1. Introduction
Many pharmacologically important peptides are bacterial or fungal in origin and are nonribosomally synthesized by multimodular enzymes, referred to as nonribosomal peptide synthetases (NRPSs) [
1,
2,
3]. Using assembly-line logic, comprising multiple modules, NRPSs utilize a thiotemplated mechanism to activate, tether, and modify amino-acid building blocks, sequentially elongating the peptide chain before releasing the complete peptide [
1,
2,
3]. In general, the order and number of modules of an NRPS system determine the sequence and length of the peptide product. In this machinery, an adenylation (A) domain in the module plays an important role in selecting and activating amino-acid building blocks as aminoacyl adenylates with ATP. Therefore, the substrate specificities of A domains determine the amino-acid components of nonribosomal peptides. Interestingly, nonribosomal peptides often contain nonproteinogenic amino acids (NPAs) in their chemical structures [
4], since the A domain can accept not only the standard amino acids but also NPAs as building blocks [
5]. This advantageous feature of NPA incorporation makes a substantial contribution to the structural diversity of peptide natural products, resulting in the potent biological activities of these compounds. Moreover, the NPA-containing peptides are more biologically stable due to their resistance to amide hydrolases, such as peptidase and protease. Consequently, screening of cryptic NPA-containing peptides has become more important for effective drug discoveries. This strategy is also supported by the fact that therapeutically important nonribosomal peptides, such as vancomycin, daptomycin, and cyclosporin, are NPA-containing peptides.
Knowledge about the biosynthetic route to the major classes of NPAs, which are incorporated into nonribosomal peptides, has steadily increased over the last two decades [
4,
5,
6]. In the present study, we focused on the biosynthesis of NPAs harboring a cyclopropane ring, as the inherent ring strain present in the small ring moiety is frequently responsible for the biological activities of these compounds [
7,
8]. The simplest cyclopropane-containing NPA is 1-aminocyclopropanecarboxylic acid (ACC) (2), which has been isolated from many fruits and plant tissues (
Figure 1A). Compound 2 is known to be the crucial and immediate precursor of the important plant hormone ethylene, which is involved in senescence, fruit ripening, and interspecies communication in plants [
8]. Furthermore, in 1979, the plant ACC scaffold was found to be derived from
S-adenosyl-L-methionine (SAM) (1) (
Figure 1A). The cyclopropanation with the production of 5′-methylthioadenosine (MTA) is mediated by pyridoxal-5′-phosphate (PLP)-dependent aminotransferases (ACC synthases) [
9]. However, very recently, it was reported that microorganisms employ alternative machineries for the ACC formation to produce ACC-containing secondary metabolites. In the colibactin biosynthesis, the ACC moiety was derived from 1, but the cyclopropanation was catalyzed not by the PLP-dependent aminotransferase, but rather by the synergic action of NRPS and polyketide synthetase (
Figure 1B) [
10]. The guangnanmycin biosynthesis employs a novel bacterial ACC synthase, GnmY, to form 2 and MTA from 1 using a PLP-dependent mechanism similar to that of the plant ACC synthases (
Figure 1C) [
11]. However, surprisingly, GnmY does not share homology with any ACC synthases from plants, although its primary structure is classified as a PLP-dependent aminotransferase.
On the other hand, the biosynthesis of the next-simplest cyclopropane-containing NPA, 1-amino-2-methylcyclopropanecarboxylic acid (MeACC) (3), remains unclear (
Figure 1D). Compound 3 (termed norcoronamic acid) was found in SW-163C [
12,
13] and its analogues. The formation of 3 in their biosynthesis is still speculative; a radical SAM protein (Swb7) and a PLP-dependent aminotransferase (Swb6) could work together closely to form 3 from L-valine via radical cyclopropanation in the SW-163 biosynthesis [
14]. However, an alternative biosynthetic route to 3 has been suggested by the fact that the amino-acid sequence of Swb6 is homologous with that of GnmY, utilizing 1 as the substrate.
Here, we report that the biosynthesis of the MeACC building block is mediated by two unique enzymes, Orf29 and Orf30, which are the Swb7 and Swb6/GnmY homologues, respectively; Orf29 (a radical SAM methyltransferase) catalyzes the C-methylation of 1, and the resulting compound is catalytically transformed to 3 by Orf30 (PLP-dependent aminotransferase) in vitro. This finding expands our knowledge of cyclopropane-containing NPA biosynthesis.
2. Materials and Methods
2.1. Chemicals
All chemicals (1-aminocyclopropanecarboxylic acid (ACC),
S-adenosyl-L-methionine (SAM),
S-adenosyl-L-homocysteine (SAH), [1-
13C]-L-methionine, and [5-
13C]-L-methionine) were purchased from Tokyo Chemical Industry (Tokyo, Japan), Sigma-Aldrich Japan Inc. (Tokyo, Japan), and Cambridge Isotope Laboratories (CIL; Tewksbury, MA, USA). The authentic standard compounds of (
1S,2R)-MeACC and (
1R,2R)-MeACC were prepared according to the procedures reported previously [
15,
16]. Oligonucleotides were obtained from Eurofines Genomics (Tokyo, Japan). All other chemicals used were of analytical grade.
2.2. Bacterial Strains, Plasmids, and Culture Media
Bacterial strains and plasmids used in this study are summarized in
Table S1.
2× SK No.2 medium, consisting of 4% (w/v) soluble starch, 1% (w/v) glucose, 1% (w/v) yeast extract (Difco Laboratories, Franklin Lakes, NJ, USA), 0.6% (w/v) beef extract (Difco), 0.6% (w/v) peptone, 0.04% (w/v) KH2PO4, and 0.12% (w/v) MgSO4 and 7H2O (pH7.6) was used for the heterologous expression experiments, using Streptomyces lividans TK23 as a host strain. The S. lividans TK23 strain was also grown in S10.3 medium, consisting of 10.3% (w/v) sucrose, 3% (w/v) glucose, 1.5% (w/v) soytone (Difco), 0.1% (w/v) glycine, 2.7 mM CaCl2, and 5 mM MgCl2 (pH 7.2). Escherichia coli EcSUF derivatives were cultured in Terrific Broth (TB) medium, containing 1% (w/v) glycerol, 2.4% (w/v) yeast extract (Difco), 1.2% (w/v) tryptone (Difco), 0.94% (w/v) K2HPO4, 0.22% (w/v) KH2PO4, 0.01% (w/v) ammonium ferric citrate, and 7.845% (w/v) FeSO4(NH4)2SO4·6H2O.
2.3. Cloning of the Biosynthetic Gene Cluster With Genes Homologous to Swb7 and Swb6/gnmY
In the draft genome database of our laboratory stock strains, antiSMASH analysis [
17] showed that the genome DNA of
Streptomyces violaceusniger 4521-SVS3 has the gene cluster carrying two genes,
orf29 (accession number, BCD33697) and
orf30 (BCD33698), which are homologous to
swb7 and
swb6/gnmY, respectively (
Figure 2). In addition, two NRPS genes (
orf22 and
orf23) were found within the gene cluster, suggesting that this gene cluster is responsible for the production of a nonribosomal peptide, containing the MeACC building block. We therefore designated the gene cluster as the MeACC cluster. After the construction of the genome library of the 4521-SVS3 strain using the BAC vector pKU518, according to a previously reported method [
18], a BAC clone containing the entire MeACC cluster was screened by PCR amplification using two sets of primers (orf21-F and orf21-R; orf30-F and orf30-R,
Table S2). The positive clone (pKU518_MeACC) carrying the whole MeACC cluster was selected, and the insert sequence was confirmed by end-sequencing (
Table S1 and
Figure 2). The 59 kbp DNA fragment was deposited in the DNA Database of Japan (DDBJ), European Molecular Biology Laboratory (EMBL), and GenBank databank under accession number LC535008.
2.4. Expression of the MeACC Cluster in a Heterologous Host Strain, S. Lividans TK23
The BAC clones, pKU518_MeACC and pKU518 (
Table S1), were respectively introduced into
S. lividans TK23 by standard procedures [
19]. The two resulting transformants, TK23_MeACC and TK23_empty (
Table S1), harboring pKU518_MeACC and pKU518, respectively, were cultured in 2×SK No.2 medium for 6 days at 28 °C. In the feeding experiments, using
13C-labeled L-methionine, 2×SK No.2 medium supplemented with 0.1% [1-
13C]-L-methionine or 0.1% [5-
13C]-L-methionine was employed. To terminate cultivation and extract the peptide compounds, an equivalent volume of acetone was added, and the culture broths were shaken for 9 h at 15 °C. After centrifugation, the resulting supernatants were evaporated to remove the acetone and were then analyzed by high-performance liquid chromatography and high-resolution electrospray ionization mass spectrometry (HPLC-HR-ESI-MS) analysis (maXis plus; Bruker) using a reversed-phase column (Sunshell RP-AQUA, 2.6 μm, 50 × 2.1 mm; ChromaNik Technologies, Osaka, Japan) at 40 °C at a flow rate of 0.3 mL/min and with a linear gradient of acetonitrile in water in 0.1% (
v/v) formic acid run over 16 min (5–100% (
v/v) acetonitrile for 16 min).
2.5. Inactivation of the orf29 and orf30 Genes
To investigate the function of
orf29 or
orf30, we constructed BAC clones carrying an MeACC cluster, in which the
orf29 or
orf30 gene was inactivated by an in-frame deletion with a PCR-targeted mutagenesis strategy [
20]. The resulting BAC vectors, pKU518_MeACC_
∆orf29 and pKU518_MeACC_
∆orf30 (
Table S1), were respectively introduced into
S. lividans TK23, and their transformants (TK23_MeACC_
∆orf29 and TK23_MeACC_
∆orf30) (
Table S1) were cultured in 2×SK No.2 medium with or without 0.2% (
w/
v) ACC for 6 days at 28 °C. To confirm the productivity of the peptide compounds from the inactivated gene clusters, the culture broths were analyzed by HPLC-HR-ESI-MS, as described above (
Section 2.4).
2.6. Overexpression and Purification of the orf30 Recombinant Enzyme
The following two PCR primers were designed and used to amplify the
orf30 gene: pHSA81_
orf30-F and pHSA81_C8His-
orf30-R (
Table S2). The PCR product was ligated with the expression vector, pHSA81 (a gift from Dr. Kobayashi, University of Tsukuba, Tsukuba, Japan). After confirmation of the DNA sequence, the resulting plasmid (pHSA81_
orf30_C8His,
Table S1) was introduced into
S. lividans TK23 for expression as a
C-terminally 8×His-tagged fusion protein by a standard procedure [
19]. The transformant (TK23_rOrf30/C8His;
Table S1) was inoculated into S10.3 medium, containing 20 μg/ml thiostrepton. After growth for 3 days at 28 °C, cells were harvested from 50 mL culture broth by centrifugation at 6000×
g for 15 min, resuspended in 5 mL Buffer A (50 mm sodium phosphate buffer (NaPB), 10% glycerol, 300 mM NaCl, 0.1 mM PLP, and pH 8.0), containing 10 mM imidazole and sonicated on ice. Insoluble material was removed by centrifugation at 12,000×
g for 15 min. The supernatant was run on a 1 mL nickel-nitriloacetic acid (Ni-NTA) Sepharose column (Qiagen) that had been pre-equilibrated with 5 mL Buffer A, containing 10 mM imidazole. The column was washed with 5 mL Buffer A, containing 20 mM imidazole, and recombinant enzymes were eluted with 1 mL Buffer A, containing 250 mM imidazole and used for in vitro enzyme reactions.
The molecular weight of the purified protein (rOrf30) was determined by SDS-PAGE and gel-exclusion chromatography, using a SunSec diol-30 column (ChromaNik Technologies).
2.7. In Vitro Enzyme Reactions with rOrf30
A reaction mixture (100 μL) consisting of 50 mM NaPB (pH 8.0), 500 μM SAM or L-methionine, and 100 μg/mL rOrf30 was incubated at 30 °C for 15 h. The enzyme reaction was quenched by heating at 100 °C for 1 min and the denatured enzyme was removed by centrifugation. The reaction product was derivatized with 3-aminopyridinyl-N-hydroxysuccinimidyl carbamate (APDS), according to the manufacturer’s instructions (FUJIFILM Wako Pure Chemical, Japan), and was then analyzed by HPLC-HR-ESI-MS using a reversed-phase column (Sunniest RP-AQUA, 3 μm, 150 × 2.1 mm; ChromaNik Technologies) at 40 °C at a flow rate of 0.3 mL/min and with a two-step linear gradient of acetonitrile in water in 0.1% (v/v) heptafluorobutyric acid (HFBA) (Wako, Japan) run over 18 min (2% (v/v) acetonitrile for 5 min, 2–50% (v/v) acetonitrile for 11 min, and 50–98% (v/v) acetonitrile for 2 min).
Kinetic assays were performed under conditions identical to those described above, except that the reaction time (60 min) was reduced to enable the measurement of steady-state kinetic parameters. All assays were carried out under linear conditions. The Km and Kcat values were calculated from curves fitting to the Michaelis–Menten equation using GraphPad Prism8 software. The kinetic analysis was performed in triplicate.
2.8. Construction of E. Coli Strains Expressing the Suf and/or Btu Operons
A plasmid pRKSUF017 [
21] carrying the suf operon (a gift from Dr. Takahashi, Saitama University, Saitama, Japan) was introduced to
E. coli C41 (DE3) for the (4Fe-4S) cluster reconstitution. The resulting strain, EcSuf (
Table S1), was used as a host strain for the heterologous co-expression experiment using two genes, orf29 and orf30 (see
Section 2.9). In addition, we constructed a plasmid, pBAD24_BtuCEDFB, which carries the cobalamin uptake genes, according to the method described by Booker et al. [
22]. The synthetic DNA fragments (
Table S3) containing five genes, btuC, btuE, btuD, btuF, and btuB, which were designed according to the plasmid map of pBAD42-BtuCEDFB, were obtained from Eurofins Genomics (Tokyo, Japan). The fragment 1 digested with NcoI and PvuI was inserted into the same restriction enzyme sites of a pRSFDuet-1 vector to obtain the plasmid pRSF_btuCE’. The fragment 2 was digested with PvuI and KpnI and then ligated into the pRSF_btuCE’ construct and digested with the same enzymes to get the plasmid pRSF_btuCEDFB’. The fragment 3 was digested with HindIII and XhoI and then ligated into the pRSF_btuCEDFB’ construct and digested with the same enzymes to get the plasmid pRSF_btuCEDFB. Finally, the NcoI–XbaI fragment of the plasmid pRSF_btuCEDFB was inserted into the same restriction enzyme sites of a pBAD24 vector [
23] (purchased from the Yale Coli Genetic Stock Center) to construct the plasmid pBAD24_BtuCEDFB.
The plasmids, pBAD24_BtuCEDFB and pRKSUF017, were introduced into
E. coli BL21(DE3). The resulting strain, EcSufBtu (
Table S1), was employed for the overexpression of rOrf29 (see
Section 2.10).
2.9. Heterologous Co-Expression of Two Genes, orf29 and orf30, in E. coli
To amplify the
orf29 and
orf30 genes, the following two sets of PCR primers were used: pETDuet-1
_orf29-F/pETDuet-1
_orf29-R and pETDuet-1
_orf30-F/pETDuet-1
_orf30-R (
Table S2). Both PCR products were ligated with a pETDuet-1 vector to yield the expression vector pETDuet-1_
orf29_
orf30 (
Table S1). In addition, pETDuet-1_
orf29 was also constructed for a control experiment. These two constructed vectors and pETDuet-1(empty) were respectively introduced into the EcSuf strain, which expressed the
suf operon for iron-sulfur cluster reconstitution (
Table S1). The resulting transformants, EcSuf_
orf29_
orf30, EcSuf_
orf29, and EcSuf_empty (
Table S1), were cultured in TB medium, supplemented with 200 μM L-cysteine, 20 μM methylcobalamine, 0.1% (
w/
v) L-methionine, and 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) for 42 h at 28 °C. Additionally, the EcSuf_
orf29 strain was grown in TB medium, supplemented with 0.2% (
w/
v) ACC (2). To terminate the cultivation and extract MeACC (3), an equivalent volume of acetone was added, and the culture broths were shaken for 9 h at 15 °C. After centrifugation, the resulting supernatants were evaporated to remove the acetone. Compound 3 in the extract was derivatized with APDS and was then analyzed by HPLC-HR-ESI-MS under the same conditions employed for the ACC analysis (
Section 2.7). (
1R,2R)-MeACC (5) and (
1S,2R)-MeACC (6) were used as authentic standards.
2.10. In Vitro Enzyme Reactions with rOrf29
The following two PCR primers were designed and used to amplify the
orf29 gene: pET28_
orf29-F and pET28_
orf29-R (
Table S2). The PCR product was ligated with the expression vector pET28. After confirmation of the DNA sequence, the resulting plasmid (pET28_
orf29;
Table S1) was introduced into the
E. coli EcSufBtu strain (see
Section 2.8) (
Table S1), which expresses the
suf and
btu operons. The resulting transformant, EcSufBtu_orf29, was inoculated into LB medium, containing 50 μg/mL kanamycin, 100 μg/mL ampicillin, and 5 μg/mL tetracycline. After growth overnight at 37 °C, the culture (1 mL) was inoculated into 200 mL of M9-ethanolamine medium [
24], containing 50 μg/mL kanamycin, 100 μg/mL ampicillin, and 5 μg/mL tetracycline, and incubated at 37 °C with 200 rpm agitation until the OD600 = 0.2. L-Arabinose was added to a final concentration of 1 mg/mL, followed by re-cultivation until the OD600 = 0.6. The culture was cooled down on ice, and then FeSO
4(NH
4)
2SO
4 and L-cysteine were added to each final concentration of 0.2 mM each. Protein expression was induced by the addition of IPTG to a final concentration of 0.2 mM. Cultivation was continued at 15 °C for 20–24 h with 80 rpm agitation. The cells were harvested by centrifugation, washed with buffer B (50 mM HEPES-Na, 10% glycerol, 300 mM NaCl, and pH 8.0), and stored at −30 °C until use. The wet cells were transferred into a glovebox and subsequent disruption and purification were conducted under anaerobic conditions ([O
2] ≤ 5 ppm). The wet cells (3 g) were suspended in degassed buffer B (30 mL). The suspension of cells was disrupted by sonication, with sonication bursts of 5 s, with a 5 s interval (total 4 min) on the cooled aluminum beads. Cell debris was removed by centrifugation (14,000×
g, 20 min, at 4 °C). The supernatant was loaded onto a TALON resin (Clontech, Mountain View, CA) column that had been pre-equilibrated with buffer B. The column was washed with buffer B, containing 10 mM imidazole. Orf29 expressed as an
N-terminally 6×His-tagged fusion protein (rOrf29) was eluted with buffer B, containing 200 mM imidazole. The protein solution was collected and desalted with a PD-10 desalting column (GE Healthcare, Buckinghamshire, UK).
To reconstitute the iron-sulfur cluster in rOrf29, the purified enzyme was incubated with 5 m dithiothreitol (DTT) for 15 min at room temperature and was then incubated with 10 equivalent mole of Na2S and 10 equivalent mole of FeSO4(NH4)2SO4 for 30 min at room temperature. After the confirmation of the reduced form of [4Fe-4S]+ by UV-VIS spectroscopic analysis, the reconstituted rOrf29 was used for enzyme assays in vitro. A reaction mixture consisting of 50 mM HEPES-Na (pH 8.0), 300 mM NaCl, 0.1 mM methylcobalamine, 1 mM methylviologen, 4 mM NADH, 10 mM DTT, 10% (v/v) glycerol, 1 mM SAM (1), and 12 μM rOrf29 was incubated at 28 °C for 16 h under an anaerobic condition. The reaction mixture was then analyzed by HPLC-HR-ESI-MS using a hydrophilic interaction chromatography (HILIC) column (TSK-gel Amide-80, 3 μm, 150 × 2.0 mm; TOSOH) at 40 °C at a flow rate of 0.2 mL/min and with a three step linear gradient of acetonitrile in water in 0.1% (v/v) formic acid and 10 mM ammonium formate run over 20 min (80% (v/v) acetonitrile for 5 min, 80–50% (v/v) acetonitrile for 10 min, and 50–10% (v/v) acetonitrile for 5 min). To confirm the production of 5′-deoxyadenosine (Ado-CH3) in the enzyme reaction, the reaction mixture was further analyzed by HPLC-ESI-MS using a reversed-phase column (TSK-gel ODS-100Z, 3 μm, 150 × 2.0 mm; TOSOH) at 40 °C at a flow rate of 0.2 mL/min and with a two-step linear gradient of acetonitrile in water run over 15 min (5% (v/v) acetonitrile for 5 min and 5–95% (v/v) acetonitrile for 10 min).
In addition, 0.5 μM rOrf30 or 0.5 mM ACC was added to the rOrf29 enzyme reaction to confirm the productivity of 3, and the absolute configuration of 3 was determined by the advanced Marfey’s method [
25,
26].
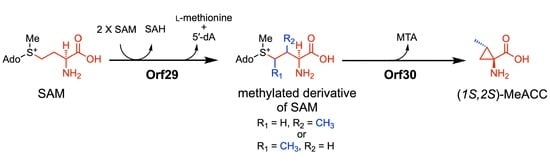
4. Discussion
In this study, from
Streptomyces genomes in our lab stock strains, we identified two genes,
orf29 and
orf30, which are involved in the biosynthesis of MeACC (3). At the beginning of the study, we hypothesized that 3 is produced from SAM (1) via ACC (2) (route A;
Figure 4), because
orf30 shared similarity (40%) with the bacterial ACC synthase GnmY, which catalyzes the cyclopropanation using 1 as the substrate (
Figure 1C). In fact, we detected the enzyme activity, but the kinetic data of rOrf30 were quite different from those of GmnY. Furthermore, our phylogenetic analysis suggested that
orf30 and GnmY formed a distinct clade (
Figure S2). These unexpected differences allowed us to think about an alternative biosynthetic route from 1 to 3 (route B;
Figure 4). Such a route was also supported by the following observations from the in vivo experiments: 1) the productivity of the putative demethyl-Q6402A was observed in the TK23_MeACC_Δ
orf30 strain, only when 0.2% ACC was added to the culture medium (
Figure S3D). 2) MeACC (3) was not produced by the EcSUF_
orf29 strain in the culture medium supplemented with AAC (2) (
Figure S5E). To confirm the alternative biosynthetic route (route B) from 1 to 3 (
Figure 4), we investigated the catalytic function of the reconstituted radical SAM enzyme, rOrf29. The in vitro analysis clearly demonstrated that SAM (1) was converted into the methylated derivative by rOrf29 (
Figure 6C) and was then converted into MeACC (3) by the rOrf30 catalysis (
Figure S9E). Because rOrf30 converts SAM (1) to ACC (2) with the low reaction rate, the methylated derivative of SAM produced by rOrf29 seems to be a better substrate of rOrf30. From these findings, we concluded that the rOrf29-mediated
C-methylation occurs in the L-methionine moiety of SAM (1) prior to the cyclopropanation reaction catalyzed by rOrf30. The production of (
1S,2S)-MeACC revealed that the
C-methylation of 1 is a stereospecific reaction by rOrf29. To determine the
C-methylation position (C3 or C4) of this unique radical, the methylated derivative of SAM produced by rOrf29, we now investigate biochemical properties of the SAM enzyme to improve the reaction efficiency to isolate the product for structural determination.
Based on a database search,
orf29 was found to have an iron-sulfur cluster-binding domain and a cobalamin-binding domain. Therefore,
orf29 belongs to the class B methyltransferase group in radical SAM methyltransferases. Class B enzymes are the most abundant radical SAM methyltransferases and appear to be the most versatile, catalyzing the methylation of both unactivated carbon and phosphorus centers [
22]. For example, CloN6 (ID no. RS42) [
30], Fms7 (ID no. RS45) [
31], and GenK (ID no. RS47) (
Table S4) [
32] catalyze the methylation of unactivated carbon. PhpK (ID no. RS50) [
33] mediates
P-methylation. In the phylogenetic analysis,
orf29 was found to form a distinct clade with these class B methyltransferases. In addition, to date, there has been no report of a class B enzyme that methylates 1. Thus,
orf29 is a novel radical SAM methyltransferase, catalyzing
C-methylation of the L-methionine moiety of SAM (
1) (
Figure 4).
We found that the
orf29 homologues (RS1 to RS6) and the
orf30 homologues (AS1 to AS6) are positioned next to each other in their gene cluster (
Figure S11), strongly suggesting that these six gene clusters are responsible for the production of peptide natural products, containing the MeACC building block (
Figure S11). In fact, the gene cluster containing RS1(
swb7)-AS1(
swb6) was reported to produce SW-163C [
14] (
Figure 1D and S11B). In addition, retimycin A, which is an SW-163C analogue compound, is also produced by the gene cluster with RS2-AS2 [
34] (
Figure S11C). In this study, we showed that the MeACC cluster from
S. violaceoniger 4521-SVS3 (
Figure 2 and
Figure S11A) is involved in the biosynthesis of putative Q6402A (C
50H
61N
7O
12, calculated for
m/z 952.445 [M + H]
+) (
Figure 3). On the other hand,
Streptomyces sp. Q6402, which is the original strain producing Q6402A [
29], was also reported to produce an analogue compound Q6402B (C
51H
63N
7O
12, calculated for
m/z 965.453 [M + H]
+) with a longer fatty acid side chain (6-methylheptanoic acid). It is possible that the MeACC cluster from
S. violaceoniger 4521-SVS3 also produces putative Q6402B, because we observed the production of not only putative demethyl-Q6402A but also putative demethyl-Q6402B in the TK23_MeACC_Δ
orf29 strain (
Figure S3B and S3D). Furthermore, the production of the demethyl compounds suggested that the A domain of
orf23 (NRPS) would accept 2 as a substrate in addition to 3.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





