
Deficiency of the Lysosomal Protein CLN5 Alters Lysosomal Function and Movement


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
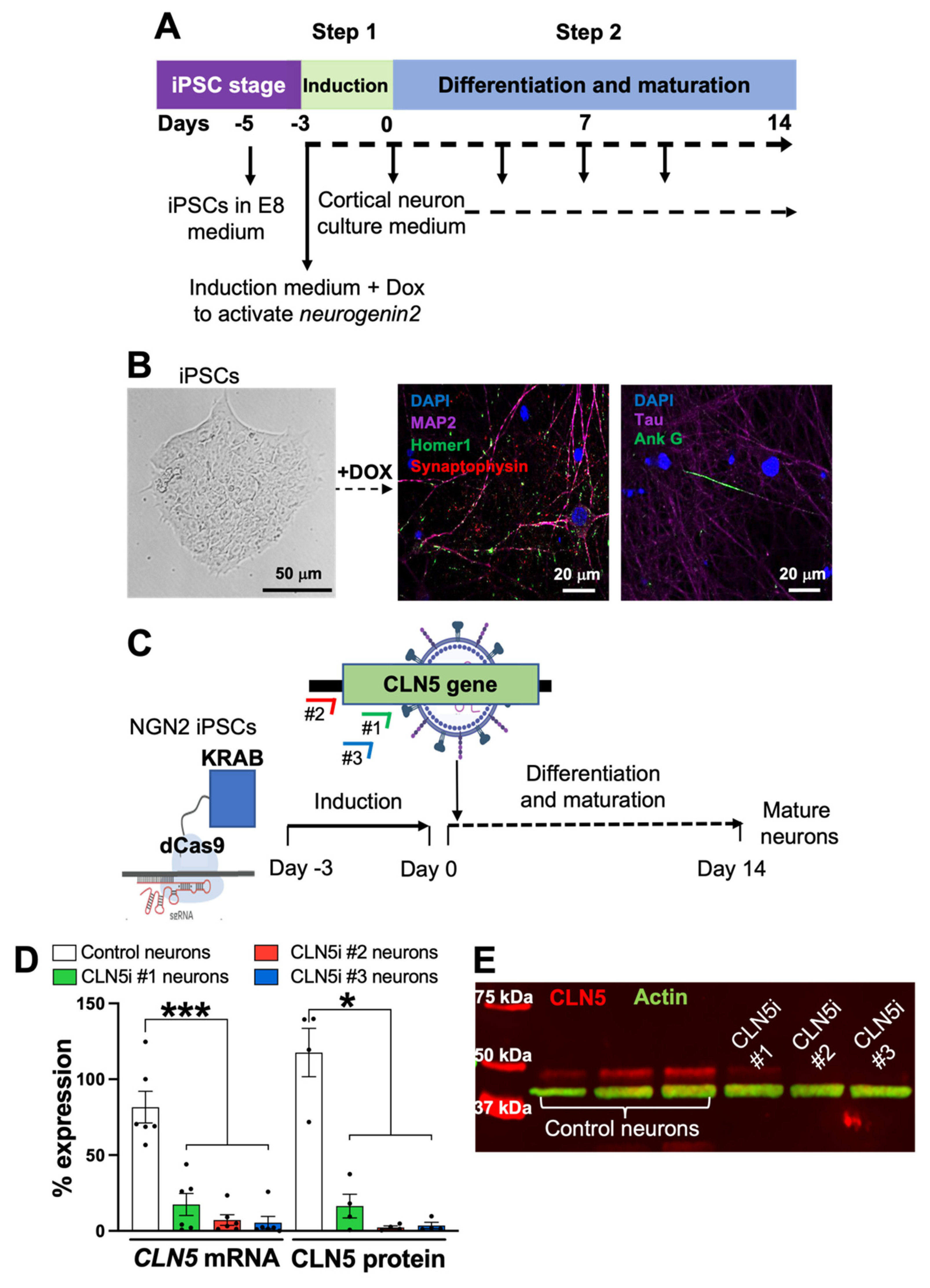
2.1. Generation of Induced Pluripotent Stem-Cell-Derived Human Neurons
2.2. CRISPR Interference in iPSC-Derived Human Neurons
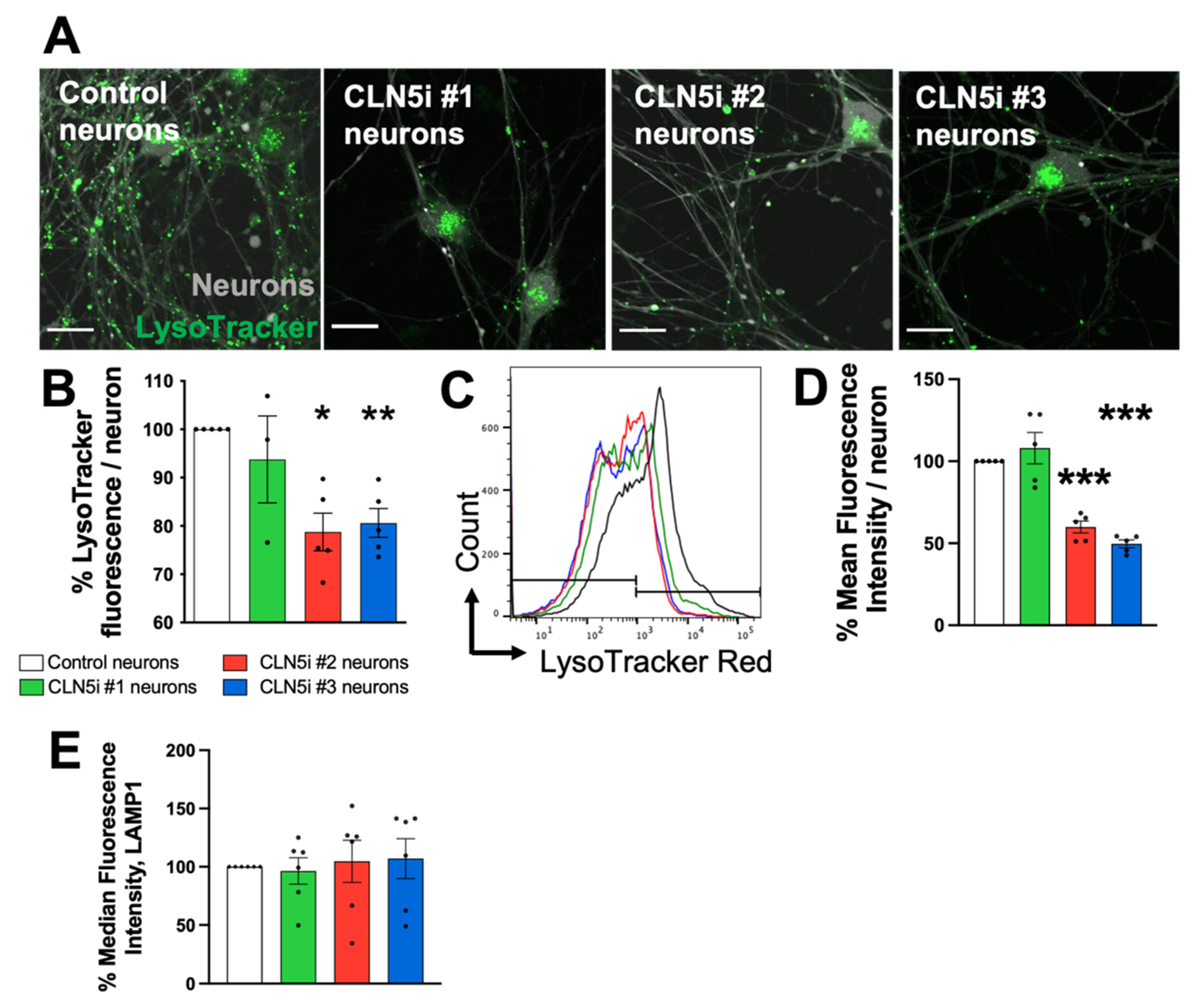
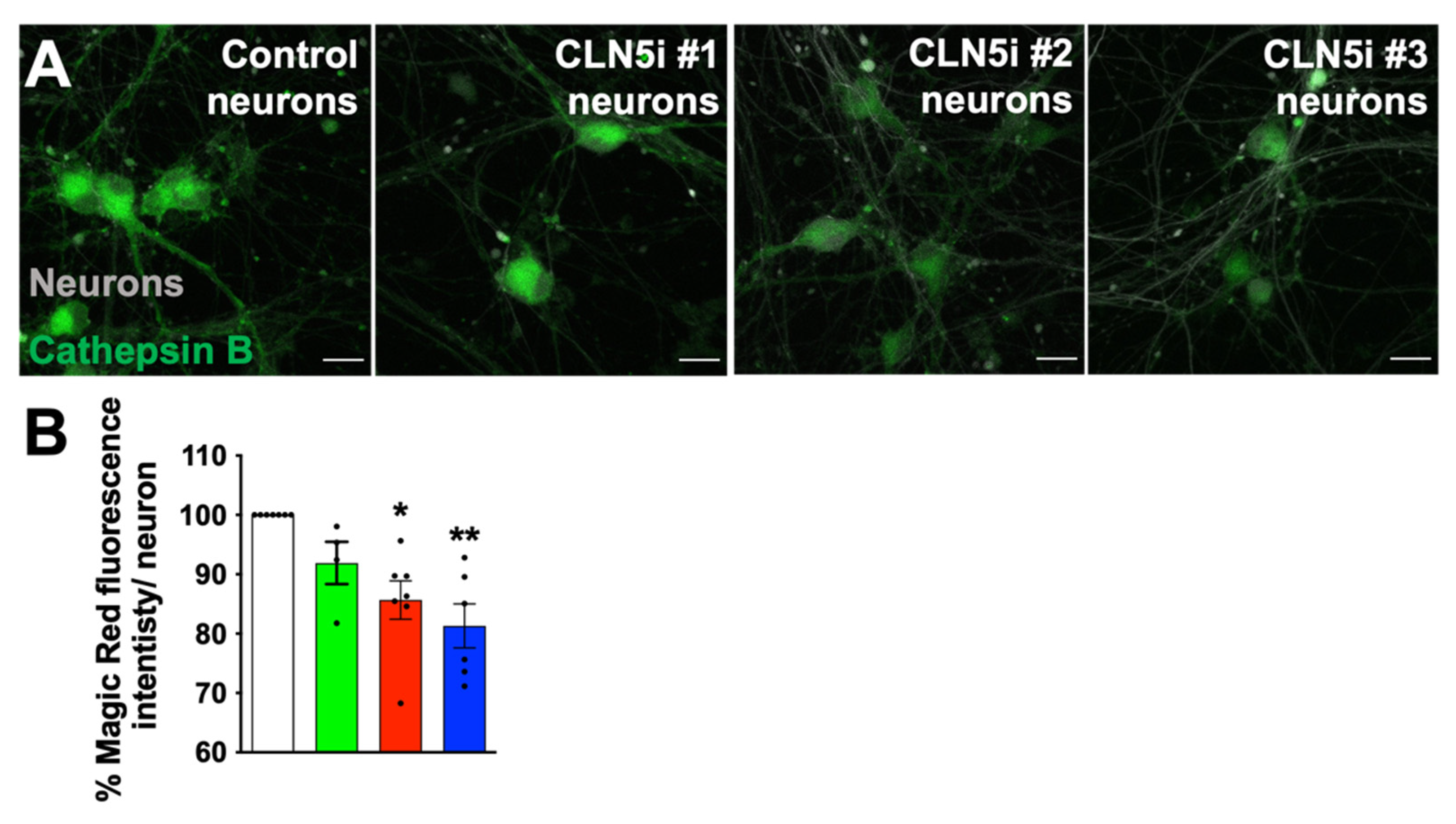
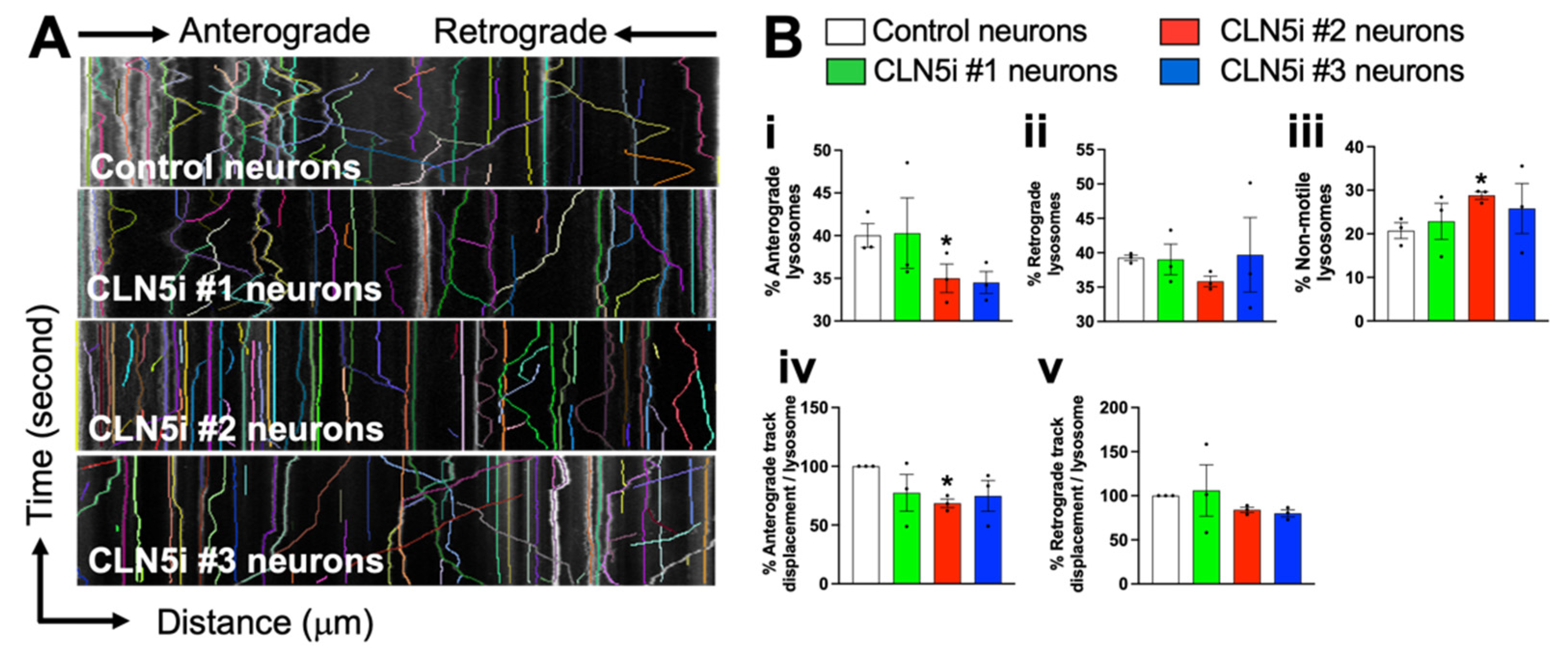
2.3. Assessment of Lysosomal Function and Movement
2.4. Statistical Analysis
3. Results
3.1. Inhibition of CLN5 in Human Neurons
3.2. Loss of CLN5 Reduces Acidic Organelles and Lysosomal Enzyme Activity in Human Neurons
3.3. Loss of CLN5 Impairs Lysosomal Movement in Human Neurons
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Basak, I.; Wicky, H.E.; McDonald, K.O.; Xu, J.B.; Palmer, J.E.; Best, H.L.; Lefrancois, S.; Lee, S.Y.; Schoderboeck, L.; Hughes, S.M. A lysosomal enigma CLN5 and its significance in understanding neuronal ceroid lipofuscinosis. Cell. Mol. Life Sci. 2021, 78, 4735–4763. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.M.; Hope, K.M.; Xu, J.B.; Mitchell, N.L.; Palmer, D.N. Inhibition of storage pathology in prenatal CLN5-deficient sheep neural cultures by lentiviral gene therapy. Neurobiol. Dis. 2014, 62, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.B.; Appu, A.P.; Sadhukhan, T.; Casey, S.; Mondal, A.; Zhang, Z.; Bagh, M.B. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol. Neurodegener. 2019, 14, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santavuori, P.; Rapola, J.; Sainio, K.; Raitta, C. A Variant of Jansky-Bielschowsky Disease. Neuropediatrics 1982, 13, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Santavuori, P.; Rapola, J.; Nuutila, A.; Raininko, R.; Lappi, M.; Launes, J.; Herva, R.; Sainio, K. The spectrum of Jan-sky-Bielschowsky disease. Neuropediatrics 1991, 22, 92–96. [Google Scholar] [CrossRef]
- Santavuori, P.; Rapola, J.; Raininko, R.; Autti, T.; Lappi, M.; Nuutila, A.; Launes, J.; Sainio, K. Early juvenile neuronal ceroid-lipofuscinosis or variant Jansky-Bielschowsky disease: Diagnostic criteria and nomenclature. J. Inherit. Metab. Dis. 1993, 16, 230–232. [Google Scholar] [CrossRef]
- Williams, R.E.; Aberg, L.; Autti, T.; Goebel, H.H.; Kohlschutter, A.; Lonnqvist, T. Diagnosis of the neuronal ceroid lipofuscinoses: An update. Biochim. Biophys. Acta. 2006, 1762, 865–872. [Google Scholar] [CrossRef] [Green Version]
- Best, H.L.; Neverman, N.J.; Wicky, H.E.; Mitchell, N.L.; Leitch, B.; Hughes, S.M. Characterisation of early changes in ovine CLN5 and CLN6 Batten disease neural cultures for the rapid screening of therapeutics. Neurobiol. Dis. 2017, 100, 62–74. [Google Scholar] [CrossRef]
- Xin, W.; Mullen, T.E.; Kiely, R.; Min, J.; Feng, X.; Cao, Y.; O’Malley, L.; Shen, Y.; Chu-Shore, C.; Mole, S.E.; et al. CLN5 mutations are frequent in juvenile and late-onset non-Finnish patients with NCL. Neurology 2010, 74, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Pineda-Trujillo, N.; Cornejo, W.; Carrizosa, J.; Wheeler, R.B.; Múnera, S.; Valencia, A.; Agudelo-Arango, J.; Cogollo, A.; Anderson, G.; Bedoya, G.; et al. A CLN5 mutation causing an atypical neuronal ceroid lipofuscinosis of juvenile onset. Neurology 2005, 64, 740–742. [Google Scholar] [CrossRef]
- Tyynelä, J.; Suopanki, J.; Santavuori, P.; Baumann, M.; Haltia, M. Variant late infantile neuronal ceroid-lipofuscinosis: Pathology and biochemistry. J. Neuropathol. Exp. Neurol. 1997, 56, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Mamo, A.; Jules, F.; Dumaresq-Doiron, K.; Costantino, S.; Lefrancois, S. The Role of Ceroid Lipofuscinosis Neuronal Protein 5 (CLN5) in Endosomal Sorting. Mol. Cell. Biol. 2012, 32, 1855–1866. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, Y.H.; Patel, V.M.; Berman, D.E.; Kothiya, M.J.; Neufeld, J.L.; Vardarajan, B.; Tang, M.; Reyes-Dumeyer, D.; Lantigua, R.; Medrano, M.; et al. An Alzheimer’s Disease-Linked Loss-of-Function CLN5 Variant Impairs Cathepsin D Maturation, Consistent with a Retromer Trafficking Defect. Mol. Cell Biol. 2018, 38, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasa, S.; Sauvageau, E.; Modica, G.; Lefrancois, S. CLN5 and CLN3 function as a complex to regulate endolysosome function. Biochem. J. 2021, 478, 2339–2357. [Google Scholar] [CrossRef]
- Schmiedt, M.L.; Blom, T.; Blom, T.; Kopra, O.; Wong, A.; von Schantz-Fant, C.; Ikonen, E.; Kuronen, M.; Jauhiainen, M.; Cooper, J.D.; et al. Cln5-deficiency in mice leads to microglial activation, defective myelination and changes in lipid metabolism. Neurobiol. Dis. 2012, 46, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Grubman, A.; Pollari, E.; Duncan, C.; Caragounis, A.; Blom, T.; Volitakis, I.; Kanninen, K.M. Deregulation of biometal homeostasis: The missing link for neuronal ceroid lipofuscinoses? Metallomics 2014, 6, 932–943. [Google Scholar] [CrossRef]
- Doccini, S.; Morani, F.; Nesti, C.; Pezzini, F.; Calza, G.; Soliymani, R.; Signore, G.; Rocchiccioli, S.; Kanninen, K.M.; Huuskonen, M.T.; et al. Proteomic and functional analyses in disease models reveal CLN5 protein involvement in mitochondrial dysfunction. Cell Death Discov. 2020, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, R.J.; Mathavarajah, S. Cln5 is secreted and functions as a glycoside hydrolase in Dictyostelium. Cell. Signal. 2018, 42, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Luebben, A.V.; Sheldrick, G.M.; Becker, S.; Wolf, A.; Kraetzner, R.; Gaertner, J.; Steinfeld, R. The CLN5 Structure Reveals a Lysosomal Protease with Important Role in Neurodegeneration. 2019. Available online: https://www.rcsb.org/structure/6R99 (accessed on 20 September 2021).
- Fernandopulle, M.S.; Prestil, R.; Grunseich, C.; Wang, C.; Gan, L.; Ward, M. Transcription Factor-Mediated Differentiation of Human iPSCs into Neurons. Curr. Protoc. Cell Biol. 2018, 79, e51. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Horlbeck, M.A.; Gilbert, L.A.; Villalta, J.E.; Adamson, B.; Pak, R.A.; Chen, Y.; Weissman, J.S. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. ELife 2016, 5, e19760. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Weissman, J.S. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, R.; Gachechiladze, M.A.; Ludwig, C.H.; Laurie, M.T.; Hong, J.Y.; Nathaniel, D.; Kampmann, M. CRISPR Interference-Based Platform for Multimodal Genetic Screens in Human iPSC-Derived Neurons. Neuron 2019, 104, 239–255. [Google Scholar] [CrossRef]
- Basak, I.; Bhatlekar, S.; Manne, B.K.; Stoller, M.; Hugo, S.; Kong, X.; Bray, P.F. miR-15a-5p regulates expression of multiple proteins in the megakaryocyte GPVI signaling pathway. J. Thromb. Haemost. 2019, 17, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Cardona, A. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangeol, P.; Prevo, B.; Peterman, E.J.G. KymographClear and KymographDirect: Two tools for the automated quantitative analysis of molecular and cellular dynamics using kymographs. Mol. Biol. Cell 2016, 27, 1948–1957. [Google Scholar] [CrossRef]
- Jakobs, M.A.; Dimitracopoulos, A.; Franze, K. KymoButler, a deep learning software for automated kymograph analysis. Elife 2019, 8, e42288. [Google Scholar] [CrossRef] [PubMed]
- Farfel-Becker, T.; Roney, J.C.; Cheng, X.-T.; Li, S.; Cuddy, S.; Sheng, Z.-H. Neuronal Soma-Derived Degradative Lysosomes Are Continuously Delivered to Distal Axons to Maintain Local Degradation Capacity. Cell Rep. 2019, 28, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Pesaola, F.; Quassollo, G.; Venier, A.C.; De Paul, A.L.; Noher, I.; Bisbal, M. The neuronal ceroid lipofuscinosis-related protein CLN8 regulates endo-lysosomal dynamics and dendritic morphology. Biol. Cell 2021, 116, 2705–2774. [Google Scholar]
- Johnson, D.E.; Ostrowski, P.; Jaumouillé, V.; Grinstein, S. The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 2016, 212, 677–692. [Google Scholar] [CrossRef] [Green Version]
- De Luca, M.; Bucci, C. A new V-ATPase regulatory mechanism mediated by the Rab interacting lysosomal protein (RILP). Commun. Integr. Biol. 2014, 7, e971572. [Google Scholar] [CrossRef]
- Ferguson, S.M. Axonal transport and maturation of lysosomes. Curr. Opin. Neurobiol. 2018, 51, 45–51. [Google Scholar] [CrossRef]
- Ferguson, S.M. Neuronal lysosomes. Neurosci. Lett. 2019, 697, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.-C.; Fernandopulle, M.S.; Wang, G.; Choi, H.; Hao, L.; Drerup, C.M.; Patel, R.; Qamar, S.; Nixon-Abell, J.; Shen, Y.; et al. RNA Granules Hitchhike on Lysosomes for Long-Distance Transport, Using Annexin A11 as a Molecular Tether. Cell 2019, 179, 147–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lie, P.P.Y.; Nixon, R.A. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol. Dis. 2019, 122, 94–105. [Google Scholar] [CrossRef]
- Overly, C.C.; Hollenbeck, P.J. Dynamic Organization of Endocytic Pathways in Axons of Cultured Sympathetic Neurons. J. Neurosci. 1996, 16, 6056–6064. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.E.; Oddo, S. Autophagic/lysosomal dysfunction in Alzheimer’s disease. Alzheimer’s Res. Ther. 2013, 5, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Schapansky, J.; Khasnavis, S.; DeAndrade, M.; Nardozzi, J.D.; Falkson, S.R.; Boyd, J.D.; Sanderson, J.B.; Bartels, T.; Melrose, H.L.; LaVoie, M.J. Familial knockin mutation of LRRK2 causes lysosomal dysfunction and accumulation of endogenous insoluble al-pha-synuclein in neurons. Neurobiol. Dis. 2018, 111, 26–35. [Google Scholar] [CrossRef]
- Uusi-Rauva, K.; Blom, T.; Schantz-Fant, V.; Blom, T.; Jalanko, A.; Kyttälä, A. Induced Pluripotent Stem Cells Derived from a CLN5 Patient Manifest Phenotypic Characteristics of Neuronal Ceroid Lipofuscinoses. Int. J. Mol. Sci. 2017, 18, 955. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basak, I.; Hansen, R.A.; Ward, M.E.; Hughes, S.M. Deficiency of the Lysosomal Protein CLN5 Alters Lysosomal Function and Movement. Biomolecules 2021, 11, 1412. https://doi.org/10.3390/biom11101412
Basak I, Hansen RA, Ward ME, Hughes SM. Deficiency of the Lysosomal Protein CLN5 Alters Lysosomal Function and Movement. Biomolecules. 2021; 11(10):1412. https://doi.org/10.3390/biom11101412
Chicago/Turabian StyleBasak, Indranil, Rachel A. Hansen, Michael E. Ward, and Stephanie M. Hughes. 2021. "Deficiency of the Lysosomal Protein CLN5 Alters Lysosomal Function and Movement" Biomolecules 11, no. 10: 1412. https://doi.org/10.3390/biom11101412
APA StyleBasak, I., Hansen, R. A., Ward, M. E., & Hughes, S. M. (2021). Deficiency of the Lysosomal Protein CLN5 Alters Lysosomal Function and Movement. Biomolecules, 11(10), 1412. https://doi.org/10.3390/biom11101412