Oxidative Stress in DNA Repeat Expansion Disorders: A Focus on NRF2 Signaling Involvement


Abstract
:1. Introduction
2. Oxidative Stress and Cellular Responses
3. NRF2 Pathway and Its Regulation
4. Oxidative Stress in Loss of Function DNA Expansion Disease
4.1. Friedreich’s Ataxia (FA)
4.2. X-Fragile (FXS)
5. Oxidative Stress in CAG/polyQ Diseases
5.1. Spinobulbar Muscular Atrophy (SBMA)
5.2. Huntington’s Disease (HD)
5.3. Spinocerebellar Ataxias (SCAs)
6. Oxidative Stress in RNA Gain of Function Expansion Disease
6.1. Fragile X–Associated Tremor Ataxia Syndrome (FXTAS)
6.2. Myotonic Dystrophy (DM)
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gur-Arie, R.; Cohen, C.J.; Eitan, Y.; Shelef, L.; Hallerman, E.M.; Kashi, Y. Simple sequence repeats in Escherichia coli: Abundance, distribution, composition, and polymorphism. Genome Res. 2000, 10, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Hamada, H.; Petrino, M.G.; Kakunaga, T. A novel repeated element with Z-DNA-forming potential is widely found in evolutionarily diverse eukaryotic genomes. Proc. Natl. Acad. Sci. USA 1982, 79, 6465–6469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckman, J.S.; Weber, J.L. Survey of human and rat microsatellites. Genomics 1992, 12, 627–631. [Google Scholar] [CrossRef]
- Toth, G.; Gaspari, Z.; Jurka, J. Microsatellites in different eukaryotic genomes: Survey and analysis. Genome Res. 2000, 10, 967–981. [Google Scholar] [CrossRef] [Green Version]
- Metzgar, D.; Bytof, J.; Wills, C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000, 10, 72–80. [Google Scholar]
- Bagshaw, A.T.M. Functional Mechanisms of Microsatellite DNA in Eukaryotic Genomes. Genome Biol. Evol. 2017, 9, 2428–2443. [Google Scholar] [CrossRef] [Green Version]
- Hui, J.; Hung, L.H.; Heiner, M.; Schreiner, S.; Neumuller, N.; Reither, G.; Haas, S.A.; Bindereif, A. Intronic CA-repeat and CA-rich elements: A new class of regulators of mammalian alternative splicing. Embo J. 2005, 24, 1988–1998. [Google Scholar] [CrossRef] [Green Version]
- Tseng, S.H.; Cheng, C.Y.; Huang, M.Z.; Chung, M.Y.; Su, T.S. Modulation of formation of the 3′-end of the human argininosuccinate synthetase mRNA by GT-repeat polymorphism. Int. J. Biochem. Mol. Biol. 2013, 4, 179–190. [Google Scholar]
- Kramer, M.; Sponholz, C.; Slaba, M.; Wissuwa, B.; Claus, R.A.; Menzel, U.; Huse, K.; Platzer, M.; Bauer, M. Alternative 5′ untranslated regions are involved in expression regulation of human heme oxygenase-1. PLoS ONE 2013, 8, e77224. [Google Scholar] [CrossRef]
- Liu, H.; Mulholland, N.; Fu, H.; Zhao, K. Cooperative activity of BRG1 and Z-DNA formation in chromatin remodeling. Mol. Cell. Biol. 2006, 26, 2550–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quilez, J.; Guilmatre, A.; Garg, P.; Highnam, G.; Gymrek, M.; Erlich, Y.; Joshi, R.S.; Mittelman, D.; Sharp, A.J. Polymorphic tandem repeats within gene promoters act as modifiers of gene expression and DNA methylation in humans. Nucleic Acids Res. 2016, 44, 3750–3762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellegren, H. Microsatellite mutations in the germline: Implications for evolutionary inference. Trends Genet. 2000, 16, 551–558. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Hellemans, B.; Volckaert, F.A.M. Microsatellites and their genomic distribution, evolution, function and applications: A review with special reference to fish genetics. Aquaculture 2006, 255, 1–29. [Google Scholar] [CrossRef]
- Weissenbach, J. Microsatellite polymorphisms and the genetic linkage map of the human genome. Curr. Opin. Genet. Dev. 1993, 3, 414–417. [Google Scholar] [CrossRef]
- López Castel, A.; Cleary, J.D.; Pearson, C.E. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat. Rev. Mol. Cell Biol. 2010, 11, 165–170. [Google Scholar] [CrossRef]
- Pearson, C.E.; Nichol Edamura, K.; Cleary, J.D. Repeat instability: Mechanisms of dynamic mutations. Nat. Rev. Genet. 2005, 6, 729–742. [Google Scholar] [CrossRef]
- Gatchel, J.R.; Zoghbi, H.Y. Diseases of unstable repeat expansion: Mechanisms and common principles. Nat. Rev. Genet. 2005, 6, 743–755. [Google Scholar] [CrossRef]
- Paulson, H. Repeat expansion diseases. Handb. Clin. Neurol. 2018, 147, 105–123. [Google Scholar] [CrossRef]
- McInnis, M.G. Anticipation: An old idea in new genes. Am. J. Hum. Genet. 1996, 59, 973–979. [Google Scholar]
- Van Mossevelde, S.; van der Zee, J.; Gijselinck, I.; Sleegers, K.; De Bleecker, J.; Sieben, A.; Vandenberghe, R.; Van Langenhove, T.; Baets, J.; Deryck, O.; et al. Clinical Evidence of Disease Anticipation in Families Segregating a C9orf72 Repeat Expansion. JAMA Neurol. 2017, 74, 445–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulson, H.L.; Fischbeck, K.H. Trinucleotide repeats in neurogenetic disorders. Annu. Rev. Neurosci. 1996, 19, 79–107. [Google Scholar] [CrossRef]
- Reetz, K.; Dogan, I.; Costa, A.S.; Dafotakis, M.; Fedosov, K.; Giunti, P.; Parkinson, M.H.; Sweeney, M.G.; Mariotti, C.; Panzeri, M.; et al. Biological and clinical characteristics of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS) cohort: A cross-sectional analysis of baseline data. Lancet Neurol. 2015, 14, 174–182. [Google Scholar] [CrossRef]
- Cook, A.; Giunti, P. Friedreich’s ataxia: Clinical features, pathogenesis and management. Br. Med Bull. 2017, 124, 19–30. [Google Scholar] [CrossRef]
- Andrew, S.E.; Goldberg, Y.P.; Kremer, B.; Telenius, H.; Theilmann, J.; Adam, S.; Starr, E.; Squitieri, F.; Lin, B.; Kalchman, M.A.; et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 1993, 4, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, K.P.; Coon, H.; Santos, N.; Velazquez, L.; Mederos, L.A.; Pulst, S.-M. Genetic analysis of age at onset variation in spinocerebellar ataxia type 2. Neurol. Genet. 2017, 3, e155. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, S.; Tanno, Y.; Onodera, O.; Yamazaki, M.; Sato, S.; Ishikawa, A.; Miyatani, N.; Nagashima, M.; Ishikawa, Y.; Sahashi, K.; et al. Strong correlation between the number of CAG repeats in androgen receptor genes and the clinical onset of features of spinal and bulbar muscular atrophy. Neurology 1992, 42, 2300–2302. [Google Scholar] [CrossRef]
- Savić, D.; Rakocvic-Stojanovic, V.; Keckarevic, D.; Culjkovic, B.; Stojkovic, O.; Mladenovic, J.; Todorovic, S.; Apostolski, S.; Romac, S. 250 CTG repeats in DMPK is a threshold for correlation of expansion size and age at onset of juvenile-adult DM1. Hum. Mutat. 2002, 19, 131–139. [Google Scholar] [CrossRef]
- Leehey, M.A.; Berry-Kravis, E.; Goetz, C.G.; Zhang, L.; Hall, D.A.; Li, L.; Rice, C.D.; Lara, R.; Cogswell, J.; Reynolds, A.; et al. FMR1 CGG repeat length predicts motor dysfunction in premutation carriers. Neurology 2008, 70, 1397–1402. [Google Scholar] [CrossRef]
- Delatycki, M.B.; Paris, D.B.; Gardner, R.J.; Nicholson, G.A.; Nassif, N.; Storey, E.; MacMillan, J.C.; Collins, V.; Williamson, R.; Forrest, S.M. Clinical and genetic study of Friedreich ataxia in an Australian population. Am. J. Med. Genet. 1999, 87, 168–174. [Google Scholar] [CrossRef]
- Cossée, M.; Dürr, A.; Schmitt, M.; Dahl, N.; Trouillas, P.; Allinson, P.; Kostrzewa, M.; Nivelon-Chevallier, A.; Gustavson, K.H.; Kohlschütter, A.; et al. Friedreich’s ataxia: Point mutations and clinical presentation of compound heterozygotes. Ann. Neurol. 1999, 45, 200–206. [Google Scholar] [CrossRef]
- De Michele, G.; Perrone, F.; Filla, A.; Mirante, E.; Giordano, M.; De Placido, S.; Campanella, G. Age of onset, sex, and cardiomyopathy as predictors of disability and survival in Friedreich’s disease: A retrospective study on 119 patients. Neurology 1996, 47, 1260–1264. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Mulder, H.; Igoillo-Esteve, M. Diabetes in Friedreich ataxia. J. Neurochem. 2013, 126 (Suppl. 1), 94–102. [Google Scholar] [CrossRef]
- Arsenault, M.E.; Prévost, C.; Lescault, A.; Laberge, C.; Puymirat, J.; Mathieu, J. Clinical characteristics of myotonic dystrophy type 1 patients with small CTG expansions. Neurology 2006, 66, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhang, F.; Lokey, L.K.; Chastain, J.L.; Lakkis, L.; Eberhart, D.; Warren, S.T. Translational suppression by trinucleotide repeat expansion at FMR1. Science 1995, 268, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.Y.; Orr, H.T. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000, 23, 217–247. [Google Scholar] [CrossRef] [PubMed]
- Nucifora, F.C., Jr.; Sasaki, M.; Peters, M.F.; Huang, H.; Cooper, J.K.; Yamada, M.; Takahashi, H.; Tsuji, S.; Troncoso, J.; Dawson, V.L.; et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 2001, 291, 2423–2428. [Google Scholar] [CrossRef] [Green Version]
- Dunah, A.W.; Jeong, H.; Griffin, A.; Kim, Y.-M.; Standaert, D.G.; Hersch, S.M.; Mouradian, M.M.; Young, A.B.; Tanese, N.; Krainc, D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science 2002, 296, 2238–2243. [Google Scholar] [CrossRef]
- Ciechanover, A.; Brundin, P. The ubiquitin proteasome system in neurodegenerative diseases: Sometimes the chicken, sometimes the egg. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barceló, J.; O’Hoy, K.; et al. Myotonic dystrophy mutation: An unstable CTG repeat in the 3′ untranslated region of the gene. Science 1992, 255, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Ranum, L.P.; Rasmussen, P.F.; Benzow, K.A.; Koob, M.D.; Day, J.W. Genetic mapping of a second myotonic dystrophy locus. Nat. Genet. 1998, 19, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.; Grigsby, J.; Zhang, L.; Brunberg, J.A.; Greco, C.; Des Portes, V.; Jardini, T.; Levine, R.; et al. Fragile X premutation tremor/ataxia syndrome: Molecular, clinical, and neuroimaging correlates. Am. J. Hum. Genet. 2003, 72, 869–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echeverria, G.V.; Cooper, T.A. Muscleblind-like 1 activates insulin receptor exon 11 inclusion by enhancing U2AF65 binding and splicing of the upstream intron. Nucleic Acids Res. 2014, 42, 1893–1903. [Google Scholar] [CrossRef] [Green Version]
- Savkur, R.S.; Philips, A.V.; Cooper, T.A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001, 29, 40–47. [Google Scholar] [CrossRef]
- Mankodi, A.; Takahashi, M.P.; Jiang, H.; Beck, C.L.; Bowers, W.J.; Moxley, R.T.; Cannon, S.C.; Thornton, C.A. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell 2002, 10, 35–44. [Google Scholar] [CrossRef]
- Sellier, C.; Rau, F.; Liu, Y.; Tassone, F.; Hukema, R.K.; Gattoni, R.; Schneider, A.; Richard, S.; Willemsen, R.; Elliott, D.J.; et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010, 29, 1248–1261. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M. Mechanisms of cell death in oxidative stress. Antioxid. Redox Signal. 2007, 9, 49–89. [Google Scholar] [CrossRef]
- Ghosh, N.; Das, A.; Chaffee, S.; Roy, S.; Sen, C.K. Chapter 4—Reactive Oxygen Species, Oxidative Damage and Cell Death. In Immunity and Inflammation in Health and Disease; Chatterjee, S., Jungraithmayr, W., Bagchi, D., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 45–55. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int. J. Mol. Sci. 2014, 16, 193–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venditti, P.; Di Stefano, L.; Di Meo, S. Mitochondrial metabolism of reactive oxygen species. Mitochondrion 2013, 13, 71–82. [Google Scholar] [CrossRef]
- Rahal, A.; Kumar, A.; Singh, V.; Yadav, B.; Tiwari, R.; Chakraborty, S.; Dhama, K. Oxidative stress, prooxidants, and antioxidants: The interplay. Biomed. Res. Int. 2014, 2014, 761264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Autreaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell. Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef] [Green Version]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Mirończuk-Chodakowska, I.; Witkowska, A.M.; Zujko, M.E. Endogenous non-enzymatic antioxidants in the human body. Adv. Med. Sci. 2018, 63, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Haenen, G.R.M.M.; Bast, A. Glutathione revisited: A better scavenger than previously thought. Front. Pharmacol. 2014, 5, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brigelius-Flohé, R. Tissue-specific functions of individual glutathione peroxidases. Free Radic. Biol. Med. 1999, 27, 951–965. [Google Scholar] [CrossRef]
- Sheehan, D.; Meade, G.; Foley, V.M.; Dowd, C.A. Structure, function and evolution of glutathione transferases: Implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 2001, 360, 1–16. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Njålsson, R.; Norgren, S. Physiological and pathological aspects of GSH metabolism. Acta Paediatr. 2005, 94, 132–137. [Google Scholar] [CrossRef]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Shaw, P.; Chattopadhyay, A. Nrf2-ARE signaling in cellular protection: Mechanism of action and the regulatory mechanisms. J. Cell. Physiol. 2019, 235, 3119–3130. [Google Scholar] [CrossRef]
- La Rosa, P.; Bertini, E.S.; Piemonte, F. The NRF2 Signaling Network Defines Clinical Biomarkers and Therapeutic Opportunity in Friedreich’s Ataxia. Int. J. Mol. Sci. 2020, 21, 916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [Green Version]
- Amoutzias, G.D.; Robertson, D.L.; Van de Peer, Y.; Oliver, S.G. Choose your partners: Dimerization in eukaryotic transcription factors. Trends Biochem. Sci. 2008, 33, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Kataoka, K.; Itoh, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature 1994, 367, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Wasserman, W.W.; Fahl, W.E. Functional antioxidant responsive elements. Proc. Natl. Acad. Sci. USA 1997, 94, 5361–5366. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [Green Version]
- Corenblum, M.J.; Ray, S.; Remley, Q.W.; Long, M.; Harder, B.; Zhang, D.D.; Barnes, C.A.; Madhavan, L. Reduced Nrf2 expression mediates the decline in neural stem cell function during a critical middle-age period. Aging Cell 2016, 15, 725–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- La Rosa, P.; Russo, M.; D’Amico, J.; Petrillo, S.; Aquilano, K.; Lettieri-Barbato, D.; Turchi, R.; Bertini, E.S.; Piemonte, F. Nrf2 Induction Re-establishes a Proper Neuronal Differentiation Program in Friedreich’s Ataxia Neural Stem Cells. Front. Cell. Neurosci. 2019, 13, 356. [Google Scholar] [CrossRef] [Green Version]
- Robledinos-Anton, N.; Rojo, A.I.; Ferreiro, E.; Nunez, A.; Krause, K.H.; Jaquet, V.; Cuadrado, A. Transcription factor NRF2 controls the fate of neural stem cells in the subgranular zone of the hippocampus. Redox Biol. 2017, 13, 393–401. [Google Scholar] [CrossRef]
- Turchi, R.; Tortolici, F.; Guidobaldi, G.; Iacovelli, F.; Falconi, M.; Rufini, S.; Faraonio, R.; Casagrande, V.; Federici, M.; De Angelis, L.; et al. Frataxin deficiency induces lipid accumulation and affects thermogenesis in brown adipose tissue. Cell Death Dis. 2020, 11, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasai, S.; Yamazaki, H.; Tanji, K.; Engler, M.J.; Matsumiya, T.; Itoh, K. Role of the ISR-ATF4 pathway and its cross talk with Nrf2 in mitochondrial quality control. J. Clin. Biochem. Nutr. 2019, 64, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Petrillo, S.; D’Amico, J.; La Rosa, P.; Bertini, E.S.; Piemonte, F. Targeting NRF2 for the Treatment of Friedreich’s Ataxia: A Comparison among Drugs. Int. J. Mol. Sci. 2019, 20, 5211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A. Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/beta-TrCP. Free Radic. Biol. Med. 2015, 88, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/beta-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Liu, Y.; Pang, Y.; Caisova, V.; Ding, J.; Yu, D.; Zhou, Y.; Huynh, T.T.; Ghayee, H.; Pacak, K.; Yang, C. Targeting NRF2-Governed Glutathione Synthesis for SDHB-Mutated Pheochromocytoma and Paraganglioma. Cancers 2020, 12, 280. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lu, Y.; Celiku, O.; Li, A.; Wu, Q.; Zhou, Y.; Yang, C. Targeting IDH1-Mutated Malignancies with NRF2 Blockade. J. Natl. Cancer Inst. 2019, 111, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Nrf2, cellular redox regulation, and neurologic implications. Neurology 2017, 88, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdalkader, M.; Lampinen, R.; Kanninen, K.M.; Malm, T.M.; Liddell, J.R. Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front. Neurosci. 2018, 12, 466. [Google Scholar] [CrossRef] [Green Version]
- Basak, P.; Sadhukhan, P.; Sarkar, P.; Sil, P.C. Perspectives of the Nrf-2 signaling pathway in cancer progression and therapy. Toxicol. Rep. 2017, 4, 306–318. [Google Scholar] [CrossRef]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef]
- Kitamura, H.; Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 2018, 109, 900–911. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Sarlette, A.; Krampfl, K.; Grothe, C.; Neuhoff, N.; Dengler, R.; Petri, S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef] [PubMed]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Opportunities. Antioxid. Redox Signal. 2019, 30, 1450–1499. [Google Scholar] [CrossRef] [PubMed]
- Cossée, M.; Schmitt, M.; Campuzano, V.; Reutenauer, L.; Moutou, C.; Mandel, J.L.; Koenig, M. Evolution of the Friedreich’s ataxia trinucleotide repeat expansion: Founder effect and premutations. Proc. Natl. Acad. Sci. USA 1997, 94, 7452–7457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Napierala, M.; Dent, S.Y. Hyperexpansion of GAA repeats affects post-initiation steps of FXN transcription in Friedreich’s ataxia. Nucleic Acids Res. 2011, 39, 8366–8377. [Google Scholar] [CrossRef] [Green Version]
- Koeppen, A.H. Friedreich’s ataxia: Pathology, pathogenesis, and molecular genetics. J. Neurol. Sci. 2011, 303, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pandolfo, M.; Pastore, A. The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J. Neurol. 2009, 256 (Suppl. 1), 9–17. [Google Scholar] [CrossRef]
- Parkinson, M.H.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Giunti, P. Clinical features of Friedreich’s ataxia: Classical and atypical phenotypes. J. Neurochem. 2013, 126 (Suppl. 1), 103–117. [Google Scholar] [CrossRef]
- Puccio, H.; Simon, D.; Cossee, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef]
- Anzovino, A.; Lane, D.J.; Huang, M.L.; Richardson, D.R. Fixing frataxin: ‘ironing out’ the metabolic defect in Friedreich’s ataxia. Br. J. Pharmacol. 2014, 171, 2174–2190. [Google Scholar] [CrossRef] [Green Version]
- Vaubel, R.A.; Isaya, G. Iron-sulfur cluster synthesis, iron homeostasis and oxidative stress in Friedreich ataxia. Mol. Cell. Neurosci. 2013, 55, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Koenig, M.; Mandel, J.L. Deciphering the cause of Friedreich ataxia. Curr. Opin. Neurobiol. 1997, 7, 689–694. [Google Scholar] [CrossRef]
- Gomes, C.M.; Santos, R. Neurodegeneration in Friedreich’s ataxia: From defective frataxin to oxidative stress. Oxid. Med. Cell. Longev. 2013, 2013, 487534. [Google Scholar] [CrossRef]
- Carletti, B.; Piemonte, F. Friedreich’s Ataxia: A Neuronal Point of View on the Oxidative Stress Hypothesis. Antioxidants 2014, 3, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Lupoli, F.; Vannocci, T.; Longo, G.; Niccolai, N.; Pastore, A. The role of oxidative stress in Friedreich’s ataxia. FEBS Lett. 2018, 592, 718–727. [Google Scholar] [CrossRef]
- Anzovino, A.; Chiang, S.; Brown, B.E.; Hawkins, C.L.; Richardson, D.R.; Huang, M.L. Molecular Alterations in a Mouse Cardiac Model of Friedreich Ataxia: An Impaired Nrf2 Response Mediated via Upregulation of Keap1 and Activation of the Gsk3β Axis. Am. J. Pathol. 2017, 187, 2858–2875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paupe, V.; Dassa, E.P.; Goncalves, S.; Auchere, F.; Lonn, M.; Holmgren, A.; Rustin, P. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS ONE 2009, 4, e4253. [Google Scholar] [CrossRef] [Green Version]
- D’Oria, V.; Petrini, S.; Travaglini, L.; Priori, C.; Piermarini, E.; Petrillo, S.; Carletti, B.; Bertini, E.; Piemonte, F. Frataxin deficiency leads to reduced expression and impaired translocation of NF-E2-related factor (Nrf2) in cultured motor neurons. Int. J. Mol. Sci. 2013, 14, 7853–7865. [Google Scholar] [CrossRef] [Green Version]
- Shan, Y.; Schoenfeld, R.A.; Hayashi, G.; Napoli, E.; Akiyama, T.; Iodi Carstens, M.; Carstens, E.E.; Pook, M.A.; Cortopassi, G.A. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxid. Redox Signal. 2013, 19, 1481–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emond, M.; Lepage, G.; Vanasse, M.; Pandolfo, M. Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology 2000, 55, 1752–1753. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.B.; Dehmer, T.; Schöls, L.; Mende, H.; Hardt, C.; Vorgerd, M.; Bürk, K.; Matson, W.; Dichgans, J.; Beal, M.F.; et al. Oxidative stress in patients with Friedreich ataxia. Neurology 2000, 55, 1719–1721. [Google Scholar] [CrossRef]
- Tozzi, G.; Nuccetelli, M.; Lo Bello, M.; Bernardini, S.; Bellincampi, L.; Ballerini, S.; Gaeta, L.M.; Casali, C.; Pastore, A.; Federici, G.; et al. Antioxidant enzymes in blood of patients with Friedreich’s ataxia. Arch. Dis. Child. 2002, 86, 376–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients 2012, 4, 1399–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotticelli, M.G.; Crabbe, A.M.; Wilson, R.B.; Shchepinov, M.S. Insights into the role of oxidative stress in the pathology of Friedreich ataxia using peroxidation resistant polyunsaturated fatty acids. Redox Biol. 2013, 1, 398–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotticelli, M.G.; Xia, S.; Lin, D.; Lee, T.; Terrab, L.; Wipf, P.; Huryn, D.M.; Wilson, R.B. Ferroptosis as a Novel Therapeutic Target for Friedreich’s Ataxia. J. Pharmacol. Exp. Ther. 2019, 369, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Abeti, R.; Parkinson, M.H.; Hargreaves, I.P.; Angelova, P.R.; Sandi, C.; Pook, M.A.; Giunti, P.; Abramov, A.Y. ‘Mitochondrial energy imbalance and lipid peroxidation cause cell death in Friedreich’s ataxia’. Cell Death Dis. 2016, 7, e2237. [Google Scholar] [CrossRef]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Esteras, N.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 activation in the treatment of neurodegenerative diseases: A focus on its role in mitochondrial bioenergetics and function. Biol. Chem. 2016, 397, 383–400. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Klomparens, E.A.; Ding, Y. The neuroprotective mechanisms and effects of sulforaphane. Brain Circ. 2019, 5, 74–83. [Google Scholar] [CrossRef]
- Uddin, M.S.; Mamun, A.A.; Jakaria, M.; Thangapandiyan, S.; Ahmad, J.; Rahman, M.A.; Mathew, B.; Abdel-Daim, M.M.; Aleya, L. Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Sci. Total Environ. 2020, 707, 135624. [Google Scholar] [CrossRef]
- Scannevin, R.H.; Chollate, S.; Jung, M.Y.; Shackett, M.; Patel, H.; Bista, P.; Zeng, W.; Ryan, S.; Yamamoto, M.; Lukashev, M.; et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther. 2012, 341, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montes Diaz, G.; Hupperts, R.; Fraussen, J.; Somers, V. Dimethyl fumarate treatment in multiple sclerosis: Recent advances in clinical and immunological studies. Autoimmun. Rev. 2018, 17, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Ranea-Robles, P.; Launay, N.; Ruiz, M.; Calingasan, N.Y.; Dumont, M.; Naudi, A.; Portero-Otin, M.; Pamplona, R.; Ferrer, I.; Beal, M.F.; et al. Aberrant regulation of the GSK-3beta/NRF2 axis unveils a novel therapy for adrenoleukodystrophy. EMBO Mol. Med. 2018, 10, e8604. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; Piermarini, E.; Pastore, A.; Vasco, G.; Schirinzi, T.; Carrozzo, R.; Bertini, E.; Piemonte, F. Nrf2-Inducers Counteract Neurodegeneration in Frataxin-Silenced Motor Neurons: Disclosing New Therapeutic Targets for Friedreich’s Ataxia. Int. J. Mol. Sci. 2017, 18, 2173. [Google Scholar] [CrossRef] [Green Version]
- Jasoliya, M.; Sacca, F.; Sahdeo, S.; Chedin, F.; Pane, C.; Brescia Morra, V.; Filla, A.; Pook, M.; Cortopassi, G. Dimethyl fumarate dosing in humans increases frataxin expression: A potential therapy for Friedreich’s Ataxia. PLoS ONE 2019, 14, e0217776. [Google Scholar] [CrossRef]
- Sahdeo, S.; Scott, B.D.; McMackin, M.Z.; Jasoliya, M.; Brown, B.; Wulff, H.; Perlman, S.L.; Pook, M.A.; Cortopassi, G.A. Dyclonine rescues frataxin deficiency in animal models and buccal cells of patients with Friedreich’s ataxia. Hum. Mol. Genet. 2014, 23, 6848–6862. [Google Scholar] [CrossRef] [Green Version]
- Clay, A.; Hearle, P.; Schadt, K.; Lynch, D.R. New developments in pharmacotherapy for Friedreich ataxia. Expert Opin. Pharmacother. 2019, 20, 1855–1867. [Google Scholar] [CrossRef]
- Hausse, A.O.; Aggoun, Y.; Bonnet, D.; Sidi, D.; Munnich, A.; Rotig, A.; Rustin, P. Idebenone and reduced cardiac hypertrophy in Friedreich’s ataxia. Heart 2002, 87, 346–349. [Google Scholar] [CrossRef]
- Di Prospero, N.A.; Baker, A.; Jeffries, N.; Fischbeck, K.H. Neurological effects of high-dose idebenone in patients with Friedreich’s ataxia: A randomised, placebo-controlled trial. Lancet Neurol. 2007, 6, 878–886. [Google Scholar] [CrossRef] [Green Version]
- Montenegro, L.; Turnaturi, R.; Parenti, C.; Pasquinucci, L. Idebenone: Novel Strategies to Improve Its Systemic and Local Efficacy. Nanomaterials 2018, 8, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, D.R.; Farmer, J.; Hauser, L.; Blair, I.A.; Wang, Q.Q.; Mesaros, C.; Snyder, N.; Boesch, S.; Chin, M.; Delatycki, M.B.; et al. Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2019, 6, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Zesiewicz, T.; Salemi, J.L.; Perlman, S.; Sullivan, K.L.; Shaw, J.D.; Huang, Y.; Isaacs, C.; Gooch, C.; Lynch, D.R.; Klein, M.B. Double-blind, randomized and controlled trial of EPI-743 in Friedreich’s ataxia. Neurodegener. Dis. Manag. 2018, 8, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef]
- Bell, M.V.; Hirst, M.C.; Nakahori, Y.; MacKinnon, R.N.; Roche, A.; Flint, T.J.; Jacobs, P.A.; Tommerup, N.; Tranebjaerg, L.; Froster-Iskenius, U.; et al. Physical mapping across the fragile X: Hypermethylation and clinical expression of the fragile X syndrome. Cell 1991, 64, 861–866. [Google Scholar] [CrossRef]
- Myrick, L.K.; Nakamoto-Kinoshita, M.; Lindor, N.M.; Kirmani, S.; Cheng, X.; Warren, S.T. Fragile X syndrome due to a missense mutation. Eur. J. Hum. Genet. 2014, 22, 1185–1189. [Google Scholar] [CrossRef] [Green Version]
- Quartier, A.; Poquet, H.; Gilbert-Dussardier, B.; Rossi, M.; Casteleyn, A.S.; Portes, V.D.; Feger, C.; Nourisson, E.; Kuentz, P.; Redin, C.; et al. Intragenic FMR1 disease-causing variants: A significant mutational mechanism leading to Fragile-X syndrome. Eur. J. Hum. Genet. 2017, 25, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiraanont, P.; Kumar, M.; Tang, H.-T.; Espinal, G.; Hagerman, P.J.; Hagerman, R.J.; Chutabhakdikul, N.; Tassone, F. Size and methylation mosaicism in males with Fragile X syndrome. Expert Rev. Mol. Diagn. 2017, 17, 1023–1032. [Google Scholar] [CrossRef] [Green Version]
- Abbeduto, L.; McDuffie, A.; Thurman, A.J. The fragile X syndrome-autism comorbidity: What do we really know? Front. Genet. 2014, 5, 355. [Google Scholar] [CrossRef] [Green Version]
- Dyer-Friedman, J.; Glaser, B.; Hessl, D.; Johnston, C.; Huffman, L.C.; Taylor, A.; Wisbeck, J.; Reiss, A.L. Genetic and environmental influences on the cognitive outcomes of children with fragile X syndrome. J. Am. Acad. Child Adolesc. Psychiatry 2002, 41, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Loesch, D.Z.; Huggins, R.M.; Hagerman, R.J. Phenotypic variation and FMRP levels in fragile X. Ment. Retard. Dev. Disabil. Res. Rev. 2004, 10, 31–41. [Google Scholar] [CrossRef]
- Bartholomay, K.L.; Lee, C.H.; Bruno, J.L.; Lightbody, A.A.; Reiss, A.L. Closing the Gender Gap in Fragile X Syndrome: Review on Females with FXS and Preliminary Research Findings. Brain Sci. 2019, 9, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardoni, B.; Schenck, A.; Mandel, J.-L. The Fragile X mental retardation protein. Brain Res. Bull. 2001, 56, 375–382. [Google Scholar] [CrossRef]
- Devys, D.; Lutz, Y.; Rouyer, N.; Bellocq, J.-P.; Mandel, J.-L. The FMR–1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat. Genet. 1993, 4, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Gutekunst, C.A.; Eberhart, D.E.; Yi, H.; Warren, S.T.; Hersch, S.M. Fragile X mental retardation protein: Nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J. Neurosci. 1997, 17, 1539–1547. [Google Scholar] [CrossRef]
- Ferrari, F.; Mercaldo, V.; Piccoli, G.; Sala, C.; Cannata, S.; Achsel, T.; Bagni, C. The fragile X mental retardation protein-RNP granules show an mGluR-dependent localization in the post-synaptic spines. Mol. Cell. Neurosci. 2007, 34, 343–354. [Google Scholar] [CrossRef]
- Dictenberg, J.B.; Swanger, S.A.; Antar, L.N.; Singer, R.H.; Bassell, G.J. A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev. Cell 2008, 14, 926–939. [Google Scholar] [CrossRef] [Green Version]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef] [Green Version]
- Nakamoto, M.; Nalavadi, V.; Epstein, M.P.; Narayanan, U.; Bassell, G.J.; Warren, S.T. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 15537–15542. [Google Scholar] [CrossRef] [Green Version]
- Eadie, B.D.; Cushman, J.; Kannangara, T.S.; Fanselow, M.S.; Christie, B.R. NMDA receptor hypofunction in the dentate gyrus and impaired context discrimination in adult Fmr1 knockout mice. Hippocampus 2012, 22, 241–254. [Google Scholar] [CrossRef]
- Centonze, D.; Rossi, S.; Mercaldo, V.; Napoli, I.; Ciotti, M.T.; De Chiara, V.; Musella, A.; Prosperetti, C.; Calabresi, P.; Bernardi, G.; et al. Abnormal striatal GABA transmission in the mouse model for the fragile X syndrome. Biol. Psychiatry 2008, 63, 963–973. [Google Scholar] [CrossRef]
- Fernandez, E.; Rajan, N.; Bagni, C. The FMRP regulon: From targets to disease convergence. Front. Neurosci. 2013, 7, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, V.; Jin, P.; Ceman, S.; Darnell, J.C.; O’Donnell, W.T.; Tenenbaum, S.A.; Jin, X.; Feng, Y.; Wilkinson, K.D.; Keene, J.D.; et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 2001, 107, 477–487. [Google Scholar] [CrossRef] [Green Version]
- Napoli, I.; Mercaldo, V.; Boyl, P.P.; Eleuteri, B.; Zalfa, F.; De Rubeis, S.; Di Marino, D.; Mohr, E.; Massimi, M.; Falconi, M.; et al. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 2008, 134, 1042–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, U.; Nalavadi, V.; Nakamoto, M.; Pallas, D.C.; Ceman, S.; Bassell, G.J.; Warren, S.T. FMRP phosphorylation reveals an immediate-early signaling pathway triggered by group I mGluR and mediated by PP2A. J. Neurosci. 2007, 27, 14349–14357. [Google Scholar] [CrossRef]
- Muddashetty, R.S.; Kelić, S.; Gross, C.; Xu, M.; Bassell, G.J. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J. Neurosci. 2007, 27, 5338–5348. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pelletier, M.R.; Perez Velazquez, J.L.; Carlen, P.L. Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol. Cell. Neurosci. 2002, 19, 138–151. [Google Scholar] [CrossRef]
- Serrano, F.; Klann, E. Reactive oxygen species and synaptic plasticity in the aging hippocampus. Ageing Res. Rev. 2004, 3, 431–443. [Google Scholar] [CrossRef]
- Sidorov, M.S.; Auerbach, B.D.; Bear, M.F. Fragile X mental retardation protein and synaptic plasticity. Mol. Brain 2013, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, A.; Chauhan, V. Oxidative stress in autism. Pathophysiology 2006, 13, 171–181. [Google Scholar] [CrossRef]
- Gingrich, J.A. Oxidative stress is the new stress. Nat. Med. 2005, 11, 1281–1282. [Google Scholar] [CrossRef]
- Bouayed, J.; Rammal, H.; Soulimani, R. Oxidative stress and anxiety: Relationship and cellular pathways. Oxid. Med. Cell. Longev. 2009, 2, 63–67. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [Green Version]
- El Bekay, R.; Romero-Zerbo, Y.; Decara, J.; Sanchez-Salido, L.; Del Arco-Herrera, I.; Rodríguez-de Fonseca, F.; de Diego-Otero, Y. Enhanced markers of oxidative stress, altered antioxidants and NADPH-oxidase activation in brains from Fragile X mental retardation 1-deficient mice, a pathological model for Fragile X syndrome. Eur. J. Neurosci. 2007, 26, 3169–3180. [Google Scholar] [CrossRef] [PubMed]
- Miyashiro, K.Y.; Beckel-Mitchener, A.; Purk, T.P.; Becker, K.G.; Barret, T.; Liu, L.; Carbonetto, S.; Weiler, I.J.; Greenough, W.T.; Eberwine, J. RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron 2003, 37, 417–431. [Google Scholar] [CrossRef] [Green Version]
- Bechara, E.G.; Didiot, M.C.; Melko, M.; Davidovic, L.; Bensaid, M.; Martin, P.; Castets, M.; Pognonec, P.; Khandjian, E.W.; Moine, H.; et al. A novel function for fragile X mental retardation protein in translational activation. PLoS Biol. 2009, 7, e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-κB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef] [Green Version]
- Marei, H.; Malliri, A. Rac1 in human diseases: The therapeutic potential of targeting Rac1 signaling regulatory mechanisms. Small GTPases 2017, 8, 139–163. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Terada, M.; Ariyoshi, K.; Morimoto, K. Monocyte chemoattractant protein-1/CC chemokine ligand 2 enhances apoptotic cell removal by macrophages through Rac1 activation. Biochem. Biophys. Res. Commun. 2010, 399, 677–682. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, F.; Chen, F.; Liu, D.; Zheng, Y.; Zhang, Y.; Dong, C.; Su, B. MEKK3 regulates IFN-gamma production in T cells through the Rac1/2-dependent MAPK cascades. J. Immunol. 2011, 186, 5791–5800. [Google Scholar] [CrossRef]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [Green Version]
- Beitel, L.; Alvarado, C.; Mokhtar, S.; Paliouras, M.; Trifiro, M. Mechanisms Mediating Spinal and Bulbar Muscular Atrophy: Investigations into Polyglutamine-Expanded Androgen Receptor Function and Dysfunction. Front. Neurol. 2013, 4, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, L.E.; Freeman, B.K.; Auh, S.; Kokkinis, A.D.; La Pean, A.; Chen, C.; Lehky, T.J.; Shrader, J.A.; Levy, E.W.; Harris-Love, M.; et al. Clinical features of spinal and bulbar muscular atrophy. Brain J. Neurol. 2009, 132, 3242–3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejager, S.; Bry-Gauillard, H.; Bruckert, E.; Eymard, B.; Salachas, F.; LeGuern, E.; Tardieu, S.; Chadarevian, R.; Giral, P.; Turpin, G. A comprehensive endocrine description of Kennedy’s disease revealing androgen insensitivity linked to CAG repeat length. J. Clin. Endocrinol. Metab. 2002, 87, 3893–3901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonini, G.; Gragnani, F.; Romaniello, A.; Pennisi, E.M.; Morino, S.; Ceschin, V.; Santoro, L.; Cruccu, G. Sensory involvement in spinal-bulbar muscular atrophy (Kennedy’s disease). Muscle Nerve 2000, 23, 252–258. [Google Scholar] [CrossRef]
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; Harding, A.E.; Fischbeck, K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991, 352, 77–79. [Google Scholar] [CrossRef]
- Pellegrini, M.; Bulzomi, P.; Lecis, M.; Leone, S.; Campesi, I.; Franconi, F.; Marino, M. Endocrine Disruptors Differently Influence Estrogen Receptor β and Androgen Receptor in Male and Female Rat VSMC. J. Cell. Physiol. 2014, 229, 1061–1068. [Google Scholar] [CrossRef]
- Rusmini, P.; Sau, D.; Crippa, V.; Palazzolo, I.; Simonini, F.; Onesto, E.; Martini, L.; Poletti, A. Aggregation and proteasome: The case of elongated polyglutamine aggregation in spinal and bulbar muscular atrophy. Neurobiol. Aging 2007, 28, 1099–1111. [Google Scholar] [CrossRef]
- Schindler, M.; Fabre, C.; de Weille, J.; Carreau, S.; Mersel, M.; Bakalara, N. Disruption of Nongenomic Testosterone Signaling in a Model of Spinal and Bulbar Muscular Atrophy. Mol. Endocrinol. 2012, 26, 1102–1116. [Google Scholar] [CrossRef] [Green Version]
- Davies, P.; Watt, K.; Kelly, S.M.; Clark, C.; Price, N.C.; McEwan, I.J. Consequences of poly-glutamine repeat length for the conformation and folding of the androgen receptor amino-terminal domain. J. Mol. Endocrinol. 2008, 41, 301–314. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, A.P.; Harmison, G.; Strand, A.D.; Olson, J.M.; Fischbeck, K.H. Altered transcriptional regulation in cells expressing the expanded polyglutamine androgen receptor. Hum. Mol. Genet. 2002, 11, 1967–1976. [Google Scholar] [CrossRef] [Green Version]
- McCampbell, A.; Taylor, J.P.; Taye, A.A.; Robitschek, J.; Li, M.; Walcott, J.; Merry, D.; Chai, Y.; Paulson, H.; Sobue, G.; et al. CREB-binding protein sequestration by expanded polyglutamine. Hum. Mol. Genet. 2000, 9, 2197–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, B.J.; Greenberg, C.R.; Allingham-Hawkins, D.J.; Spriggs, E.L. Expression of X-linked bulbospinal muscular atrophy (Kennedy disease) in two homozygous women. Neurology 2002, 59, 770–772. [Google Scholar] [CrossRef] [PubMed]
- Takeyama, K.; Ito, S.; Yamamoto, A.; Tanimoto, H.; Furutani, T.; Kanuka, H.; Miura, M.; Tabata, T.; Kato, S. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron 2002, 35, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Katsuno, M.; Adachi, H.; Kume, A.; Li, M.; Nakagomi, Y.; Niwa, H.; Sang, C.; Kobayashi, Y.; Doyu, M.; Sobue, G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 2002, 35, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Cary, G.A.; La Spada, A.R. Androgen Receptor Function in Motor Neuron Survival and Degeneration. Phys. Med. Rehabil. Clin. N. Am. 2008, 19, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Mitsuma, N.; Inukai, A.; Ito, Y.; Li, M.; Mitsuma, T.; Sobue, G. Expression of GDNF and GDNFR-alpha mRNAs in muscles of patients with motor neuron diseases. Neurochem. Res. 1999, 24, 785–790. [Google Scholar] [CrossRef]
- Yu, Z.; Dadgar, N.; Albertelli, M.; Gruis, K.; Jordan, C.; Robins, D.M.; Lieberman, A.P. Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J. Clin. Investig. 2006, 116, 2663–2672. [Google Scholar] [CrossRef]
- Sopher, B.L.; Thomas, P.S., Jr.; LaFevre-Bernt, M.A.; Holm, I.E.; Wilke, S.A.; Ware, C.B.; Jin, L.W.; Libby, R.T.; Ellerby, L.M.; La Spada, A.R. Androgen receptor YAC transgenic mice recapitulate SBMA motor neuronopathy and implicate VEGF164 in the motor neuron degeneration. Neuron 2004, 41, 687–699. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, S.; Harmison, G.G.; Meyertholen, K.; Pennuto, M.; Burnett, B.G.; Fischbeck, K.H. Mitochondrial abnormalities in spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2009, 18, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Finsterer, J.; Mishra, A.; Wakil, S.; Pennuto, M.; Soraru, G. Mitochondrial implications in bulbospinal muscular atrophy (Kennedy disease). Amyotroph. Lateral Scler. Frontotemporal Degener. 2015, 17, 112–118. [Google Scholar] [CrossRef]
- Gavrilova-Jordan, L.P.; Price, T.M. Actions of steroids in mitochondria. Semin. Reprod. Med. 2007, 25, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Stenoien, D.L.; Cummings, C.J.; Adams, H.P.; Mancini, M.G.; Patel, K.; DeMartino, G.N.; Marcelli, M.; Weigel, N.L.; Mancini, M.A. Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum. Mol. Genet. 1999, 8, 731–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeoni, S.; Mancini, M.A.; Stenoien, D.L.; Marcelli, M.; Weigel, N.L.; Zanisi, M.; Martini, L.; Poletti, A. Motoneuronal cell death is not correlated with aggregate formation of androgen receptors containing an elongated polyglutamine tract. Hum. Mol. Genet. 2000, 9, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisodia, S.S. Nuclear inclusions in glutamine repeat disorders: Are they pernicious, coincidental, or beneficial? Cell 1998, 95, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Piccioni, F.; Pinton, P.; Simeoni, S.; Pozzi, P.; Fascio, U.; Vismara, G.; Martini, L.; Rizzuto, R.; Poletti, A. Androgen receptor with elongated polyglutamine tract forms aggregates that alter axonal trafficking and mitochondrial distribution in motor neuronal processes. FASEB J. 2002, 16, 1418–1420. [Google Scholar] [CrossRef] [Green Version]
- Beauchemin, A.M.J.; Gottlieb, B.; Beitel, L.K.; Elhaji, Y.A.; Pinsky, L.; Trifiro, M.A. Cytochrome c oxidase subunit Vb interacts with human androgen receptor: A potential mechanism for neurotoxicity in spinobulbar muscular atrophy. Brain Res. Bull. 2001, 56, 285–297. [Google Scholar] [CrossRef]
- Malik, B.; Devine, H.; Patani, R.; La Spada, A.R.; Hanna, M.G.; Greensmith, L. Gene expression analysis reveals early dysregulation of disease pathways and links Chmp7 to pathogenesis of spinal and bulbar muscular atrophy. Sci. Rep. 2019, 9, 3539. [Google Scholar] [CrossRef] [Green Version]
- Alavez, S.; Vantipalli, M.C.; Zucker, D.J.S.; Klang, I.M.; Lithgow, G.J. Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 2011, 472, 226–229. [Google Scholar] [CrossRef] [Green Version]
- Calamini, B.; Silva, M.C.; Madoux, F.; Hutt, D.M.; Khanna, S.; Chalfant, M.A.; Saldanha, S.A.; Hodder, P.; Tait, B.D.; Garza, D.; et al. Small-molecule proteostasis regulators for protein conformational diseases. Nat. Chem. Biol. 2012, 8, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Porat, Y.; Abramowitz, A.; Gazit, E. Inhibition of amyloid fibril formation by polyphenols: Structural similarity and aromatic interactions as a common inhibition mechanism. Chem. Biol. Drug Des. 2006, 67, 27–37. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Massiah, M.A.; Bozak, R.E.; Hicks, R.J.; Talalay, P. Potency of Michael reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulfhydryl groups. Proc. Natl. Acad. Sci. USA 2001, 98, 3404–3409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, P.; Paital, B.; Jena, S.; Swain, S.S.; Kumar, S.; Yadav, M.K.; Chainy, G.B.N.; Samanta, L. Possible activation of NRF2 by Vitamin E/Curcumin against altered thyroid hormone induced oxidative stress via NFκB/AKT/mTOR/KEAP1 signalling in rat heart. Sci. Rep. 2019, 9, 7408. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Chang, Y.-J.; Yu, I.C.; Yeh, S.; Wu, C.-C.; Miyamoto, H.; Merry, D.E.; Sobue, G.; Chen, L.-M.; Chang, S.-S.; et al. ASC-J9 ameliorates spinal and bulbar muscular atrophy phenotype via degradation of androgen receptor. Nat. Med. 2007, 13, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Bott, L.C.; Badders, N.M.; Chen, K.L.; Harmison, G.G.; Bautista, E.; Shih, C.C.; Katsuno, M.; Sobue, G.; Taylor, J.P.; Dantuma, N.P.; et al. A small-molecule Nrf1 and Nrf2 activator mitigates polyglutamine toxicity in spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2016, 25, 1979–1989. [Google Scholar] [CrossRef] [Green Version]
- Furtado, S.; Suchowersky, O.; Rewcastle, B.; Graham, L.; Klimek, M.L.; Garber, A. Relationship between trinucleotide repeats and neuropathological changes in Huntington’s disease. Ann. Neurol. 1996, 39, 132–136. [Google Scholar] [CrossRef]
- Snell, R.G.; MacMillan, J.C.; Cheadle, J.P.; Fenton, I.; Lazarou, L.P.; Davies, P.; MacDonald, M.E.; Gusella, J.F.; Harper, P.S.; Shaw, D.J. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat. Genet. 1993, 4, 393–397. [Google Scholar] [CrossRef] [PubMed]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Ravina, B.; Romer, M.; Constantinescu, R.; Biglan, K.; Brocht, A.; Kieburtz, K.; Shoulson, I.; McDermott, M.P. The relationship between CAG repeat length and clinical progression in Huntington’s disease. Mov. Disord. 2008, 23, 1223–1227. [Google Scholar] [CrossRef]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894. [Google Scholar] [CrossRef] [Green Version]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Shacham, T.; Sharma, N.; Lederkremer, G.Z. Protein Misfolding and ER Stress in Huntington’s Disease. Front. Mol. Biosci. 2019, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallardo-Orihuela, A.; Hervás-Corpión, I.; Hierro-Bujalance, C.; Sanchez-Sotano, D.; Jiménez-Gómez, G.; Mora-López, F.; Campos-Caro, A.; Garcia-Alloza, M.; Valor, L.M. Transcriptional correlates of the pathological phenotype in a Huntington’s disease mouse model. Sci. Rep. 2019, 9, 18696. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Baranov, S.V.; Baranova, O.V.; Kim, J.; Pan, Y.; Yablonska, S.; Carlisle, D.L.; Ferrante, R.J.; Kim, A.H.; Friedlander, R.M. Inhibition of mitochondrial protein import by mutant huntingtin. Nat. Neurosci. 2014, 17, 822–831. [Google Scholar] [CrossRef]
- Stack, E.C.; Matson, W.R.; Ferrante, R.J. Evidence of oxidant damage in Huntington’s disease: Translational strategies using antioxidants. Ann. N. Y. Acad. Sci. 2008, 1147, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Fox, J.H. Novel proteomic changes in brain mitochondria provide insights into mitochondrial dysfunction in mouse models of Huntington’s disease. Mitochondrion 2019, 47, 318–329. [Google Scholar] [CrossRef]
- Bogdanov, M.B.; Ferrante, R.J.; Kuemmerle, S.; Klivenyi, P.; Beal, M.F. Increased vulnerability to 3-nitropropionic acid in an animal model of Huntington’s disease. J. Neurochem. 1998, 71, 2642–2644. [Google Scholar] [CrossRef] [Green Version]
- Brouillet, E. The 3-NP Model of Striatal Neurodegeneration. Curr. Protoc. Neurosci. 2014, 67, 9.48.1–9.48.14. [Google Scholar] [CrossRef]
- Rosenstock, T.R.; Carvalho, A.C.; Jurkiewicz, A.; Frussa-Filho, R.; Smaili, S.S. Mitochondrial calcium, oxidative stress and apoptosis in a neurodegenerative disease model induced by 3-nitropropionic acid. J. Neurochem. 2004, 88, 1220–1228. [Google Scholar] [CrossRef]
- Browne, S.E.; Beal, M.F. Oxidative damage in Huntington’s disease pathogenesis. Antioxid. Redox Signal. 2006, 8, 2061–2073. [Google Scholar] [CrossRef]
- Maiuri, T.; Bowie, L.E.; Truant, R. DNA Repair Signaling of Huntingtin: The Next Link Between Late-Onset Neurodegenerative Disease and Oxidative DNA Damage. DNA Cell Biol. 2019, 38, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Wu, Y.R.; Cheng, M.L.; Liu, J.L.; Lee, Y.M.; Lee, P.W.; Soong, B.W.; Chiu, D.T. Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem. Biophys. Res. Commun. 2007, 359, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-López, F.; Tasset, I.; Agüera, E.; Feijóo, M.; Fernández-Bolaños, R.; Sánchez, F.M.; Ruiz, M.C.; Cruz, A.H.; Gascón, F.; Túnez, I. Oxidative stress and inflammation biomarkers in the blood of patients with Huntington’s disease. Neurol. Res. 2012, 34, 721–724. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Fox, J.; Thyagarajan, B.; Fox, J.H. Brain mitochondrial iron accumulates in Huntington’s disease, mediates mitochondrial dysfunction, and can be removed pharmacologically. Free Radic. Biol. Med. 2018, 120, 317–329. [Google Scholar] [CrossRef]
- Peyser, C.E.; Folstein, M.; Chase, G.A.; Starkstein, S.; Brandt, J.; Cockrell, J.R.; Bylsma, F.; Coyle, J.T.; McHugh, P.R.; Folstein, S.E. Trial of d-alpha-tocopherol in Huntington’s disease. Am. J. Psychiatry 1995, 152, 1771–1775. [Google Scholar] [CrossRef]
- Kasparová, S.; Sumbalová, Z.; Bystrický, P.; Kucharská, J.; Liptaj, T.; Mlynárik, V.; Gvozdjáková, A. Effect of coenzyme Q10 and vitamin E on brain energy metabolism in the animal model of Huntington’s disease. Neurochem. Int. 2006, 48, 93–99. [Google Scholar] [CrossRef]
- Rebec, G.V.; Barton, S.J.; Marseilles, A.M.; Collins, K. Ascorbate treatment attenuates the Huntington behavioral phenotype in mice. Neuroreport 2003, 14, 1263–1265. [Google Scholar] [CrossRef]
- Wright, D.J.; Gray, L.J.; Finkelstein, D.I.; Crouch, P.J.; Pow, D.; Pang, T.Y.; Li, S.; Smith, Z.M.; Francis, P.S.; Renoir, T.; et al. N-acetylcysteine modulates glutamatergic dysfunction and depressive behavior in Huntington’s disease. Hum. Mol. Genet. 2016, 25, 2923–2933. [Google Scholar] [CrossRef] [Green Version]
- Wright, D.J.; Renoir, T.; Smith, Z.M.; Frazier, A.E.; Francis, P.S.; Thorburn, D.R.; McGee, S.L.; Hannan, A.J.; Gray, L.J. N-Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease. Transl. Psychiatry 2015, 5, e492. [Google Scholar] [CrossRef] [Green Version]
- Sandhir, R.; Sood, A.; Mehrotra, A.; Kamboj, S.S. N-Acetylcysteine reverses mitochondrial dysfunctions and behavioral abnormalities in 3-nitropropionic acid-induced Huntington’s disease. Neurodegener. Dis. 2012, 9, 145–157. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Ferrante, R.J.; Dedeoglu, A.; Beal, M.F. Lipoic acid improves survival in transgenic mouse models of Huntington’s disease. Neuroreport 2001, 12, 3371–3373. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hettinger, C.L.; Zhang, D.; Rezvani, K.; Wang, X.; Wang, H. Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington’s disease. J. Neurochem. 2014, 129, 539–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinti, L.; Dayalan Naidu, S.; Träger, U.; Chen, X.; Kegel-Gleason, K.; Llères, D.; Connolly, C.; Chopra, V.; Low, C.; Moniot, S.; et al. KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc. Natl. Acad. Sci. USA 2017, 114, E4676–E4685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- Ellrichmann, G.; Petrasch-Parwez, E.; Lee, D.H.; Reick, C.; Arning, L.; Saft, C.; Gold, R.; Linker, R.A. Efficacy of fumaric acid esters in the R6/2 and YAC128 models of Huntington’s disease. PLoS ONE 2011, 6, e16172. [Google Scholar] [CrossRef]
- Jang, M.; Choi, J.H.; Chang, Y.; Lee, S.J.; Nah, S.Y.; Cho, I.H. Gintonin, a ginseng-derived ingredient, as a novel therapeutic strategy for Huntington’s disease: Activation of the Nrf2 pathway through lysophosphatidic acid receptors. Brain Behav. Immun. 2019, 80, 146–162. [Google Scholar] [CrossRef]
- Taroni, F.; DiDonato, S. Pathways to motor incoordination: The inherited ataxias. Nat. Rev. Neurosci. 2004, 5, 641–655. [Google Scholar] [CrossRef]
- Koeppen, A.H. The pathogenesis of spinocerebellar ataxia. Cerebellum 2005, 4, 62–73. [Google Scholar] [CrossRef]
- Paulson, H.L. The spinocerebellar ataxias. J. Neuroophthalmol. 2009, 29, 227–237. [Google Scholar] [CrossRef] [Green Version]
- Harding, A.E. Clinical features and classification of inherited ataxias. Adv. Neurol. 1993, 61, 1–14. [Google Scholar] [PubMed]
- Duenas, A.M.; Goold, R.; Giunti, P. Molecular pathogenesis of spinocerebellar ataxias. Brain 2006, 129, 1357–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, T. Hereditary Ataxia Overview. Available online: http://www.geneclinics.org/servlet/access?db=geneclinics&site=gt&id=8888891&key=Fkx-A35oEHZ2G&gry=&fcn=y&fw=g8bn&filename=/profiles/ataxias/index.html (accessed on 1 February 2020).
- Ashizawa, T.; Oz, G.; Paulson, H.L. Spinocerebellar ataxias: Prospects and challenges for therapy development. Nat. Rev. Neurol. 2018, 14, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Paulson, H. Machado-Joseph disease/spinocerebellar ataxia type 3. Handb. Clin. Neurol. 2012, 103, 437–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Lee, Y.; Choi, S.; Song, J.-J. Structural basis of the phosphorylation dependent complex formation of neurodegenerative disease protein Ataxin-1 and RBM17. Biochem. Biophys. Res. Commun. 2014, 449, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Rosas, S.; Weber, C.; Fievet, L.; Messaddeq, N.; Karam, A.; Trottier, Y. Loss of zebrafish Ataxin-7, a SAGA subunit responsible for SCA7 retinopathy, causes ocular coloboma and malformation of photoreceptors. Hum. Mol. Genet. 2018, 28, 912–927. [Google Scholar] [CrossRef]
- Johnson, S.A.; Dubeau, L.; White, R.J.; Johnson, D.L. The TATA-binding protein as a regulator of cellular transformation. Cell Cycle 2003, 2, 442–444. [Google Scholar] [CrossRef]
- Wang, L.; Tsai, C.-C. Atrophin proteins: An overview of a new class of nuclear receptor corepressors. Nucl. Recept Signal 2008, 6, e009. [Google Scholar] [CrossRef] [Green Version]
- Lastres-Becker, I.; Nonis, D.; Eich, F.; Klinkenberg, M.; Gorospe, M.; Kotter, P.; Klein, F.A.; Kedersha, N.; Auburger, G. Mammalian ataxin-2 modulates translation control at the pre-initiation complex via PI3K/mTOR and is induced by starvation. Biochim. Biophys. Acta 2016, 1862, 1558–1569. [Google Scholar] [CrossRef]
- Blount, J.R.; Tsou, W.-L.; Ristic, G.; Burr, A.A.; Ouyang, M.; Galante, H.; Scaglione, K.M.; Todi, S.V. Ubiquitin-binding site 2 of ataxin-3 prevents its proteasomal degradation by interacting with Rad23. Nat. Commun. 2014, 5, 4638. [Google Scholar] [CrossRef] [Green Version]
- Kordasiewicz, H.B.; Gomez, C.M. Molecular pathogenesis of spinocerebellar ataxia type 6. Neurotherapeutics 2007, 4, 285–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespo-Barreto, J.; Fryer, J.D.; Shaw, C.A.; Orr, H.T.; Zoghbi, H.Y. Partial loss of ataxin-1 function contributes to transcriptional dysregulation in spinocerebellar ataxia type 1 pathogenesis. PLoS Genet. 2010, 6, e1001021. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Hao, T.; Shaw, C.; Patel, A.J.; Szabo, G.; Rual, J.F.; Fisk, C.J.; Li, N.; Smolyar, A.; Hill, D.E.; et al. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell 2006, 125, 801–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.; Lindenberg, K.S.; Krebs, A.; Schols, L.; Laccone, F.; Herms, J.; Rechsteiner, M.; Riess, O.; Landwehrmeyer, G.B. Protein surveillance machinery in brains with spinocerebellar ataxia type 3: Redistribution and differential recruitment of 26S proteasome subunits and chaperones to neuronal intranuclear inclusions. Ann. Neurol. 2002, 51, 302–310. [Google Scholar] [CrossRef]
- Cummings, C.J.; Mancini, M.A.; Antalffy, B.; DeFranco, D.B.; Orr, H.T.; Zoghbi, H.Y. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 1998, 19, 148–154. [Google Scholar] [CrossRef]
- Park, Y.; Hong, S.; Kim, S.J.; Kang, S. Proteasome function is inhibited by polyglutamine-expanded ataxin-1, the SCA1 gene product. Mol. Cells 2005, 19, 23–30. [Google Scholar]
- Hong, S.; Kim, S.-J.; Ka, S.; Choi, I.; Kang, S. USP7, a Ubiquitin-Specific Protease, Interacts with Ataxin-1, the SCA1 Gene Product. Mol. Cell. Neurosci. 2002, 20, 298–306. [Google Scholar] [CrossRef]
- Yu, X.; Ajayi, A.; Boga, N.R.; Ström, A.-L. Differential degradation of full-length and cleaved ataxin-7 fragments in a novel stable inducible SCA7 model. J. Mol. Neurosci. 2012, 47, 219–233. [Google Scholar] [CrossRef] [Green Version]
- Chai, Y.; Berke, S.S.; Cohen, R.E.; Paulson, H.L. Poly-ubiquitin binding by the polyglutamine disease protein ataxin-3 links its normal function to protein surveillance pathways. J. Biol. Chem. 2004, 279, 3605–3611. [Google Scholar] [CrossRef] [Green Version]
- Zeviani, M.; Simonati, A.; Bindoff, L.A. Chapter 22—Ataxia in mitochondrial disorders. In Handbook of Clinical Neurology; Subramony, S.H., Dürr, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 103, pp. 359–372. [Google Scholar]
- Stucki, D.M.; Ruegsegger, C.; Steiner, S.; Radecke, J.; Murphy, M.P.; Zuber, B.; Saxena, S. Mitochondrial impairments contribute to Spinocerebellar ataxia type 1 progression and can be ameliorated by the mitochondria-targeted antioxidant MitoQ. Free Radic. Biol. Med. 2016, 97, 427–440. [Google Scholar] [CrossRef] [Green Version]
- Qi, M.-L.; Tagawa, K.; Enokido, Y.; Yoshimura, N.; Wada, Y.-I.; Watase, K.; Ishiura, S.-I.; Kanazawa, I.; Botas, J.; Saitoe, M.; et al. Proteome analysis of soluble nuclear proteins reveals that HMGB1/2 suppress genotoxic stress in polyglutamine diseases. Nat. Cell Biol. 2007, 9, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Fujita, K.; Tagawa, K.; Chen, X.; Homma, H.; Sasabe, T.; Shimizu, J.; Shimizu, S.; Tamura, T.; Muramatsu, S.-i.; et al. HMGB1 facilitates repair of mitochondrial DNA damage and extends the lifespan of mutant ataxin-1 knock-in mice. EMBO Mol. Med. 2015, 7, 78–101. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, H.; Shen, R.; Fang, J.; Yang, Y.; Dai, W.; Zhu, Y.; Zhou, M. Mitochondrial-targeted antioxidant MitoQ provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury possibly via the Nrf2-ARE pathway. Am. J. Transl. Res. 2018, 10, 1887–1899. [Google Scholar] [PubMed]
- Cornelius, N.; Wardman, J.H.; Hargreaves, I.P.; Neergheen, V.; Bie, A.S.; Tumer, Z.; Nielsen, J.E.; Nielsen, T.T. Evidence of oxidative stress and mitochondrial dysfunction in spinocerebellar ataxia type 2 (SCA2) patient fibroblasts: Effect of coenzyme Q10 supplementation on these parameters. Mitochondrion 2017, 34, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.Y.; Figueroa, K.P.; Pulst, S.M.; Lin, C.-Y.; Perlman, S.; Wilmot, G.; Gomez, C.; Schmahmann, J.; Paulson, H.; Shakkottai, V.G.; et al. Coenzyme Q10 and spinocerebellar ataxias. Mov. Disord. 2015, 30, 214–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulson, H.L. Dominantly inherited ataxias: Lessons learned from Machado-Joseph disease/spinocerebellar ataxia type 3. Semin. Neurol. 2007, 27, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.C.; Kuo, C.L.; Cheng, W.L.; Liu, C.S.; Hsieh, M. Decreased antioxidant enzyme activity and increased mitochondrial DNA damage in cellular models of Machado-Joseph disease. J. Neurosci. Res. 2009, 87, 1884–1891. [Google Scholar] [CrossRef]
- Laço, M.N.; Oliveira, C.R.; Paulson, H.L.; Rego, A.C. Compromised mitochondrial complex II in models of Machado–Joseph disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, L.S.; da Silveira, A.F.; Trott, A.; Houenou, L.J.; Algarve, T.D.; Belló, C.; Lenz, A.F.; Mânica-Cattani, M.F.; da Cruz, I.B.M. Association between Machado–Joseph disease and oxidative stress biomarkers. Mutat. Res. Genet. Toxicol. Environ. Mutagenesis 2013, 757, 99–103. [Google Scholar] [CrossRef]
- De Assis, A.M.; Saute, J.A.M.; Longoni, A.; Haas, C.B.; Torrez, V.R.; Brochier, A.W.; Souza, G.N.; Furtado, G.V.; Gheno, T.C.; Russo, A.; et al. Peripheral Oxidative Stress Biomarkers in Spinocerebellar Ataxia Type 3/Machado-Joseph Disease. Front. Neurol. 2017, 8, 485. [Google Scholar] [CrossRef] [Green Version]
- Reina, C.P.; Zhong, X.; Pittman, R.N. Proteotoxic stress increases nuclear localization of ataxin-3. Hum. Mol. Genet. 2009, 19, 235–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saudou, F.; Finkbeiner, S.; Devys, D.; Greenberg, M.E. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 1998, 95, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Peters, M.F.; Nucifora, F.C.; Kushi, J.; Seaman, H.C.; Cooper, J.K.; Herring, W.J.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Nuclear Targeting of Mutant Huntingtin Increases Toxicity. Mol. Cell. Neurosci. 1999, 14, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Fujigasaki, H.; Uchihara, T.; Koyano, S.; Iwabuchi, K.; Yagishita, S.; Makifuchi, T.; Nakamura, A.; Ishida, K.; Toru, S.; Hirai, S.; et al. Ataxin-3 is translocated into the nucleus for the formation of intranuclear inclusions in normal and Machado-Joseph disease brains. Exp. Neurol. 2000, 165, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Nucifora, F.C., Jr.; Ellerby, L.M.; Wellington, C.L.; Wood, J.D.; Herring, W.J.; Sawa, A.; Hayden, M.R.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Nuclear localization of a non-caspase truncation product of atrophin-1, with an expanded polyglutamine repeat, increases cellular toxicity. J. Biol. Chem. 2003, 278, 13047–13055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bichelmeier, U.; Schmidt, T.; Hübener, J.; Boy, J.; Rüttiger, L.; Häbig, K.; Poths, S.; Bonin, M.; Knipper, M.; Schmidt, W.J.; et al. Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3: In vivo evidence. J. Neurosci. 2007, 27, 7418–7428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Klotz, L.-O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [Green Version]
- Araujo, J.; Breuer, P.; Dieringer, S.; Krauss, S.; Dorn, S.; Zimmermann, K.; Pfeifer, A.; Klockgether, T.; Wuellner, U.; Evert, B.O. FOXO4-dependent upregulation of superoxide dismutase-2 in response to oxidative stress is impaired in spinocerebellar ataxia type 3. Hum. Mol. Genet. 2011, 20, 2928–2941. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-M.; Weng, Y.-T.; Chen, W.-L.; Lin, T.-H.; Chao, C.-Y.; Lin, C.-H.; Chen, I.C.; Lee, L.-C.; Lin, H.-Y.; Wu, Y.-R.; et al. Aqueous extract of Glycyrrhiza inflata inhibits aggregation by upregulating PPARGC1A and NFE2L2–ARE pathways in cell models of spinocerebellar ataxia 3. Free Radic. Biol. Med. 2014, 71, 339–350. [Google Scholar] [CrossRef]
- Chang, K.-H.; Chen, W.-L.; Wu, Y.-R.; Lin, T.-H.; Wu, Y.-C.; Chao, C.-Y.; Lin, J.-Y.; Lee, L.-C.; Chen, Y.-C.; Lee-Chen, G.-J.; et al. Aqueous extract of Gardenia jasminoides targeting oxidative stress to reduce polyQ aggregation in cell models of spinocerebellar ataxia 3. Neuropharmacology 2014, 81, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Chang, J.C.; Lin, W.Y.; Li, C.C.; Hsieh, M.; Chen, H.W.; Wang, T.S.; Wu, W.T.; Liu, C.S.; Liu, K.L. Caffeic acid and resveratrol ameliorate cellular damage in cell and Drosophila models of spinocerebellar ataxia type 3 through upregulation of Nrf2 pathway. Free Radic. Biol. Med. 2018, 115, 309–317. [Google Scholar] [CrossRef]
- Ajayi, A.; Yu, X.; Lindberg, S.; Langel, U.; Ström, A.L. Expanded ataxin-7 cause toxicity by inducing ROS production from NADPH oxidase complexes in a stable inducible Spinocerebellar ataxia type 7 (SCA7) model. BMC Neurosci. 2012, 13, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukhtouche, F.; Vodjdani, G.; Jarvis, C.I.; Bakouche, J.; Staels, B.; Mallet, J.; Mariani, J.; Lemaigre-Dubreuil, Y.; Brugg, B. Human retinoic acid receptor-related orphan receptor alpha1 overexpression protects neurones against oxidative stress-induced apoptosis. J. Neurochem. 2006, 96, 1778–1789. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M.J.; Shah, A.G.; Fang, Z.H.; Ward, E.G.; Warren, S.T.; Li, S.; Li, X.J. Polyglutamine domain modulates the TBP-TFIIB interaction: Implications for its normal function and neurodegeneration. Nat. Neurosci. 2007, 10, 1519–1528. [Google Scholar] [CrossRef]
- Wyttenbach, A.; Sauvageot, O.; Carmichael, J.; Diaz-Latoud, C.; Arrigo, A.P.; Rubinsztein, D.C. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum. Mol. Genet. 2002, 11, 1137–1151. [Google Scholar] [CrossRef]
- Chen, C.-M.; Lee, L.-C.; Soong, B.-W.; Fung, H.-C.; Hsu, W.-C.; Lin, P.-Y.; Huang, H.-J.; Chen, F.-L.; Lin, C.-Y.; Lee-Chen, G.-J.; et al. SCA17 repeat expansion: Mildly expanded CAG/CAA repeat alleles in neurological disorders and the functional implications. Clin. Chim. Acta 2010, 411, 375–380. [Google Scholar] [CrossRef]
- Lee, L.C.; Weng, Y.T.; Wu, Y.R.; Soong, B.W.; Tseng, Y.C.; Chen, C.M.; Lee-Chen, G.J. Downregulation of proteins involved in the endoplasmic reticulum stress response and Nrf2-ARE signaling in lymphoblastoid cells of spinocerebellar ataxia type 17. J. Neural Transm. 2014, 121, 601–610. [Google Scholar] [CrossRef]
- Koriyama, Y.; Chiba, K.; Yamazaki, M.; Suzuki, H.; Muramoto, K.; Kato, S. Long-acting genipin derivative protects retinal ganglion cells from oxidative stress models in vitro and in vivo through the Nrf2/antioxidant response element signaling pathway. J. Neurochem. 2010, 115, 79–91. [Google Scholar] [CrossRef]
- Miyata, R.; Hayashi, M.; Tanuma, N.; Shioda, K.; Fukatsu, R.; Mizutani, S. Oxidative stress in neurodegeneration in dentatorubral-pallidoluysian atrophy. J. Neurol. Sci. 2008, 264, 133–139. [Google Scholar] [CrossRef]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Hall, D.A.; Coffey, S.; Leehey, M.; Bourgeois, J.; Gould, J.; Zhang, L.; Seritan, A.; Berry-Kravis, E.; Olichney, J.; et al. Treatment of fragile X-associated tremor ataxia syndrome (FXTAS) and related neurological problems. Clin. Interv. Aging 2008, 3, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquemont, S.; Hagerman, R.J.; Hagerman, P.J.; Leehey, M.A. Fragile-X syndrome and fragile X-associated tremor/ataxia syndrome: Two faces of FMR1. Lancet Neurol. 2007, 6, 45–55. [Google Scholar] [CrossRef]
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.A.; Hall, D.A.; Levine, R.A.; Brunberg, J.A.; Zhang, L.; Jardini, T.; Gane, L.W.; Harris, S.W.; et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA 2004, 291, 460–469. [Google Scholar] [CrossRef] [Green Version]
- Tassone, F.; Hagerman, R.J.; Chamberlain, W.D.; Hagerman, P.J. Transcription of the FMR1 gene in individuals with fragile X syndrome. Am. J. Med. Genet. 2000, 97, 195–203. [Google Scholar] [CrossRef]
- Greco, C.M.; Hagerman, R.J.; Tassone, F.; Chudley, A.E.; Del Bigio, M.R.; Jacquemont, S.; Leehey, M.; Hagerman, P.J. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 2002, 125, 1760–1771. [Google Scholar] [CrossRef]
- Tassone, F.; Iwahashi, C.; Hagerman, P.J. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS). RNA Biol. 2004, 1, 103–105. [Google Scholar] [CrossRef] [Green Version]
- Iwahashi, C.K.; Yasui, D.H.; An, H.J.; Greco, C.M.; Tassone, F.; Nannen, K.; Babineau, B.; Lebrilla, C.B.; Hagerman, R.J.; Hagerman, P.J. Protein composition of the intranuclear inclusions of FXTAS. Brain 2006, 129, 256–271. [Google Scholar] [CrossRef]
- Jin, P.; Duan, R.; Qurashi, A.; Qin, Y.; Tian, D.; Rosser, T.C.; Liu, H.; Feng, Y.; Warren, S.T. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 2007, 55, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Sofola, O.A.; Jin, P.; Qin, Y.; Duan, R.; Liu, H.; de Haro, M.; Nelson, D.L.; Botas, J. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 2007, 55, 565–571. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Herren, A.W.; Espinal, G.; Randol, J.; McLaughlin, B.; Martinez-Cerdeno, V.; Pessah, I.N.; Hagerman, R.J.; Hagerman, P.J. Composition of the Intranuclear Inclusions of Fragile X-associated Tremor/Ataxia Syndrome. Acta Neuropathol. Commun. 2019, 7, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iijima, T.; Wu, K.; Witte, H.; Hanno-Iijima, Y.; Glatter, T.; Richard, S.; Scheiffele, P. SAM68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell 2011, 147, 1601–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iijima, Y.; Tanaka, M.; Suzuki, S.; Hauser, D.; Tanaka, M.; Okada, C.; Ito, M.; Ayukawa, N.; Sato, Y.; Ohtsuka, M.; et al. SAM68-Specific Splicing Is Required for Proper Selection of Alternative 3′ UTR Isoforms in the Nervous System. iScience 2019, 22, 318–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Rosa, P.; Bielli, P.; Compagnucci, C.; Cesari, E.; Volpe, E.; Farioli Vecchioli, S.; Sette, C. Sam68 promotes self-renewal and glycolytic metabolism in mouse neural progenitor cells by modulating Aldh1a3 pre-mRNA 3′-end processing. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Mora, M.I.; Rodriguez-Revenga, L.; Madrigal, I.; Guitart-Mampel, M.; Garrabou, G.; Mila, M. Impaired Mitochondrial Function and Dynamics in the Pathogenesis of FXTAS. Mol. Neurobiol. 2017, 54, 6896–6902. [Google Scholar] [CrossRef]
- Robin, G.; Lopez, J.R.; Espinal, G.M.; Hulsizer, S.; Hagerman, P.J.; Pessah, I.N. Calcium dysregulation and Cdk5-ATM pathway involved in a mouse model of fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet. 2017, 26, 2649–2666. [Google Scholar] [CrossRef] [Green Version]
- Ross-Inta, C.; Omanska-Klusek, A.; Wong, S.; Barrow, C.; Garcia-Arocena, D.; Iwahashi, C.; Berry-Kravis, E.; Hagerman, R.J.; Hagerman, P.J.; Giulivi, C. Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem. J. 2010, 429, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Napoli, E.; Wong, S.; Hagerman, R.; Liu, S.; Tassone, F.; Giulivi, C. Altered redox mitochondrial biology in the neurodegenerative disorder fragile X-tremor/ataxia syndrome: Use of antioxidants in precision medicine. Mol. Med. 2016, 22, 548–559. [Google Scholar] [CrossRef] [Green Version]
- Napoli, E.; Ross-Inta, C.; Wong, S.; Omanska-Klusek, A.; Barrow, C.; Iwahashi, C.; Garcia-Arocena, D.; Sakaguchi, D.; Berry-Kravis, E.; Hagerman, R.; et al. Altered zinc transport disrupts mitochondrial protein processing/import in fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet. 2011, 20, 3079–3092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, E.S.; Cao, Z.; Hulsizer, S.; Tassone, F.; Berman, R.F.; Hagerman, P.J.; Pessah, I.N. Early mitochondrial abnormalities in hippocampal neurons cultured from Fmr1 pre-mutation mouse model. J. Neurochem. 2012, 123, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Hatch, A.L.; Gurel, P.S.; Higgs, H.N. Novel roles for actin in mitochondrial fission. J. Cell. Sci. 2014, 127, 4549–4560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastore, A.; Tozzi, G.; Gaeta, L.M.; Bertini, E.; Serafini, V.; Di Cesare, S.; Bonetto, V.; Casoni, F.; Carrozzo, R.; Federici, G.; et al. Actin glutathionylation increases in fibroblasts of patients with Friedreich’s ataxia: A potential role in the pathogenesis of the disease. J. Biol. Chem. 2003, 278, 42588–42595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.I.; Kobayashi, A.; Wakabayashi, N.; Kim, S.G.; Yamamoto, M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2046–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gakh, O.; Cavadini, P.; Isaya, G. Mitochondrial processing peptidases. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2002, 1592, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Condò, I.; Ventura, N.; Malisan, F.; Rufini, A.; Tomassini, B.; Testi, R. In vivo maturation of human frataxin. Hum. Mol. Genet. 2007, 16, 1534–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, D.M.; Shi, Q.; Dancis, A.; Pain, D. Maturation of Frataxin Within Mammalian and Yeast Mitochondria: One-Step Processing by Matrix Processing Peptidase. Hum. Mol. Genet. 1999, 8, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Schoser, B.; Timchenko, L. Myotonic dystrophies 1 and 2: Complex diseases with complex mechanisms. Curr. Genom. 2010, 11, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Winblad, S.; Samuelsson, L.; Lindberg, C.; Meola, G. Cognition in myotonic dystrophy type 1: A 5-year follow-up study. Eur. J. Neurol. 2016, 23, 1471–1476. [Google Scholar] [CrossRef]
- Sansone, V.; Gandossini, S.; Cotelli, M.; Calabria, M.; Zanetti, O.; Meola, G. Cognitive impairment in adult myotonic dystrophies: A longitudinal study. Neurol. Sci. 2007, 28, 9–15. [Google Scholar] [CrossRef]
- Schara, U.; Schoser, B.G. Myotonic dystrophies type 1 and 2: A summary on current aspects. Semin. Pediatr. Neurol. 2006, 13, 71–79. [Google Scholar] [CrossRef]
- Machuca-Tzili, L.; Brook, D.; Hilton-Jones, D. Clinical and molecular aspects of the myotonic dystrophies: A review. Muscle Nerve 2005, 32, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, J.W.; Ricker, K.; Jacobsen, J.F.; Rasmussen, L.J.; Dick, K.A.; Kress, W.; Schneider, C.; Koch, M.C.; Beilman, G.J.; Harrison, A.R.; et al. Myotonic dystrophy type 2: Molecular, diagnostic and clinical spectrum. Neurology 2003, 60, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Yum, K.; Wang, E.T.; Kalsotra, A. Myotonic dystrophy: Disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes. Curr. Opin. Genet. Dev. 2017, 44, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Ziegler, A.; Ricker, K.; Grimm, T.; Kress, W.; Reimers, C.D.; Meinck, H.; Reiners, K.; Toyka, K.V. Proximal myotonic myopathy: Evidence for anticipation in families with linkage to chromosome 3q. Neurology 2000, 55, 383–388. [Google Scholar] [CrossRef]
- Reddy, S.; Smith, D.B.; Rich, M.M.; Leferovich, J.M.; Reilly, P.; Davis, B.M.; Tran, K.; Rayburn, H.; Bronson, R.; Cros, D.; et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat. Genet. 1996, 13, 325–335. [Google Scholar] [CrossRef]
- Wang, J.; Pegoraro, E.; Menegazzo, E.; Gennarelli, M.; Hoop, R.C.; Angelini, C.; Hoffman, E.P. Myotonic dystrophy: Evidence for a possible dominant-negative RNA mutation. Hum. Mol. Genet. 1995, 4, 599–606. [Google Scholar] [CrossRef]
- Warf, M.B.; Berglund, J.A. MBNL binds similar RNA structures in the CUG repeats of myotonic dystrophy and its pre-mRNA substrate cardiac troponin T. RNA 2007, 13, 2238–2251. [Google Scholar] [CrossRef] [Green Version]
- Pagliarini, V.; La Rosa, P.; Sette, C. Faulty RNA splicing: Consequences and therapeutic opportunities in brain and muscle disorders. Hum. Genet. 2017, 136, 1215–1235. [Google Scholar] [CrossRef]
- Kuyumcu-Martinez, N.M.; Wang, G.S.; Cooper, T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell 2007, 28, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Charlet, B.N.; Savkur, R.S.; Singh, G.; Philips, A.V.; Grice, E.A.; Cooper, T.A. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell 2002, 10, 45–53. [Google Scholar] [CrossRef]
- Mateos-Aierdi, A.J.; Goicoechea, M.; Aiastui, A.; Fernández-Torrón, R.; Garcia-Puga, M.; Matheu, A.; López de Munain, A. Muscle wasting in myotonic dystrophies: A model of premature aging. Front. Aging Neurosci. 2015, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Usuki, F.; Ishiura, S. Expanded CTG repeats in myotonin protein kinase increase susceptibility to oxidative stress. Neuroreport 1998, 9, 2291–2296. [Google Scholar] [CrossRef] [PubMed]
- Usuki, F.; Takahashi, N.; Sasagawa, N.; Ishiura, S. Differential signaling pathways following oxidative stress in mutant myotonin protein kinase cDNA-transfected C2C12 cell lines. Biochem. Biophys. Res. Commun. 2000, 267, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Ihara, Y.; Mori, A.; Hayabara, T.; Namba, R.; Nobukuni, K.; Sato, K.; Miyata, S.; Edamatsu, R.; Liu, J.; Kawai, M. Free radicals, lipid peroxides and antioxidants in blood of patients with myotonic dystrophy. J. Neurol. 1995, 242, 119–122. [Google Scholar] [CrossRef]
- Head, E. Oxidative damage and cognitive dysfunction: Antioxidant treatments to promote healthy brain aging. Neurochem. Res. 2009, 34, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Baierle, M.; Nascimento, S.N.; Moro, A.M.; Brucker, N.; Freitas, F.; Gauer, B.; Durgante, J.; Bordignon, S.; Zibetti, M.; Trentini, C.M.; et al. Relationship between inflammation and oxidative stress and cognitive decline in the institutionalized elderly. Oxid. Med. Cell. Longev. 2015, 2015, 804198. [Google Scholar] [CrossRef] [Green Version]
- Michel, T.M.; Pülschen, D.; Thome, J. The role of oxidative stress in depressive disorders. Curr. Pharm. Des. 2012, 18, 5890–5899. [Google Scholar] [CrossRef] [Green Version]
- Rawdin, B.J.; Mellon, S.H.; Dhabhar, F.S.; Epel, E.S.; Puterman, E.; Su, Y.; Burke, H.M.; Reus, V.I.; Rosser, R.; Hamilton, S.P.; et al. Dysregulated relationship of inflammation and oxidative stress in major depression. Brain Behav. Immun. 2013, 31, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Ramon-Duaso, C.; Gener, T.; Consegal, M.; Fernández-Avilés, C.; Gallego, J.J.; Castarlenas, L.; Swanson, M.S.; de la Torre, R.; Maldonado, R.; Puig, M.V.; et al. Methylphenidate Attenuates the Cognitive and Mood Alterations Observed in Mbnl2 Knockout Mice and Reduces Microglia Overexpression. Cereb. Cortex 2018, 29, 2978–2997. [Google Scholar] [CrossRef]
- Bruna, B.; Lobos, P.; Herrera-Molina, R.; Hidalgo, C.; Paula-Lima, A.; Adasme, T. The signaling pathways underlying BDNF-induced Nrf2 hippocampal nuclear translocation involve ROS, RyR-Mediated Ca2+ signals, ERK and PI3K. Biochem. Biophys. Res. Commun. 2018, 505, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Zhang, J.C.; Ishima, T.; Dong, C.; Yang, C.; Ren, Q.; Ma, M.; Han, M.; Wu, J.; Suganuma, H.; et al. Role of Keap1-Nrf2 signaling in depression and dietary intake of glucoraphanin confers stress resilience in mice. Sci. Rep. 2016, 6, 30659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, W.; Zhang, J.-C.; Ishima, T.; Ren, Q.; Yang, C.; Dong, C.; Ma, M.; Saito, A.; Honda, T.; Hashimoto, K. Antidepressant effects of TBE-31 and MCE-1, the novel Nrf2 activators, in an inflammation model of depression. Eur. J. Pharmacol. 2016, 793, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Winblad, S.; Jensen, C.; Månsson, J.-E.; Samuelsson, L.; Lindberg, C. Depression in Myotonic Dystrophy type 1: Clinical and neuronal correlates. Behav. Brain Funct. 2010, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Wegner, M.; Araszkiewicz, A.; Piorunska-Stolzmann, M.; Wierusz-Wysocka, B.; Zozulinska-Ziolkiewicz, D. Association between IL-6 concentration and diabetes-related variables in DM1 patients with and without microvascular complications. Inflammation 2013, 36, 723–728. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.E. Anticipation in hereditary disease: The history of a biomedical concept. Hum. Genet. 2011, 130, 705–714. [Google Scholar] [CrossRef]





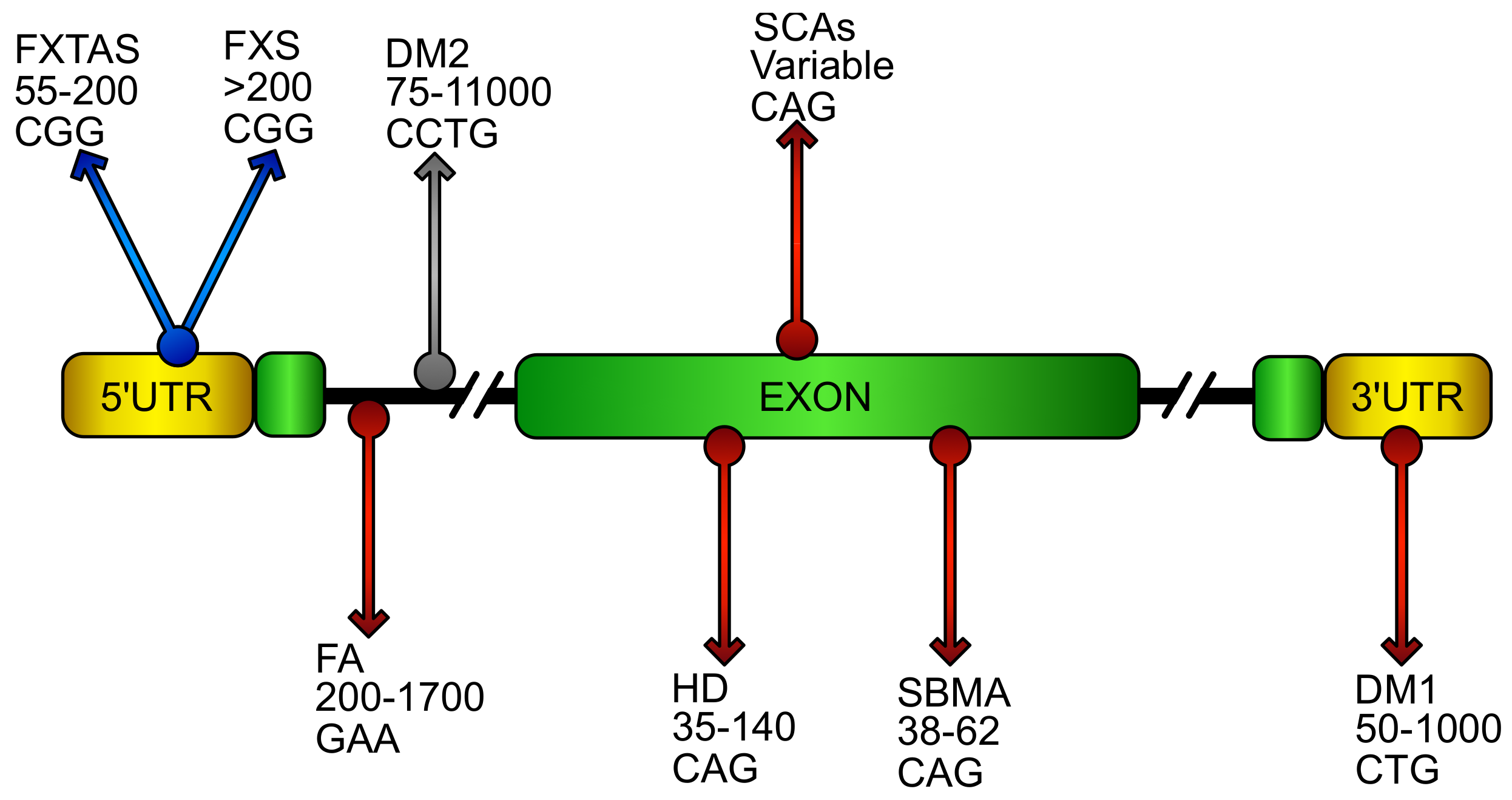{kind=link}
{kind=link}
{kind=link}
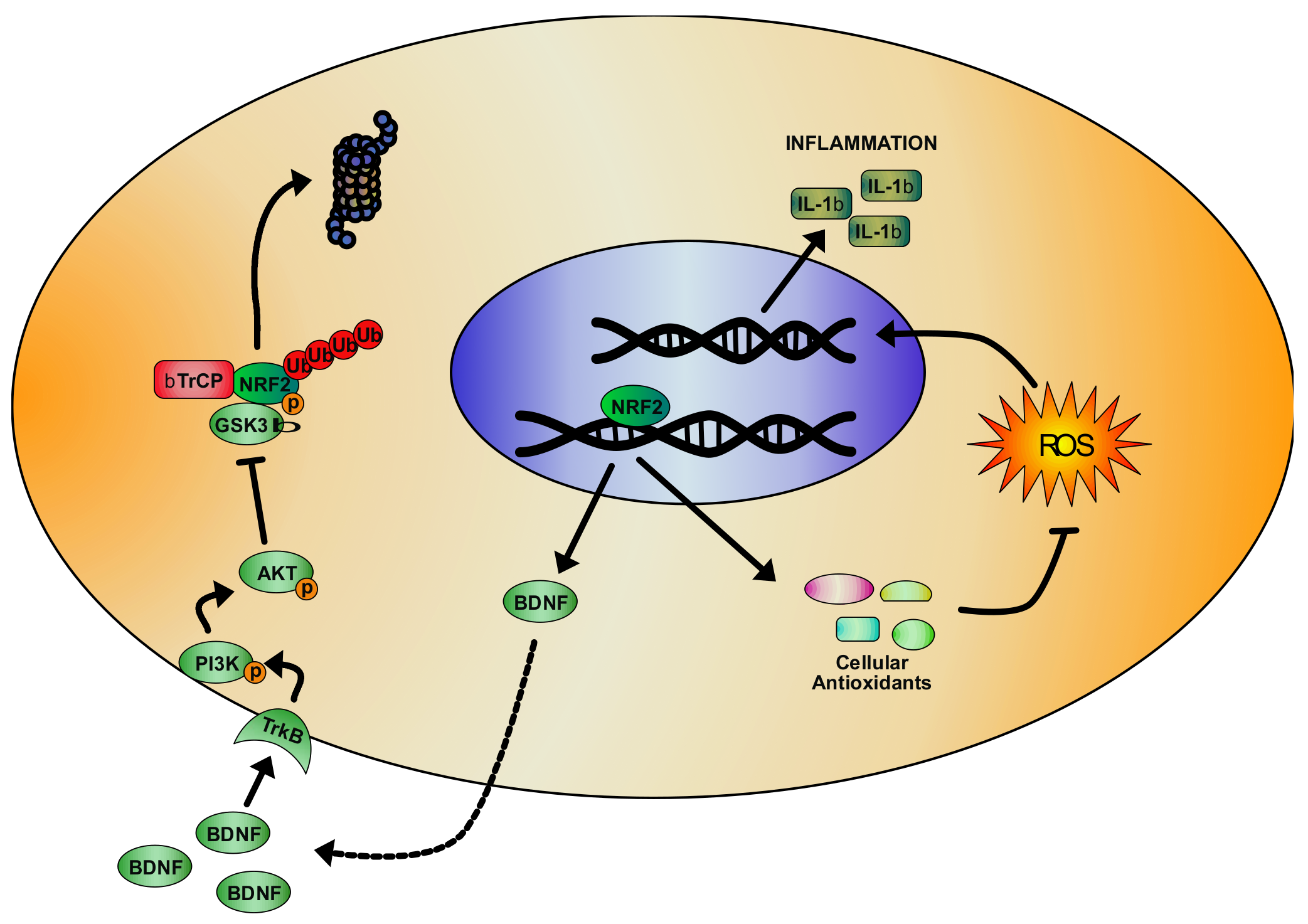{kind=link}
| Disease | Compound | Model | Effect of Treatment | Ref. |
| FA | SFN, DMF, NAC, EPI-743, RTA408, Idebenone | FA patients’ fibroblasts | Increase of GSH content; enhancement of FXN, NRF2 and down-stream genes mRNA. | [111] |
| SFN, DMF | shFXN NSC34 motor neurons | Rebalance of GSH/GSSG ratio; increase of FXN, NRF2 and down-stream genes expression. | [124] | |
| Idebenone | Patients | Reduction of cardiac hypertrophy. | [128] | |
| EPI-743 | Patients | Improvement of neurological functions. | [132] | |
| SBMA | ASC-J9 | AR-112Q PC12 cells; AR-97Q mice | Reduction of AR aggregates; rescue of motor defects and muscular atrophy; increase of VEGF expression. | [203] |
| ASC-J17 | SBMA patients’ fibroblasts; AR97Q mouse; AR52Q drosophila | Increase of NRF2 down-stream genes; suppression of polyQ toxicity in mutant flies; amelioration of mutant mice phenotype and decrease of mutant AR accumulation. | [204] | |
| HD | SFN | mHtt-94Q Hek293 | Increase of mHtt degradation and reduction of mHtt-induced toxicity. | [232] |
| MIND4-17 | HD patients’ primary monocytes | Reduction of inflammatory cytokines expression. | [233] | |
| DMF | R6/2 and YAC128 mice | Increased survival and motor functions; preservation of striatal neurons morphology; increase of NRF2 expression. | [236] | |
| SCA1 | MitoQ | Sca1 154Q/2Q mice | Improvement of motor coordination defects; reduction of mitochondrial morphological abnormalities and ETC activity defects. | [262] |
| SCA3 | Glycyrrhiza inflata extract, AMGZ, Licochalcone A | ATXN3/Q75-GFP Hek293 and SH-SY5Y cells | Decrease of Ataxin3 aggregates; up-regulation of NRF2 and down-stream genes; reduction of GSSG and ROS levels. | [282] |
| Gardenia jasminoides extract, genipin, geniposide, crocin | ATXN3/Q75-GFP Hek293 and SH-SY5Y cells | Reduction of Ataxin3 aggregates and Caspase3 activity; increase of NRF2 and its target genes; decrease of ROS concentration. | [283] | |
| SCA17 | Resveratrol, genipin | SCA17 lynfoblastoid cells | Increase of NRF2 antioxidant target genes and cell viability; decrease of ROS. | [290] |
| DM1 | MPH | Mbnl2 KO mice. | Increase of NRF2 and BDNF expression; rescue of behavioral deficits; decrease of inflammation. | [344] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Rosa, P.; Petrillo, S.; Bertini, E.S.; Piemonte, F. Oxidative Stress in DNA Repeat Expansion Disorders: A Focus on NRF2 Signaling Involvement. Biomolecules 2020, 10, 702. https://doi.org/10.3390/biom10050702
La Rosa P, Petrillo S, Bertini ES, Piemonte F. Oxidative Stress in DNA Repeat Expansion Disorders: A Focus on NRF2 Signaling Involvement. Biomolecules. 2020; 10(5):702. https://doi.org/10.3390/biom10050702
Chicago/Turabian StyleLa Rosa, Piergiorgio, Sara Petrillo, Enrico Silvio Bertini, and Fiorella Piemonte. 2020. "Oxidative Stress in DNA Repeat Expansion Disorders: A Focus on NRF2 Signaling Involvement" Biomolecules 10, no. 5: 702. https://doi.org/10.3390/biom10050702
APA StyleLa Rosa, P., Petrillo, S., Bertini, E. S., & Piemonte, F. (2020). Oxidative Stress in DNA Repeat Expansion Disorders: A Focus on NRF2 Signaling Involvement. Biomolecules, 10(5), 702. https://doi.org/10.3390/biom10050702