
Transcriptomic and Epigenomic Landscape in Rett Syndrome





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
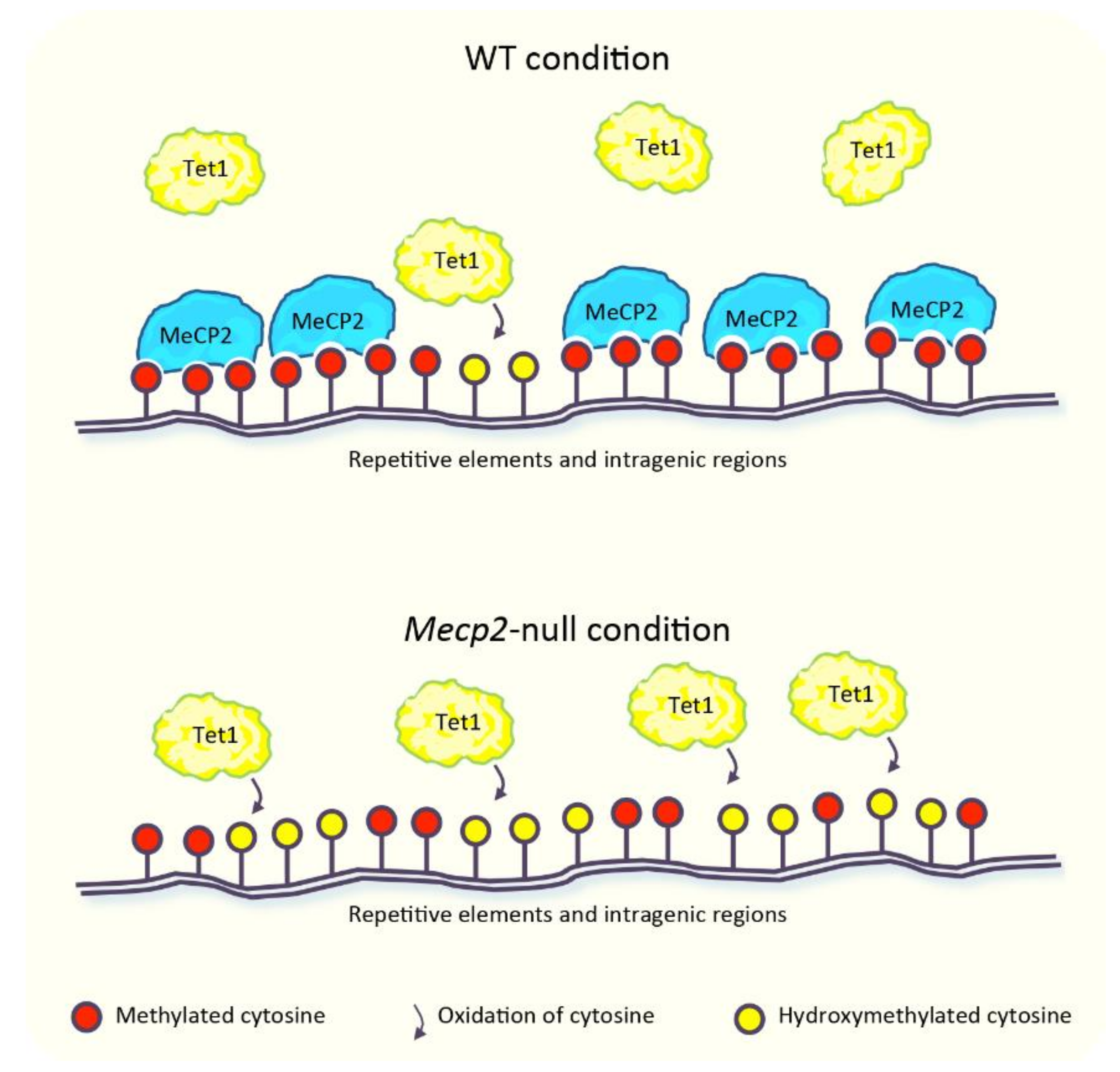
2. MeCP2: A Reader or a Regulator of DNA Methylome?
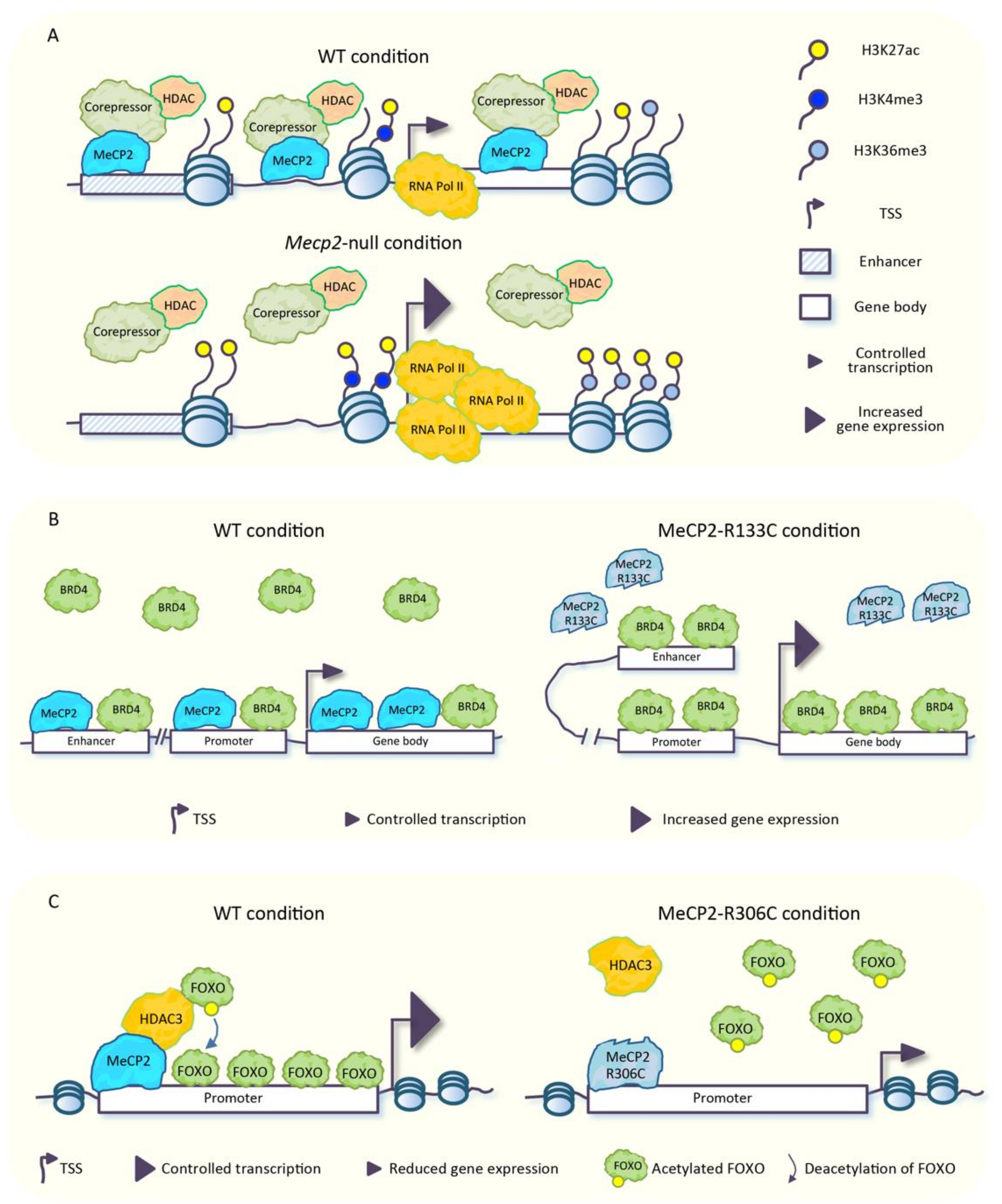
3. MeCP2: “The Guardian” of Neuronal Epigenome
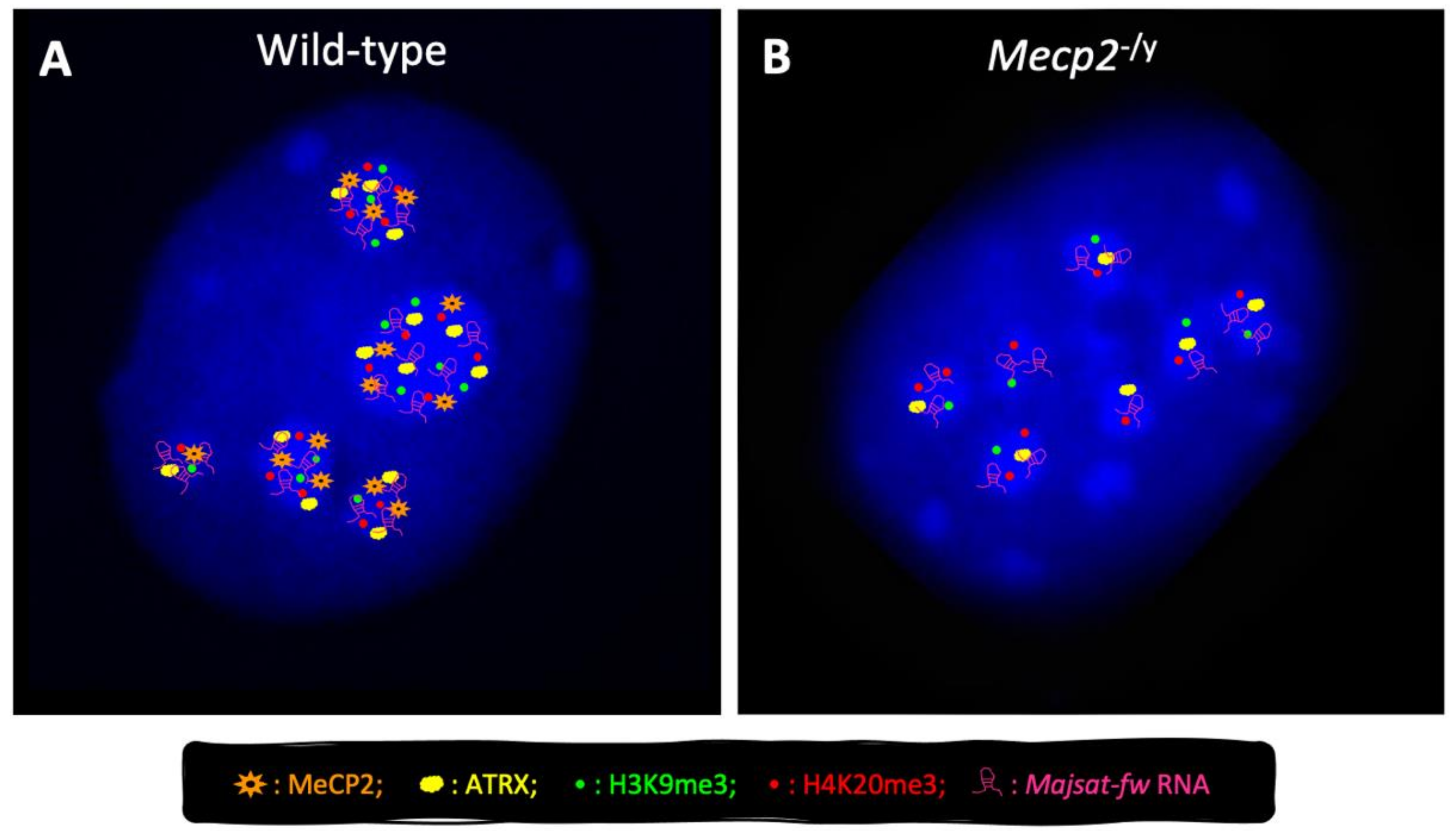
4. MeCP2 Is a Genome-Wide Modulator of Chromatin Architecture in Neurons
5. The Transcriptomic Landscape Is Globally Altered in RTT
5.1. MeCP2 Is a Fine-Tuning Modulator of Transcription
5.2. Transcriptional Deregulation Affects Many Biological Processes in RTT Brain
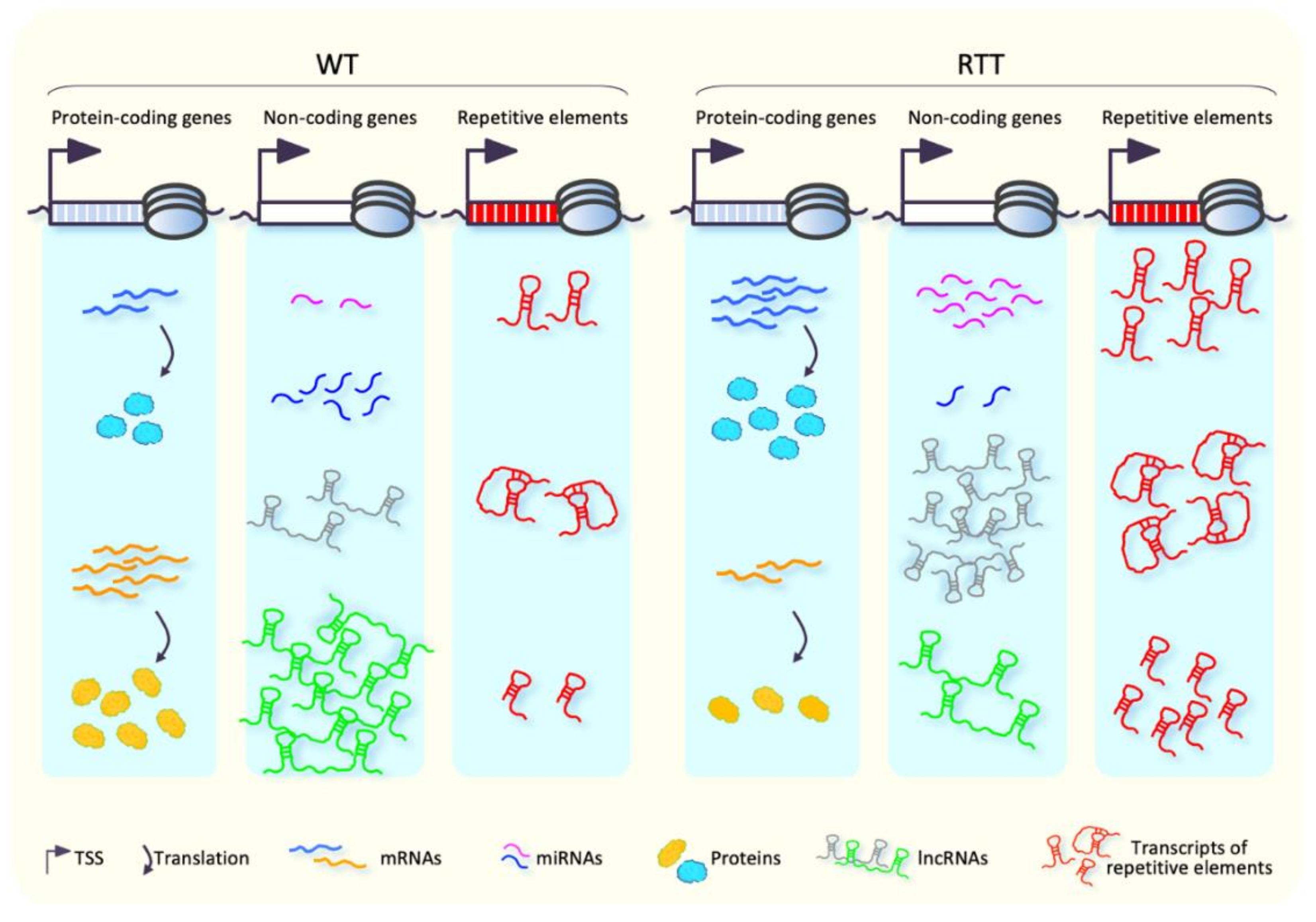
5.3. Levels of Non-Coding RNAs Are Widely Altered in RTT
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rett, A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien. Med. Wochenschr. 1966, 116, 723–726. [Google Scholar] [PubMed]
- Hagberg, B. Rett’s syndrome: Prevalence and impact on progressive severe mental retardation in girls. Acta Paediatr. Scand. 1985, 74, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, B.; Aicardi, J.; Dias, K.; Ramos, O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: Report of 35 cases. Ann. Neurol. 1983, 14, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Ben Zeev Ghidoni, B. Rett syndrome. Child. Adolesc. Psychiatr. Clin. N. Am. 2007, 16, 723–743. [Google Scholar] [CrossRef]
- Gonzales, M.L.; LaSalle, J.M. The role of MeCP2 in brain development and neurodevelopmental disorders. Curr. Psychiatry Rep. 2010, 12, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Williamson, S.L.; Christodoulou, J. Rett syndrome: New clinical and molecular insights. Eur. J. Hum. Genet. 2006, 14, 896–903. [Google Scholar] [CrossRef] [Green Version]
- Neul, j.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [Green Version]
- Percy, A.K.; Neul, J.L.; Glaze, D.G.; Motil, K.J.; Skinner, S.A.; Khwaja, O.; Lee, H.S.; Lane, J.B.; Barrish, J.O.; Annese, F.; et al. Rett syndrome diagnostic criteria: Lessons from the Natural History Study. Ann. Neurol. 2010, 68, 951–955. [Google Scholar] [CrossRef] [Green Version]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef] [Green Version]
- Ross, P.D.; Guy, J.; Selfridge, J.; Kamal, B.; Bahey, N.; Tanner, K.E.; Gillingwater, T.H.; Jones, R.A.; Loughrey, C.M.; McCarroll, C.S.; et al. Exclusive expression of MeCP2 in the nervous system distinguishes between brain and peripheral Rett syndrome-like phenotypes. Hum. Mol. Genet. 2016, 25, 4389–4404. [Google Scholar] [CrossRef] [Green Version]
- Bienvenu, T.; Chelly, J. Molecular genetics of Rett syndrome: When DNA methylation goes unrecognized. Nat. Rev. Genet. 2006, 7, 415–426. [Google Scholar] [CrossRef]
- Chahrour, M.; Zoghbi, H.Y. The story of Rett syndrome: From clinic to neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [Green Version]
- Roux, J.C.; Villard, L. Biogenic amines in Rett syndrome: The usual suspects. Behav. Genet. 2010, 40, 59–75. [Google Scholar] [CrossRef]
- Lewis, J.D.; Meehan, R.R.; Henzel, W.J.; Maurer-Fogy, I.; Jeppesen, P.; Klein, F.; Bird, A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992, 69, 905–914. [Google Scholar] [CrossRef]
- Kishi, N.; Macklis, J.D. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol. Cell. Neurosci. 2004, 27, 306–321. [Google Scholar] [CrossRef]
- Skene, P.J.; Illingworth, R.S.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Andrews, R.; Bird, A.P. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 2010, 37, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.Z.; Akbarian, S.; Tudor, M.; Jaenisch, R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 2001, 27, 327–331. [Google Scholar] [CrossRef]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef]
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007, 315, 1143–1147. [Google Scholar] [CrossRef] [Green Version]
- Adams, V.H.; McBryant, S.J.; Wade, P.A.; Woodcock, C.L.; Hansen, J.C. Intrinsic disorder and autonomous domain function in the multifunctional nuclear protein, MeCP2. J. Biol. Chem. 2007, 282, 15057–15064. [Google Scholar] [CrossRef] [Green Version]
- Hite, K.C.; Kalashnikova, A.A.; Hansen, J.C. Coil-to-helix transitions in intrinsically disordered methyl CpG binding protein 2 and its isolated domains. Protein Sci. 2012, 21, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Alarcon, D.; Claveria-Gimeno, R.; Vega, S.; Jorge-Torres, O.C.; Esteller, M.; Abian, O.; Velazquez-Campoy, A. Molecular Context-Dependent Effects Induced by Rett Syndrome-Associated Mutations in MeCP2. Biomolecules 2020, 10, 1533. [Google Scholar] [CrossRef]
- Adkins, N.L.; Georgel, P.T. MeCP2: Structure and function. Biochem. Cell Biol. 2011, 89, 1–11. [Google Scholar] [CrossRef]
- Ghosh, R.P.; Nikitina, T.; Horowitz-Scherer, R.A.; Gierasch, L.M.; Uversky, V.N.; Hite, K.; Hansen, J.C.; Woodcock, C.L. Unique physical properties and interactions of the domains of methylated DNA binding protein 2. Biochemistry 2010, 49, 4395–4410. [Google Scholar] [CrossRef] [Green Version]
- Kudo, S.; Nomura, Y.; Segawa, M.; Fujita, N.; Nakao, M.; Schanen, C.; Tamura, M. Heterogeneity in residual function of MeCP2 carrying missense mutations in the methyl CpG binding domain. J. Med. Genet. 2003, 40, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Nan, X.; Campoy, F.J.; Bird, A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 1997, 88, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Lyst, M.J.; Ekiert, R.; Ebert, D.H.; Merusi, C.; Nowak, J.; Selfridge, J.; Guy, J.; Kastan, N.R.; Robinson, N.D.; de Lima Alves, F.; et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 2013, 16, 898–902. [Google Scholar] [CrossRef] [Green Version]
- Tillotson, R.; Selfridge, J.; Koerner, M.V.; Gadalla, K.K.E.; Guy, J.; De Sousa, D.; Hector, R.D.; Cobb, S.R.; Bird, A. Radically truncated MeCP2 rescues Rett syndrome-like neurological defects. Nature 2017, 550, 398–401. [Google Scholar] [CrossRef]
- Della Ragione, F.; Vacca, M.; Fioriniello, S.; Pepe, G.; D’Esposito, M. MECP2, a multi-talented modulator of chromatin architecture. Brief. Funct. Genom. 2016, 15, 420–431. [Google Scholar] [CrossRef] [Green Version]
- Dragich, J.M.; Kim, Y.H.; Arnold, A.P.; Schanen, N.C. Differential distribution of the MeCP2 splice variants in the postnatal mouse brain. J. Comp. Neurol. 2007, 501, 526–542. [Google Scholar] [CrossRef] [PubMed]
- Gianakopoulos, P.J.; Zhang, Y.; Pencea, N.; Orlic-Milacic, M.; Mittal, K.; Windpassinger, C.; White, S.J.; Kroisel, P.M.; Chow, E.W.; Saunders, C.J.; et al. Mutations in MECP2 exon 1 in classical Rett patients disrupt MECP2_e1 transcription, but not transcription of MECP2_e2. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159B, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Mnatzakanian, G.N.; Lohi, H.; Munteanu, I.; Alfred, S.E.; Yamada, T.; MacLeod, P.J.; Jones, J.R.; Scherer, S.W.; Schanen, N.C.; Friez, M.J.; et al. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat. Genet. 2004, 36, 339–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasui, D.H.; Gonzales, M.L.; Aflatooni, J.O.; Crary, F.K.; Hu, D.J.; Gavino, B.J.; Golub, M.S.; Vincent, J.B.; Carolyn Schanen, N.; Olson, C.O.; et al. Mice with an isoform-ablating Mecp2 exon 1 mutation recapitulate the neurologic deficits of Rett syndrome. Hum. Mol. Genet. 2014, 23, 2447–2458. [Google Scholar] [CrossRef]
- Martinez de Paz, A.; Khajavi, L.; Martin, H.; Claveria-Gimeno, R.; Tom Dieck, S.; Cheema, M.S.; Sanchez-Mut, J.V.; Moksa, M.M.; Carles, A.; Brodie, N.I.; et al. MeCP2-E1 isoform is a dynamically expressed, weakly DNA-bound protein with different protein and DNA interactions compared to MeCP2-E2. Epigenetics Chromatin 2019, 12, 63. [Google Scholar] [CrossRef] [Green Version]
- Kerr, B.; Soto, C.J.; Saez, M.; Abrams, A.; Walz, K.; Young, J.I. Transgenic complementation of MeCP2 deficiency: Phenotypic rescue of Mecp2-null mice by isoform-specific transgenes. Eur. J. Hum. Genet. 2012, 20, 69–76. [Google Scholar] [CrossRef]
- Klose, R.; Bird, A. Molecular biology. MeCP2 repression goes nonglobal. Science 2003, 302, 793–795. [Google Scholar] [CrossRef] [Green Version]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef]
- Tillotson, R.; Bird, A. The Molecular Basis of MeCP2 Function in the Brain. J. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Ben-Shachar, S.; Chahrour, M.; Thaller, C.; Shaw, C.A.; Zoghbi, H.Y. Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum. Mol. Genet. 2009, 18, 2431–2442. [Google Scholar] [CrossRef] [Green Version]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [Green Version]
- Sugino, K.; Hempel, C.M.; Okaty, B.W.; Arnson, H.A.; Kato, S.; Dani, V.S.; Nelson, S.B. Cell-type-specific repression by methyl-CpG-binding protein 2 is biased toward long genes. J. Neurosci. 2014, 34, 12877–12883. [Google Scholar] [CrossRef] [Green Version]
- Maunakea, A.K.; Chepelev, I.; Cui, K.; Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013. [Google Scholar] [CrossRef] [Green Version]
- Young, J.I.; Hong, E.P.; Castle, J.C.; Crespo-Barreto, J.; Bowman, A.B.; Rose, M.F.; Kang, D.; Richman, R.; Johnson, J.M.; Berget, S.; et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. USA 2005, 102, 17551–17558. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Dong, Q.; Yuan, X.; Zeng, X.; Gao, Y.; Chiao, C.; Li, H.; Zhao, X.; Keles, S.; Wang, Z.; et al. Misregulation of Alternative Splicing in a Mouse Model of Rett Syndrome. PLoS Genet. 2016, 12, e1006129. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.L.; Chen, J.; Wan, H.; Tang, B.; Tian, W.; Liao, L.; Qiu, Z. Regulation of mRNA splicing by MeCP2 via epigenetic modifications in the brain. Sci Rep. 2017, 7, 42790. [Google Scholar] [CrossRef] [Green Version]
- Osenberg, S.; Karten, A.; Sun, J.; Li, J.; Charkowick, S.; Felice, C.A.; Kritzer, M.; Nguyen, M.V.C.; Yu, P.; Ballas, N. Activity-dependent aberrations in gene expression and alternative splicing in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2018, 115, E5363–E5372. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.L.; Wang, Z.; Liao, Q.; Zhu, Y.; Zhou, W.H.; Xu, W.; Qiu, Z. MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev. Cell 2014, 28, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Della Ragione, F.; Filosa, S.; Scalabri, F.; D’Esposito, M. MeCP2 as a genome-wide modulator: The renewal of an old story. Front. Genet. 2012, 3, 181. [Google Scholar] [CrossRef] [Green Version]
- Lagger, S.; Connelly, J.C.; Schweikert, G.; Webb, S.; Selfridge, J.; Ramsahoye, B.H.; Yu, M.; He, C.; Sanguinetti, G.; Sowers, L.C.; et al. MeCP2 recognizes cytosine methylated tri-nucleotide and di-nucleotide sequences to tune transcription in the mammalian brain. PLoS Genet. 2017, 13, e1006793. [Google Scholar] [CrossRef]
- Chen, L.; Chen, K.; Lavery, L.A.; Baker, S.A.; Shaw, C.A.; Li, W.; Zoghbi, H.Y. MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, 5509–5514. [Google Scholar] [CrossRef] [Green Version]
- Gabel, H.W.; Kinde, B.; Stroud, H.; Gilbert, C.S.; Harmin, D.A.; Kastan, N.R.; Hemberg, M.; Ebert, D.H.; Greenberg, M.E. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 2015, 522, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, N.L.; Heaven, M.R.; Holt, L.M.; Crossman, D.K.; Boggio, K.J.; Shaffer, S.A.; Flint, D.L.; Olsen, M.L. RNA sequencing and proteomics approaches reveal novel deficits in the cortex of Mecp2-deficient mice, a model for Rett syndrome. Mol. Autism 2017, 8, 56. [Google Scholar] [CrossRef]
- Renthal, W.; Boxer, L.D.; Hrvatin, S.; Li, E.; Silberfeld, A.; Nagy, M.A.; Griffith, E.C.; Vierbuchen, T.; Greenberg, M.E. Characterization of human mosaic Rett syndrome brain tissue by single-nucleus RNA sequencing. Nat. Neurosci. 2018, 21, 1670–1679. [Google Scholar] [CrossRef]
- Fioriniello, S.; Csukonyi, E.; Marano, D.; Brancaccio, A.; Madonna, M.; Zarrillo, C.; Romano, A.; Marracino, F.; Matarazzo, M.R.; D’Esposito, M.; et al. MeCP2 and Major Satellite Forward RNA Cooperate for Pericentric Heterochromatin Organization. Stem Cell Rep. 2020, 15, 1317–1332. [Google Scholar] [CrossRef]
- Fioriniello, S.; Marano, D.; Fiorillo, F.; D’Esposito, M.; Della Ragione, F. Epigenetic Factors That Control Pericentric Heterochromatin Organization in Mammals. Genes 2020, 11, 595. [Google Scholar] [CrossRef]
- Linhoff, M.W.; Garg, S.K.; Mandel, G. A high-resolution imaging approach to investigate chromatin architecture in complex tissues. Cell 2015, 163, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Marano, D.; Fioriniello, S.; Fiorillo, F.; Gibbons, R.J.; D’Esposito, M.; Della Ragione, F. ATRX Contributes to MeCP2-Mediated Pericentric Heterochromatin Organization during Neural Differentiation. Int. J. Mol. Sci. 2019, 20, 5371. [Google Scholar] [CrossRef] [Green Version]
- Bertulat, B.; De Bonis, M.L.; Della Ragione, F.; Lehmkuhl, A.; Milden, M.; Storm, C.; Jost, K.L.; Scala, S.; Hendrich, B.; D’Esposito, M.; et al. MeCP2 dependent heterochromatin reorganization during neural differentiation of a novel Mecp2-deficient embryonic stem cell reporter line. PLoS ONE 2012, 7, e47848. [Google Scholar] [CrossRef] [Green Version]
- Boxer, L.D.; Renthal, W.; Greben, A.W.; Whitwam, T.; Silberfeld, A.; Stroud, H.; Li, E.; Yang, M.G.; Kinde, B.; Griffith, E.C.; et al. MeCP2 Represses the Rate of Transcriptional Initiation of Highly Methylated Long Genes. Mol. Cell 2020, 77, 294–309 e299. [Google Scholar] [CrossRef] [PubMed]
- Nikitina, T.; Shi, X.; Ghosh, R.P.; Horowitz-Scherer, R.A.; Hansen, J.C.; Woodcock, C.L. Multiple modes of interaction between the methylated DNA binding protein MeCP2 and chromatin. Mol. Cell. Biol. 2007, 27, 864–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellen, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [Green Version]
- Good, K.V.; Vincent, J.B.; Ausio, J. MeCP2: The Genetic Driver of Rett Syndrome Epigenetics. Front. Genet. 2021, 12, 620859. [Google Scholar] [CrossRef]
- Lavery, L.A.; Ure, K.; Wan, Y.W.; Luo, C.; Trostle, A.J.; Wang, W.; Jin, H.; Lopez, J.; Lucero, J.; Durham, M.A.; et al. Losing Dnmt3a dependent methylation in inhibitory neurons impairs neural function by a mechanism impacting Rett syndrome. Elife 2020, 9. [Google Scholar] [CrossRef]
- Tillotson, R.; Cholewa-Waclaw, J.; Chhatbar, K.; Connelly, J.C.; Kirschner, S.A.; Webb, S.; Koerner, M.V.; Selfridge, J.; Kelly, D.A.; De Sousa, D.; et al. Neuronal non-CG methylation is an essential target for MeCP2 function. Mol. Cell 2021, 81, 1260–1275. [Google Scholar] [CrossRef]
- Chhatbar, K.; Cholewa-Waclaw, J.; Shah, R.; Bird, A.; Sanguinetti, G. Quantitative analysis questions the role of MeCP2 as a global regulator of alternative splicing. PLoS Genet. 2020, 16, e1009087. [Google Scholar] [CrossRef]
- Kimura, H.; Shiota, K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem. 2003, 278, 4806–4812. [Google Scholar] [CrossRef] [Green Version]
- Rajavelu, A.; Lungu, C.; Emperle, M.; Dukatz, M.; Brohm, A.; Broche, J.; Hanelt, I.; Parsa, E.; Schiffers, S.; Karnik, R.; et al. Chromatin-dependent allosteric regulation of DNMT3A activity by MeCP2. Nucleic Acids Res. 2018, 46, 9044–9056. [Google Scholar] [CrossRef]
- Aldinger, K.A.; Plummer, J.T.; Levitt, P. Comparative DNA methylation among females with neurodevelopmental disorders and seizures identifies TAC1 as a MeCP2 target gene. J. Neurodev. Disord. 2013, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Miyake, K.; Yang, C.; Minakuchi, Y.; Ohori, K.; Soutome, M.; Hirasawa, T.; Kazuki, Y.; Adachi, N.; Suzuki, S.; Itoh, M.; et al. Comparison of Genomic and Epigenomic Expression in Monozygotic Twins Discordant for Rett Syndrome. PLoS ONE 2013, 8, e66729. [Google Scholar] [CrossRef] [Green Version]
- Vogel Ciernia, A.; Laufer, B.I.; Hwang, H.; Dunaway, K.W.; Mordaunt, C.E.; Coulson, R.L.; Yasui, D.H.; LaSalle, J.M. Epigenomic Convergence of Neural-Immune Risk Factors in Neurodevelopmental Disorder Cortex. Cereb. Cortex 2020, 30, 640–655. [Google Scholar] [CrossRef]
- Ludwig, A.K.; Zhang, P.; Hastert, F.D.; Meyer, S.; Rausch, C.; Herce, H.D.; Muller, U.; Lehmkuhl, A.; Hellmann, I.; Trummer, C.; et al. Binding of MBD proteins to DNA blocks Tet1 function thereby modulating transcriptional noise. Nucleic Acids Res. 2017, 45, 2438–2457. [Google Scholar] [CrossRef] [Green Version]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011, 14, 1607–1616. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [Green Version]
- Eleftheriou, M.; Pascual, A.J.; Wheldon, L.M.; Perry, C.; Abakir, A.; Arora, A.; Johnson, A.D.; Auer, D.T.; Ellis, I.O.; Madhusudan, S.; et al. 5-Carboxylcytosine levels are elevated in human breast cancers and gliomas. Clin. Epigenetics 2015, 7, 88. [Google Scholar] [CrossRef]
- Guo, M.; Li, X.; Zhang, L.; Liu, D.; Du, W.; Yin, D.; Lyu, N.; Zhao, G.; Guo, C.; Tang, D. Accurate quantification of 5-Methylcytosine, 5-Hydroxymethylcytosine, 5-Formylcytosine, and 5-Carboxylcytosine in genomic DNA from breast cancer by chemical derivatization coupled with ultra performance liquid chromatography- electrospray quadrupole time of flight mass spectrometry analysis. Oncotarget 2017, 8, 91248–91257. [Google Scholar] [CrossRef] [Green Version]
- Pearson, E.C.; Bates, D.L.; Prospero, T.D.; Thomas, J.O. Neuronal nuclei and glial nuclei from mammalian cerebral cortex. Nucleosome repeat lengths, DNA contents and H1 contents. Eur. J. Biochem. 1984, 144, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Ito-Ishida, A.; Yamalanchili, H.K.; Shao, Y.; Baker, S.A.; Heckman, L.D.; Lavery, L.A.; Kim, J.Y.; Lombardi, L.M.; Sun, Y.; Liu, Z.; et al. Genome-wide distribution of linker histone H1.0 is independent of MeCP2. Nat. Neurosci. 2018, 21, 794–798. [Google Scholar] [CrossRef]
- Clemens, A.W.; Wu, D.Y.; Moore, J.R.; Christian, D.L.; Zhao, G.; Gabel, H.W. MeCP2 Represses Enhancers through Chromosome Topology-Associated DNA Methylation. Mol. Cell 2020, 77, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Devaiah, B.N.; Case-Borden, C.; Gegonne, A.; Hsu, C.H.; Chen, Q.; Meerzaman, D.; Dey, A.; Ozato, K.; Singer, D.S. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat. Struct. Mol. Biol. 2016, 23, 540–548. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Tanaka, Y.; Patterson, B.; Hwang, S.M.; Hysolli, E.; Cakir, B.; Kim, K.Y.; Wang, W.; Kang, Y.J.; Clement, E.M.; et al. Dysregulation of BRD4 Function Underlies the Functional Abnormalities of MeCP2 Mutant Neurons. Mol. Cell 2020, 79, 84–98 e89. [Google Scholar] [CrossRef]
- Nott, A.; Cheng, J.; Gao, F.; Lin, Y.T.; Gjoneska, E.; Ko, T.; Minhas, P.; Zamudio, A.V.; Meng, J.; Zhang, F.; et al. Histone deacetylase 3 associates with MeCP2 to regulate FOXO and social behavior. Nat. Neurosci. 2016, 19, 1497–1505. [Google Scholar] [CrossRef]
- Daitoku, H.; Hatta, M.; Matsuzaki, H.; Aratani, S.; Ohshima, T.; Miyagishi, M.; Nakajima, T.; Fukamizu, A. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10042–10047. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, H.; Daitoku, H.; Hatta, M.; Aoyama, H.; Yoshimochi, K.; Fukamizu, A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. USA 2005, 102, 11278–11283. [Google Scholar] [CrossRef] [Green Version]
- Mihaylova, M.M.; Vasquez, D.S.; Ravnskjaer, K.; Denechaud, P.D.; Yu, R.T.; Alvarez, J.G.; Downes, M.; Evans, R.M.; Montminy, M.; Shaw, R.J. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 2011, 145, 607–621. [Google Scholar] [CrossRef] [Green Version]
- Gemelli, T.; Berton, O.; Nelson, E.D.; Perrotti, L.I.; Jaenisch, R.; Monteggia, L.M. Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol. Psychiatry 2006, 59, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Politz, J.C.; Scalzo, D.; Groudine, M. Something silent this way forms: The functional organization of the repressive nuclear compartment. Annu. Rev. Cell Dev. Biol. 2013, 29, 241–270. [Google Scholar] [CrossRef] [Green Version]
- Wijchers, P.J.; Geeven, G.; Eyres, M.; Bergsma, A.J.; Janssen, M.; Verstegen, M.; Zhu, Y.; Schell, Y.; Vermeulen, C.; de Wit, E.; et al. Characterization and dynamics of pericentromere-associated domains in mice. Genome Res. 2015, 25, 958–969. [Google Scholar] [CrossRef] [Green Version]
- Almouzni, G.; Probst, A.V. Heterochromatin maintenance and establishment: Lessons from the mouse pericentromere. Nucleus 2011, 2, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Brero, A.; Easwaran, H.P.; Nowak, D.; Grunewald, I.; Cremer, T.; Leonhardt, H.; Cardoso, M.C. Methyl CpG-binding proteins induce large-scale chromatin reorganization during terminal differentiation. J. Cell Biol. 2005, 169, 733–743. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, N.; Becker, A.; Jost, K.L.; Haase, S.; Thakur, B.K.; Brero, A.; Hardt, T.; Kudo, S.; Leonhardt, H.; Cardoso, M.C. MeCP2 Rett mutations affect large scale chromatin organization. Hum. Mol. Genet. 2011, 20, 4187–4195. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, R.J.; Picketts, D.J.; Higgs, D.R. Syndromal mental retardation due to mutations in a regulator of gene expression. Hum. Mol. Genet. 1995, 4, 1705–1709. [Google Scholar] [CrossRef]
- Gibbons, R.J.; Picketts, D.J.; Villard, L.; Higgs, D.R. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome). Cell 1995, 80, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Casas-Delucchi, C.S.; Becker, A.; Bolius, J.J.; Cardoso, M.C. Targeted manipulation of heterochromatin rescues MeCP2 Rett mutants and re-establishes higher order chromatin organization. Nucleic Acids Res. 2012, 40, e176. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, T.I.; Ausio, J.; Faghfoury, H.; Silver, J.; Lane, J.B.; Eubanks, J.H.; MacLeod, P.; Percy, A.K.; Vincent, J.B. From Function to Phenotype: Impaired DNA Binding and Clustering Correlates with Clinical Severity in Males with Missense Mutations in MECP2. Sci. Rep. 2016, 6, 38590. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, N.; Hardt, T.; Brero, A.; Nowak, D.; Rothbauer, U.; Becker, A.; Leonhardt, H.; Cardoso, M.C. MeCP2 interacts with HP1 and modulates its heterochromatin association during myogenic differentiation. Nucleic Acids Res. 2007, 35, 5402–5408. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.L.; Yewdell, W.T.; Bell, J.C.; McNulty, S.M.; Duda, Z.; O’Neill, R.J.; Sullivan, B.A.; Straight, A.F. RNA-dependent stabilization of SUV39H1 at constitutive heterochromatin. Elife 2017, 6. [Google Scholar] [CrossRef]
- Velazquez Camacho, O.; Galan, C.; Swist-Rosowska, K.; Ching, R.; Gamalinda, M.; Karabiber, F.; De La Rosa-Velazquez, I.; Engist, B.; Koschorz, B.; Shukeir, N.; et al. Major satellite repeat RNA stabilize heterochromatin retention of Suv39h enzymes by RNA-nucleosome association and RNA:DNA hybrid formation. Elife 2017, 6. [Google Scholar] [CrossRef]
- Fan, C.; Zhang, H.; Fu, L.; Li, Y.; Du, Y.; Qiu, Z.; Lu, F. Rett mutations attenuate phase separation of MeCP2. Cell Discov. 2020, 6, 38. [Google Scholar] [CrossRef]
- Wang, L.; Hu, M.; Zuo, M.Q.; Zhao, J.; Wu, D.; Huang, L.; Wen, Y.; Li, Y.; Chen, P.; Bao, X.; et al. Rett syndrome-causing mutations compromise MeCP2-mediated liquid-liquid phase separation of chromatin. Cell Res. 2020, 30, 393–407. [Google Scholar] [CrossRef]
- Hyman, A.A.; Weber, C.A.; Julicher, F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef] [Green Version]
- Elbaum-Garfinkle, S.; Kim, Y.; Szczepaniak, K.; Chen, C.C.; Eckmann, C.R.; Myong, S.; Brangwynne, C.P. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl. Acad. Sci. USA 2015, 112, 7189–7194. [Google Scholar] [CrossRef] [Green Version]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Gibson, B.A.; Doolittle, L.K.; Schneider, M.W.G.; Jensen, L.E.; Gamarra, N.; Henry, L.; Gerlich, D.W.; Redding, S.; Rosen, M.K. Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 2019, 179, 470–484. [Google Scholar] [CrossRef]
- Larson, A.G.; Elnatan, D.; Keenen, M.M.; Trnka, M.J.; Johnston, J.B.; Burlingame, A.L.; Agard, D.A.; Redding, S.; Narlikar, G.J. Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature 2017, 547, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, A.G.; Narlikar, G.J. The Role of Phase Separation in Heterochromatin Formation, Function, and Regulation. Biochemistry 2018, 57, 2540–2548. [Google Scholar] [CrossRef] [PubMed]
- Strom, A.R.; Emelyanov, A.V.; Mir, M.; Fyodorov, D.V.; Darzacq, X.; Karpen, G.H. Phase separation drives heterochromatin domain formation. Nature 2017, 547, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gao, Y.; Zheng, X.; Liu, C.; Dong, S.; Li, R.; Zhang, G.; Wei, Y.; Qu, H.; Li, Y.; et al. Histone Modifications Regulate Chromatin Compartmentalization by Contributing to a Phase Separation Mechanism. Mol. Cell 2019, 76, 646–659. [Google Scholar] [CrossRef]
- Chadwick, L.H.; Wade, P.A. MeCP2 in Rett syndrome: Transcriptional repressor or chromatin architectural protein? Curr. Opin. Genet. Dev. 2007, 17, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Sanfeliu, A.; Kaufmann, W.E.; Gill, M.; Guasoni, P.; Tropea, D. Transcriptomic Studies in Mouse Models of Rett Syndrome: A Review. Neuroscience 2019, 413, 183–205. [Google Scholar] [CrossRef]
- Tudor, M.; Akbarian, S.; Chen, R.Z.; Jaenisch, R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc. Natl. Acad. Sci. USA 2002, 99, 15536–15541. [Google Scholar] [CrossRef] [Green Version]
- Vacca, M.; Tripathi, K.P.; Speranza, L.; Aiese Cigliano, R.; Scalabri, F.; Marracino, F.; Madonna, M.; Sanseverino, W.; Perrone-Capano, C.; Guarracino, M.R.; et al. Effects of Mecp2 loss of function in embryonic cortical neurons: A bioinformatics strategy to sort out non-neuronal cells variability from transcriptome profiling. BMC Bioinform. 2016, 17 (Suppl. 2), 14. [Google Scholar] [CrossRef] [Green Version]
- Kinde, B.; Wu, D.Y.; Greenberg, M.E.; Gabel, H.W. DNA methylation in the gene body influences MeCP2-mediated gene repression. Proc. Natl. Acad. Sci. USA 2016, 113, 15114–15119. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, H.; Muffat, J.; Cheng, A.W.; Orlando, D.A.; Loven, J.; Kwok, S.M.; Feldman, D.A.; Bateup, H.S.; Gao, Q.; et al. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell 2013, 13, 446–458. [Google Scholar] [CrossRef] [Green Version]
- Cholewa-Waclaw, J.; Shah, R.; Webb, S.; Chhatbar, K.; Ramsahoye, B.; Pusch, O.; Yu, M.; Greulich, P.; Waclaw, B.; Bird, A.P. Quantitative modelling predicts the impact of DNA methylation on RNA polymerase II traffic. Proc. Natl. Acad. Sci. USA 2019, 116, 14995–15000. [Google Scholar] [CrossRef] [Green Version]
- Lyst, M.J.; Bird, A. Rett syndrome: A complex disorder with simple roots. Nat. Rev. Genet. 2015, 16, 261–275. [Google Scholar] [CrossRef]
- Ebert, D.H.; Gabel, H.W.; Robinson, N.D.; Kastan, N.R.; Hu, L.S.; Cohen, S.; Navarro, A.J.; Lyst, M.J.; Ekiert, R.; Bird, A.P.; et al. Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature 2013, 499, 341–345. [Google Scholar] [CrossRef] [Green Version]
- Shahbazian, M.D.; Antalffy, B.; Armstrong, D.L.; Zoghbi, H.Y. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 2002, 11, 115–124. [Google Scholar] [CrossRef]
- Johnson, B.S.; Zhao, Y.T.; Fasolino, M.; Lamonica, J.M.; Kim, Y.J.; Georgakilas, G.; Wood, K.H.; Bu, D.; Cui, Y.; Goffin, D.; et al. Biotin tagging of MeCP2 in mice reveals contextual insights into the Rett syndrome transcriptome. Nat. Med. 2017, 23, 1203–1214. [Google Scholar] [CrossRef]
- Raman, A.T.; Pohodich, A.E.; Wan, Y.W.; Yalamanchili, H.K.; Lowry, W.E.; Zoghbi, H.Y.; Liu, Z. Apparent bias toward long gene misregulation in MeCP2 syndromes disappears after controlling for baseline variations. Nat. Commun. 2018, 9, 3225. [Google Scholar] [CrossRef]
- Bedogni, F.; Rossi, R.L.; Galli, F.; Cobolli Gigli, C.; Gandaglia, A.; Kilstrup-Nielsen, C.; Landsberger, N. Rett syndrome and the urge of novel approaches to study MeCP2 functions and mechanisms of action. Neurosci. Biobehav. Rev. 2014, 46 Pt. 2, 187–201. [Google Scholar] [CrossRef]
- Krishnaraj, R.; Haase, F.; Coorey, B.; Luca, E.J.; Wong, I.; Boyling, A.; Ellaway, C.; Christodoulou, J.; Gold, W.A. Genome-wide transcriptomic and proteomic studies of Rett syndrome mouse models identify common signaling pathways and cellular functions as potential therapeutic targets. Hum. Mutat. 2019, 40, 2184–2196. [Google Scholar] [CrossRef]
- Buchovecky, C.M.; Turley, S.D.; Brown, H.M.; Kyle, S.M.; McDonald, J.G.; Liu, B.; Pieper, A.A.; Huang, W.; Katz, D.M.; Russell, D.W.; et al. A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat. Genet. 2013, 45, 1013–1020. [Google Scholar] [CrossRef] [Green Version]
- Lekman, A.Y.; Hagberg, B.A.; Svennerholm, L.T. Membrane cerebral lipids in Rett syndrome. Pediatr. Neurol. 1991, 7, 186–190. [Google Scholar] [CrossRef]
- Lekman, A.Y.; Hagberg, B.A.; Svennerholm, L.T. Altered cerebellar ganglioside pattern in rett syndrome. Neurochem. Intern. 1991, 19, 505–509. [Google Scholar] [CrossRef]
- Lekman, A.Y.; Hagberg, B.A.; Svennerholm, L.T. Cerebrospinal fluid gangliosides in patients with Rett syndrome and infantile neuronal ceroid lipofuscinosis. Eur. J. Paediatr. Neurol. 1999, 3, 119–123. [Google Scholar] [CrossRef]
- Pecorelli, A.; Belmonte, G.; Meloni, I.; Cervellati, F.; Gardi, C.; Sticozzi, C.; De Felice, C.; Signorini, C.; Cortelazzo, A.; Leoncini, S.; et al. Alteration of serum lipid profile, SRB1 loss, and impaired Nrf2 activation in CDKL5 disorder. Free Radic. Biol. Med. 2015, 86, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Segatto, M.; Trapani, L.; Di Tunno, I.; Sticozzi, C.; Valacchi, G.; Hayek, J.; Pallottini, V. Cholesterol metabolism is altered in Rett syndrome: A study on plasma and primary cultured fibroblasts derived from patients. PLoS ONE 2014, 9, e104834. [Google Scholar] [CrossRef] [Green Version]
- Sticozzi, C.; Belmonte, G.; Pecorelli, A.; Cervellati, F.; Leoncini, S.; Signorini, C.; Ciccoli, L.; De Felice, C.; Hayek, J.; Valacchi, G. Scavenger receptor B1 post-translational modifications in Rett syndrome. FEBS Lett. 2013, 587, 2199–2204. [Google Scholar] [CrossRef] [Green Version]
- Colak, D.; Al-Dhalaan, H.; Nester, M.; Albakheet, A.; Al-Younes, B.; Al-Hassnan, Z.; Al-Dosari, M.; Chedrawi, A.; Al-Owain, M.; Abudheim, N.; et al. Genomic and transcriptomic analyses distinguish classic Rett and Rett-like syndrome and reveals shared altered pathways. Genomics 2011, 97, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Lopez, A.M.; Chuang, J.C.; Posey, K.S.; Turley, S.D. Suppression of brain cholesterol synthesis in male Mecp2-deficient mice is age dependent and not accompanied by a concurrent change in the rate of fatty acid synthesis. Brain Res. 2017, 1654, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Luoni, M.; Giannelli, S.; Indrigo, M.T.; Niro, A.; Massimino, L.; Iannielli, A.; Passeri, L.; Russo, F.; Morabito, G.; Calamita, P.; et al. Whole brain delivery of an instability-prone Mecp2 transgene improves behavioral and molecular pathological defects in mouse models of Rett syndrome. Elife 2020, 9. [Google Scholar] [CrossRef]
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Imran, A.; Qasim, M.; Zafar, S.; Kamran, S.K.S.; Razzaq, A.; Aziz, N.; et al. Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis. 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Park, M.J.; Aja, S.; Li, Q.; Degano, A.L.; Penati, J.; Zhuo, J.; Roe, C.R.; Ronnett, G.V. Anaplerotic triheptanoin diet enhances mitochondrial substrate use to remodel the metabolome and improve lifespan, motor function, and sociability in MeCP2-null mice. PLoS ONE 2014, 9, e109527. [Google Scholar] [CrossRef] [Green Version]
- Villani, C.; Sacchetti, G.; Bagnati, R.; Passoni, A.; Fusco, F.; Carli, M.; Invernizzi, R.W. Lovastatin fails to improve motor performance and survival in methyl-CpG-binding protein2-null mice. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Zhao, L.; Wang, X.; Tong, D.; Hoover, C.; Wu, F.; Liu, Y.; Wang, L.; Liu, L.; Ni, L.; et al. MeCP2 regulated glycogenes contribute to proliferation and apoptosis of gastric cancer cells. Glycobiology 2017, 27, 306–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhoff, R.; Sandhoff, K. Emerging concepts of ganglioside metabolism. FEBS Lett. 2018, 592, 3835–3864. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, T.N.; Heinecke, K.A.; Mantis, J.G.; Denny, C.A. Brain lipid analysis in mice with Rett syndrome. Neurochem. Res. 2009, 34, 1057–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Yoo, Y.; Lim, B.C.; Kim, K.J.; Song, J.; Choi, M.; Chae, J.H. GM3 synthase deficiency due to ST3GAL5 variants in two Korean female siblings: Masquerading as Rett syndrome-like phenotype. Am. J. Med. Genet. A 2016. [Google Scholar] [CrossRef]
- Cappuccio, G.; Donti, T.; Pinelli, M.; Bernardo, P.; Bravaccio, C.; Elsea, S.H.; Brunetti-Pierri, N. Sphingolipid Metabolism Perturbations in Rett Syndrome. Metabolites 2019, 9, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulmez Karaca, K.; Brito, D.V.C.; Oliveira, A.M.M. MeCP2: A Critical Regulator of Chromatin in Neurodevelopment and Adult Brain Function. Int. J. Mol. Sci. 2019, 20, 4577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belichenko, P.V.; Wright, E.E.; Belichenko, N.P.; Masliah, E.; Li, H.H.; Mobley, W.C.; Francke, U. Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: Evidence for disruption of neuronal networks. J. Comp. Neurol. 2009, 514, 240–258. [Google Scholar] [CrossRef]
- Palmer, A.; Qayumi, J.; Ronnett, G. MeCP2 mutation causes distinguishable phases of acute and chronic defects in synaptogenesis and maintenance, respectively. Mol. Cell. Neurosci. 2008, 37, 794–807. [Google Scholar] [CrossRef]
- Ehrhart, F.; Coort, S.L.; Cirillo, E.; Smeets, E.; Evelo, C.T.; Curfs, L.M. Rett syndrome—Biological pathways leading from MECP2 to disorder phenotypes. Orphanet J. Rare Dis. 2016, 11, 158. [Google Scholar] [CrossRef] [Green Version]
- Jordan, C.; Li, H.H.; Kwan, H.C.; Francke, U. Cerebellar gene expression profiles of mouse models for Rett syndrome reveal novel MeCP2 targets. BMC Med. Genet. 2007, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Pozzo-Miller, L. BDNF deregulation in Rett syndrome. Neuropharmacology 2014, 76 Pt. C, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Chang, Q.; Khare, G.; Dani, V.; Nelson, S.; Jaenisch, R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 2006, 49, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Deng, V.; Matagne, V.; Banine, F.; Frerking, M.; Ohliger, P.; Budden, S.; Pevsner, J.; Dissen, G.A.; Sherman, L.S.; Ojeda, S.R. FXYD1 is an MeCP2 target gene overexpressed in the brains of Rett syndrome patients and Mecp2-null mice. Hum. Mol. Genet. 2007, 16, 640–650. [Google Scholar] [CrossRef]
- Ehinger, Y.; Bruyere, J.; Panayotis, N.; Abada, Y.S.; Borloz, E.; Matagne, V.; Scaramuzzino, C.; Vitet, H.; Delatour, B.; Saidi, L.; et al. Huntingtin phosphorylation governs BDNF homeostasis and improves the phenotype of Mecp2 knockout mice. EMBO Mol. Med. 2020, 12, e10889. [Google Scholar] [CrossRef]
- Hsu, W.L.; Ma, Y.L.; Liu, Y.C.; Tai, D.J.C.; Lee, E.H.Y. Restoring Wnt6 signaling ameliorates behavioral deficits in MeCP2 T158A mouse model of Rett syndrome. Sci. Rep. 2020, 10, 1074. [Google Scholar] [CrossRef]
- Vicario-Abejon, C.; Owens, D.; McKay, R.; Segal, M. Role of neurotrophins in central synapse formation and stabilization. Nat. Rev. Neurosci. 2002, 3, 965–974. [Google Scholar] [CrossRef]
- Calfa, G.; Li, W.; Rutherford, J.M.; Pozzo-Miller, L. Excitation/inhibition imbalance and impaired synaptic inhibition in hippocampal area CA3 of Mecp2 knockout mice. Hippocampus 2015, 25, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Kishi, N.; MacDonald, J.L.; Ye, J.; Molyneaux, B.J.; Azim, E.; Macklis, J.D. Reduction of aberrant NF-kappaB signalling ameliorates Rett syndrome phenotypes in Mecp2-null mice. Nat. Commun. 2016, 7, 10520. [Google Scholar] [CrossRef] [Green Version]
- O’Driscoll, C.M.; Lima, M.P.; Kaufmann, W.E.; Bressler, J.P. Methyl CpG binding protein 2 deficiency enhances expression of inflammatory cytokines by sustaining NF-kappaB signaling in myeloid derived cells. J. Neuroimmunol. 2015, 283, 23–29. [Google Scholar] [CrossRef]
- Urdinguio, R.G.; Lopez-Serra, L.; Lopez-Nieva, P.; Alaminos, M.; Diaz-Uriarte, R.; Fernandez, A.F.; Esteller, M. Mecp2-null mice provide new neuronal targets for Rett syndrome. PLoS ONE 2008, 3, e3669. [Google Scholar] [CrossRef] [Green Version]
- Delepine, C.; Nectoux, J.; Letourneur, F.; Baud, V.; Chelly, J.; Billuart, P.; Bienvenu, T. Astrocyte Transcriptome from the Mecp2(308)-Truncated Mouse Model of Rett Syndrome. Neuromol. Med. 2015, 17, 353–363. [Google Scholar] [CrossRef]
- Pecorelli, A.; Cervellati, C.; Cordone, V.; Hayek, J.; Valacchi, G. Compromised immune/inflammatory responses in Rett syndrome. Free Radic. Biol. Med. 2020, 152, 100–106. [Google Scholar] [CrossRef]
- Cronk, J.C.; Derecki, N.C.; Ji, E.; Xu, Y.; Lampano, A.E.; Smirnov, I.; Baker, W.; Norris, G.T.; Marin, I.; Coddington, N.; et al. Methyl-CpG Binding Protein 2 Regulates Microglia and Macrophage Gene Expression in Response to Inflammatory Stimuli. Immunity 2015, 42, 679–691. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Mokhtari, R.; Pedrosa, E.; Birnbaum, R.; Zheng, D.; Lachman, H.M. Transcriptome analysis of microglia in a mouse model of Rett syndrome: Differential expression of genes associated with microglia/macrophage activation and cellular stress. Mol. Autism 2017, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Derecki, N.C.; Cronk, J.C.; Lu, Z.; Xu, E.; Abbott, S.B.; Guyenet, P.G.; Kipnis, J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 2012, 484, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Maezawa, I.; Swanberg, S.; Harvey, D.; LaSalle, J.M.; Jin, L.W. Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J. Neurosci. 2009, 29, 5051–5061. [Google Scholar] [CrossRef]
- Gibson, J.H.; Slobedman, B.; Harikrishnan, K.N.; Williamson, S.L.; Minchenko, D.; El-Osta, A.; Stern, J.L.; Christodoulou, J. Downstream targets of methyl CpG binding protein 2 and their abnormal expression in the frontal cortex of the human Rett syndrome brain. BMC Neurosci. 2010, 11, 53. [Google Scholar] [CrossRef] [Green Version]
- Kriaucionis, S.; Paterson, A.; Curtis, J.; Guy, J.; Macleod, N.; Bird, A. Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol. Cell. Biol. 2006, 26, 5033–5042. [Google Scholar] [CrossRef] [Green Version]
- Filosa, S.; Pecorelli, A.; D’Esposito, M.; Valacchi, G.; Hajek, J. Exploring the possible link between MeCP2 and oxidative stress in Rett syndrome. Free Radic. Biol. Med. 2015, 88, 81–90. [Google Scholar] [CrossRef]
- Shovlin, S.; Tropea, D. Transcriptome level analysis in Rett syndrome using human samples from different tissues. Orphanet J. Rare Dis. 2018, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kim, K.Y.; Zhong, M.; Pan, X.; Weissman, S.M.; Park, I.H. Transcriptional regulation in pluripotent stem cells by methyl CpG-binding protein 2 (MeCP2). Hum. Mol. Genet. 2014, 23, 1045–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecorelli, A.; Leoni, G.; Cervellati, F.; Canali, R.; Signorini, C.; Leoncini, S.; Cortelazzo, A.; De Felice, C.; Ciccoli, L.; Hayek, J.; et al. Genes related to mitochondrial functions, protein degradation, and chromatin folding are differentially expressed in lymphomonocytes of Rett syndrome patients. Mediat. Inflamm. 2013, 2013, 137629. [Google Scholar] [CrossRef] [Green Version]
- Gold, W.A.; Williamson, S.L.; Kaur, S.; Hargreaves, I.P.; Land, J.M.; Pelka, G.J.; Tam, P.P.; Christodoulou, J. Mitochondrial dysfunction in the skeletal muscle of a mouse model of Rett syndrome (RTT): Implications for the disease phenotype. Mitochondrion 2014, 15, 10–17. [Google Scholar] [CrossRef]
- Esposito, L.A.; Melov, S.; Panov, A.; Cottrell, B.A.; Wallace, D.C. Mitochondrial disease in mouse results in increased oxidative stress. Proc. Natl. Acad. Sci. USA 1999, 96, 4820–4825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Felice, C.; Della Ragione, F.; Signorini, C.; Leoncini, S.; Pecorelli, A.; Ciccoli, L.; Scalabri, F.; Marracino, F.; Madonna, M.; Belmonte, G.; et al. Oxidative brain damage in Mecp2-mutant murine models of Rett syndrome. Neurobiol. Dis. 2014, 68, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Signorini, C.; De Felice, C.; Leoncini, S.; Giardini, A.; D’Esposito, M.; Filosa, S.; Della Ragione, F.; Rossi, M.; Pecorelli, A.; Valacchi, G.; et al. F(4)-neuroprostanes mediate neurological severity in Rett syndrome. Clin. Chim. Acta 2011, 412, 1399–1406. [Google Scholar] [CrossRef]
- Cheng, T.L.; Qiu, Z. MeCP2: Multifaceted roles in gene regulation and neural development. Neurosci. Bull. 2014, 30, 601–609. [Google Scholar] [CrossRef] [Green Version]
- Mellios, N.; Feldman, D.A.; Sheridan, S.D.; Ip, J.P.K.; Kwok, S.; Amoah, S.K.; Rosen, B.; Rodriguez, B.A.; Crawford, B.; Swaminathan, R.; et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol. Psychiatry 2018, 23, 1051–1065. [Google Scholar] [CrossRef] [Green Version]
- Sheinerman, K.; Djukic, A.; Tsivinsky, V.G.; Umansky, S.R. Brain-enriched microRNAs circulating in plasma as novel biomarkers for Rett syndrome. PLoS ONE 2019, 14, e0218623. [Google Scholar] [CrossRef] [Green Version]
- Szulwach, K.E.; Li, X.; Smrt, R.D.; Li, Y.; Luo, Y.; Lin, L.; Santistevan, N.J.; Li, W.; Zhao, X.; Jin, P. Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J. Cell Biol. 2010, 189, 127–141. [Google Scholar] [CrossRef] [Green Version]
- Urdinguio, R.G.; Fernandez, A.F.; Lopez-Nieva, P.; Rossi, S.; Huertas, D.; Kulis, M.; Liu, C.G.; Croce, C.M.; Calin, G.A.; Esteller, M. Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics 2010, 5, 656–663. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Tao, J.; Chen, P.J.; Shahab, A.; Ge, W.; Hart, R.P.; Ruan, X.; Ruan, Y.; Sun, Y.E. Genome-wide analysis reveals methyl-CpG-binding protein 2-dependent regulation of microRNAs in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 18161–18166. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Yu, X.; Hu, S.; Yu, J. A brief review on the mechanisms of miRNA regulation. Genom. Proteom. Bioinform. 2009, 7, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Della Ragione, F.; Gagliardi, M.; D’Esposito, M.; Matarazzo, M.R. Non-coding RNAs in chromatin disease involving neurological defects. Front. Cell Neurosci. 2014, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Fineberg, S.K.; Kosik, K.S.; Davidson, B.L. MicroRNAs potentiate neural development. Neuron 2009, 64, 303–309. [Google Scholar] [CrossRef] [Green Version]
- Kawase-Koga, Y.; Otaegi, G.; Sun, T. Different timings of Dicer deletion affect neurogenesis and gliogenesis in the developing mouse central nervous system. Dev. Dyn. 2009, 238, 2800–2812. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science 2007, 317, 1220–1224. [Google Scholar] [CrossRef] [Green Version]
- Kosik, K.S. The neuronal microRNA system. Nat. Rev. Neurosci. 2006, 7, 911–920. [Google Scholar] [CrossRef]
- Schratt, G.M.; Tuebing, F.; Nigh, E.A.; Kane, C.G.; Sabatini, M.E.; Kiebler, M.; Greenberg, M.E. A brain-specific microRNA regulates dendritic spine development. Nature 2006, 439, 283–289. [Google Scholar] [CrossRef]
- Wayman, G.A.; Davare, M.; Ando, H.; Fortin, D.; Varlamova, O.; Cheng, H.Y.; Marks, D.; Obrietan, K.; Soderling, T.R.; Goodman, R.H.; et al. An activity-regulated microRNA controls dendritic plasticity by down-regulating p250GAP. Proc. Natl. Acad. Sci. USA 2008, 105, 9093–9098. [Google Scholar] [CrossRef] [Green Version]
- Fiore, R.; Khudayberdiev, S.; Christensen, M.; Siegel, G.; Flavell, S.W.; Kim, T.K.; Greenberg, M.E.; Schratt, G. Mef2-mediated transcription of the miR379-410 cluster regulates activity-dependent dendritogenesis by fine-tuning Pumilio2 protein levels. EMBO J. 2009, 28, 697–710. [Google Scholar] [CrossRef]
- Hebert, S.S.; Horre, K.; Nicolai, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; De Strooper, B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef] [Green Version]
- Jiao, J.; Herl, L.D.; Farese, R.V.; Gao, F.B. MicroRNA-29b regulates the expression level of human progranulin, a secreted glycoprotein implicated in frontotemporal dementia. PLoS ONE 2010, 5, e10551. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shin, B.C.; Thamotharan, S.; Devaskar, S.U. Differential methylation of the micro-RNA 7b gene targets postnatal maturation of murine neuronal Mecp2 gene expression. Dev. Neurobiol. 2014, 74, 407–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Su, J.; Guo, W.; Polich, E.D.; Magyar, D.P.; Xing, Y.; Li, H.; Smrt, R.D.; Chang, Q.; Zhao, X. Inhibition of miR-15a Promotes BDNF Expression and Rescues Dendritic Maturation Deficits in MeCP2-Deficient Neurons. Stem Cells 2015, 33, 1618–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, T.; Kimura, M.; Horii, T.; Morita, S.; Soejima, H.; Kudo, S.; Hatada, I. MeCP2-dependent repression of an imprinted miR-184 released by depolarization. Hum. Mol. Genet. 2008, 17, 1192–1199. [Google Scholar] [CrossRef] [Green Version]
- Glaich, O.; Parikh, S.; Bell, R.E.; Mekahel, K.; Donyo, M.; Leader, Y.; Shayevitch, R.; Sheinboim, D.; Yannai, S.; Hollander, D.; et al. DNA methylation directs microRNA biogenesis in mammalian cells. Nat. Commun. 2019, 10, 5657. [Google Scholar] [CrossRef] [Green Version]
- Tsujimura, K.; Irie, K.; Nakashima, H.; Egashira, Y.; Fukao, Y.; Fujiwara, M.; Itoh, M.; Uesaka, M.; Imamura, T.; Nakahata, Y.; et al. miR-199a Links MeCP2 with mTOR Signaling and Its Dysregulation Leads to Rett Syndrome Phenotypes. Cell Rep. 2015, 12, 1887–1901. [Google Scholar] [CrossRef] [Green Version]
- Smrt, R.D.; Szulwach, K.E.; Pfeiffer, R.L.; Li, X.; Guo, W.; Pathania, M.; Teng, Z.Q.; Luo, Y.; Peng, J.; Bordey, A.; et al. MicroRNA miR-137 regulates neuronal maturation by targeting ubiquitin ligase mind bomb-1. Stem Cells 2010, 28, 1060–1070. [Google Scholar] [CrossRef] [Green Version]
- Klein, M.E.; Lioy, D.T.; Ma, L.; Impey, S.; Mandel, G.; Goodman, R.H. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat. Neurosci. 2007, 10, 1513–1514. [Google Scholar] [CrossRef]
- Vo, N.; Klein, M.E.; Varlamova, O.; Keller, D.M.; Yamamoto, T.; Goodman, R.H.; Impey, S. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 16426–16431. [Google Scholar] [CrossRef] [Green Version]
- Petazzi, P.; Sandoval, J.; Szczesna, K.; Jorge, O.C.; Roa, L.; Sayols, S.; Gomez, A.; Huertas, D.; Esteller, M. Dysregulation of the long non-coding RNA transcriptome in a Rett syndrome mouse model. RNA Biol. 2013, 10, 1197–1203. [Google Scholar] [CrossRef] [Green Version]
- Harvey, V.L.; Duguid, I.C.; Krasel, C.; Stephens, G.J. Evidence that GABA rho subunits contribute to functional ionotropic GABA receptors in mouse cerebellar Purkinje cells. J. Physiol. 2006, 577, 127–139. [Google Scholar] [CrossRef]
- Muotri, A.R.; Marchetto, M.C.; Coufal, N.G.; Oefner, R.; Yeo, G.; Nakashima, K.; Gage, F.H. L1 retrotransposition in neurons is modulated by MeCP2. Nature 2010, 468, 443–446. [Google Scholar] [CrossRef]
- Zhao, B.; Wu, Q.; Ye, A.Y.; Guo, J.; Zheng, X.; Yang, X.; Yan, L.; Liu, Q.R.; Hyde, T.M.; Wei, L.; et al. Somatic LINE-1 retrotransposition in cortical neurons and non-brain tissues of Rett patients and healthy individuals. PLoS Genet. 2019, 15, e1008043. [Google Scholar] [CrossRef] [Green Version]
- Han, J.S.; Szak, S.T.; Boeke, J.D. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature 2004, 429, 268–274. [Google Scholar] [CrossRef]
- Kazazian, H.H., Jr. Mobile elements and disease. Curr. Opin. Genet. Dev. 1998, 8, 343–350. [Google Scholar] [CrossRef]
- Perepelitsa-Belancio, V.; Deininger, P. RNA truncation by premature polyadenylation attenuates human mobile element activity. Nat. Genet. 2003, 35, 363–366. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marano, D.; Fioriniello, S.; D’Esposito, M.; Della Ragione, F. Transcriptomic and Epigenomic Landscape in Rett Syndrome. Biomolecules 2021, 11, 967. https://doi.org/10.3390/biom11070967
Marano D, Fioriniello S, D’Esposito M, Della Ragione F. Transcriptomic and Epigenomic Landscape in Rett Syndrome. Biomolecules. 2021; 11(7):967. https://doi.org/10.3390/biom11070967
Chicago/Turabian StyleMarano, Domenico, Salvatore Fioriniello, Maurizio D’Esposito, and Floriana Della Ragione. 2021. "Transcriptomic and Epigenomic Landscape in Rett Syndrome" Biomolecules 11, no. 7: 967. https://doi.org/10.3390/biom11070967
APA StyleMarano, D., Fioriniello, S., D’Esposito, M., & Della Ragione, F. (2021). Transcriptomic and Epigenomic Landscape in Rett Syndrome. Biomolecules, 11(7), 967. https://doi.org/10.3390/biom11070967