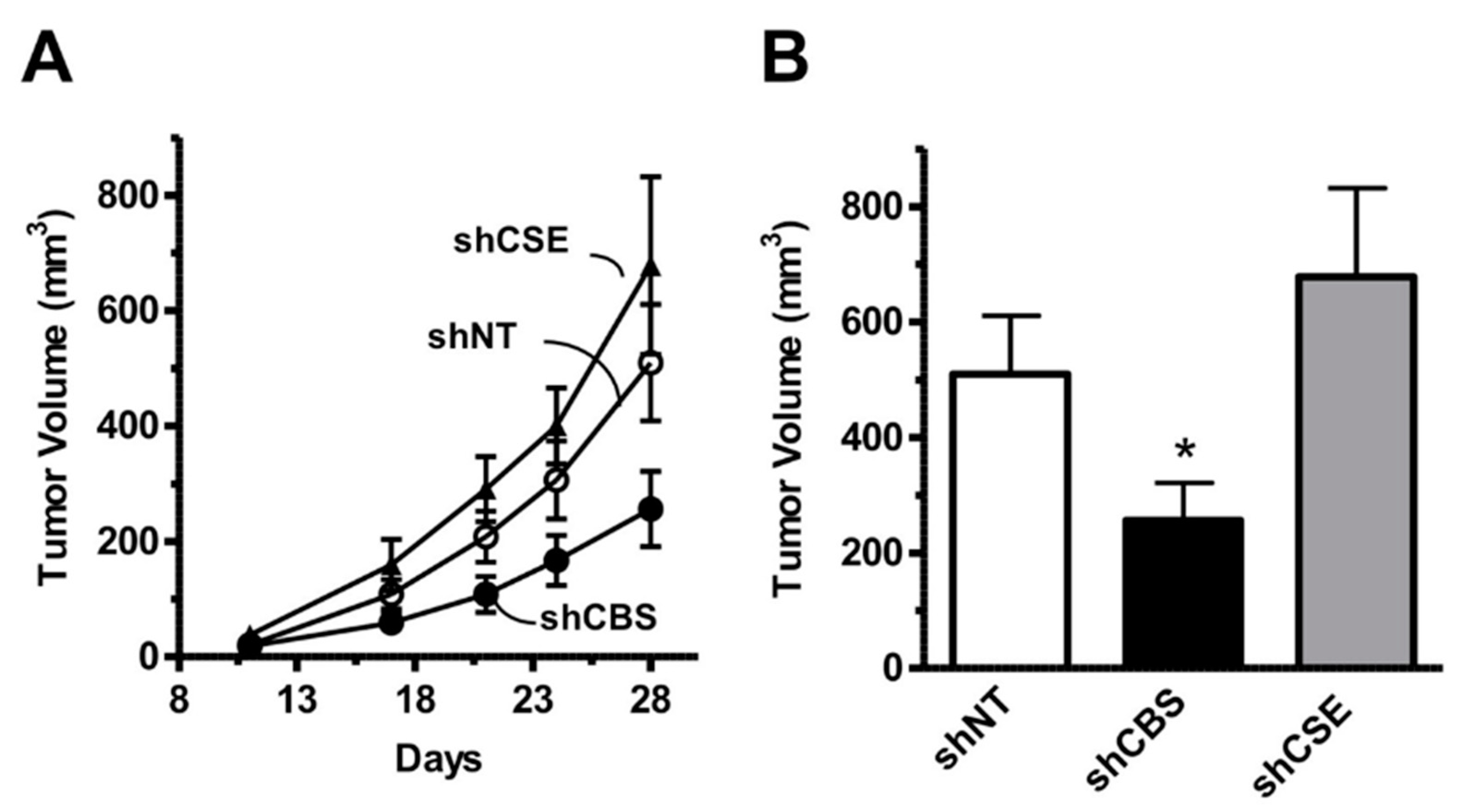{kind=link}
{kind=link}
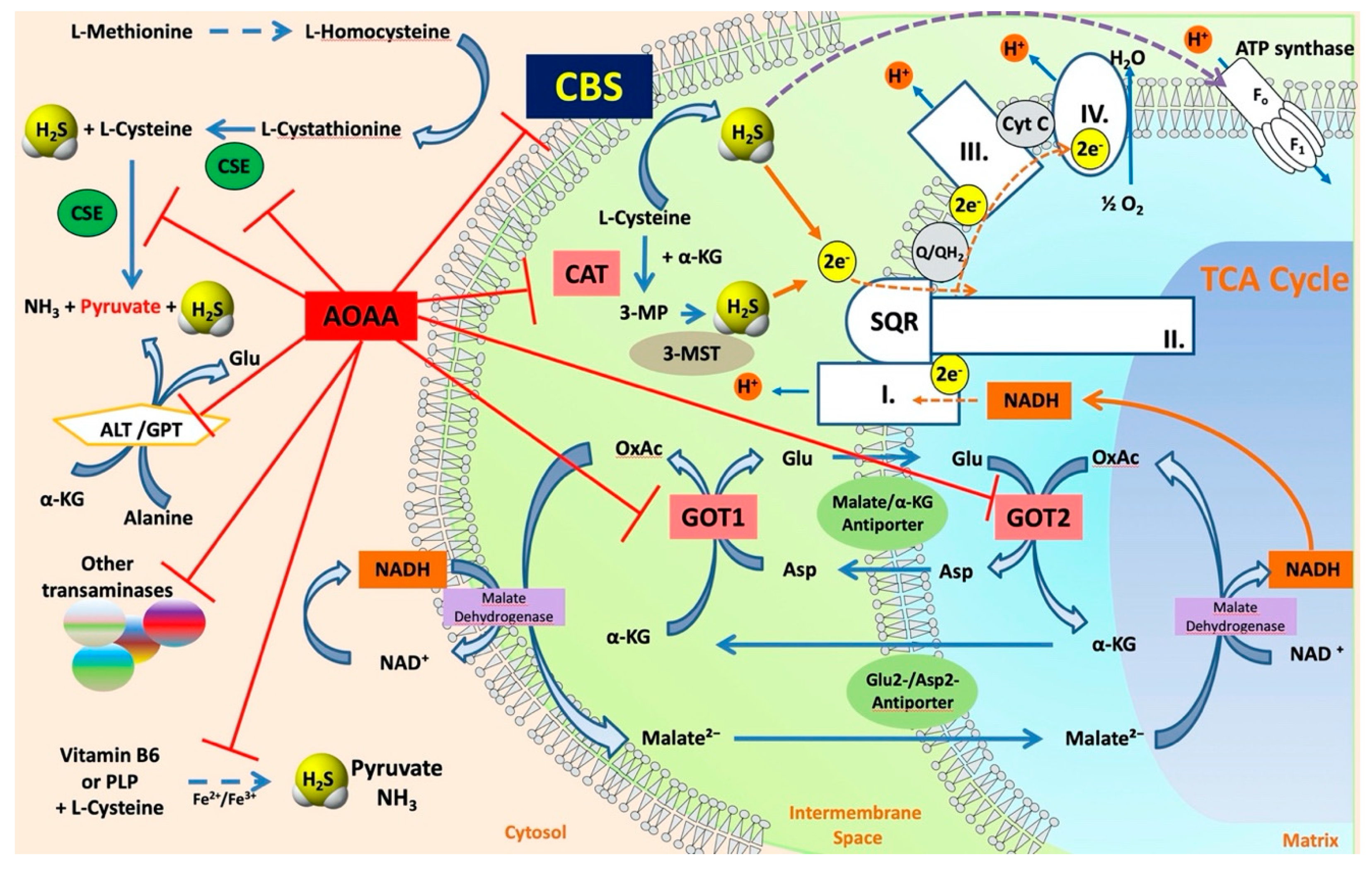{kind=link}
| Principal Approach | Effect on CBS | Pharmacological Modulators | Potential Off-Target Effects |
|---|---|---|---|
| Inhibition of SAM binding to CBS | Partial inhibition of CBS catalytic activity. Destabilization of the CBS tetramer | MAT inhibitors, competitors of SAM binding to CBS (SAM analogs) | Inhibition of other SAM-dependent enzymes and processes |
| Stimulation of CO or NO binding to CBS | Partial inhibition of CBS catalytic activity | NO or CO donors | Activation other NO- or CO-dependent enzymes and processes (e.g., vasodilatation, hypotension) |
| Inhibition of CBS phosphorylation | Partial inhibition of CBS catalytic activity | Kinase inhibitors | Inhibition of other enzymes phosphorylated by the same kinase; modulation of multiple downstream processes |
| Inhibition of CBS S-glutathionylation | Partial inhibition of CBS catalytic activity | Glutathione S-transferase inhibitors | Inhibition of other enzymes glutathionylated by the same S-transferase |
| Stimulation of SUMOylation | Proteolytic degradation of CBS; reduced total cellular CBS activity | Possible approach may be the modulation of upstream processes, e.g., E1 activating enzyme (the heterodimer SAE1/2) or E2 conjugase (Ubc9). No known pharmacological agents | Broad dysregulation of protein processing and protein degradation |
| Stimulation of ubiquitination | Proteasomal degradation of CBS; reduced total cellular CBS activity | Pharmacological activation of E1 activating, E2 conjugating and E3 ligating enzymes (only theoretical; no known inhibitors). Or: pharmacological inhibition of deubiquitinases (this approach has been proposed to degrade undruggable targets for cancer therapy) | Broad dysregulation of protein processing and protein degradation |
| Inhibition of CBS proteolytic cleavage | Inhibition of the proteolytic conversion of CBS into the highly active 45-kDa form; inhibition of cellular CBS activity | Proteolysis inhibitors (not suitable as a practical approach; selective intracellular delivery of protease inhibitors is not feasible) | Broad dysregulation of protein processing and protein degradation |
| Activation of Lon protease | Proteolytic degradation of CBS into inactive forms | Lon activators (e.g., Heat Shock Protein Q) exist but only as experimental tools | Broad dysregulation of mitochondrial protein homoeostasis |
| Inhibiting substrate availability by blocking cystine transport into the cell | Lower CBS activity | Cystine/glutamate antiporter system blockers | Broad dysregulation of sulfur-containing amino acid homeostasis |
| Cancer Type | Evidence for CBS Up-regulation | Effect of CBS Silencing | Reference |
|---|---|---|---|
| Biliary tract carcinoma | CBS cRNA hybridization levels measured on oligonucleotide microarray are higher in gallbladder carcinoma, intrahepatic cholangiocarcinoma, distal bile duct carcinomas, EGI-1, TFK-1, HUH28, HUCCT-1, SNU 245, SNU 308, SNU 1079, GB-H3, and GB-D1 cells than in normal extrahepatic biliary and gallbladder epithelial scrapings | Not tested | [287] |
| Breast cancer | • CBS mRNA levels detected by RT-qPCR and protein levels detected by immunoblotting are both higher in Hs 578T, MCF-7, and MDA-MB-468 cells than in HMEC and MCF-10A cells • CBS protein levels detected by immunohistochemistry are increased with the disease progression on tissue microarray with 60 human breast cancer tissue, and compared to the controls human breast epithelial tissue • CBS protein levels detected by immunoblotting are higher in doxorubicin-resistant MCF-7 cells that in normal MCF-7 cells | • Silencing of CBS in MCF-7 and MDA-MB-468 cells reduces cell viability in the presence of GOx, DOX or activated macrophages • Silencing of CBS in MCF-7 cells reduces xenograft growth in female Balb/c nude mice • Silencing of CBS in MCF-7 and MDA-MB-468 cells causes dilation of the ER and increases cytosolic calcium concentrations • Silencing of CBS in MCF-7 and MDA-MB-468 cells decreases cristae formation and increases vacuole formation in mitochondria, increases MPTP opening, and decreases mitochondrial reserve capacity | [82,289,294] |
| Colon cancer | • CBS protein levels detected by immunoblotting are higher in human colorectal tumor tissues compared to respective normal mucosa tissues • CBS protein levels detected by immunoblotting are higher in LoVo, HCT116, and HT29 cells compared to NCM356 cells • CBS protein levels detected by immunoblotting are higher in premalignant polyps compared to normal mucosa tissues • CBS protein levels detected by immunohistochemistry are increased in hyperplastic polyps compared to normal crypt cells • CBS protein levels detected by immunohistochemistry on tissue microarray are increased in 40 human colon cancer tissues compared to paired adjacent tissues over 52 colorectal cancer cases • CBS protein levels are increased in the colon cancer cell derived circulating tumor cell population CTC-MCC-41 • The development of multi-drug resistance is associated with an up-regulation of CBS protein in HCT116 cells | Silencing of CBS in HCT116 cells decreases cell proliferation and cellular bioenergetics in vitro and attenuates HCT116 xenograft growth and vascularization in female Balb/c nude mice | [88,290,296,300,302,303,308,309,310,313,315,318] |
| Glioma | CBS mRNA levels detected by PCR and protein levels detected by immunoblotting are both higher in U-87 MG cells than in SHSY5Y cells | • Not tested | [301] |
| Liver cancer | • CBS protein levels detected by immunoblotting are higher in HepG2 cells and SMMC-7721 than in HL-7702 cells a • In HepG2 cells stress conditions (e.g., oxidative stress, chemotherapeutics, irradiation) induces cancer cell stemness and multi-drug resistance and this is associated with up-regulation of CBS protein | • Silencing of CBS in SMMC-7721 decreases cell viability and proliferation, increases ROS levels and apoptosis | [291,293,297,312,314] |
| Lung cancer | • CBS mRNA levels detected by RT-qPCR and CBS protein levels detected by immunoblotting are higher in human primary tumor tissues compared to matched human normal tissues • CBS protein levels detected by immunoblotting are higher in human lung adenocarcinoma tumors than in normal adjacent tissues • CBS protein levels detected by immunoblotting are higher in A549, H522, and H1944 cells compared to BEAS 2B cells | • Down-regulation of CBS by rpL3 enhances Calu-6 cells apoptosis and reduces cell migration and invasion • Transient CBS depletion represses mtDNA repair and increases CPT-induced necrosis in A549 cells | [298,299,319] |
| Multiple myeloma | • cRNA hybridization levels for CBS, measured on oligonucleotide microarray, are higher in human malignant plasma cells from patients with multiple myeloma than in normal plasma cells • mRNA levels detected by RT-qPCR are higher in HMCL than in PPCL | Not tested | [286] |
| Ovarian cancer | • Human ovarian tumor tissues exhibited moderate-strong CBS protein expression detected by immunohistochemistry on tissue microarray • CBS mRNA levels detected by RT-qPCR and protein levels detected by immunoblotting are both higher in OV167, OV202, SKOV3, and A2780 cells than in OSE cells • CBS protein levels detected by immunoblotting are higher in OV90, CP20, OVSAHO, Kuramochi, and TykNu and cisplatin-resistant TykNu cells than in OSE, FTE188 and HOSE cells • The ferroptosis inducer small-molecule erastin induced an up-regulation of CBS protein and yielded an erastin-resistant version of ovarian cancer cell lines SKOV3 and OVCA429. | • Silencing of CBS in OV202, SKOV3, A2780, and cisplatin-resistant A2780 decreases total cellular glutathione level and cell proliferation • Silencing of CBS in A2780 cells increases cellular and mitochondrial ROS levels, down-regulates NF-κB, decreases cellular bioenergetics, and sensitizes to cisplatin • Silencing of CBS in cisplatin-resistant A2780 reduces xenograft growth and vascularization, and nodules formation in female nude mice, enhances sensitivity to cisplatin, and decreases MFN2 expression • Silencing of CBS in CP20 and OV90 decreases cell proliferation, mitochondrial membrane potential, and network by promoting mitochondrial fission, cellular bioenergetics and promotes MFN2 degradation • In the erastin-resistant version of ovarian cancer cell lines SKOV3 and OVCA429, CBS silencing induces cell death via induction of ferroptosis | [89,292,306,307,317] |
| Prostate cancer | • CBS protein levels detected by immunoblotting are higher in BPH-1, LNCaP, and DU145 cells than in RWPE-1 and WPMY-1 cells • CBS protein levels measured by immunofluorescence are higher in LNCaP cells than in RWPE-1 cells | Not tested | [83] |
| Renal carcinoma | • CBS protein levels detected by immunohistochemistry are increased with the disease progression on tissue microarray, and in 53 renal urothelial carcinomas and 9 renal clear cell carcinomas at Fuhrman grade IV compared to 11 benign renal cortex tissues • cRNA hybridization levels of CBS, measured on oligonucleotide microarray are higher in 2 angiomyolipoma and 3 papillary carcinoma tissues compared to respective unaffected part of kidney tissues | Not tested | [304,305] |
| Bladder cancer | CBS protein levels were detected in bladder tissue specimens (gallbladder squamous cell/adenosquamous carcinomas and adenocarcinomas) and in the bladder carcinoma cell lines 5637, EJ, and UM-UC-3 | Not tested | [311] |
| Thyroid cancer | Increased CBS protein levels were detected in thyroid carcinomas compared to benign thyroid tissue (but not in thyroid follicular adenomas or oncocytomas) | Not tested | [73] |
| Animal Model | Dose of AOAA | Effects of AOAA; Proposed Mechanism of Action | Reference |
|---|---|---|---|
| Methionine sulfoximine or thiosemicarbazide induced seizures in mice, Sprague-Dawley rats, and cats | 23–50 mg/kg i.p. single dose | AOAA dose-dependently decreased the incidence of convulsions and improved survival. The mechanism of action was proposed to be inhibition by AOAA of GABA-T activity in the CNS and subsequent elevation of brain GABA content; in support of this hypothesis, brain GABA levels were measured and were found to be increased at the same doses of AOAA where functional benefits were also noted. | [408] |
| Endocochlear potentials in response to 6 kHz tone bursts in anesthetized guinea pigs | 10–80 mg/kg i.v. single dose | AOAA dose-dependently attenuated the generation of endocochlear potentials. The mechanism of action was not identified, but observations of this type have subsequently led to clinical trials with AOAA in patients with tinnitus. | [409] |
| Isonicotinic acid hydrazide-induced seizures in male Swiss albino mice | 23 mg/kg i.p. single dose | AOAA dose-dependently decreased the incidence of convulsions. The mechanism of action was proposed to be due to a combined inhibition by AOAA of GABA-T activity (which inhibits GABA degradation) and of glutamate decarboxylase activity (which catalyzes GABA production from glutamate), and the resulting changes in the brain GABA content are the function of these two combined enzymatic effects. | [410,411] |
| Pentobarbital metabolism in mice | 30 mg/kg i.v. single dose | AOAA increased pentobarbital plasma levels and decreased the plasma levels of pentobarbital metabolites. The mechanism of action was not identified, but it was suggested to relate to an AOAA-induced broad suppression of cellular bioenergetics. | [412] |
| Cobalt-induced epilepsy in male piebald rats | 2.5–10 mg/kg i.p. single dose | AOAA reduced the frequency of epileptic spikes in the secondary foci of cobalt epileptic rats. The mechanism of action was proposed to be inhibition of GABA-T activity in the brain; however, the protective effect of AOAA was more pronounced at the lower dose (5 mg/kg) while the enhancement of CNS GABA-T levels was more pronounced at higher doses, where the functional benefit of AOAA was less pronounced. | [413] |
| Memory consolidation in male Sprague-Dawley rats | 25 mg/kg/day i.p. for 8 days | In the shuttlebox shock avoidance used, controls animals showed learning both within and across sessions, while AOAA-treated only showed learning within sessions but exhibited a lack of consolidation across sessions. Because GABA plays a role in memory consolidation, the mechanism was hypothesized to relate to the inhibitory effect of AOAA on GABA-T, but no pharmacological mechanism was investigated in the study. | [414] |
| Hyperbaric oxygen induced seizures in chicken | 2.5 mg/kg s.q. single dose | AOAA decreased the onset and duration of the convulsions. The mechanism of action was proposed to be inhibition by AOAA of the GABA-T activity in the CNS and an elevation of central GABA levels, but no biochemical markers were measured. | [415] |
| Dichlorovinylcysteine induced nephrotoxicity model in male NMRI mice | 40 mg/kg i.p. single dose | AOAA attenuated the generation of various lipid peroxidation markers. The mechanism of action was not directly explored but was presumed to be related to an antioxidant effect of AOAA. | [416] |
| Circulating glucose and insulin and glucagon levels in control and streptozotocin-diabetic female Wistar rats | 30 mg/kg i.p. single dose | In control animals, AOAA significantly increased circulating insulin levels (but not glucose or glucagon levels). In the diabetic animals, AOAA protected against the development of streptozotocin-induced hyperglycemia. Streptozotocin caused a 50% drop in plasma insulin levels in the rats; this effect was largely absent in the AOAA-treated streptozotocin animals. The proposed mechanism relates to AOAA’s effect on some peripheral GABA-T system and subsequent increases in peripheral GABA levels, but no direct measurements were provided. | [417] |
| Male Wistar rats subjected to stroke (transient middle cerebral artery occlusion) | 2.5, 5, 10 or 50 mg/kg i.p. single dose | AOAA at 10 and 50 mg/kg significantly reduced stroke volume and brain edema and improved neurological scores, without affecting post-ischemic cerebral blood flow, brain malondialdehyde content, SOD, or glutathione peroxidase activity. The mechanism of action was proposed to be inhibition of CBS activity by AOAA in the brain, but no biochemical markers were measured. | [418] |
| Hypoxia-induced central apneas in ventilated C57BL/6J mice | 30 mg/kg i.p. single dose | AOAA reduced the percentage of animals expressing one or more apneas during reoxygenation. AOAA-treated mice also exhibited a smaller coefficient of variation for frequency during reoxygenation, suggesting improved respiratory stability. The mechanism of action was proposed to be inhibition of CBS activity in the CNS, but no biochemical markers were measured. | [419] |
| Cisplatin nephrotoxicity in male C57BL/6 mice or F344 rats | 100 mg/kg p.o., single dose | AOAA protected against the biochemical (plasma BUN) and histological (renal tubular alterations) damage induced by cisplatin. The mechanism of action was proposed to be inhibition of cysteine S-conjugate b-lyase activity by AOAA (and/or an inhibitory effect of AOAA on some other PLP-dependent enzyme, most likely a transaminase). However, no experiments were conducted to delineate the molecular mechanism of AOAA’s action. | [420,421] |
| Tumor growth in female BALB/c nude mice bearing MDA-MB-231 human breast cancer subcutaneous xenografts | 10 mg/kg/day i.p. for 14 days | AOAA significantly inhibited tumor growth. Based on complementary in vitro studies, the mechanism of AOAA’s action was proposed to relate to the suppression of tumor cell bioenergetics, in particular due to AOAA-mediated inhibition of tumor cell aspartate aminotransferase activity (an enzyme which functions in tandem with malate dehydrogenase to regulate mitochondrial electron transport). | [394] |
| Complete Freund adjuvant (CFA)-induced mechanical hyperalgesia model in adult Sprague-Dawley rats | 5, 15 or 45 mg/kg/day i.p. single dose | AOAA dose-dependently attenuated mechanical hyperalgesia due to an inhibition of the hyperexcitability of dorsal root ganglion neurons. In these neurons, CFA up-regulated CBS mRNA transcription and subsequent translation of CBS protein. The mode of AOAA’s action was proposed to be related to inhibition of CBS activity, and the consequent prevention of the H2S-mediated opening of tetrodotoxin-resistant voltage-gated sodium channels. | [422] |
| Tumor growth in female athymic nude mice bearing subcutaneous xenografts of HCT116 colon cancer cells or human patient-derived colon cancer xenografts (PDTX). Liver metastasis model (nude mice, intracecal HCT116 implantation) | 1, 3 or 9 mg/kg/day i.p. for 2 weeks | AOAA (at 9 mg/kg/day, but not at the lower doses) suppressed tumor growth. The underlying mechanisms was proposed to relate to the AOAA-induced inhibition of intratumor CBS, inhibition of intratumor H2S production, which, in turn, inhibits cellular bioenergetics and reduces tumor angiogenesis. The effect of AOAA was independent of the tumor’s K-ras status. The effects of AOAA were reproduced by the AOAA prodrug YD0171, which, however, was more potent (effective at 0.5 and 1 mg/kg/day). YD0171 (at 3 mg/kg/day for 3 weeks), caused the regression of established HCT116 subcutaneous xenografts. YD0171 also inhibited liver metastasis formation in an intracecal HCT116 implantation model. | [88,296,320] |
| Athymic Balb/c mice bearing SUM149, SUM159, or HCC1954 MDA-MB-231 xenografts; MMTV-rTtA-TetO-myc mouse mammary tumor model | 5 mg/kg/day i.p. or 0.5 mg/kg/day i.p. in the TetO-myc model | AOAA suppressed the growth of the UM149, SUM159 xenografts, but did not affect the growth of HCC1954 xenografts. AOAA was also effective in the TetO-myc model. In the MDA-MB-231 xenografts, AOAA did not inhibit tumor growth alone, but potentiated the growth-suppressant effect of paclitaxel. The underlying mechanisms was proposed to relate to the inhibition of intratumor GOT activity, as it is associated with increased C-MYC expression in the tumors and the subsequent increased reliance of the tumor cells on glutaminolysis. | [369] |
| Male BALB/c mice subjected to burn injury | 10 mg/kg/day i.p. for 6 days | AOAA attenuated the degree of burn-induced oxidative stress in various tissues. It also reduced plasma levels of various circulating mediators (IL-6, IL-10). It improved various plasma markers of multiorgan failure. The effects were attributed to AOAA’s effect as an inhibitor of CBS. | [175] |
| Female athymic nude mice bearing subcutaneous xenografts of various human colon cancer tumor lines | 5 or 10 mg/kg/day i.p. for 2–4 weeks (depending on the growth of the particular cell line graft) | AOAA dose-dependently reduced tumor growth of the HCT116, DLD1, RKO, and HT29 xenografts, but did not affect the growth of SW40 or LoVo xenografts). The underlying mechanisms was proposed to relate to the inhibition by AOAA of glutamate pyruvate transaminase 2 (GPT2) in the tumor cells. This hypothesis was supported by the findings that the growth of PIK3CA mutant xenograft tumors (which express GPT2) were inhibited by AOAA, but GPT2 knockdown tumors were not. (It should be noted, however that the latter tumors showed a significantly slower baseline proliferation rate in the absence of AOAA). | [423] |
| Male Wistar rats subjected to experimental subarachnoid hemorrhage induced by double blood injection; effect of L-cysteine | 5 mg/kg i.p. single dose | AOAA suppressed the neuroprotective effect of L-cysteine. Its mechanism of action was proposed to be inhibition of CBS-induced H2S production. The authors’ working hypothesis is that L-cysteine increases CBS-derived H2S production, and this produces neuroprotective effects. Unfortunately, the effect of AOAA (in the absence of L-cysteine) was not tested in the study. | [424] |
| Female athymic nude mice bearing subcutaneous xenografts of NCM356 colon epithelial cells overexpressing CBS | 9 mg/kg/day i.p. for 2 weeks | AOAA significantly decreased the size of established tumors. The underlying mechanisms was proposed to relate to the inhibition of intratumor CBS activity by AOAA and the consequent inhibition of intratumor H2S production. Metabolomic and pharmacological studies also implicated a role for the pentose phosphate pathway in the CBS-mediated enhancement of tumor growth. | [303] |
| Experimental allergic encephalomyelitis model in C57BL/6 mice induced by a myelin oligodendrocyte glycoprotein peptide fragment | 35 mg/kg/day i.p. for 7 days | Disease severity was suppressed by AOAA. The effect of AOAA was associated with significant changes in immune cell populations. The percentage of IL-17-producing T cells was reduced while the percentage of FOXP3+ T cells increased, while the percentage of IFNγ + cells was unaffected in the central nervous system. The ratio of FOXP3+ cells to IL-17+ cells increased by AOAA. AOAA markedly reduced the total number of mononuclear cells infiltrating into the central nervous system. Based on complementary in vitro and in vivo studies, the mechanism proposed to underlie AOAA’s action was proposed to relate to the suppression of immune cell bioenergetics, in particular due to AOAA-mediated inhibition of GOT1 activity, which produces an increase in 2-hydroxyglutarate levels in differentiating TH17 cells, which in turn results in the hypermethylation of the Foxp3 gene locus and inhibited Foxp3 transcription, which ultimately regulates the differentiation towards TH17. | [178] |
| Male Sprague-Dawley rats subjected to an experimental model of chronic alcoholism (chronic ethanol consumption) | 5 mg/kg/day i.p. for 2 weeks | Alcoholism produced learning and memory deficits (assessed by the Morris water maze test). AOAA improved latency and swimming distance parameters and improved the animals’ performance in the spatial probe test. AOAA also prevented the down-regulation of myelin basic protein expression and protected against the deterioration of mitochondrial ultrastructure. The mechanism of action was proposed to be inhibition of CBS activity by AOAA in the brain; the AOAA-induced normalization of hippocampal H2S levels provided some experimental support for this theory. AOAA also induced complex changes in gene expression and antioxidant levels in the brain of the animals. | [425,426] |
| Male Swiss albino mice subjected to stroke (transient middle cerebral artery occlusion) in combination with remote ischemic preconditioning | 50 mg/kg i.p. single dose | AOAA suppressed the neuroprotective effect of remote ischemic preconditioning. Its mechanism of action was proposed to be inhibition of CBS-induced H2S production. The authors’ working hypothesis is that stroke down-regulates CBS expression in the CNS, and this down-regulation is prevented by preconditioning. Unfortunately, the effect of AOAA on stroke (in the absence of preconditioning) was not tested in the study. | [427] |
| Male and female SOD1G93A mice, a model of familial ALS | 8.75 mg/kg/day i.p. for 100 days | AOAA significantly improved motor performance (Rotarod test) in the female (but not male) animals and tended to extend survival. The underlying mechanisms was proposed to relate to an up-regulation of CBS in ALS, which, in turn, elevates H2S to cytotoxic concentrations. Thus, it was hypothesized that inhibition of CBS activity with AOAA reduces neuronal and glial H2S levels to physiological (cytoprotective) levels. The gender difference was proposed to relate to higher levels of CNS H2S levels in females with ALS than males with ALS. | [428] |
| Male athymic nude mice bearing subcutaneous human colon cancer cell line xenografts | 9 mg/kg/day i.p. 5 days per week for 4 weeks | AOAA potentiated the inhibitory effect of oxaliplatin on tumor growth, but on its own, did not exert a significant inhibitory effect. The underlying mechanisms was proposed to relate to the AOAA-induced inhibition of intratumor CBS and the subsequent inhibition of intratumor H2S production, with a consequent suppression of cellular bioenergetics and of tumor angiogenesis. The potentiation of oxaliplatin’s antitumor effect was hypothesized to be related to an enhancement by AOAA of oxaliplatin-induced tumor cell apoptosis. | [318] |
| Classification | Enzyme | IC50 (µM) | Ki (µM) | Reference |
|---|---|---|---|---|
| EC 2.6.1.19 | 4-aminobutyrate-2-oxoglutarate transaminase (GABA-T) | 1.8 | [353,371,430] | |
| EC 2.6.1.2 | alanine transaminase (ALT) (aka glutamate pyruvate transaminase, GPT) (aka alanine:oxalacetate transaminase) | 0.5 | [431,432] | |
| EC 2.6.1.6 | D-amino acid transaminase | 0.1 | [433] | |
| EC 2.7.1.35 | pyridoxal kinase | 10 | [434] | |
| EC 4.1.1.22 | histidine decarboxylase | 5 | [357,435] | |
| EC 4.1.1.19 | arginine decarboxylase | 500 | [435] | |
| EC 4.2.1.22 | cystathionine β-synthase (CBS) | 3–8 | [329,363,399,437] | |
| EC 2.6.1.1 | aspartate transaminase (AST) a (aka glutamic oxaloacetic transaminase, GOT) | <0.1 | [405,438,439] | |
| EC 2.6.1.3 | cysteine transaminase (CAT) b | <0.1 | [405,438,439] | |
| EC 4.1.2.5 | threonine aldolase | 1000–5000 | [440] | |
| EC 2.1.2.1 | serine hydroxymethyltransferase | 1000 | [441,442] | |
| EC 5.1.1.1 | alanine racemase | >10 | [358,443] | |
| EC 4.4.1.9 | beta-cyano-L-alanine synthase | 10–100 | [444] | |
| EC 4.4.1.1 | cystathionine γ-lyase (CSE) | 1 | [364,399] | |
| EC 4.3.1.5 | phenylalanine ammonia-lyase | 0.45 | [445] | |
| EC 2.6.1.7 | kynurenine-oxoglutarate transaminase | 25 | [445,446] | |
| EC 4.1.1.20 | diaminopimelate decarboxylase | 2500 c | [447] | |
| EC 2.6.1.42 | branched-chain amino acid transaminase | 21 | [383,448] | |
| EC 2.6.1.5 | tyrosine transaminase | ∼20 | [449] | |
| EC 4.1.1.28 | DOPA decarboxylase | 50 c | [362,450] | |
| EC 2.6.1.44 | alanine-glyoxylate transaminase | 0.15 | [451] | |
| EC 4.4.1.14 | 1-aminocyclopropanecarboxylate synthase | 0.8 | [452] | |
| EC 4.4.1.4 | alliin lyase | 250 c | [453] | |
| EC 4.4.1.15 | D-cysteine desulfhydrase | 3.3 | [400,454] | |
| EC 4.1.1.11 | cysteine sulfinic acid decarboxylase | 30 | [455,456,457] | |
| EC 2.6.1.45 | serine-glyoxylate aminotransferase | 0.01 | [442,458] | |
| EC 2.6.1.37 | (2-aminoethyl)phosphonate-pyruvate transaminase | 1000 c | [459] | |
| EC 2.6.1.43 | aminolevulinate transaminase | 0.03 | [460] | |
| EC 4.1.1.12 | L-aspartate 4-carboxy-lyase | 1.6 | [461] | |
| EC 4.4.1.13 | cysteine-S-conjugate beta-lyase (mitochondrial) | 8 | [462] | |
| EC 4.4.1.13 | cysteine-S-conjugate beta-lyase (cytosolic) | 0.8 | [462] | |
| EC 4.1.1.15 | glutamate decarboxylase | 1 | [463,464] | |
| EC 5.1.1.18 | serine racemase | ∼100 | [465] |
| Inhibitor Structure | Name | IC50 | Selectivity | Reference |
|---|---|---|---|---|
 | Aminooxyacetic acid | 1–8.5 μM | The compound is a potent CBS inhibitor which works by reacting with its PLP prosthetic group. Although it is commonly referred to as a “CBS inhibitor”, it is an even more potent inhibitor of CSE (IC50: 1 µM) | [296,302,329] |
 | Benserazide | 30 μΜ | Relatively potent CBS inhibitor that reacts with its PLP prosthetic group. It has some selectivity for CBS (CSE is inhibited 16% at 100 µM benserazide and 3-MST is inhibited 50% at 300 µM benserazide) | [327,329] |
 | 2,3,4-Trihydroxy-benzylhydrazine | 30 μΜ | It inhibits CBS by reacting with its PLP prosthetic group. It may be responsible for some of the CBS-inhibitory effect of benserazide in vivo. Its effect on CSE has not been tested | [329] |
 | 3-Hydroxy-benzylhydrazine | 60 µM | It inhibits CBS by reacting with its PLP prosthetic group. Its effect on CSE has not been tested. It is known to inhibit GABA-T and other PLP-dependent enzymes | [329] |
 | Disulfiram | Not a direct inhibitor | In yeast assays and in Down syndrome mice, it has biological effects consistent with cell-based CBS inhibition | [259] |
 | Hydroxylamine | 20–400 μM | The compound inhibits CBS, but it inhibits CSE more potently (IC50: 5 µM) | [328] |
| Cu2+ | Copper | 0.2–10 µM | The assessment of true CBS-inhibitory potency is made difficult by the fact that it also reacts with H2S, the product of the CBS reaction measured in the assay | [329,542,543] |
 | NSC 67078 1,6-dimethyl-pyrimido[5,4-e]-1,2,4-triazine-5,7(1H,6H)-dione | 12–30 µM | It preferentially inhibits CBS; it also inhibits CSE, but with lower potency (IC50: 30 µM) | [328,329] |
 | NSC11041 | 4 µM | Approximately equipotent on inhibitor of CBS and CSE (IC50 ~3–4 µM) | [328] |
 | Sikokianin C | 3.1 µM | Potent CBS inhibitor; its potency on CSE is weaker (IC50: 40 µM) | [302,562] |
 | Tannic acid | 40 µM | CBS inhibitor; its effect on other H2S producing enzymes has not been tested | [329] |
 | Hypericin | 3.1 µM | Potent CBS inhibitor; its potency on CSE is weaker (IC50: 40 µM) | [562] |
 | Caraphenol A | 5.9 µM | Fairly potent CBS inhibitor; its potency on CSE is almost comparable (IC50: 12 µM) | [562] |
 | 2′′,4′′-Di-O-(Z-p-coumaroyl) afzelin | 6.2 µM | Potent CBS inhibitor; its potency on CSE is very weaker (IC50 > 400 µM) | [562] |
 | 3′-Hydroxy-volkensiflavon | 7.8 µM | Potent CBS inhibitor; its potency on CSE is very weaker (IC50 > 400 µM) | [562] |
 | Cupressuflavone | 11.5 µM | Potent CBS inhibitor; its potency on CSE is very weaker (IC50 > 400 µM) | [562] |
 | Podocarpusflavone A | 8.9 µM | Potent CBS inhibitor; its potency on CSE is very weaker (IC50 > 400 µM) | [562] |
 | Agathisflavone | 17.1 µM | Potent CBS inhibitor; its potency on CSE is very weaker (IC50 > 400 µM) | [562] |
 | Tangeritin | IC25: 46 µM | CBS inhibitor; its effect on other H2S producing enzymes has not been tested | [327] |
 | Myricetin | 18.8 µM | Fairly potent CBS inhibitor; its potency on CSE is similar (IC50: 14.4 µM) | [562] |
 | Apigenin | 83 µM | CBS inhibitor; its effect on other H2S producing enzymes has not been tested | [327] |
 | 12α-hydroxy-5-deoxydehydro-munduserone | 56 µM | CBS inhibitor; its effect on other H2S producing enzymes has not been tested | [327] |
 | Rutin | 116 µM | CBS inhibitor; its effect on other H2S producing enzymes has not been tested | [327] |
 | Fraxetin | 134 µM | CBS inhibitor; its effect on other H2S producing enzymes has not been tested | [327] |
 | CH004 | 0.6–1.7 µM | A highly potent CBS inhibitor, with some selectivity towards CBS over CSE (IC50 ~ 30 µM) | [256] |
 | 6S | Ki = 48 µM | It inhibits CBS inhibitor via interacting with its PLP group. Its effect on other H2S-producing enzymes or other PLP-dependent enzymes has not been characterized | [566] |
 | Aurintricarboxylic acid | 3–80 µM | CBS inhibitor with considerable potency; it is even more potent as a CSE inhibitor (IC50 0.6–3 µM) | [329,570] |
 | Hexachlorophene | 60 µM | CBS inhibitor with average potency | [329] |
 | Trifluoroalanine | 66 μΜ | It does not have a high potency as a CBS inhibitor, but it exhibits some selectivity for CBS over CSE (IC50 ~ 300 µM) | [399] |
 | JHU-8555 | 8–12 µM | Approximately equipotent on inhibitor of CBS and CSE, with some preference for CBS (IC50 ~ 10–25 µM) | [328] |
 | MBSEW03275 | 15 µM | It does not have a high inhibitory potency as a CBS inhibitor, but it does have some selectivity for CBS over CSE (IC50 ~ 200 µM) | [328] |
 | SP14311008 | 20 µM | Approximately equipotent on inhibitor of CBS and CSE, with some preference for CBS (IC50 ~ 40 µM for CSE) | [328] |
 | 1,4-Naphtoquinone | 35 µM | CBS inhibitor; its effect on other H2S producing enzymes has not been tested | [327] |
 | 2,4-Dinitrophenol | 56 µM | CBS inhibitor; its effect on other H2S producing enzymes has not been tested | [327] |
 | Piperine | 61 µM | CBS inhibitor; its effect on other H2S producing enzymes has not been tested | [327] |
 | Amiloride | 89 µM | CBS inhibitor; its effect on other H2S producing enzymes has not been tested | [327] |
 | MNP2-A6 | 83 µM | CBS inhibitor; its effect on other H2S producing enzymes has not been tested | [571] |
 | MNP2-B7 | 87 µM | CBS inhibitor; its effect on other H2S producing enzymes has not been tested | [571] |
 | NP-014428 | 7.4 µM | Fairly potent CBS inhibitor; its potency on CSE is weaker (IC50: 62 µM) | [562] |
 | NP-003872 | 8.1 µM | Fairly potent CBS inhibitor; its potency on CSE is weaker (IC50: 122 µM) | [562] |
 | β-cyano-alanine | 40% inhibition at 10 mM | The compound is a weak CBS inhibitor, but it is a potent inhibitor of CSE (IC50: 14 µM) | [399] |
 | “Compound #1” 3-MST inhibitor | 25% inhibition at 100 µM | The compound was identified as a potent 3-MST inhibitor (IC50: 1.7 µM), but it also exerts a weak inhibitory effect on CBS and CSE | [572] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuhra, K.; Augsburger, F.; Majtan, T.; Szabo, C. Cystathionine-β-synthase: Molecular Regulation and Pharmacological Inhibition. Biomolecules 2020, 10, 697. https://doi.org/10.3390/biom10050697
Zuhra K, Augsburger F, Majtan T, Szabo C. Cystathionine-β-synthase: Molecular Regulation and Pharmacological Inhibition. Biomolecules. 2020; 10(5):697. https://doi.org/10.3390/biom10050697
Chicago/Turabian StyleZuhra, Karim, Fiona Augsburger, Tomas Majtan, and Csaba Szabo. 2020. "Cystathionine-β-synthase: Molecular Regulation and Pharmacological Inhibition" Biomolecules 10, no. 5: 697. https://doi.org/10.3390/biom10050697
APA StyleZuhra, K., Augsburger, F., Majtan, T., & Szabo, C. (2020). Cystathionine-β-synthase: Molecular Regulation and Pharmacological Inhibition. Biomolecules, 10(5), 697. https://doi.org/10.3390/biom10050697