Phytochemical Targeting of STAT3 Orchestrated Lipid Metabolism in Therapy-Resistant Cancers



Abstract
:1. Introduction
1.1. Lipid Metabolism in Cancer
1.2. Adipokines
1.3. IL-6R/JAK/STAT3 Signalling
2. The Role of STAT3 in Lipid Metabolism
2.1. JAK/STAT3 Pathway as a Therapeutic Target
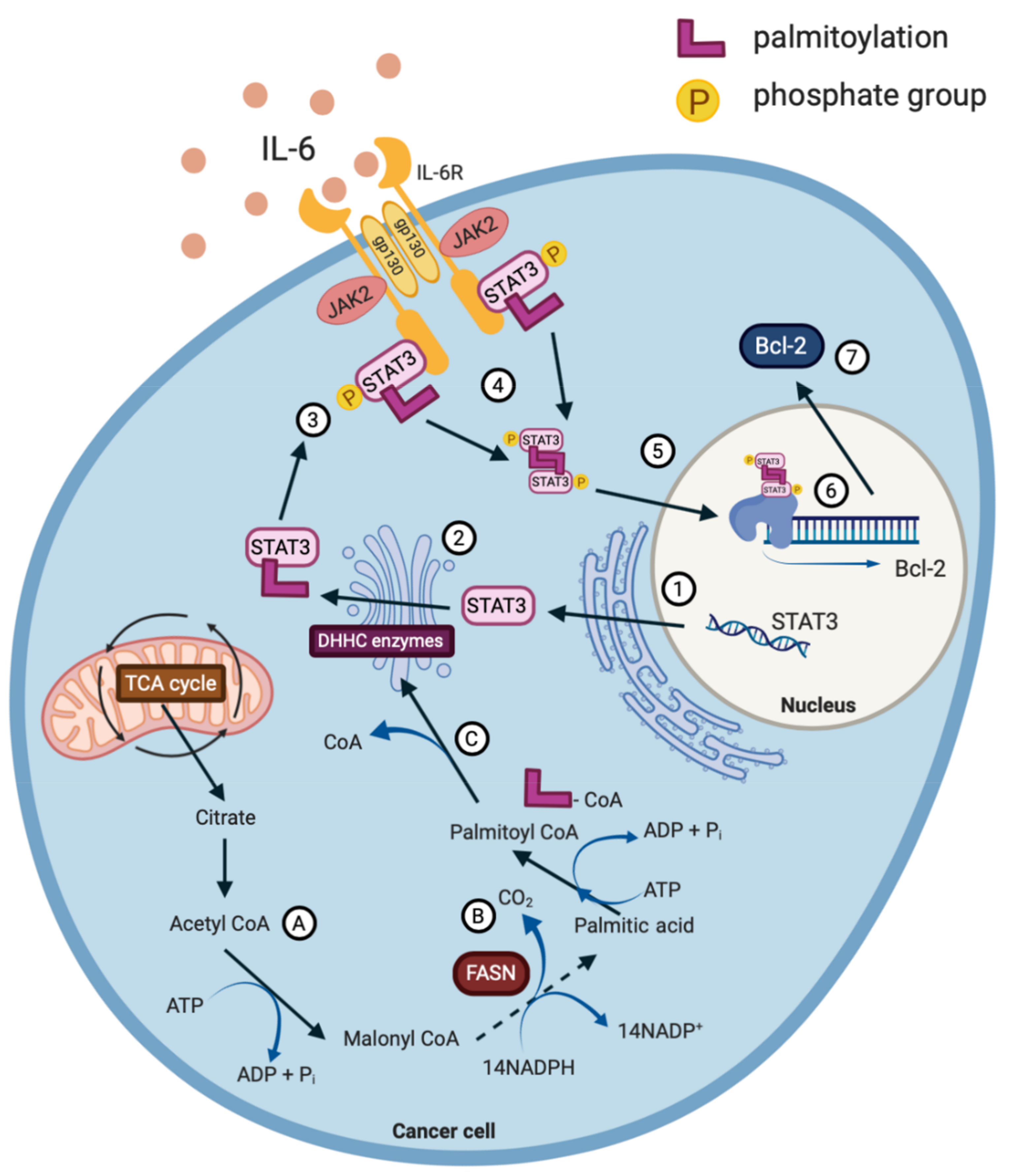
2.2. Post-Translational Modification of STAT3 as a Therapeutic Target
3. The Role of STAT3 and Lipid Metabolism in Therapy Resistance
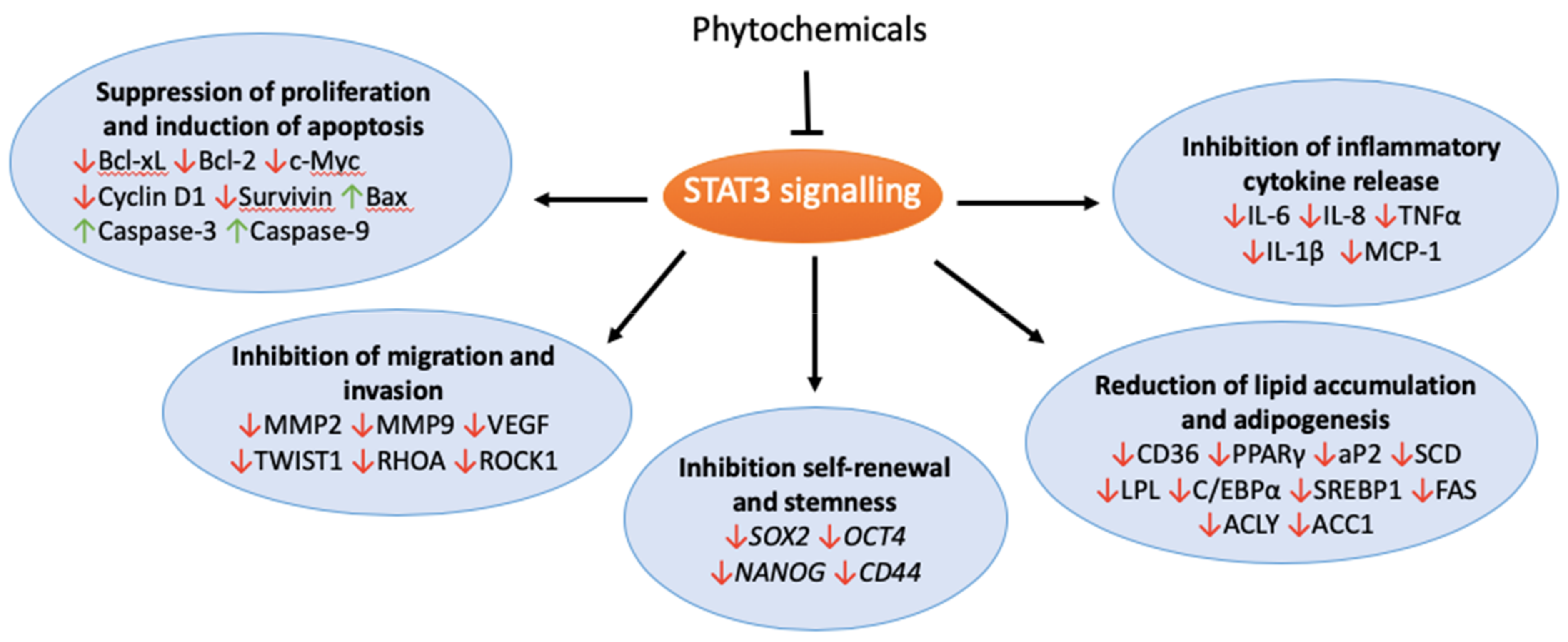
4. Phytochemicals
4.1. Apigenin
4.2. Cucurbitacin B and I
4.3. Curcumin
4.4. Epigallocatechin Gallate (EGCG)
4.5. Resveratrol
4.6. Silibinin
4.7. Palmitoylation and Phytochemicals
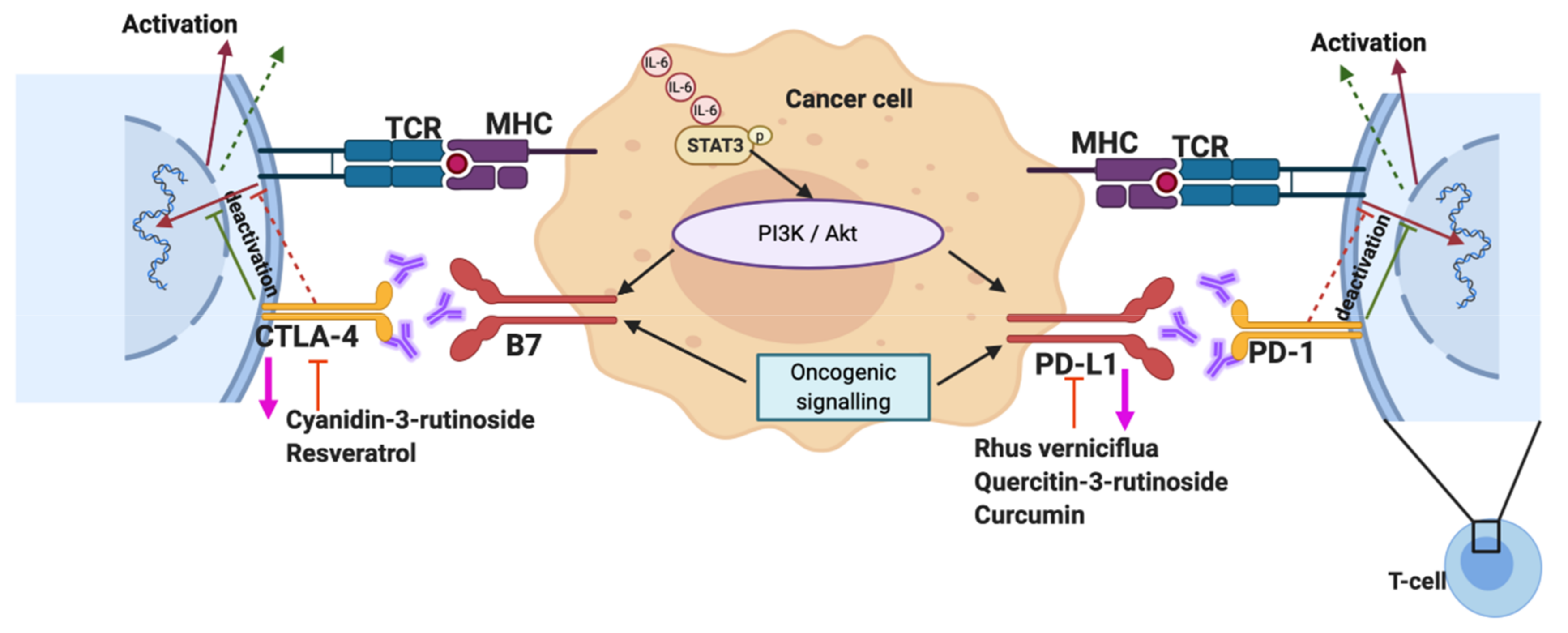
4.8. Phytochemicals and Immunotherapy
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, Q.; Luo, Q.; Halim, A.; Song, G. Targeting lipid metabolism of cancer cells: A promising therapeutic strategy for cancer. Cancer Lett. 2017, 401, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Kadye, R.; Stoffels, M.; Fanucci, S.; Mbanxa, S.; Prinsloo, E. A STAT3 of addiction: Adipose tissue, adipocytokine signalling and STAT3 as mediators of metabolic remodelling in the tumour microenvironment. Cells 2020, 9, 1043. [Google Scholar] [CrossRef] [Green Version]
- Gyamfi, J.; Lee, Y.-H.; Eom, M.; Choi, J. Interleukin-6/STAT3 signalling regulates adipocyte induced epithelial-mesenchymal transition in breast cancer cells. Sci. Rep. 2018, 8, 8859. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Vizza, D.; Panza, S.; Barone, I.; Bonofiglio, D.; Lanzino, M.; Sisci, D.; De Amicis, F.; Fuqua, S.A.W.; Catalano, S.; et al. Leptin increases HER2 protein levels through a STAT3-mediated up-regulation of Hsp90 in breast cancer cells. Mol. Oncol. 2013, 7, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, S.; Xu, L.; Lin, J.; Zhao, C.; Huang, X. Novel activators and small-molecule inhibitors of STAT3 in cancer. Cytokine Growth Factor Rev. 2019, 49, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ouyang, Y.; Liu, J.; Zhu, M.; Zhao, G.; Bao, W.; Hu, F.B. Fruit and vegetable consumption and mortality from all causes, cardiovascular disease, and cancer: Systematic review and dose-response meta-analysis of prospective cohort studies. BMJ 2014, 349, g4490. [Google Scholar] [CrossRef] [Green Version]
- Chikara, S.; Nagaprashantha, L.D.; Singhal, J.; Horne, D.; Awasthi, S.; Singhal, S.S. Oxidative stress and dietary phytochemicals: Role in cancer chemoprevention and treatment. Cancer Lett. 2018, 413, 122–134. [Google Scholar] [CrossRef]
- Ferguson, L.R.; Philpott, M. Cancer prevention by dietary bioactive components that target the immune response. Curr. Cancer Drug Targets 2007, 7, 459–464. [Google Scholar] [CrossRef]
- Gómez-Zorita, S.; Trepiana, J.; González-Arceo, M.; Aguirre, L.; Milton-Laskibar, I.; González, M.; Eseberri, I.; Fernández-Quintela, A.; Portillo, M.P. Anti-Obesity Effects of Microalgae. Int. J. Mol. Sci. 2019, 21, 41. [Google Scholar] [CrossRef] [Green Version]
- Nagaraju, G.P.C.; Sharma, D. Anti-cancer role of SPARC, an inhibitor of adipogenesis. Cancer Treat. Rev. 2011, 37, 559–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisková, T.; Kassayová, M. Resveratrol action on lipid metabolism in cancer. Int. J. Mol. Sci. 2019, 20, 2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Attane, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-regulated fatty acid beta-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 2018, 27, 1357. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.A.; Xing, X.; Harris, J.W.; Zaytseva, Y.Y.; Mitov, M.I.; Napier, D.L.; Weiss, H.L.; Mark Evers, B.; Gao, T. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017, 8, e2593. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem. Cell 2016, 19, 23–37. [Google Scholar] [CrossRef] [Green Version]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the wheels of the cancer machine: The role of lipid metabolism in cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef]
- Valli, A.; Rodriguez, M.; Moutsianas, L.; Fischer, R.; Fedele, V.; Huang, H.L.; Van Stiphout, R.; Jones, D.; McCarthy, M.; Vinaxia, M.; et al. Hypoxia induces a lipogenic cancer cell phenotype via HIF1alpha-dependent and -independent pathways. Oncotarget 2015, 6, 1920–1941. [Google Scholar] [CrossRef] [Green Version]
- Caro, P.; Kishan, A.U.; Norberg, E.; Stanley, I.A.; Chapuy, B.; Ficarro, S.B.; Polak, K.; Tondera, D.; Gounarides, J.; Yin, H.; et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 2012, 22, 547–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Daniels, G.; Lee, P.; Monaco, M.E. Lipid metabolism in prostate cancer. Am. J. Clin. Exp. Urol. 2014, 2, 111–120. [Google Scholar] [PubMed]
- Oben, J.E.; Enyegue, D.M.; Fomekong, G.I.; Soukontoua, Y.B.; Agbor, G.A. The effect of Cissus quadrangularis (CQR-300) and a Cissus formulation (CORE) on obesity and obesity-induced oxidative stress. Lipids Health Dis. 2007, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abidov, M.T.; del Rio, M.J.; Ramazanov, T.Z.; Klimenov, A.L.; Dzhamirze, S.; Kalyuzhin, O.V. Effects of Aralia mandshurica and Engelhardtia chrysolepis extracts on some parameters of lipid metabolism in women with nondiabetic obesity. Bull. Exp. Biol. Med. 2006, 141, 343–346. [Google Scholar] [CrossRef]
- Ahn, I.S.; Do, M.S.; Kim, S.O.; Jung, H.S.; Kim, Y.I.; Kim, H.J.; Park, K.Y. Antiobesity effect of Kochujang (Korean fermented red pepper paste) extract in 3T3-L1 adipocytes. J. Med. Food 2006, 9, 15–21. [Google Scholar] [CrossRef]
- Moreno, L.A.; Tresaco, B.; Bueno, G.; Fleta, J.; Rodriguez, G.; Garagorri, J.M.; Bueno, M. Psyllium fibre and the metabolic control of obese children and adolescents. J. Physiol. Biochem. 2003, 59, 235–242. [Google Scholar] [CrossRef]
- Han, L.K.; Sumiyoshi, M.; Zhang, J.; Liu, M.X.; Zhang, X.F.; Zheng, Y.N.; Okuda, H.; Kimura, Y. Anti-obesity action of Salix matsudana leaves (Part 1). Anti-obesity action by polyphenols of Salix matsudana in high fat-diet treated rodent animals. Phytother. Res. 2003, 17, 1188–1194. [Google Scholar] [CrossRef]
- Moreno, D.A.; Ilic, N.; Poulev, A.; Raskin, I. Effects of Arachis hypogaea nutshell extract on lipid metabolic enzymes and obesity parameters. Life Sci. 2006, 78, 2797–2803. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. The role of prostaglandin E(2) in tumor-associated immunosuppression. Trends Mol. Med. 2016, 22, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Kerkhoff, C.; Sorg, C.; Tandon, N.N.; Nacken, W. Interaction of S100A8/S100A9-arachidonic acid complexes with the scavenger receptor CD36 may facilitate fatty acid uptake by endothelial cells. Biochemistry 2001, 40, 241–248. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zaman, M.M.; Vlasakov, I.; Roy, R.; Huang, L.; Martin, C.R.; Freedman, S.D.; Serhan, C.N.; Moses, M.A. Adipocytes promote ovarian cancer chemoresistance. Sci. Rep. 2019, 9, 13316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.; Zhao, Y.; Pan, Y.; Zheng, X.; Shao, D.; Jia, Y.; He, K.; Li, K.; Chen, L. Chemotherapy induces ovarian cancer cell repopulation through the caspase 3-mediated arachidonic acid metabolic pathway. Onco. Targets 2017, 10, 5817–5826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiraide, T.; Ikegami, K.; Sakaguchi, T.; Morita, Y.; Hayasaka, T.; Masaki, N.; Waki, M.; Sugiyama, E.; Shinriki, S.; Takeda, M.; et al. Accumulation of arachidonic acid-containing phosphatidylinositol at the outer edge of colorectal cancer. Sci. Rep. 2016, 6, 29935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, O.; Kan, M.; Zhang, M.; Shao, D.; Pan, Y.; Zheng, H.; Zhang, X.; Chen, L.; Liu, S. Berberine induces apoptosis by suppressing the arachidonic acid metabolic pathway in hepatocellular carcinoma. Mol. Med. Rep. 2015, 12, 4572–4577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Cui, L.; Pan, Y.; Shao, D.; Zheng, X.; Zhang, F.; Zhang, H.; He, K.; Chen, L. Berberine inhibits the chemotherapy-induced repopulation by suppressing the arachidonic acid metabolic pathway and phosphorylation of FAK in ovarian cancer. Cell Prolif. 2017, 50. [Google Scholar] [CrossRef] [Green Version]
- Siriwardhana, N.; Kalupahana, N.S.; Fletcher, S.; Xin, W.; Claycombe, K.J.; Quignard-Boulange, A.; Zhao, L.; Saxton, A.M.; Moustaid-Moussa, N. N-3 and n-6 polyunsaturated fatty acids differentially regulate adipose angiotensinogen and other inflammatory adipokines in part via NF-kappaB-dependent mechanisms. J. Nutr. Biochem. 2012, 23, 1661–1667. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [Green Version]
- Maccio, A.; Madeddu, C. Inflammation and ovarian cancer. Cytokine 2012, 58, 133–147. [Google Scholar] [CrossRef] [Green Version]
- Rozovski, U.; Grgurevic, S.; Bueso-Ramos, C.; Harris, D.M.; Li, P.; Liu, Z.; Wu, J.Y.; Jain, P.; Wierda, W.; Burger, J.; et al. Aberrant LPL expression, driven by STAT3, mediates free fatty acid metabolism in CLL cells. Mol. Cancer Res. 2015, 13, 944–953. [Google Scholar] [CrossRef] [Green Version]
- Burke, W.M.; Jin, X.; Lin, H.J.; Huang, M.; Liu, R.; Reynolds, R.K.; Lin, J. Inhibition of constitutively active Stat3 suppresses growth of human ovarian and breast cancer cells. Oncogene 2001, 20, 7925–7934. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Yin, C.; Wang, S.; Xiao, Y. JAK-STAT in lipid metabolism of adipocytes. JaK-Stat 2013, 2, e27203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Balaban, S.; Shearer, R.F.; Lee, L.S.; van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017, 5, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Niu, X.; Du, Y.; Chen, Y.; Liu, X.; Xu, L.; Iwakura, Y.; Ma, X.; Li, Y.; Yao, Z.; et al. IL-17A promotes fatty acid uptake through the IL-17A/IL-17RA/p-STAT3/FABP4 axis to fuel ovarian cancer growth in an adipocyte-rich microenvironment. Cancer Immunol. Immunother. 2020, 69, 115–126. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martin, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, H.; Xu, R.; Zhao, Y.; Chinnaswamy, K.; McEachern, D.; Chen, J.; Yang, C.Y.; Liu, Z.; Wang, M.; et al. A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell 2019, 36, 498–511.e17. [Google Scholar] [CrossRef]
- Milligan, G.; Parenti, M.; Magee, A.I. The dynamic role of palmitoylation in signal transduction. Trends Biochem. Sci. 1995, 20, 181–187. [Google Scholar] [CrossRef]
- Resh, M.D. Palmitoylation of proteins in cancer. Biochem. Soc. Trans. 2017, 45, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.A.; Gilman, A.G. Autoacylation of G protein alpha subunits. J. Biol. Chem. 1996, 271, 23594–23600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemonidis, K.; Werno, M.W.; Greaves, J.; Diez-Ardanuy, C.; Sanchez-Perez, M.C.; Salaun, C.; Thomson, D.M.; Chamberlain, L.H. The zDHHC family of S-acyltransferases. Biochem. Soc. Trans. 2015, 43, 217–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, P.J.; Dixon, S.J. Protein palmitoylation and cancer. Embo Rep. 2018, 19. [Google Scholar] [CrossRef]
- Yeste-Velasco, M.; Mao, X.; Grose, R.; Kudahetti, S.C.; Lin, D.; Marzec, J.; Vasiljevic, N.; Chaplin, T.; Xue, L.; Xu, M.; et al. Identification of ZDHHC14 as a novel human tumour suppressor gene. J. Pathol. 2014, 232, 566–577. [Google Scholar] [CrossRef] [Green Version]
- Ren, W.; Jhala, U.S.; Du, K. Proteomic analysis of protein palmitoylation in adipocytes. Adipocyte 2013, 2, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, F.; Corral-Escariz, M.; Perez-Gil, J.; Rodriguez-Crespo, I. Palmitoylation of R-Ras by human DHHC19, a palmitoyl transferase with a CaaX box. Biochim. Biophys. Acta 2010, 1798, 592–604. [Google Scholar] [CrossRef] [Green Version]
- Rocks, O.; Peyker, A.; Kahms, M.; Verveer, P.J.; Koerner, C.; Lumbierres, M.; Kuhlmann, J.; Waldmann, H.; Wittinghofer, A.; Bastiaens, P.I. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science 2005, 307, 1746–1752. [Google Scholar] [CrossRef]
- Eelen, G.; Dubois, C.; Cantelmo, A.R.; Goveia, J.; Bruning, U.; DeRan, M.; Jarugumilli, G.; van Rijssel, J.; Saladino, G.; Comitani, F.; et al. Role of glutamine synthetase in angiogenesis beyond glutamine synthesis. Nature 2018, 561, 63–69. [Google Scholar] [CrossRef]
- Chen, B.; Sun, Y.; Niu, J.; Jarugumilli, G.K.; Wu, X. Protein lipidation in cell signaling and diseases: Function, regulation, and therapeutic opportunities. Cell Chem. Biol. 2018, 25, 817–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leventis, R.; Juel, G.; Knudsen, J.K.; Silvius, J.R. Acyl-CoA binding proteins inhibit the nonenzymic S-acylation of cysteinyl-containing peptide sequences by long-chain acyl-CoAs. Biochemistry 1997, 36, 5546–5553. [Google Scholar] [CrossRef] [PubMed]
- Dunphy, J.T.; Schroeder, H.; Leventis, R.; Greentree, W.K.; Knudsen, J.K.; Silvius, J.R.; Linder, M.E. Differential effects of acyl-CoA binding protein on enzymatic and non-enzymatic thioacylation of protein and peptide substrates. Biochim. Biophys. Acta 2000, 1485, 185–198. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 therapies in cancer: Mechanisms of action, efficacy, and limitations. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Zerdes, I.; Wallerius, M.; Sifakis, E.G.; Wallmann, T.; Betts, S.; Bartish, M.; Tsesmetzis, N.; Tobin, N.P.; Coucoravas, C.; Bergh, J.; et al. STAT3 activity promotes programmed-death ligand 1 expression and suppresses immune responses in breast cancer. Cancers 2019, 11, 1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; Mihm, M.C.; Soiffer, R.J.; Haluska, F.G.; Butler, M.; Seiden, M.V.; Davis, T.; Henry-Spires, R.; MacRae, S.; Willman, A.; et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl. Acad. Sci. USA 2003, 100, 4712–4717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, G.Q.; Yang, J.C.; Sherry, R.M.; Hwu, P.; Topalian, S.L.; Schwartzentruber, D.J.; Restifo, N.P.; Haworth, L.R.; Seipp, C.A.; Freezer, L.J.; et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 2003, 100, 8372–8377. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yue, C.; Herrmann, A.; Song, J.; Egelston, C.; Wang, T.; Zhang, Z.; Li, W.; Lee, H.; Aftabizadeh, M.; et al. STAT3 activation-induced fatty acid oxidation in CD8(+) T effector cells is critical for obesity-promoted breast tumor growth. Cell Metab. 2020, 31, 148–161. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Li, H.; Lin, H.J.; Yang, S.; Lin, J.; Liang, G. Feedback activation of STAT3 as a cancer drug-resistance mechanism. Trends Pharm. Sci. 2016, 37, 47–61. [Google Scholar] [CrossRef]
- Ji, T.; Gong, D.; Han, Z.; Wei, X.; Yan, Y.; Ye, F.; Ding, W.; Wang, J.; Xia, X.; Li, F.; et al. Abrogation of constitutive Stat3 activity circumvents cisplatin resistant ovarian cancer. Cancer Lett. 2013, 341, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Rozovski, U.; Harris, D.M.; Li, P.; Liu, Z.; Jain, P.; Ferrajoli, A.; Burger, J.; Thompson, P.; Jain, N.; Wierda, W.; et al. STAT3-activated CD36 facilitates fatty acid uptake in chronic lymphocytic leukemia cells. Oncotarget 2018, 9, 21268–21280. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.T.; Yun, S.J.; Yan, C.; Jeong, P.; Kim, Y.H.; Lee, I.S.; Kang, H.W.; Park, S.; Moon, S.K.; Choi, Y.H.; et al. Metabolic pathway signatures associated with urinary metabolite biomarkers differentiate bladder cancer patients from healthy controls. Yonsei Med. J. 2016, 57, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Medina, E.A.; Oberheu, K.; Polusani, S.R.; Ortega, V.; Velagaleti, G.V.; Oyajobi, B.O. PKA/AMPK signaling in relation to adiponectin’s antiproliferative effect on multiple myeloma cells. Leukemia 2014, 28, 2080–2089. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Joshi, A.; Zhang, Y.; Jaeger, S.A.; Bulyk, M.; Tsichlis, P.N.; Shirley Liu, X.; Struhl, K. A transcriptional signature and common gene networks link cancer with lipid metabolism and diverse human diseases. Cancer Cell 2010, 17, 348–361. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Banaszak, L.; Fracci, S.; Basali, D.; Dunlap, S.M.; Hursting, S.D.; Rich, J.N.; Hjlemeland, A.B.; Vasanji, A.; Berger, N.A.; et al. Leptin receptor maintains cancer stem-like properties in triple negative breast cancer cells. Endocr. Relat. Cancer 2013, 20, 797–808. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Schroeder, A.; Herrmann, A.; Cherryholmes, G.; Kowolik, C.; Buettner, R.; Pal, S.; Yu, H.; Muller-Newen, G.; Jove, R. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 2014, 74, 1227–1237. [Google Scholar] [CrossRef] [Green Version]
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, S.A.; Montag, A.; et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018, 37, 2285–2301. [Google Scholar] [CrossRef]
- Bochet, L.; Meulle, A.; Imbert, S.; Salles, B.; Valet, P.; Muller, C. Cancer-associated adipocytes promotes breast tumor radioresistance. Biochem. Biophys. Res. Commun. 2011, 411, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, R.; Hou, Y.; Sun, W.; Yu, J.; Liu, X.; Niu, Y.; Lu, J.-J.; Chen, X. Natural products to prevent drug resistance in cancer chemotherapy: A review. Ann. N. Y. Acad. Sci. 2017, 1401, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Zubair, H.; Azim, S.; Ahmad, A.; Khan, M.A.; Patel, G.K.; Singh, S.; Singh, A.P. Cancer chemoprevention by phytochemicals: Nature’s healing touch. Molecules 2017, 22, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.-H.; Chu, J.-H.; Kwan, H.-Y.; Su, T.; Yu, H.; Cheng, C.-Y.; Fu, X.-Q.; Guo, H.; Li, T.; Tse, A.K.-W.; et al. Inhibition of the STAT3 signaling pathway contributes to apigenin-mediated anti-metastatic effect in melanoma. Sci. Rep. 2016, 6, 21731. [Google Scholar] [CrossRef] [Green Version]
- Thoennissen, N.H.; Iwanski, G.B.; Doan, N.B.; Okamoto, R.; Lin, P.; Abbassi, S.; Song, J.H.; Yin, D.; Toh, M.; Xie, W.D.; et al. Cucurbitacin B induces apoptosis by inhibition of the JAK/STAT pathway and potentiates antiproliferative effects of gemcitabine on pancreatic cancer cells. Cancer Res. 2009, 69, 5876–5884. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.; Wu, S.; Wang, X.; Zhu, G.; Chen, X.; Ding, Y.; Jiang, W. Cucurbitacin I induces pro-death autophagy in A549 cells via the ERK-mTOR-STAT3 signaling pathway. J. Cell. Biochem. 2018, 119, 6104–6112. [Google Scholar] [CrossRef]
- Li, Y.; Sun, W.; Han, N.; Zou, Y.; Yin, D. Curcumin inhibits proliferation, migration, invasion and promotes apoptosis of retinoblastoma cell lines through modulation of miR-99a and JAK/STAT pathway. BMC Cancer 2018, 18, 1230. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ren, X.; Deng, C.; Yang, L.; Yan, E.; Guo, T.; Li, Y.; Xu, M.X. Mechanism of the inhibition of the STAT3 signaling pathway by EGCG. Oncol. Rep. 2013, 30, 2691–2696. [Google Scholar] [CrossRef]
- Duan, J.; Yue, W.; JianYu, E.; Malhotra, J.; Lu, S.-E.; Gu, J.; Xu, F.; Tan, X.-L. In vitro comparative studies of resveratrol and triacetylresveratrol on cell proliferation, apoptosis, and STAT3 and NFκB signaling in pancreatic cancer cells. Sci. Rep. 2016, 6, 31672. [Google Scholar] [CrossRef]
- Suh, J.; Kim, D.-H.; Surh, Y.-J. Resveratrol suppresses migration, invasion and stemness of human breast cancer cells by interfering with tumor-stromal cross-talk. Arch. Biochem. Biophys. 2018, 643, 62–71. [Google Scholar] [CrossRef]
- Shi, Z.; Zhou, Q.; Gao, S.; Li, W.; Li, X.; Liu, Z.; Jin, P.; Jiang, J. Silibinin inhibits endometrial carcinoma via blocking pathways of STAT3 activation and SREBP1-mediated lipid accumulation. Life Sci. 2019, 217, 70–80. [Google Scholar] [CrossRef]
- Agarwal, C.; Tyagi, A.; Kaur, M.; Agarwal, R. Silibinin inhibits constitutive activation of Stat3, and causes caspase activation and apoptotic death of human prostate carcinoma DU145 cells. Carcinogenesis 2007, 28, 1463–1470. [Google Scholar] [CrossRef] [Green Version]
- Sung, B.; Chung, H.Y.; Kim, N.D. Role of apigenin in cancer prevention via the induction of apoptosis and autophagy. J. Cancer Prev. 2016, 21, 216–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Qi, M.; Li, P.; Zhan, Y.; Shao, H. Apigenin in cancer therapy: Anti-cancer effects and mechanisms of action. Cell Biosci. 2017, 7, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.-Y.; Nie, J.; Huang, J.-P.; Zheng, G.-J.; Feng, B. Targeting STAT3 inhibition to reverse cisplatin resistance. Biomed. Pharm. 2019, 117, 109135. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.-S.; Ku, J.M.; Choi, H.S.; Woo, J.-K.; Lee, B.H.; Kim, D.S.; Song, H.J.; Jang, B.-H.; Shin, Y.C.; Ko, S.-G. Apigenin overcomes drug resistance by blocking the signal transducer and activator of transcription 3 signaling in breast cancer cells. Oncol. Rep. 2017, 38, 715–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, X.-Y.; Qin, Y.; Liu, H.-J.; Cui, Z.-H.; Li, M.; Yang, J.-H.; Zhong, W.-L.; Liu, Y.-R.; Chen, S.; Sun, T.; et al. Apigenin inhibits colonic inflammation and tumorigenesis by suppressing STAT3-NF-κB signaling. Oncotarget 2017, 8, 100216–100226. [Google Scholar] [CrossRef] [Green Version]
- Ono, M.; Fujimori, K. Antiadipogenic effect of dietary apigenin through activation of AMPK in 3T3-L1 cells. J. Agric. Food Chem. 2011, 59, 13346–13352. [Google Scholar] [CrossRef]
- Su, T.; Huang, C.; Yang, C.; Jiang, T.; Su, J.; Chen, M.; Fatima, S.; Gong, R.; Hu, X.; Bian, Z.; et al. Apigenin inhibits STAT3/CD36 signaling axis and reduces visceral obesity. Pharm. Res. 2020, 152, 104586. [Google Scholar] [CrossRef]
- Guo, H.; Kuang, S.; Song, Q.-L.; Liu, M.; Sun, X.-X.; Yu, Q. Cucurbitacin I inhibits STAT3, but enhances STAT1 signaling in human cancer cells in vitro through disrupting actin filaments. Acta Pharm. Sin. 2018, 39, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Yin, D.; Wakimoto, N.; Xing, H.; Lu, D.; Huynh, T.; Wang, X.; Black, K.L.; Koeffler, H.P. Cucurbitacin B markedly inhibits growth and rapidly affects the cytoskeleton in glioblastoma multiforme. Int. J. Cancer 2008, 123, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, N.; Yin, D.; O’Kelly, J.; Haritunians, T.; Karlan, B.; Said, J.; Xing, H.; Koeffler, H.P. Cucurbitacin B has a potent antiproliferative effect on breast cancer cells in vitro and in vivo. Cancer Sci. 2008, 99, 1793–1797. [Google Scholar] [CrossRef] [PubMed]
- Oi, T.; Asanuma, K.; Matsumine, A.; Matsubara, T.; Nakamura, T.; Iino, T.; Asanuma, Y.; Goto, M.; Okuno, K.; Kakimoto, T.; et al. STAT3 inhibitor, cucurbitacin I, is a novel therapeutic agent for osteosarcoma. Int. J. Oncol. 2016, 49, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, Y.; Yang, R.; Zhou, T.; Ke, W.; Si, Y.; Yang, S.; Zhang, T.; Liu, X.; Zhang, L.; et al. Cucurbitacin B inhibits gastric cancer progression by suppressing STAT3 activity. Arch. Biochem. Biophys. 2020, 684, 108314. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Liu, J.; Yang, M.; Huang, N.; Zhong, Y.; Zeng, T.; Wei, R.; Wu, Z.; Xiao, C.; Cao, X.; et al. Cucurbitacin B exerts anti-cancer activities in human multiple myeloma cells in vitro and in vivo by modulating multiple cellular pathways. Oncotarget 2017, 8, 5800–5813. [Google Scholar] [CrossRef] [Green Version]
- Ishdorj, G.; Johnston, J.B.; Gibson, S.B. Inhibition of constitutive activation of STAT3 by Curcurbitacin-I (JSI-124) sensitized human B-leukemia cells to apoptosis. Mol. Cancer Ther. 2010, 9, 3302–3314. [Google Scholar] [CrossRef] [Green Version]
- Seo, C.-R.; Yang, D.K.; Song, N.-J.; Yun, U.J.; Gwon, A.R.; Jo, D.-G.; Cho, J.Y.; Yoon, K.; Ahn, J.-Y.; Nho, C.W.; et al. Cucurbitacin B and cucurbitacin I suppress adipocyte differentiation through inhibition of STAT3 signaling. Food Chem. Toxicol. 2014, 64, 217–224. [Google Scholar] [CrossRef]
- Meydani, M.; Hasan, S.T. Dietary polyphenols and obesity. Nutrients 2010, 2, 737–751. [Google Scholar] [CrossRef]
- Jang, E.M.; Choi, M.S.; Jung, U.J.; Kim, M.J.; Kim, H.J.; Jeon, S.M.; Shin, S.K.; Seong, C.N.; Lee, M.K. Beneficial effects of curcumin on hyperlipidemia and insulin resistance in high-fat-fed hamsters. Metabolism 2008, 57, 1576–1583. [Google Scholar] [CrossRef]
- Ejaz, A.; Wu, D.; Kwan, P.; Meydani, M. Curcumin inhibits adipogenesis in 3T3-L1 adipocytes and angiogenesis and obesity in C57/BL mice. J. Nutr. 2009, 139, 919–925. [Google Scholar] [CrossRef]
- Seo, J.H.; Jeong, K.J.; Oh, W.J.; Sul, H.J.; Sohn, J.S.; Kim, Y.K.; Cho, D.Y.; Kang, J.K.; Park, C.G.; Lee, H.Y. Lysophosphatidic acid induces STAT3 phosphorylation and ovarian cancer cell motility: Their inhibition by curcumin. Cancer Lett. 2010, 288, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Syng-ai, C.; Kumari, A.L.; Khar, A. Effect of curcumin on normal and tumor cells: Role of glutathione and bcl-2. Mol. Cancer Ther. 2004, 3, 1101–1108. [Google Scholar]
- Kamat, A.M.; Sethi, G.; Aggarwal, B.B. Curcumin potentiates the apoptotic effects of chemotherapeutic agents and cytokines through down-regulation of nuclear factor-κB and nuclear factor-κB–regulated gene products in IFN-α–sensitive and IFN-α–resistant human bladder cancer cells. Mol. Cancer Ther. 2007, 6, 1022–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Liu, L.; Wang, Y.; He, A.; Hu, H.; Zhang, J.; Han, M.; Huang, Y. Curcumin inhibits the proliferation and invasion of MG-63 cells through inactivation of the p-JAK2/p-STAT3 pathway. Oncotargets Ther. 2019, 12, 2011–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawat, N.; Alhamdani, A.; McAdam, E.; Cronin, J.; Eltahir, Z.; Lewis, P.; Griffiths, P.; Baxter, J.N.; Jenkins, G.J. Curcumin abrogates bile-induced NF-kappaB activity and DNA damage in vitro and suppresses NF-kappaB activity whilst promoting apoptosis in vivo, suggesting chemopreventative potential in Barrett’s oesophagus. Clin. Transl. Oncol. 2012, 14, 302–311. [Google Scholar] [CrossRef]
- Zingg, J.-M.; Hasan, S.T.; Meydani, M. Molecular mechanisms of hypolipidemic effects of curcumin. BioFactors 2013, 39, 101–121. [Google Scholar] [CrossRef]
- Jung, J.H.; Yun, M.; Choo, E.-J.; Kim, S.-H.; Jeong, M.-S.; Jung, D.-B.; Lee, H.; Kim, E.-O.; Kato, N.; Kim, B.; et al. A derivative of epigallocatechin-3-gallate induces apoptosis via SHP-1-mediated suppression of BCR-ABL and STAT3 signalling in chronic myelogenous leukaemia. Br. J. Pharm. 2015, 172, 3565–3578. [Google Scholar] [CrossRef]
- Tang, W.; Song, H.; Cai, W.; Shen, X. Real time monitoring of inhibition of adipogenesis and angiogenesis by (-)-epigallocatechin-3-gallate in 3T3-L1 adipocytes and human umbilical vein endothelial cells. Nutrients 2015, 7, 8871–8886. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wu, K.; Xu, T.; Wu, J.; Li, P.; Wang, H.; Wu, H.; Wu, G. Epigallocatechin-3-gallate enhances the osteoblastogenic differentiation of human adipose-derived stem cells. Drug Des. Dev. Ther. 2019, 13, 1311–1321. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Mao, L.; Xu, P.; Wang, Y. Effects of (-)-Epigallocatechin Gallate (EGCG) on energy expenditure and microglia-mediated hypothalamic inflammation in mice fed a high-fat diet. Nutrients 2018, 10, 1681. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.-H.; Chen, H.-Y.; Zhan, W.-H.; Wang, C.-Y.; Cai, S.-R.; Wang, Z.; Zhang, C.-H.; He, Y.-L. (-)-Epigallocatechin-3-gallate inhibits VEGF expression induced by IL-6 via Stat3 in gastric cancer. World J. Gastroenterol. 2011, 17, 2315–2325. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Della-Fera, M.A.; Baile, C.A. Green tea polyphenol epigallocatechin gallate inhibits adipogenesis and induces apoptosis in 3T3-L1 adipocytes. Obes. Res. 2005, 13, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Hochstetter, D.; Yao, L.; Zhao, Y.; Zhou, J.; Wang, Y.; Xu, P. Green tea polyphenol (-)-Epigallocatechin Gallate (EGCG) attenuates neuroinflammation in palmitic acid-stimulated BV-2 microglia and high-fat diet-induced obese mice. Int. J. Mol. Sci. 2019, 20, 5081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotha, A.; Sekharam, M.; Cilenti, L.; Siddiquee, K.; Khaled, A.; Zervos, A.S.; Carter, B.; Turkson, J.; Jove, R. Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol. Cancer Ther. 2006, 5, 621–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, P.R.; Okuda, H.; Watabe, M.; Pai, S.K.; Liu, W.; Kobayashi, A.; Xing, F.; Fukuda, K.; Hirota, S.; Sugai, T.; et al. Resveratrol suppresses growth of cancer stem-like cells by inhibiting fatty acid synthase. Breast Cancer Res. Treat. 2011, 130, 387–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, P.R.; Xing, F.; Sharma, S.; Watabe, M.; Pai, S.K.; Iiizumi-Gairani, M.; Fukuda, K.; Hirota, S.; Mo, Y.Y.; Watabe, K. Elevated lipogenesis in epithelial stem-like cell confers survival advantage in ductal carcinoma in situ of breast cancer. Oncogene 2013, 32, 5111–5122. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Chen, B.; Jiang, R.; Li, J.; Wang, B. Resveratrol inhibits lung cancer growth by suppressing M2-like polarization of tumor associated macrophages. Cell. Immunol. 2017, 311, 86–93. [Google Scholar] [CrossRef]
- Zhang, P.; Li, H.; Yang, B.; Yang, F.; Zhang, L.-L.; Kong, Q.-Y.; Chen, X.-Y.; Wu, M.-L.; Liu, J. Biological significance and therapeutic implication of resveratrol-inhibited Wnt, Notch and STAT3 signaling in cervical cancer cells. Genes Cancer 2014, 5, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.X.; Zhang, Y.; Wu, M.L.; Liu, Y.N.; Zhang, P.; Chen, X.Y.; Kong, Q.Y.; Liu, J.; Li, H. Resveratrol and STAT inhibitor enhance autophagy in ovarian cancer cells. Cell Death Discov. 2016, 2, 15071. [Google Scholar] [CrossRef]
- Bosch-Barrera, J.; Queralt, B.; Menendez, J.A. Targeting STAT3 with silibinin to improve cancer therapeutics. Cancer Treat. Rev. 2017, 58, 61–69. [Google Scholar] [CrossRef]
- Cuyàs, E.; Pérez-Sánchez, A.; Micol, V.; Menendez, J.A.; Bosch-Barrera, J. STAT3-targeted treatment with silibinin overcomes the acquired resistance to crizotinib in ALK-rearranged lung cancer. Cell Cycle 2016, 15, 3413–3418. [Google Scholar] [CrossRef] [PubMed]
- Chittezhath, M.; Deep, G.; Singh, R.P.; Agarwal, C.; Agarwal, R. Silibinin inhibits cytokine-induced signaling cascades and down-regulates inducible nitric oxide synthase in human lung carcinoma A549 cells. Mol. Cancer Ther. 2008, 7, 1817–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.K.; Dasgupta, A.; Mehla, K.; Gunda, V.; Vernucci, E.; Souchek, J.; Goode, G.; King, R.; Mishra, A.; Rai, I.; et al. Silibinin-mediated metabolic reprogramming attenuates pancreatic cancer-induced cachexia and tumor growth. Oncotarget 2015, 6, 41146–41161. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.P.; Raina, K.; Deep, G.; Chan, D.; Agarwal, R. Silibinin suppresses growth of human prostate carcinoma PC-3 orthotopic xenograft via activation of extracellular signal-regulated kinase 1/2 and inhibition of signal transducers and activators of transcription signaling. Clin. Cancer Res. 2009, 15, 613–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilley, C.; Deep, G.; Agarwal, C.; Wempe, M.F.; Biedermann, D.; Valentová, K.; Kren, V.; Agarwal, R. Silibinin and its 2,3-dehydro-derivative inhibit basal cell carcinoma growth via suppression of mitogenic signaling and transcription factors activation. Mol. Carcinog. 2016, 55, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, A.; Singh, R.P.; Ramasamy, K.; Raina, K.; Redente, E.F.; Dwyer-Nield, L.D.; Radcliffe, R.A.; Malkinson, A.M.; Agarwal, R. Growth inhibition and regression of lung tumors by silibinin: Modulation of angiogenesis by macrophage-associated cytokines and nuclear factor-kappaB and signal transducers and activators of transcription 3. Cancer Prev. Res. 2009, 2, 74–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, R.; Ma, J.; Wang, D.; Dong, W.; Wang, S.; Liu, T.; Xie, R.; Liu, L.; Wang, B.; Cao, H. Chemopreventive effects of silibinin on colitis-associated tumorigenesis by inhibiting IL-6/STAT3 signaling pathway. Mediat. Inflamm. 2018, 2018, 1562010. [Google Scholar] [CrossRef]
- Verdura, S.; Cuyàs, E.; Llorach-Parés, L.; Pérez-Sánchez, A.; Micol, V.; Nonell-Canals, A.; Joven, J.; Valiente, M.; Sánchez-Martínez, M.; Bosch-Barrera, J.; et al. Silibinin is a direct inhibitor of STAT3. Food Chem. Toxicol. 2018, 116, 161–172. [Google Scholar] [CrossRef]
- Hentschel, A.; Zahedi, R.P.; Ahrends, R. Protein lipid modifications—More than just a greasy ballast. Proteomics 2016, 16, 759–782. [Google Scholar] [CrossRef]
- Coleman, D.T.; Soung, Y.H.; Surh, Y.J.; Cardelli, J.A.; Chung, J. Curcumin prevents palmitoylation of integrin beta4 in breast cancer cells. PLoS ONE 2015, 10, e0125399. [Google Scholar] [CrossRef] [Green Version]
- Gulvady, A.A.; Ciolino, H.P.; Cabrera, R.M.; Jolly, C.A. Resveratrol inhibits the deleterious effects of diet-induced obesity on thymic function. J. Nutr. Biochem. 2013, 24, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Chen, Y.; Wang, Y.; Zhang, H.; Fan, S.; Gao, Y.; Jiao, T.; Fu, K.; Sun, J.; Yu, A.; et al. Effects of carnitine palmitoyltransferases on cancer cellular senescence. J. Cell Physiol. 2019, 234, 1707–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Wang, H.Y.; Marzec, M.; Raghunath, P.N.; Nagasawa, T.; Wasik, M.A. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 6948–6953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holliday, R. DNA methylation and epigenetic defects in carcinogenesis. Mutat. Res. 1987, 181, 215–217. [Google Scholar] [CrossRef]
- Schlake, T.; Klehr-Wirth, D.; Yoshida, M.; Beppu, T.; Bode, J. Gene expression within a chromatin domain: The role of core histone hyperacetylation. Biochemistry 1994, 33, 4197–4206. [Google Scholar] [CrossRef]
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar]
- Lewis, K.A.; Jordan, H.R.; Tollefsbol, T.O. Effects of SAHA and EGCG on growth potentiation of triple-negative breast cancer cells. Cancers 2018, 11, 23. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Wu, R.; Gaspar, J.M.; Sargsyan, D.; Su, Z.Y.; Zhang, C.; Gao, L.; Cheng, D.; Li, W.; Wang, C.; et al. DNA methylome and transcriptome alterations and cancer prevention by curcumin in colitis-accelerated colon cancer in mice. Carcinogenesis 2018, 39, 669–680. [Google Scholar] [CrossRef]
- Greaves, J.; Chamberlain, L.H. New links between S-acylation and cancer. J. Pathol. 2014, 233, 4–6. [Google Scholar] [CrossRef]
- Chen, H.M.; Wang, P.H.; Chen, S.S.; Wen, C.C.; Chen, Y.H.; Yang, W.C.; Yang, N.S. Shikonin induces immunogenic cell death in tumor cells and enhances dendritic cell-based cancer vaccine. Cancer Immunol. Immunother. 2012, 61, 1989–2002. [Google Scholar] [CrossRef]
- Lin, T.J.; Lin, H.T.; Chang, W.T.; Mitapalli, S.P.; Hsiao, P.W.; Yin, S.Y.; Yang, N.S. Shikonin-enhanced cell immunogenicity of tumor vaccine is mediated by the differential effects of DAMP components. Mol. Cancer 2015, 14, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Hsieh, S.Y.; Fan, Y.T.; Wei, W.C.; Hsiao, P.W.; Tsai, D.H.; Wu, T.S.; Yang, N.S. Necroptosis promotes autophagy-dependent upregulation of DAMP and results in immunosurveillance. Autophagy 2018, 14, 778–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Xu, X.; Gao, X.; Chen, H.; Geng, L. Shikonin suppresses IL-17-induced VEGF expression via blockage of JAK2/STAT3 pathway. Int. Immunopharmacol. 2014, 19, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.Y.; Zhu, X.; Luo, Y.L.; Lin, H.Y.; Tang, C.Y.; Qi, J.L.; Pang, Y.J.; Yang, R.W.; Lu, G.H.; Wang, X.M.; et al. Identification of new shikonin derivatives as antitumor agents targeting STAT3 SH2 domain. Sci. Rep. 2017, 7, 2863. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.C.; Ren, Y.G.; Zhao, J.; Long, F.; Chen, J.Y.; Jiang, Z. Shikonin enhances sensitization of gefitinib against wild-type EGFR non-small cell lung cancer via inhibition PKM2/stat3/cyclinD1 signal pathway. Life Sci. 2018, 204, 71–77. [Google Scholar] [CrossRef]
- Guo, Z.L.; Li, J.Z.; Ma, Y.Y.; Qian, D.; Zhong, J.Y.; Jin, M.M.; Huang, P.; Che, L.Y.; Pan, B.; Wang, Y.; et al. Shikonin sensitizes A549 cells to TRAIL-induced apoptosis through the JNK, STAT3 and AKT pathways. BMC Cell Biol. 2018, 19, 29. [Google Scholar] [CrossRef]
- Sun, L.X.; Li, W.D.; Lin, Z.B.; Duan, X.S.; Li, X.F.; Yang, N.; Lan, T.F.; Li, M.; Sun, Y.; Yu, M.; et al. Protection against lung cancer patient plasma-induced lymphocyte suppression by Ganoderma lucidum polysaccharides. Cell Physiol. Biochem. 2014, 33, 289–299. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Mandal, D.; Saha, B.; Sen, G.S.; Das, T.; Sa, G. Curcumin prevents tumor-induced T cell apoptosis through Stat-5a-mediated Bcl-2 induction. J. Biol. Chem. 2007, 282, 15954–15964. [Google Scholar] [CrossRef] [Green Version]
- Mace, T.A.; King, S.A.; Ameen, Z.; Elnaggar, O.; Young, G.; Riedl, K.M.; Schwartz, S.J.; Clinton, S.K.; Knobloch, T.J.; Weghorst, C.M.; et al. Bioactive compounds or metabolites from black raspberries modulate T lymphocyte proliferation, myeloid cell differentiation and Jak/STAT signaling. Cancer Immunol. Immunother. 2014, 63, 889–900. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Sun, J.; Li, X.H.; Zhou, Q.Q.; Bai, J.; Shi, Y.H.; Le, G.W. Resveratrol prevents suppression of regulatory T-cell production, oxidative stress, and inflammation of mice prone or resistant to high-fat diet-induced obesity. Nutr. Res. 2013, 33, 971–981. [Google Scholar] [CrossRef]
- Read, S.; Malmstrom, V.; Powrie, F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J. Exp. Med. 2000, 192, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.Y.; Hu, B.; Xu, D.M.; Liew, F.Y. CD4+CD25+ regulatory T cells cure murine colitis: The role of IL-10, TGF-beta, and CTLA4. J. Immunol. 2003, 171, 5012–5017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Kim, T.I.; Kim, J.H.; Chung, H.-S. Immune checkpoint PD-1/PD-L1 CTLA-4/CD80 are blocked by rhus verniciflua dtokes and its active compounds. Molecules 2019, 24, 4062. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; Warshauer, J.T.; Bluestone, J.A. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat. Med. 2017, 23, 540–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-W.; Um, E.-S.; Jung, B.-B.; Choi, E.-S.; Kim, E.-Y.; Lee, S.; Jang, E.; Lee, J.-H.; Kim, Y. Rhus verniciflua Stokes extract induces inhibition of cell growth and apoptosis in human chronic myelogenous leukemia K562 cells. Oncol. Rep. 2018, 39, 1141–1147. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Phytochemical | Cell Line/Model | Molecular Mechanism | Effect | Ref |
|---|---|---|---|---|
| Apigenin | A375 | ↓pSTAT3 ↓MMP2 ↓MMP9 ↓VEGF ↓TWIST1 ↓E-cad ↓Keratin-8 ↑N-cad ↑Fibronectin |
| [84] |
| Cucurbitacin B | Panc-1 | ↓pSTAT3 ↓pSTAT5 ↓pJAK2 ↓Cyclin A ↓Cyclin B1 ↓Bcl-xL ↑Caspase-3 ↑Caspase-9 |
| [85] |
| Cucurbitacin I | A549 | ↓pSTAT3 ↓p-mTOR ↓pERK ↑LC3II |
| [86] |
| Curcumin | SO-Rb50, Y79 | ↑MIR99a ↓pJAK1 ↓pSTAT1 ↓pSTAT3 ↓MMP2 ↓RHOA ↓ROCK1 ↓Bcl-2 ↓Vimentin ↑Bax |
| [87] |
| Epigallocatechin Gallate (EGCG) | BEL-7402, OGY-7703 | ↓pSTAT3 ↓Bcl-xL ↓c-Myc ↓VEGF ↓Cyclin D1 |
| [88] |
| Resveratrol | PANC-1, BxPC-3 | ↓pSTAT3 ↓pNF-κB ↓Mcl-1 ↑BIM ↑PUMA |
| [89] |
| MCF7, MDA-MB-231 | ↓pSTAT3 ↓pAkt ↓MYC ↓MMP2 ↓MMP9 ↓SOX2 ↓BMI1 ↓CD44 |
| [90] | |
| Silibinin | Ishikawa, RL-952 | ↓pSTAT3 ↓SREBP1 ↓FAS ↓pACLY ↓Survivin ↓Bcl-2 ↓Caspase-3 ↓Ki67 ↓Cyclin D1 |
| [91] |
| DU145 | ↓pSTAT3 ↓Mcl-1 ↓Cyclin D1 ↓Bcl-xL ↓Survivin |
| [92] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tse, C.; Warner, A.; Farook, R.; Cronin, J.G. Phytochemical Targeting of STAT3 Orchestrated Lipid Metabolism in Therapy-Resistant Cancers. Biomolecules 2020, 10, 1118. https://doi.org/10.3390/biom10081118
Tse C, Warner A, Farook R, Cronin JG. Phytochemical Targeting of STAT3 Orchestrated Lipid Metabolism in Therapy-Resistant Cancers. Biomolecules. 2020; 10(8):1118. https://doi.org/10.3390/biom10081118
Chicago/Turabian StyleTse, Carmen, Ashleigh Warner, Rufaik Farook, and James G Cronin. 2020. "Phytochemical Targeting of STAT3 Orchestrated Lipid Metabolism in Therapy-Resistant Cancers" Biomolecules 10, no. 8: 1118. https://doi.org/10.3390/biom10081118
APA StyleTse, C., Warner, A., Farook, R., & Cronin, J. G. (2020). Phytochemical Targeting of STAT3 Orchestrated Lipid Metabolism in Therapy-Resistant Cancers. Biomolecules, 10(8), 1118. https://doi.org/10.3390/biom10081118