Secondary Metabolites from Plants Possessing Inhibitory Properties against Beta-Amyloid Aggregation as Revealed by Thioflavin-T Assay and Correlations with Investigations on Transgenic Mouse Models of Alzheimer’s Disease

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Secondary Metabolites from Plants Identified as Inhibitors of Amyloid-Beta Fibrillogenesis
2.1. Gallic Acid
2.2. Rosmarinic Acid
2.3. Salvianolic Acid B
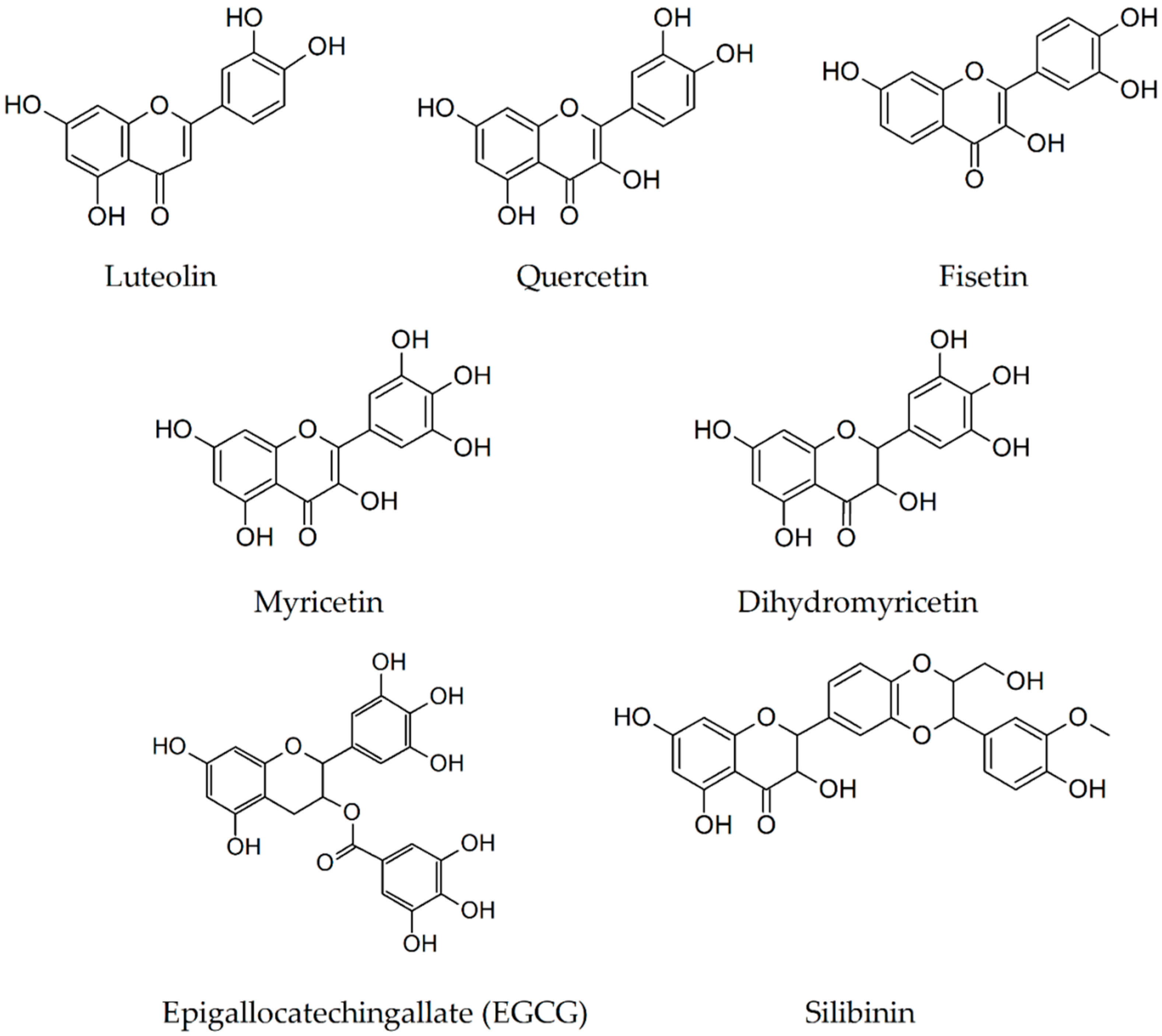
2.4. Luteolin
2.5. Quercetin
2.6. Fisetin
2.7. Myricetin
2.8. Dihydromyricetin
2.9. Epigallocatechin-3-Gallate
2.10. Silibinin
2.11. Oleuropein
2.12. Rutin
2.13. Curcumin
2.14. Crocin
2.15. Cryptotanshinone
2.16. Tabersonine
2.17. Other Plant Secondary Metabolites
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | Amyloid-beta |
| APPswe/PS1dE9 mice | referred to as APP/PS1, double transgenic mouse model of Alzheimer’s disease over expressing amyloid precursor protein (APPswe), encoding the Swedish mutations at amino acids 595/596 and an exon-9-deleted human PS1 (PS1dE9) |
| TG-SwDI mice | triple transgenic mouse model of Alzheimer’s disease, express an AβPP with Aβ flanking, double Swedish mutations (Lys670→Asn/ Met671→Leu), the Dutch (Glu693→Gln), and the Iowa (Asp694→Asn) mutations (sequence numbering in the AβPP770 isoform notation) |
| TG2576 mice | transgenic mouse model, which express a 695-aa residue splice form of human amyloid precursor protein modified by the Swedish Familial AD double mutation K670N-M671L |
| TgCRND8 mice | transgenic mouse model of Alzheimer’s disease |
| GABA | gamma-aminobutyric acid |
| 5XFAD mice | transgenic mice overexpress mutant human APP(695) with the Swedish (K670N, M671L), Florida (I716V), and London (V717I) Familial Alzheimer’s Disease (FAD) mutations along with human PS1 harboring two FAD mutations, M146L and L286V |
| APP | Amyloid Precursor Protein |
| PSEN1 | presenilin 1 |
| APOE | apolipoprotein E |
| MAPT | microtubule-associated protein tau |
| Trem2 | triggering receptor expressed on myeloid cells 2 |
| BACE1 | Beta-Secretase 1 |
| EGCG | Epigallocatechin-3-gallate |
| CTF | carboxyterminal fragments generated by α-secretase |
| sAPPα | generated when α-secretase cleaves APP |
| TNF-a | Tumor Necrosis Factor alpha |
| JNK | c-Jun N-terminal kinases |
| A11 | oligomer-specific antibody |
| W20 | oligomer-specific single chain variable fragment |
| TBS | tris-buffered saline |
| PBS | phosphate-buffered saline |
| DMSO | dimethyl sulfoxide |
| PCR | polymerase chain reaction |
| PMDs | protein misfolding disorders |
| LTP | long-term potentiation |
| LTD | long-term depression of excitatory synaptic transmission |
References
- Nie, Q.; Du, X.G.; Geng, M.Y. Small molecule inhibitors of amyloid β peptide aggregation as a potential therapeutic strategy for Alzheimer’s disease. Acta Pharmacol. Sin. 2011, 32, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Weidner, W.S.; Barbarino, P. World Alzheimer Report 2018—The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International (ADI): London, UK, 2018. [Google Scholar]
- Ma, L.; Yang, C.; Zheng, J.; Chen, Y.; Xiao, Y.; Huang, K. Non-polyphenolic natural inhibitors of amyloid aggregation. Eur. J. Med. Chem. 2020, 192. [Google Scholar] [CrossRef]
- Velander, P.; Wu, L.; Henderson, F.; Zhang, S.; Bevan, D.R.; Xu, B. Natural product-based amyloid inhibitors. Biochem. Pharmacol. 2017, 139, 40–55. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, P.R.; Dubey, A.K.; Masters, C.L.; Martins, R.N.; Macreadie, I.G. Aβ aggregation and possible implications in Alzheimer’s disease pathogenesis. J. Cell. Mol. Med. 2008, 13, 412–421. [Google Scholar] [CrossRef]
- Zhang, X.; Fu, Z.; Meng, L.; He, M.; Zhang, Z. The Early Events That Initiate β-Amyloid Aggregation in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Stanciu, G.D.; Luca, A.; Rusu, R.N.; Bild, V.; Chiriac, S.I.B.; Solcan, C.; Bild, W.; Ababei, D.C. Alzheimer’s disease pharmacotherapy in relation to cholinergic system involvement. Biomolecules 2020, 10, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, P.; Patel, B.; Makwana, V.; Jadhav, H.R.; Kiefel, M.; Davey, A.; Reekie, T.A.; Rudrawar, S.; Kassiou, M. Peptides, Peptidomimetics, and Carbohydrate-Peptide Conjugates as Amyloidogenic Aggregation Inhibitors for Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 1530–1551. [Google Scholar] [CrossRef] [PubMed]
- Lührs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Döbeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid-β(1-42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef] [Green Version]
- Dhouafli, Z.; Cuanalo-Contreras, K.; Hayouni, E.A.; Mays, C.E.; Soto, C.; Moreno-Gonzalez, I. Inhibition of protein misfolding and aggregation by natural phenolic compounds. Cell. Mol. Life Sci. 2018, 75, 3521–3538. [Google Scholar] [CrossRef] [PubMed]
- Yoshiike, Y.; Tanemura, K.; Murayama, O.; Akagi, T.; Murayama, M.; Sato, S.; Sun, X.; Tanaka, N.; Takashima, A. New Insights on How Metals Disrupt Amyloid β-Aggregation and Their Effects on Amyloid-β Cytotoxicity. J. Biol. Chem. 2001, 276, 32293–32299. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [Green Version]
- Biancalana, M.; Koide, S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta - Proteins Proteomics 2010, 1804, 1405–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maezawa, I.; Hong, H.S.; Liu, R.; Wu, C.Y.; Cheng, R.H.; Kung, M.P.; Kung, H.F.; Lam, K.S.; Oddo, S.; LaFerla, F.M.; et al. Congo red and thioflavin-T analogs detect Aβ oligomers. J. Neurochem. 2008, 104, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Levine, H. Thioflavine T interaction with synthetic Alzheimer’s disease β-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 1993, 2, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Groenning, M. Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils-current status. J. Chem. Biol. 2010, 3, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 1–22. [Google Scholar] [CrossRef]
- Alzheimer Association Early Signs and Symptoms of Alzheimer’s. Alzheimer’s Dement. 2019, 1–88.
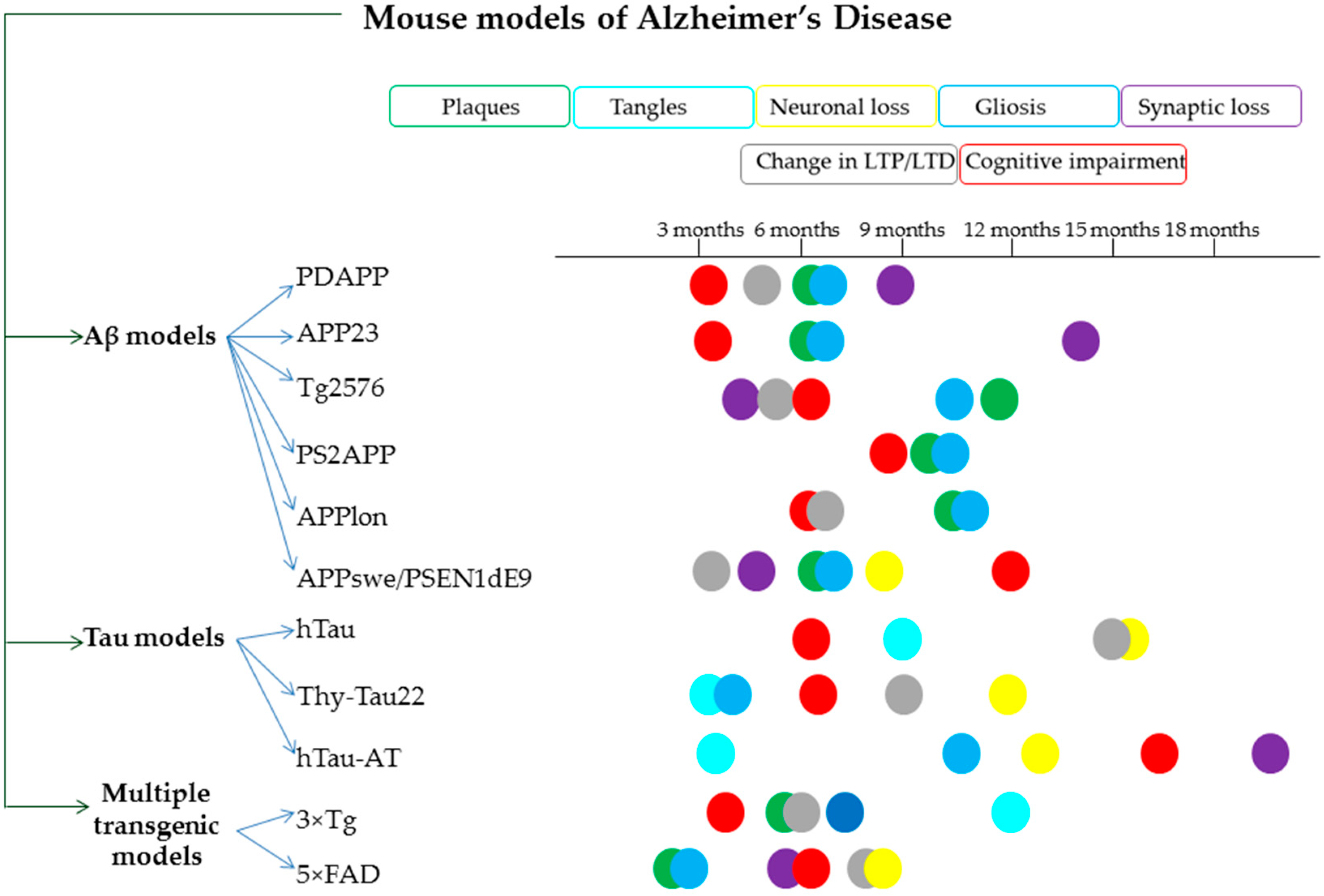
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Borthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef]
- Calhoun, M.E.; Wiederhold, K.H.; Abramowski, D.; Phinney, A.L.; Probst, A.; Sturchler-Pierrat, C.; Staufenbiel, M.; Sommer, B.; Jucker, M. Neuron loss in APP transgenic mice [7]. Nature 1998, 395, 755–756. [Google Scholar] [CrossRef]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.A.; et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, L.; Gordon, M.N.; Mcgowan, E.; Yu, X.; Benkovic, S.; Jantzen, P.; Wright, K.; Saad, I.; Mueller, R.; Morgan, D.; et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 1998, 4, 97–100. [Google Scholar] [CrossRef]
- Richards, J.G.; Higgins, G.A.; Ouagazzal, A.M.; Ozmen, L.; Kew, J.N.C.; Bohrmann, B.; Malherbe, P.; Brockhaus, M.; Loetscher, H.; Czech, C.; et al. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J. Neurosci. 2003, 23, 8989–9003. [Google Scholar] [CrossRef] [Green Version]
- Moechars, D.; Dewachter, I.; Lorent, K.; Reversé, D.; Baekelandt, V.; Naidu, A.; Tesseur, I.; Spittaels, K.; Van Den Haute, C.; Checler, F.; et al. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J. Biol. Chem. 1999, 274, 6483–6492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankowsky, J.L.; Slunt, H.H.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Borchelt, D.R. APP processing and amyloid deposition in mice haplo-insufficient for presenilin 1. Neurobiol. Aging 2004, 25, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Andorfer, C.; Kress, Y.; Espinoza, M.; De Silva, R.; Tucker, K.L.; Barde, Y.A.; Duff, K.; Davies, P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 2003, 86, 582–590. [Google Scholar] [CrossRef]
- Schindowski, K.; Bretteville, A.; Leroy, K.; Bégard, S.; Brion, J.P.; Hamdane, M.; Buée, L. Alzheimer’s disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am. J. Pathol. 2006, 169, 599–616. [Google Scholar] [CrossRef] [Green Version]
- Decker, J.M.; Krüger, L.; Sydow, A.; Dennissen, F.J.; Siskova, Z.; Mandelkow, E.; Mandelkow, E. The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR 2B receptor-mediated excitotoxicity. EMBO Rep. 2016, 17, 552–569. [Google Scholar] [CrossRef] [Green Version]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s Disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Mullane, K.; Williams, M. Preclinical Models of Alzheimer’s Disease: Relevance and Translational Validity. Curr. Protoc. Pharmacol. 2019, 84, 1–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 5, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, A.; McGonigle, P. Overview of Transgenic Mouse Models for Alzheimer’s Disease. Curr. Protoc. Neurosci. 2019, 89. [Google Scholar] [CrossRef] [PubMed]
- Lippi, S.L.P.; Smith, M.L.; Flinn, J.M. A Novel hAPP/htau Mouse Model of Alzheimer’s Disease: Inclusion of APP With Tau Exacerbates Behavioral Deficits and Zinc Administration Heightens Tangle Pathology. Front. Aging Neurosci. 2018, 10, 382. [Google Scholar] [CrossRef] [Green Version]
- Foidl, B.; Humpel, C. Can mouse models mimic sporadic Alzheimer’s disease? Neural Regen. Res. 2020, 15, 401. [Google Scholar]
- Ştefănescu, R.; Stanciu, G.D.; Luca, A.; Caba, I.C.; Tamba, B.I.; Mihai, C.T. Contributions of mass spectrometry to the identification of low molecular weight molecules able to reduce the toxicity of amyloid-β peptide to cell cultures and transgenic mouse models of Alzheimer’s disease. Molecules 2019, 24, 1167. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Pukala, T.L.; Musgrave, I.F.; Williams, D.M.; Dehle, F.C.; Carver, J.A. Gallic acid is the major component of grape seed extract that inhibits amyloid fibril formation. Bioorganic Med. Chem. Lett. 2013, 23, 6336–6340. [Google Scholar] [CrossRef]
- Wong, D.Y.S.; Musgrave, I.F.; Harvey, B.S.; Smid, S.D. Açaí (Euterpe oleraceae Mart.) berry extract exerts neuroprotective effects against β-amyloid exposure in vitro. Neurosci. Lett. 2013, 556, 221–226. [Google Scholar] [CrossRef]
- Porzoor, A.; Alford, B.; Hügel, H.M.; Grando, D.; Caine, J.; Macreadie, I. Anti-amyloidogenic properties of some phenolic compounds. Biomolecules 2015, 5, 505–527. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Chen, X.; Liu, J.; Ma, Q.; Zhuo, Z.; Chen, H.; Zhou, L.; Yang, S.; Zheng, L.; Ning, C.; et al. Gallic acid disruption of Aβ1–42 aggregation rescues cognitive decline of APP/PS1 double transgenic mouse. Neurobiol. Dis. 2019, 124, 67–80. [Google Scholar] [CrossRef]
- Sun, J.; Jiang, G.; Shigemori, H. Inhibitory Activity on Amyloid Aggregation of Rosmarinic Acid and Its Substructures from Isodon japonicus. Nat. Prod. Commun. 2019. [Google Scholar] [CrossRef] [Green Version]
- Hamaguchi, T.; Ono, K.; Murase, A.; Yamada, M. Phenolic Compounds Prevent Alzheimer’s Pathology through Different Effects on the Amyloid-β Aggregation Pathway. Am. J. Pathol. 2009, 175, 2557–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hase, T.; Shishido, S.; Yamamoto, S.; Yamashita, R.; Nukima, H.; Taira, S. Rosmarinic acid suppresses Alzheimer ’ s disease development by reducing amyloid β aggregation by increasing monoamine secretion. Sci. Rep. 2019, 1–13. [Google Scholar]
- Durairajan, S.S.K.; Yuan, Q.; Xie, L.; Chan, W.S.; Kum, W.F.; Koo, I.; Liu, C.; Song, Y.; Huang, J.D.; Klein, W.L.; et al. Salvianolic acid B inhibits Aβ fibril formation and disaggregates preformed fibrils and protects against Aβ-induced cytotoxicty. Neurochem. Int. 2008, 52, 741–750. [Google Scholar] [CrossRef]
- Shen, L.; Han, B.; Geng, Y.; Wang, J.; Wang, Z.; Wang, M. Amelioration of cognitive impairments in APPswe/PS1dE9 mice is associated with metabolites alteration induced by total salvianolic acid. PLoS ONE 2017, 12, e0174763. [Google Scholar] [CrossRef]
- Akaishi, T.; Morimoto, T.; Shibao, M.; Watanabe, S.; Sakai-Kato, K.; Utsunomiya-Tate, N.; Abe, K. Structural requirements for the flavonoid fisetin in inhibiting fibril formation of amyloid β protein. Neurosci. Lett. 2008, 444, 280–285. [Google Scholar] [CrossRef]
- Churches, Q.I.; Caine, J.; Cavanagh, K.; Epa, V.C.; Waddington, L.; Tranberg, C.E.; Meyer, A.G.; Varghese, J.N.; Streltsov, V.; Duggan, P.J. Naturally occurring polyphenolic inhibitors of amyloid beta aggregation. Bioorganic Med. Chem. Lett. 2014, 24, 3108–3112. [Google Scholar] [CrossRef]
- Rezai-Zadeh, K.; Douglas Shytle, R.; Bai, Y.; Tian, J.; Hou, H.; Mori, T.; Zeng, J.; Obregon, D.; Town, T.; Tan, J. Flavonoid-mediated presenilin-1 phosphorylation reduces Alzheimer’s disease β-amyloid production. J. Cell. Mol. Med. 2009, 13, 574–588. [Google Scholar] [CrossRef]
- Sawmiller, D.; Li, S.; Shahaduzzaman, M.; Smith, A.J.; Obregon, D.; Giunta, B.; Borlongan, C.V.; Sanberg, P.R.; Tan, J. Luteolin reduces Alzheimer’s disease pathologies induced by traumatic brain injury. Int. J. Mol. Sci. 2014, 15, 895–904. [Google Scholar] [CrossRef] [Green Version]
- Pérez Corredor, P.; Sabogal Guáqueta, A.; Hormaza, C.; Cardona Gómez, G. Preventive effect of quercetin in a triple transgenic Alzheimer’s disease mice model. Molecules 2019, 24, 2287. [Google Scholar] [CrossRef] [Green Version]
- Sabogal-Guáqueta, A.M.; Muñoz-Manco, J.I.; Ramírez-Pineda, J.R.; Lamprea-Rodriguez, M.; Osorio, E.; Cardona-Gómez, G.P. The flavonoid quercetin ameliorates Alzheimer’s disease pathology and protects cognitive and emotional function in aged triple transgenic Alzheimer’s disease model mice. Neuropharmacology 2015, 93, 134–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Hu, J.; Zhong, L.; Wang, N.; Yang, L.; Liu, C.C.; Li, H.; Wang, X.; Zhou, Y.; Zhang, Y.; et al. Quercetin stabilizes apolipoprotein e and reduces brain Aβ levels in amyloid model mice. Neuropharmacology 2016, 108, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Currais, A.; Prior, M.; Dargusch, R.; Armando, A.; Ehren, J.; Schubert, D.; Quehenberger, O.; Maher, P. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell 2014, 13, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Multifunction of myricetin on Aβ: Neuroprotection via a conformational change of Aβ and reduction of Aβ via the interference of secretases. J. Neurosci. Res. 2008, 86, 368–377. [Google Scholar] [CrossRef]
- Jia, L.; Zhao, W.; Sang, J.; Wang, W.; Wei, W.; Wang, Y.; Zhao, F.; Lu, F.; Liu, F. Inhibitory Effect of a Flavonoid Dihydromyricetin against Aβ40 Amyloidogenesis and Its Associated Cytotoxicity. ACS Chem. Neurosci. 2019, 10, 4696–4703. [Google Scholar] [CrossRef]
- Liang, J.; Lindemeyer, A.K.; Shen, Y.; López-Valdés, H.E.; Martínez-Coria, H.; Shao, X.M.; Olsen, R.W. Dihydromyricetin ameliorates behavioral deficits and reverses neuropathology of transgenic mouse models of Alzheimer’s disease. Neurochem. Res. 2014, 39, 1171–1181. [Google Scholar] [CrossRef]
- Huang, Q.; Zhao, Q.; Peng, J.; Yu, Y.; Wang, C.; Zou, Y.; Su, Y.; Zhu, L.; Wang, C.; Yang, Y. Peptide-Polyphenol (KLVFF/EGCG) binary modulators for inhibiting aggregation and neurotoxicity of amyloid-β peptide. ACS Omega 2019, 4, 4233–4242. [Google Scholar] [CrossRef]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezai-Zadeh, K.; Shytle, D.; Sun, N.; Mori, T.; Hou, H.; Jeanniton, D.; Ehrhart, J.; Townsend, K.; Zeng, J.; Morgan, D.; et al. Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J. Neurosci. 2005, 25, 8807–8814. [Google Scholar] [CrossRef] [Green Version]
- Rezai-Zadeh, K.; Arendash, G.W.; Hou, H.; Fernandez, F.; Jensen, M.; Runfeldt, M.; Shytle, R.D.; Tan, J. Green tea epigallocatechin-3-gallate (EGCG) reduces β-amyloid mediated cognitive impairment and modulates tau pathology in Alzheimer transgenic mice. Brain Res. 2008, 1214, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Gordon, M.; Tan, J.; Morgan, D. Oral administration of green tea epigallocatechin-3-gallate (EGCG) reduces amyloid beta deposition in transgenic mouse model of Alzheimer’s disease. Exp. Neurol. 2006, 198, 575–576. [Google Scholar] [CrossRef]
- Jia, N.; Han, K.; Kong, J.J.; Zhang, X.M.; Sha, S.; Ren, G.R.; Cao, Y.P. (-)-Epigallocatechin-3-gallate alleviates spatial memory impairment in APP/PS1 mice by restoring IRS-1 signaling defects in the hippocampus. Mol. Cell. Biochem. 2013, 380, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Perreau, V.M.; Pop, V.; Cotman, C.W. Brief Communication Voluntary Exercise Decreases Amyloid Load in a Transgenic Model of Alzheimer’s Disease. J. Neurosci. 2005, 25, 4217–4221. [Google Scholar] [CrossRef]
- Yin, F.; Liu, J.; Ji, X.; Wang, Y.; Zidichouski, J.; Zhang, J. Silibinin: A novel inhibitor of Aβ aggregation. Neurochem. Int. 2011, 58, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Duan, S.; Guan, X.; Lin, R.; Liu, X.; Yan, Y.; Lin, R.; Zhang, T.; Chen, X.; Huang, J.; Sun, X.; et al. Silibinin inhibits acetylcholinesterase activity and amyloid β peptide aggregation: A dual-target drug for the treatment of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 1792–1807. [Google Scholar] [CrossRef]
- Omar, S.H.; Scott, C.J.; Hamlin, A.S.; Obied, H.K. Olive biophenols reduces alzheimer’s pathology in SH-SY5Y cells and APPswe mice. Int. J. Mol. Sci. 2019, 20, 125. [Google Scholar] [CrossRef] [Green Version]
- Grossi, C.; Rigacci, S.; Ambrosini, S.; Ed Dami, T.; Luccarini, I.; Traini, C.; Failli, P.; Berti, A.; Casamenti, F.; Stefani, M. The Polyphenol Oleuropein Aglycone Protects TgCRND8 Mice against Aß Plaque Pathology. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Wang, S.W.; Wang, Y.J.; Su, Y.J.; Zhou, W.W.; Yang, S.G.; Zhang, R.; Zhao, M.; Li, Y.N.; Zhang, Z.P.; Zhan, D.W.; et al. Rutin inhibits β-amyloid aggregation and cytotoxicity, attenuates oxidative stress, and decreases the production of nitric oxide and proinflammatory cytokines. Neurotoxicology 2012, 33, 482–490. [Google Scholar] [CrossRef]
- Xu, P.X.; Wang, S.W.; Yu, X.L.; Su, Y.J.; Wang, T.; Zhou, W.W.; Zhang, H.; Wang, Y.J.; Liu, R.T. Rutin improves spatial memory in Alzheimer’s disease transgenic mice by reducing Aβ oligomer level and attenuating oxidative stress and neuroinflammation. Behav. Brain Res. 2014, 264, 173–180. [Google Scholar] [CrossRef]
- Hu, B.; Dai, F.; Fan, Z.; Ma, G.; Tang, Q.; Zhang, X. Nanotheranostics: Congo Red/Rutin-MNPs with Enhanced Magnetic Resonance Imaging and H2O2-Responsive Therapy of Alzheimer’s Disease in APPswe/PS1dE9 Transgenic Mice. Adv. Mater. 2015, 27, 5499–5505. [Google Scholar] [CrossRef]
- Jiang, T.; Zhi, X.L.; Zhang, Y.H.; Pan, L.F.; Zhou, P. Inhibitory effect of curcumin on the Al(III)-induced Aβ42 aggregation and neurotoxicity in vitro. Biochim. Biophys. Acta - Mol. Basis Dis. 2012, 1822, 1207–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Alloza, M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacskai, B.J. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem. 2007, 102, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. 2001, 21, 8370–8377. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Su, C.; Li, R.; Wang, H.; Ren, Y.; Sun, H.; Yang, J.; Sun, J.; Shi, J.; Tian, J.; et al. Mechanisms and effects of curcumin on spatial learning and memory improvement in APPswe/PS1dE9 mice. J. Neurosci. Res. 2014, 92, 218–231. [Google Scholar] [CrossRef]
- Ghahghaei, A.; Bathaie, S.Z.; Bahraminejad, E. Mechanisms of the effects of crocin on aggregation and deposition of ab1-40 fibrils in Alzheimer’s Disease. Int. J. Pept. Res. Ther. 2012, 18, 347–351. [Google Scholar] [CrossRef]
- Ghahghaei, A.; Bathaie, S.Z.; Kheirkhah, H.; Bahraminejad, E. The protective effect of crocin on the amyloid fibril formation of aβ42 peptide in vitro. Cell. Mol. Biol. Lett. 2013, 18, 328–339. [Google Scholar] [CrossRef] [Green Version]
- Batarseh, Y.S.; Bharate, S.S.; Kumar, V.; Kumar, A.; Vishwakarma, R.A.; Bharate, S.B.; Kaddoumi, A.; States, U.; Road, C. Crocus sativus Extract Tightens the Blood-Brain Barrier, Reduces Amyloid β Load and Related Toxicity in 5XFAD Mice. ACS Chem. Neurosci. 2018, 8, 1756–1766. [Google Scholar] [CrossRef]
- Mei, Z.; Yan, P.; Situ, B.; Mou, Y.; Liu, P. Cryptotanshinione inhibits β-amyloid aggregation and protects damage from β-amyloid in SH-SY5Y cells. Neurochem. Res. 2012, 37, 622–628. [Google Scholar] [CrossRef]
- Mei, Z.; Zhang, F.; Tao, L.; Zheng, W.; Cao, Y.; Wang, Z.; Tang, S.; Le, K.; Chen, S.; Pi, R.; et al. Cryptotanshinone, a compound from Salvia miltiorrhiza modulates amyloid precursor protein metabolism and attenuates β-amyloid deposition through upregulating α-secretase in vivo and in vitro. Neurosci. Lett. 2009. [Google Scholar] [CrossRef]
- Kai, T.; Zhang, L.; Wang, X.; Jing, A.; Zhao, B.; Yu, X.; Zheng, J.; Zhou, F. Tabersonine Inhibits Amyloid Fibril Formation and Cytotoxicity of Aβ(1-42). ACS Chem. Neurosci. 2015, 6, 879–888. [Google Scholar] [CrossRef]
- Xu, J.; Wei, K.; Zhang, G.; Lei, L.; Yang, D.; Wang, W.; Han, Q.; Xia, Y.; Bi, Y.; Yang, M.; et al. Ethnopharmacology, phytochemistry, and pharmacology of Chinese Salvia species: A review. J. Ethnopharmacol. 2018, 225, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Ullah, H.; Aschner, M.; Cheang, W.S.; Akkol, E.K. Neuroprotective Effects of Quercetin in Alzheimer’s Disease. Biomolecules 2019, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Perret, D.; Luo, Z.D. Targeting voltage-gated calcium channels for neuropathic pain management. Neurotherapeutics 2009, 6, 679–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q.; Cai, S.; Jia, Y.; Sun, X.; Yi, J.; Du, J. Effects of hot-water extract from vine tea (Ampelopsis grossedentata) on acrylamide formation, quality and consumer acceptability of bread. Foods 2020, 9, 373. [Google Scholar] [CrossRef] [Green Version]
- Adler, B.L.; Yarchoan, M.; Hwang, H.M.; Louneva, N.; Blair, J.A.; Palm, R.; Smith, M.A.; Lee, H.G.; Arnold, S.E.; Casadesus, G. Neuroprotective effects of the amylin analogue pramlintide on Alzheimer’s disease pathogenesis and cognition. Neurobiol. Aging 2014, 35, 793–801. [Google Scholar] [CrossRef]
- Hussain, H.; Hussain, J.; Al-Harrasi, A.; Green, I.R. Chemistry and biology of the genus Voacanga. Pharm. Biol. 2012, 50, 1183–1193. [Google Scholar] [CrossRef]
- Ștefănescu, R.; Lupu, L.; Manea, M.; Iacob, R.E.; Przybylski, M. Molecular characterization of the β-amyloid(4-10) epitope of plaque specific Aβ antibodies by affinity-mass spectrometry using alanine site mutation. J. Pept. Sci. 2018, 24, e3047. [Google Scholar] [CrossRef]
- Bazoti, F.N.; Bergquist, J.; Markides, K.; Tsarbopoulos, A. Localization of the noncovalent binding site between amyloid-beta-peptide and oleuropein using electrospray ionization FT-ICR mass spectrometry. J. Am. Soc. Mass Spectrom. 2008, 19, 1078–1085. [Google Scholar] [CrossRef] [Green Version]
- Bazoti, F.N.; Bergquist, J.; Markides, K.E.; Tsarbopoulos, A. Noncovalent interaction between amyloid-beta-peptide (1-40) and oleuropein studied by electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 2006, 17, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Marek, P.; Potter, K.J.; Verchere, C.B.; Raleigh, D.P. Rifampicin does not prevent amyloid fibril formation by human islet amyloid polypeptide but does inhibit fibril thioflavin-T interactions: Implications for mechanistic studies of β-cell death. Biochemistry 2008, 47, 6016–6024. [Google Scholar] [CrossRef]
- Kroes-Nijboer, A.; Lubbersen, Y.S.; Venema, P.; van der Linden, E. Thioflavin T fluorescence assay for β-lactoglobulin fibrils hindered by DAPH. J. Struct. Biol. 2009, 165, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Hudson, S.A.; Ecroyd, H.; Kee, T.W.; Carver, J.A. The thioflavin T fluorescence assay for amyloid fibril detection can be biased by the presence of exogenous compounds. FEBS J. 2009, 276, 5960–5972. [Google Scholar] [CrossRef] [Green Version]
- Molino, S.; Dossena, M.; Buonocore, D.; Ferrari, F.; Venturini, L.; Ricevuti, G.; Verri, M. Polyphenols in dementia: From molecular basis to clinical trials. Life Sci. 2016, 161, 69–77. [Google Scholar] [CrossRef]
- Mandel, S.; Amit, T.; Reznichenko, L.; Weinreb, O.; Youdim, M.B.H. Green tea catechins as brain-permeable, natural iron chelators-antioxidants for the treatment of neurodegenerative disorders. Mol. Nutr. Food Res. 2006, 50, 229–234. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, M.; Lu, F.; Luo, N.; He, Z.P.; Yang, H. Involvement of α7 nAChR signaling cascade in epigallocatechin gallate suppression of β-Amyloid-Induced apoptotic cortical neuronal insults. Mol. Neurobiol. 2014, 49, 66–77. [Google Scholar] [CrossRef]
- De Oliveira, M.R.; Nabavi, S.F.; Daglia, M.; Rastrelli, L.; Nabavi, S.M. Epigallocatechin gallate and mitochondria - A story of life and death. Pharmacol. Res. 2016, 104, 70–85. [Google Scholar] [CrossRef]
- Lee, J.H.; Moon, J.H.; Kim, S.W.; Jeong, J.K.; Nazim, U.M.D.; Lee, Y.J.; Seol, J.W.; Park, S.Y. EGCG-mediated autophagy flux has a neuroprotection effect via a class III histone deacetylase in primary neuron cells. Oncotarget 2015, 6, 9701–9717. [Google Scholar] [CrossRef] [Green Version]
- Pogačnik, L.; Pirc, K.; Palmela, I.; Skrt, M.; Kwang, K.S.; Brites, D.; Brito, M.A.; Ulrih, N.P.; Silva, R.F.M. Potential for brain accessibility and analysis of stability of selected flavonoids in relation to neuroprotection in vitro. Brain Res. 2016, 1651, 17–26. [Google Scholar] [CrossRef]
- Noble, W. Challenges in neurodegeneration research. Front. Psychiatry 2010, 1, 7. [Google Scholar] [CrossRef] [Green Version]
- Serban, D.; Anton, E.; Chirita, R.; Bild, V.; Ciobica, A.; Alexinschi, O.; Arcan, O.; Popescu, R.; Paduraru, L.; Timofte, D. Current aspects of the interactions between dementia, the brain renin-angiotensin system and oxidative stress. Arch. Biol. Sci. 2015, 67, 903–907. [Google Scholar] [CrossRef]
- Patel, S.S.; Acharya, A.; Ray, R.S.; Agrawal, R.; Raghuwanshi, R.; Jain, P. Cellular and molecular mechanisms of curcumin in prevention and treatment of disease. Crit. Rev. Food Sci. Nutr. 2020, 60, 887–939. [Google Scholar] [CrossRef] [PubMed]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beigi, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G.; et al. Oral curcumin for Alzheimer’s disease: Tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimer’s Res. Ther. 2012, 4, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baum, L.; Lam, C.W.K.; Cheung, S.K.K.; Kwok, T.; Lui, V.; Tsoh, J.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; et al. Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease [7]. J. Clin. Psychopharmacol. 2008, 28, 110–113. [Google Scholar] [CrossRef] [Green Version]
- Barril, X. Druggability predictions: Methods, limitations, and applications. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 1–12. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Secondary Metabolite | Scientific Name of the Plant (Family) | Effects Observed Using Thioflavin T Assay | In Vivo Findings |
|---|---|---|---|
| Gallic acid [38,41] | Vitis vinifera (Vitaceae) | diminishes/blocks fibril formation disaggregates preformed fibrils | reduction of Aβ(1-42) plaques size, improvement of the spatial reference and working memories of 4-month-old transgenic mice; reduction of cognitive deficits in 9-month-old AD mice |
| Rosmarinic acid [40,42,43,44] | Rosmarinus officinalis (Lamiaceae) | diminishes/blocks fibril formation in dose-dependent manner | a significant reduction of Aβ plaque number memory improvement increase of TBS-soluble Aβ monomers and reduction of A11-positive oligomers |
| Salvianolic acid B [45,82] | Salvia miltiorrhiza (Lamiaceae) | diminishes fibril formation in dose-dependent manner | decrease of Aβ(1-42) and Aβ(1-40) levels in the hippocampus reduction of spatial cognitive impairments |
| Luteolin [47,48,60] | Daucus carota (Apiaceae) | diminishes fibril formation | inhibition of soluble Aβ(1–40) and Aβ(1-42) generation by 25% and 49%, respectively, attenuation of the cognitive impairments |
| Quercetin [47,52,53,83,84] | Malus domestica (Rosaceae) | diminishes fibril formation | reduction of tauopathy and extracellular amyloidosis |
| Fisetin [47,54] | Fragaria moschata (Rosaceae) | diminishes fibril formation | prevention of progressive memory loss and learning impairments |
| Myricetin [43,47,55] | Vitis vinifera (Vitaceae) | diminishes fibril formation in dose-dependent manner | reduction of the A11-positive oligomers and a tendency to attenuate Aβ plaque deposition |
| Dihydromyricetin [56,57,85] | Ampelopsis grossedentata (Vitaceae) | diminishes/blocks fibril formation in dose-dependent manner, disaggregates preformed fibrils in dose-dependent manner | reduction of Aβ(1-42) and Aβ(1-40) levels, amelioration of behavioral deficits, reduction of learning and cognitive impairments |
| EGCG [39,48,49,59,62,63,86] | Thea sinensis (Theaceae) | diminishes/blocks fibril formation in dose-dependent manner disaggregates preformed fibrils | reduction of the plaque formation, decrease soluble and insoluble Aβ(1-40) and Aβ(1-42), improvement of working memory |
| Silibinin [65,66] | Silybum marianum (Asteraceae) | diminishes fibril formation in dose-dependent manner | a remarkable reduction in the surface area of Aβ plaque, a decrease in the activity and quantity of acetylcholinesterase, and an increase in synaptic protection, gliogenesis, and neurogenesis amelioration of cognitive deficits |
| Oleuropein [67,68] | Olea europea (Oleaceae) | diminishes fibril formation in dose-dependent manner | reduction of Aβ levels and plaque areas in the cortex and hippocampus |
| Rutin [69,70,71] | Malus domestica (Rosaceae) | diminishes/blocks fibril formation in dose-dependent manner | a reduction in Aβ oligomers levels, attenuation of memory deficits, reduction of microgliosis, astrocytosis, glutathione peroxidase, malondialdehyde, interleukin-6, and interleukin-1β levels; increase of glutathione/glutathione disulfide ratio |
| Curcumin [12,72,74] | Curcuma longa (Zingiberaceae) | diminishes/blocks fibril formation in dose-dependent manner, disaggregates preformed fibrils | reduction of soluble and insoluble Aβ, plaque burden, and the astrocytic marker GFAP using low-dose TBS-soluble, reduction of A11-positive oligomers |
| Crocin [76,77,78] | Crocus sativus (Iridaceae) | diminishes fibril formation | decrease of Aβ(1-40) by 25% and Aβ(1-42) levels by 29%, respectively |
| Cryptotanshinone [79] | Salvia miltiorrhiza (Lamiaceae) | diminishes fibril formation | attenuation of Aβ deposits amelioration of spatial learning and memory deficits |
| Tabersonine [81,87] | Voacanga africana (Apocynaceae) | diminishes fibril formation disaggregates preformed fibrils | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stefanescu, R.; Stanciu, G.D.; Luca, A.; Paduraru, L.; Tamba, B.-I. Secondary Metabolites from Plants Possessing Inhibitory Properties against Beta-Amyloid Aggregation as Revealed by Thioflavin-T Assay and Correlations with Investigations on Transgenic Mouse Models of Alzheimer’s Disease. Biomolecules 2020, 10, 870. https://doi.org/10.3390/biom10060870
Stefanescu R, Stanciu GD, Luca A, Paduraru L, Tamba B-I. Secondary Metabolites from Plants Possessing Inhibitory Properties against Beta-Amyloid Aggregation as Revealed by Thioflavin-T Assay and Correlations with Investigations on Transgenic Mouse Models of Alzheimer’s Disease. Biomolecules. 2020; 10(6):870. https://doi.org/10.3390/biom10060870
Chicago/Turabian StyleStefanescu, Raluca, Gabriela Dumitriṭa Stanciu, Andrei Luca, Luminita Paduraru, and Bogdan-Ionel Tamba. 2020. "Secondary Metabolites from Plants Possessing Inhibitory Properties against Beta-Amyloid Aggregation as Revealed by Thioflavin-T Assay and Correlations with Investigations on Transgenic Mouse Models of Alzheimer’s Disease" Biomolecules 10, no. 6: 870. https://doi.org/10.3390/biom10060870
APA StyleStefanescu, R., Stanciu, G. D., Luca, A., Paduraru, L., & Tamba, B. -I. (2020). Secondary Metabolites from Plants Possessing Inhibitory Properties against Beta-Amyloid Aggregation as Revealed by Thioflavin-T Assay and Correlations with Investigations on Transgenic Mouse Models of Alzheimer’s Disease. Biomolecules, 10(6), 870. https://doi.org/10.3390/biom10060870