
Measuring Treatment Response in Progressive Multiple Sclerosis—Considerations for Adapting to an Era of Multiple Treatment Options



Abstract
:1. Introduction
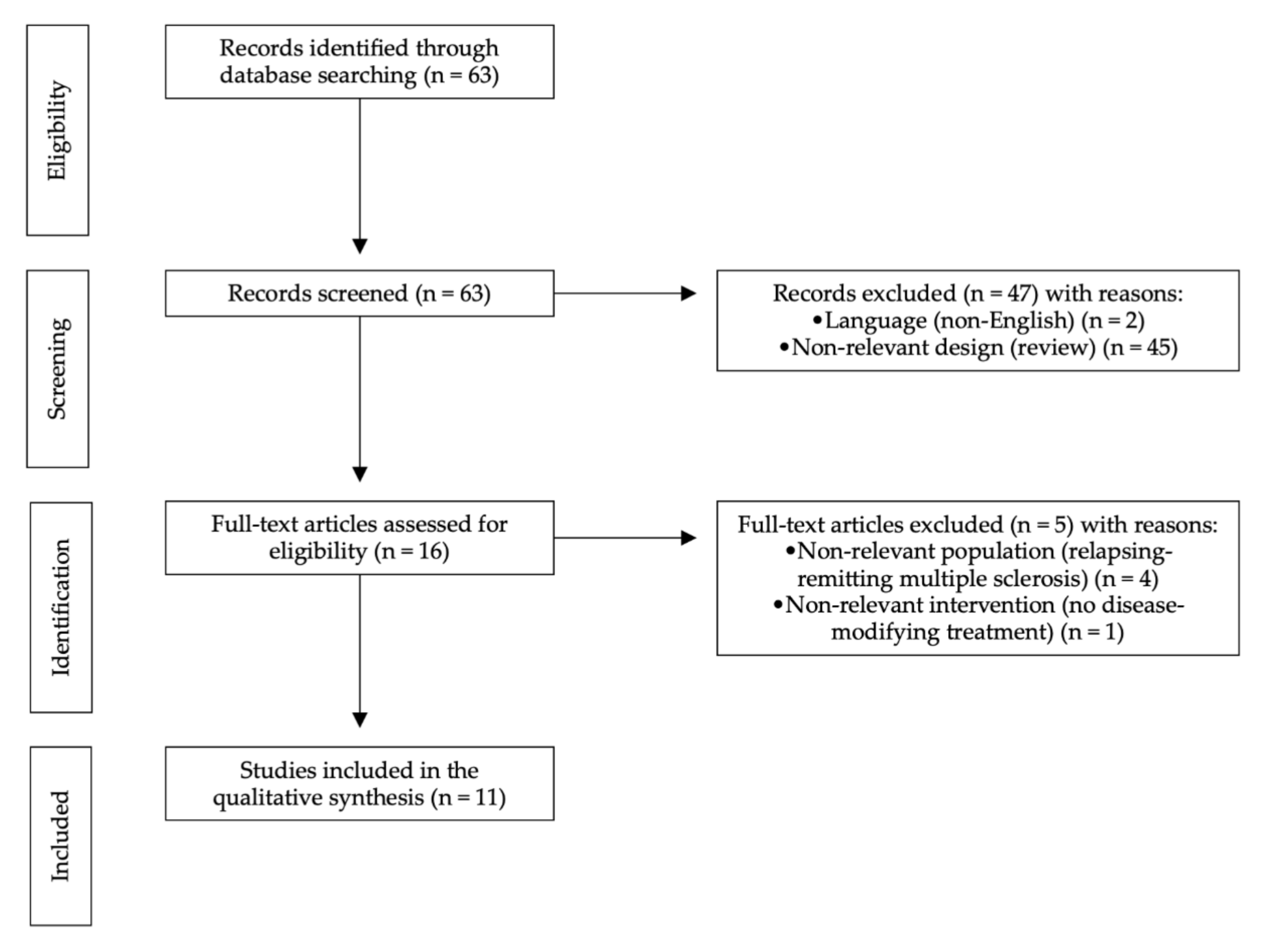
2. Methods
2.1. Search Methods
2.2. Selection Criteria
3. Results
3.1. Expanded Disability Status Scale
3.2. Timed 25-Foot Walk Test
3.3. 9-Hole Peg Test
3.4. Paced Auditory Serial Addition Test
3.5. Symbol Digit Modalities Test
3.6. Low-Contrast Letter Acuity
3.7. Scripps Neurologic Rating Scale
3.8. Magnetic Resonance Imaging
3.9. Composite Outcome Measures
4. Discussion
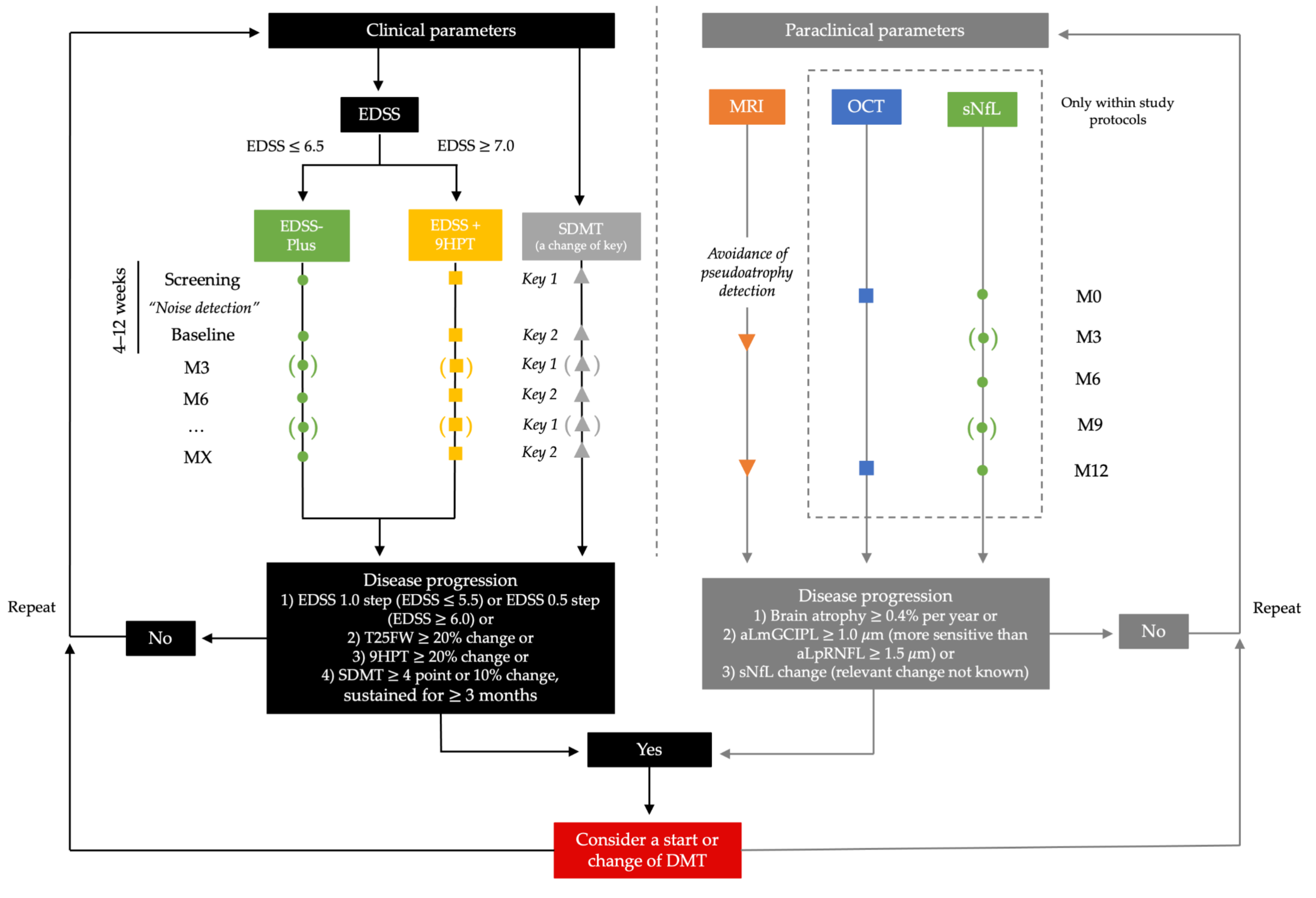
4.1. Detection of Disability Progression Depends on the Level of Disability
4.2. The Absence of Worsening vs. Improvement of Disability
4.3. Time Frame for Objectivation of Treatment Response
4.4. Other Outcome Measures
4.5. Recommendations for Future Study Trials and Everyday Clinical Practice
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oh, J.; Vidal-Jordana, A.; Montalban, X. Multiple sclerosis: Clinical aspects. Curr. Opin. Neurol. 2018, 31, 752–759. [Google Scholar] [CrossRef]
- Miller, D.H.; Leary, S.M. Primary-progressive multiple sclerosis. Lancet Neurol. 2007, 6, 903–912. [Google Scholar] [CrossRef]
- Kister, I.; Bacon, T.E.; Chamot, E.; Salter, A.R.; Cutter, G.R.; Kalina, J.T.; Herbert, J. Natural History of Multiple Sclerosis Symptoms. Int. J. MS Care 2013, 15, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Tutuncu, M.; Tang, J.; Zeid, N.A.; Kale, N.; Crusan, D.J.; Atkinson, E.J.; Siva, A.; Pittock, S.J.; Pirko, I.; Keegan, B.M.; et al. Onset of progressive phase is an age-dependent clinical milestone in multiple sclerosis. Mult. Scler. J. 2012, 19, 188–198. [Google Scholar] [CrossRef]
- Weinshenker, B.G.; Bass, B.; Rice, G.P.; Noseworthy, J.; Carriere, W.; Baskerville, J.; Ebers, G.C. The natural history of multiple sclerosis: A geographically based study. I. Clinical course and disability. Brain 1989, 112, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Confavreux, C.; Vukusic, S.; Adeleine, P. Early clinical predictors and progression of irreversible disability in multiple sclerosis: An amnesic process. Brain 2003, 126, 770–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runmarker, B.; Andersen, O. Prognostic factors in a multiple sclerosis incidence cohort with twenty-five years of follow-up. Brain 1993, 116, 117–134. [Google Scholar] [CrossRef]
- Bsteh, G.; Ehling, R.; Lutterotti, A.; Hegen, H.; Di Pauli, F.; Auer, M.; Deisenhammer, F.; Reindl, M.; Berger, T. Long Term Clinical Prognostic Factors in Relapsing-Remitting Multiple Sclerosis: Insights from a 10-Year Observational Study. PLoS ONE 2016, 11, e0158978. [Google Scholar] [CrossRef]
- Scott, T.F.; Hackett, C.T.; Quigley, M.R.; Schramke, C.J. Relapsing multiple sclerosis patients treated with disease modifying therapy exhibit highly variable disease progression: A predictive model. Clin. Neurol. Neurosurg. 2014, 127, 86–92. [Google Scholar] [CrossRef]
- Brown, W.; Coles, A.; Horakova, D.; Havrdova, E.; Izquierdo, G.; Prat, A.; Girard, M.; Duquette, P.; Trojano, M.; Lugaresi, A.; et al. Association of Initial Disease-Modifying Therapy with Later Conversion to Secondary Progressive Multiple Sclerosis. JAMA 2019, 321, 175–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; De Seze, J.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Bar-Or, A.; Cree, B.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Kurtzke, J.F. Rating neurologic impairment in multiple sclerosis: An expanded disability status scale (EDSS). Neurology 1983, 33, 1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawker, K.; O’Connor, P.; Freedman, M.S.; Calabresi, P.A.; Antel, J.; Simon, J.; Hauser, S.; Waubant, E.; Vollmer, T.; Panitch, H.; et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol. 2009, 66, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L. Placebo-controlled multicentre randomised trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis. European Study Group on interferon beta-1b in secondary progressive MS. Lancet 1998, 352, 1491–1497. [Google Scholar] [CrossRef]
- Wolinsky, J.S.; Narayana, P.A.; O’Connor, P.; Coyle, P.K.; Ford, C.; Johnson, K.; Miller, A.; Pardo, L.; Kadosh, S.; Ladkani, D.; et al. Glatiramer acetate in primary progressive multiple sclerosis: Results of a multinational, multicenter, double-blind, placebo-controlled trial. Ann. Neurol. 2007, 61, 14–24. [Google Scholar] [CrossRef]
- SPECTRIMS Study Group. Secondary Progressive Efficacy Clinical Trial of Recombinant Interferon-Beta-1a in MSSG. Randomized controlled trial of interferon- beta-1a in secondary progressive MS: Clinical results. Neurology 2001, 56, 1496–1504. [Google Scholar] [CrossRef]
- Lublin, F.; Miller, D.H.; Freedman, M.S.; Cree, B.; Wolinsky, J.S.; Weiner, H.; Lubetzki, C.; Hartung, H.-P.; Montalban, X.; Uitdehaag, B.M.J.; et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet 2016, 387, 1075–1084. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Hartung, H.-P.; Gonsette, R.; Konig, N.; Kwiecinski, H.; Guseo, A.; Morrissey, S.P.; Krapf, H.; Zwingers, T. Mitoxantrone in progressive multiple sclerosis: A placebo-controlled, double-blind, randomised, multicentre trial. Lancet 2002, 360, 2018–2025. [Google Scholar] [CrossRef]
- Lepore, V.; Bosetti, C.; Santucci, C.; Iaffaldano, P.; Trojano, M.; Mosconi, P.; Totaro, R.; Coniglio, M.G.; Bossio, R.B.; Valentino, P.; et al. Detection of disability worsening in relapsing-remitting multiple sclerosis patients: A real-world roving Expanded Disability Status Scale reference analysis from the Italian Multiple Sclerosis Register. Eur. J. Neurol. 2020, 28, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Butzkueven, H.; Wiendl, H.; Spelman, T.; Pellegrini, F.; Chen, Y.; Dong, Q.; Koendgen, H.; Belachew, S.; Trojano, M.; et al. Greater sensitivity to multiple sclerosis disability worsening and progression events using a roving versus a fixed reference value in a prospective cohort study. Mult. Scler. J. 2017, 24, 963–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremenchutzky, M.; Rice, G.P.A.; Baskerville, J.; Wingerchuk, D.M.; Ebers, G.C. The natural history of multiple sclerosis: A geographically based study 9: Observations on the progressive phase of the disease. Brain 2006, 129, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Cinar, B.P.; Yorgun, Y.G. What We Learned from The History of Multiple Sclerosis Measurement: Expanded Disability Status Scale. Noro. Psikiyatr. Ars. 2018, 55, S69–S75. [Google Scholar]
- Sharrack, B.; Hughes, R.A.C.; Soudain, S.; Dunn, G. The psychometric properties of clinical rating scales used in multiple sclerosis. Brain 1999, 122, 141–159. [Google Scholar] [CrossRef] [Green Version]
- Cadavid, D.; Tang, Y.; O’Neill, G. Responsiveness of the Expanded Disability Status Scale (EDSS) to disease progression and therapeutic intervention in progressive forms of multiple sclerosis. Rev. Neurol. 2010, 51, 321–329. [Google Scholar]
- Hyland, M.; Rudick, R.A. Challenges to clinical trials in multiple sclerosis: Outcome measures in the era of disease-modifying drugs. Curr. Opin. Neurol. 2011, 24, 255–261. [Google Scholar] [CrossRef]
- Kragt, J.J.; Thompson, A.; Montalban, X.; Tintore, M.; Rio, J.; Polman, C.H.; Uitdehaag, B. Responsiveness and predictive value of EDSS and MSFC in primary progressive MS. Neurology 2008, 70, 1084–1091. [Google Scholar] [CrossRef]
- Brissart, H.; Sauvée, M.; Latarche, C.; Dillier, C.; Debouverie, M. Integration of Cognitive Impairment in the Expanded Disability Status Scale of 215 Patients with Multiple Sclerosis. Eur. Neurol. 2010, 64, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Amato, M.P.; Fratiglioni, L.; Groppi, C.; Siracusa, G.; Amaducci, L. Interrater Reliability in Assessing Functional Systems and Disability on the Kurtzke Scale in Multiple Sclerosis. Arch. Neurol. 1988, 45, 746–748. [Google Scholar] [CrossRef]
- Goodkin, D.E.; Cookfair, D.; Wende, K.; Bourdette, D.; Pullicino, P.; Scherokman, B.; Whitham, R. Inter- and intrarater scoring agreement using grades 1.0 to 3.5 of the Kurtzke Expanded Disability Status Scale (EDSS). Neurology 1992, 42, 859. [Google Scholar] [CrossRef]
- Noseworthy, J.H.; Vandervoort, M.K.; Wong, C.J.; Ebers, G. Interrater variability with the Expanded Disability Status Scale (EDSS) and Functional Systems (FS) in a multiple sclerosis clinical trial. The Canadian Cooperation MS Study Group. Neurology 1990, 40, 971. [Google Scholar] [CrossRef]
- Meyer-Moock, S.; Feng, Y.-S.; Maeurer, M.; Dippel, F.-W.; Kohlmann, T. Systematic literature review and validity evaluation of the Expanded Disability Status Scale (EDSS) and the Multiple Sclerosis Functional Composite (MSFC) in patients with multiple sclerosis. BMC Neurol. 2014, 14, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieseier, B.C.; Pozzilli, C. Assessing walking disability in multiple sclerosis. Mult. Scler. J. 2012, 18, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Motl, R.W.; Cohen, J.A.; Benedict, R.; Phillips, G.; LaRocca, N.; Hudson, L.D.; Rudick, R.; Multiple Sclerosis Outcome Assessments Consortium. Validity of the timed 25-foot walk as an ambulatory performance outcome measure for multiple sclerosis. Mult. Scler. J. 2017, 23, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Bosma, L.V.; Kragt, J.J.; Knol, D.L.; Polman, C.H.; Uitdehaag, B.M. Clinical scales in progressive MS: Predicting long-term disability. Mult. Scler. J. 2011, 18, 345–350. [Google Scholar] [CrossRef]
- Orbach, R.; Zhao, Z.; Wang, Y.-C.; O’Neill, G.; Cadavid, D. Comparison of Disease Activity in SPMS and PPMS in the Context of Multicenter Clinical Trials. PLoS ONE 2012, 7, e45409. [Google Scholar] [CrossRef] [Green Version]
- Phan-Ba, R.; Pace, A.; Calay, P.; Grodent, P.; Douchamps, F.; Hyde, R.; Hotermans, C.; Delvaux, V.; Hansen, I.; Moonen, G.; et al. Comparison of the Timed 25-Foot and the 100-Meter Walk as Performance Measures in Multiple Sclerosis. Neurorehabilit. Neural Repair 2011, 25, 672–679. [Google Scholar] [CrossRef]
- Kapoor, R.; Ho, P.-R.; Campbell, N.; Chang, I.; Deykin, A.; Forrestal, F.; Lucas, N.; Yu, B.; Arnold, D.L.; Freedman, M.S.; et al. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): A phase 3, randomised, double-blind, placebo-controlled trial with an open-label extension. Lancet Neurol. 2018, 17, 405–415. [Google Scholar] [CrossRef]
- Kaufman, M.; Moyer, D.; Norton, J. The significant change for the Timed 25-foot Walk in the multiple sclerosis functional composite. Mult. Scler. J. 2000, 6, 286–290. [Google Scholar] [CrossRef]
- Coleman, C.I.; Sobieraj, D.; Marinucci, L.N. Minimally important clinical difference of the Timed 25-Foot Walk Test: Results from a randomized controlled trial in patients with multiple sclerosis. Curr. Med. Res. Opin. 2011, 28, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Kragt, J.J.; Linden, F.A.H.V.D.; Nielsen, J.M.; Uitdehaag, B.M.J.; Polman, C.H. Clinical impact of 20% worsening on Timed 25-foot Walk and 9-hole Peg Test in multiple sclerosis. Mult. Scler. J. 2006, 12, 594–598. [Google Scholar] [CrossRef] [PubMed]
- Bosma, L.V.; Kragt, J.J.; Brieva, L.; Khaleeli, Z.; Montalban, X.; Polman, C.H.; Thompson, A.J.; Tintoré, M.; Uitdehaag, B.M.J. Progression on the Multiple Sclerosis Functional Composite in multiple sclerosis: What is the optimal cut-off for the three components? Mult. Scler. J. 2010, 16, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Hobart, J.; Blight, A.R.; Goodman, A.; Lynn, F.; Putzki, N. Timed 25-Foot Walk: Direct evidence that improving 20% or greater is clinically meaningful in MS. Neurology 2013, 80, 1509–1517. [Google Scholar] [CrossRef]
- Koch, M.; Mostert, J.; Uitdehaag, B.; Cutter, G. Clinical outcome measures in SPMS trials: An analysis of the IMPACT and ASCEND original trial data sets. Mult. Scler. J. 2019, 26, 1540–1549. [Google Scholar] [CrossRef] [PubMed]
- Sikes, E.M.; Cederberg, K.L.; Sandroff, B.M.; Bartolucci, A.; Motl, R.W. Quantitative Synthesis of Timed 25-Foot Walk Performance in Multiple Sclerosis. Arch. Phys. Med. Rehabil. 2019, 101, 524–534. [Google Scholar] [CrossRef]
- Goldman, M.D.; LaRocca, N.G.; Rudick, R.A.; Hudson, L.D.; Chin, P.S.; Francis, G.S.; Jacobs, A.; Kapoor, R.; Matthews, P.M.; Mowry, E.M.; et al. Evaluation of multiple sclerosis disability outcome measures using pooled clinical trial data. Neurology 2019, 93, e1921–e1931. [Google Scholar] [CrossRef] [Green Version]
- Cutter, G.R.; Baier, M.L.; Rudick, R.A.; Cookfair, D.L.; Fischer, J.S.; Petkau, J.; Syndulko, K.; Weinshenker, B.G.; Antel, J.; Confavreux, C.; et al. Development of a multiple sclerosis functional composite as a clinical trial outcome measure. Brain 1999, 122, 871–882. [Google Scholar] [CrossRef]
- Goodkin, D.E.; Hertsgaard, D.; Seminary, J. Upper extremity function in multiple sclerosis: Improving assessment sensitivity with box-and-block and nine-hole peg tests. Arch. Phys. Med. Rehabil. 1988, 69, 850–854. [Google Scholar] [PubMed]
- Bertoni, R.; Lamers, I.; Chen, C.C.; Feys, P.; Cattaneo, D. Unilateral and bilateral upper limb dysfunction at body functions, activity and participation levels in people with multiple sclerosis. Mult. Scler. J. 2015, 21, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Lamers, I.; Cattaneo, D.; Chen, C.C.; Bertoni, R.; Van Wijmeersch, B.; Feys, P. Associations of Upper Limb Disability Measures on Different Levels of the International Classification of Functioning, Disability and Health in People with Multiple Sclerosis. Phys. Ther. 2015, 95, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Solaro, C.; Cattaneo, D.; Brichetto, G.; Castelli, L.; Tacchino, A.; Gervasoni, E.; Prosperini, L. Clinical correlates of 9-hole peg test in a large population of people with multiple sclerosis. Mult. Scler. Relat. Disord. 2019, 30, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rosti-Otajärvi, E.; Hämäläinen, P.; Koivisto, K.; Hokkanen, L. The reliability of the MSFC and its components. Acta Neurol. Scand. 2007, 117, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Feys, P.; Lamers, I.; Francis, G.; Benedict, R.; Phillips, G.; LaRocca, N.; Hudson, L.D.; Rudick, R.; Multiple Sclerosis Outcome Assessments Consortium. The Nine-Hole Peg Test as a manual dexterity performance measure for multiple sclerosis. Mult. Scler. J. 2017, 23, 711–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tombaugh, T.N. A comprehensive review of the Paced Auditory Serial Addition Test (PASAT). Arch. Clin. Neuropsychol. 2006, 21, 53–76. [Google Scholar] [CrossRef] [Green Version]
- Gronwall, D.M.A. Paced Auditory Serial-Addition Task: A Measure of Recovery from Concussion. Percept. Mot. Ski. 1977, 44, 367–373. [Google Scholar] [CrossRef]
- Rudick, R.A.; Cutter, G.; Reingold, S. The multiple sclerosis functional composite: A new clinical outcome measure for multiple sderosis trials. Mult. Scler. J. 2002, 8, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Brooks, J.B.B.; Giraud, V.O.; Saleh, Y.J.; Rodrigues, S.J.; Daia, L.A.; Fragoso, Y.D. Paced auditory serial addition test (PASAT): A very difficult test even for individuals with high intellectual capability. Arq. Neuro-Psiquiatria 2011, 69, 482–484. [Google Scholar] [CrossRef] [Green Version]
- Van Schependom, J.; D’Hooghe, M.B.; Cleynhens, K.; D’hooge, M.; Haelewyck, M.C.; De Keyser, J.; Nagels, G. The Symbol Digit Modalities Test as sentinel test for cognitive impairment in multiple sclerosis. Eur. J. Neurol. 2014, 21, 1219-e72. [Google Scholar] [CrossRef]
- Kalb, R.; Beier, M.; Benedict, R.H.; Charvet, L.; Costello, K.; Feinstein, A.; Gingold, J.; Goverover, Y.; Halper, J.; Harris, C.; et al. Recommendations for cognitive screening and management in multiple sclerosis care. Mult. Scler. J. 2018, 24, 1665–1680. [Google Scholar] [CrossRef] [Green Version]
- Strober, L.; DeLuca, J.; Benedict, R.H.; Jacobs, A.; Cohen, J.A.; Chiaravalloti, N.; Hudson, L.D.; Rudick, R.A.; LaRocca, N.G.; Multiple Sclerosis Outcome Assessments Consortium (MSOAC). Symbol Digit Modalities Test: A valid clinical trial endpoint for measuring cognition in multiple sclerosis. Mult. Scler. J. 2018, 25, 1781–1790. [Google Scholar] [CrossRef]
- Ontaneda, D.; LaRocca, N.; Coetzee, T.; Rudick, R. Revisiting the Multiple Sclerosis Functional Composite: Proceedings from the National Multiple Sclerosis Society (NMSS) Task Force on Clinical Disability Measures. Mult. Scler. J. 2012, 18, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- López-Góngora, M.; Querol, L.; Escartín, A. A one-year follow-up study of the Symbol Digit Modalities Test (SDMT) and the Paced Auditory Serial Addition Test (PASAT) in relapsing-remitting multiple sclerosis: An appraisal of comparative longitudinal sensitivity. BMC Neurol. 2015, 15, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Benedict, R.H.B.; Tomic, D.; Cree, B.A.; Fox, R.; Giovannoni, G.; Bar-Or, A.; Gold, R.; Vermersch, P.; Pohlmann, H.; Wright, I.; et al. Siponimod and cognition in secondary progressive multiple sclerosis: EXPAND secondary analyses. Neurology 2021, 96, e376–e386. [Google Scholar] [CrossRef] [PubMed]
- Benedict, R.H.; DeLuca, J.; Phillips, G.; LaRocca, N.; Hudson, L.D.; Rudick, R.; Multiple Sclerosis Outcome Assessments Consortium. Validity of the Symbol Digit Modalities Test as a cognition performance outcome measure for multiple sclerosis. Mult. Scler. J. 2017, 23, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Drake, A.; Weinstock-Guttman, B.; Morrow, S.A.; Hojnacki, D.; Munschauer, F.E.; Benedict, R.H.B. Psychometrics and normative data for the Multiple Sclerosis Functional Composite: Replacing the PASAT with the Symbol Digit Modalities Test. Mult. Scler. J. 2009, 16, 228–237. [Google Scholar] [CrossRef]
- Kiely, K.M.; Butterworth, P.; Watson, N.; Wooden, M. The Symbol Digit Modalities Test: Normative Data from a Large Nationally Representative Sample of Australians. Arch. Clin. Neuropsychol. 2014, 29, 767–775. [Google Scholar] [CrossRef]
- Roar, M.; Illes, Z.; Sejbaek, T. Practice effect in Symbol Digit Modalities Test in multiple sclerosis patients treated with natalizumab. Mult. Scler. Relat. Disord. 2016, 10, 116–122. [Google Scholar] [CrossRef]
- Galetta, K.M.; Balcer, L.J. Measures of visual pathway structure and function in MS: Clinical usefulness and role for MS trials. Mult. Scler. Relat. Disord. 2013, 2, 172–182. [Google Scholar] [CrossRef] [Green Version]
- Balcer, L.J.; Raynowska, J.; Nolan, R.; Galetta, S.L.; Kapoor, R.; Benedict, R.; Phillips, G.; LaRocca, N.; Hudson, L.; Rudick, R.; et al. Validity of low-contrast letter acuity as a visual performance outcome measure for multiple sclerosis. Mult. Scler. J. 2017, 23, 734–747. [Google Scholar] [CrossRef]
- Talman, L.S.; Bisker, E.R.; Sackel, D.J.; Long, D.A., Jr.; Galetta, K.M.; Ratchford, J.N.; Lile, D.J.; Farrell, S.K.; Loguidice, M.J.; Remington, G.; et al. Longitudinal study of vision and retinal nerve fiber layer thickness in multiple sclerosis. Ann. Neurol. 2010, 67, 749–760. [Google Scholar]
- Walter, S.D.; Ishikawa, H.; Galetta, K.M.; Sakai, R.E.; Feller, D.; Henderson, S.B.; Wilson, J.A.; Maguire, M.G.; Galetta, S.L.; Frohman, E.; et al. Ganglion Cell Loss in Relation to Visual Disability in Multiple Sclerosis. Ophthalmology 2012, 119, 1250–1257. [Google Scholar] [CrossRef] [Green Version]
- Reich, D.S.; Smith, S.A.; Gordon-Lipkin, E.; Ozturk, A.; Caffo, B.S.; Balcer, L.J.; Calabresi, P.A. Damage to the Optic Radiation in Multiple Sclerosis Is Associated With Retinal Injury and Visual Disability. Arch. Neurol. 2009, 66, 998–1006. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.A.; Cutter, G.R.; Fischer, J.S.; Goodman, A.D.; Heidenreich, F.R.; Kooijmans, M.F.; Sandrock, A.W.; Rudick, R.A.; Simon, J.H.; Simonian, N.A.; et al. Benefit of interferon beta-1a on MSFC progression in secondary progressive MS. Neurology 2002, 59, 679–687. [Google Scholar] [CrossRef]
- Balcer, L.J.; Baier, M.L.; Pelak, V.S.; Fox, R.J.; Shuwairi, S.; Galetta, S.L.; Cutter, G.R.; Maguire, M.G. New low-contrast vision charts: Reliability and test characteristics in patients with multiple sclerosis. Mult. Scler. 2000, 6, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Balcer, L.J.; Galetta, S.L.; Polman, C.H.; Eggenberger, E.; Calabresi, P.A.; Zhang, A.; Scanlon, J.V.; Hyde, R. Low-contrast acuity measures visual improvement in phase 3 trial of natalizumab in relapsing MS. J. Neurol. Sci. 2012, 318, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Beck, R.W.; Maguire, M.G.; Bressler, N.M.; Glassman, A.R.; Lindblad, A.S.; Ferris, F.L. Visual Acuity as an Outcome Measure in Clinical Trials of Retinal Diseases. Ophthalmology 2007, 114, 1804–1809. [Google Scholar] [CrossRef]
- Jakimovski, D.; Benedict, R.H.B.; Weinstock-Guttman, B.; Ozel, O.; Fuchs, T.A.; Lincoff, N.; Bergsland, N.; Dwyer, M.G.; Zivadinov, R. Visual deficits and cognitive assessment of multiple sclerosis: Confounder, correlate, or both? J. Neurol. 2021, 268, 2578–2588. [Google Scholar] [CrossRef] [PubMed]
- Balcer, L.J.; Miller, D.H.; Reingold, S.C.; Cohen, J.A. Vision and vision-related outcome measures in multiple sclerosis. Brain 2014, 138, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Sipe, J.C.; Knobler, R.L.; Braheny, S.L.; Rice, G.P.; Panitch, H.S.; Oldstone, M.B. A neurologic rating scale (NRS) for use in multiple sclerosis. Neurology 1984, 34, 1368. [Google Scholar] [CrossRef]
- Koziol, J.A.; Lucero, A.; Sipe, J.C.; Romine, J.S.; Beutler, E. Responsiveness of the Scripps neurologic rating scale during a multiple sclerosis clinical trial. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 1999, 26, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Sipe, J.C.; Romine, J.S.; Koziol, J.A.; McMillan, R.; Beutler, E.; Sipe, J.C.; Romine, J.S.; Zyroff, J. Cladribine in treatment of chronic progressive multiple sclerosis. Lancet 1994, 344, 9–13. [Google Scholar] [CrossRef]
- Eshaghi, A.; Prados, F.; Brownlee, W.J.; Altmann, D.R.; Tur, C.; Cardoso, M.J.; De Angelis, F.; van de Pavert, S.H.; Cawley, N.; De Stefano, N.; et al. Deep gray matter volume loss drives disability worsening in multiple sclerosis. Ann. Neurol. 2018, 83, 210–222. [Google Scholar] [CrossRef] [Green Version]
- De Stefano, N.; Stromillo, M.L.; Giorgio, A.; Bartolozzi, M.L.; Battaglini, M.; Baldini, M.; Portaccio, E.; Amato, M.P.; Sormani, M.P. Establishing pathological cut-offs of brain atrophy rates in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2016, 87, 93–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eijlers, A.J.; Dekker, I.; Steenwijk, M.D.; Meijer, K.A.; Hulst, H.E.; Pouwels, P.J.; Uitdehaag, B.M.; Barkhof, F.; Vrenken, H.; Schoonheim, M.M.; et al. Cortical atrophy accelerates as cognitive decline worsens in multiple sclerosis. Neurology 2019, 93, e1348–e1359. [Google Scholar] [CrossRef]
- Duning, T.; Kloska, S.; Steinstrater, O.; Kugel, H.; Heindel, W.; Knecht, S. Dehydration confounds the assessment of brain atrophy. Neurology 2005, 64, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Brown, R.A.; Narayanan, S.; Collins, D.L.; Arnold, D.L. Diurnal fluctuations in brain volume: Statistical analyses of MRI from large populations. NeuroImage 2015, 118, 126–132. [Google Scholar] [CrossRef]
- Enzinger, C.; Fazekas, F.; Matthews, P.M.; Ropele, S.; Schmidt, H.; Smith, S. Risk factors for progression of brain atrophy in aging: Six-year follow-up of normal subjects. Neurology 2005, 64, 1704–1711. [Google Scholar] [CrossRef]
- De Stefano, N.; Airas, L.; Grigoriadis, N.; Mattle, H.P.; O’Riordan, J.; Oreja-Guevara, C.; Sellebjerg, F.; Stankoff, B.; Walczak, A.; Wiendl, H.; et al. Clinical relevance of brain volume measures in multiple sclerosis. CNS Drugs 2014, 28, 147–156. [Google Scholar]
- Zivadinov, R.; Raj, B.; Ramanathan, M.; Teter, B.; Durfee, J.; Dwyer, M.; Bergsland, N.; Kolb, C.; Hojnacki, D.; Benedict, R.; et al. Autoimmune Comorbidities Are Associated with Brain Injury in Multiple Sclerosis. Am. J. Neuroradiol. 2016, 37, 1010–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zivadinov, R.; Reder, A.T.; Filippi, M.; Minagar, A.; Stuve, O.; Lassmann, H.; Racke, M.K.; Dwyer, M.; Frohman, E.M.; Khan, O. Mechanisms of action of disease-modifying agents and brain volume changes in multiple sclerosis. Neurology 2008, 71, 136–144. [Google Scholar] [CrossRef]
- Song, X.; Li, D.; Qiu, Z.; Su, S.; Wu, Y.; Wang, J.; Liu, Z.; Dong, H. Correlation between EDSS scores and cervical spinal cord atrophy at 3T MRI in multiple sclerosis: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2019, 37, 101426. [Google Scholar] [CrossRef]
- Casserly, C.; Seyman, E.; Alcaide-Leon, P.; Guenette, M.; Lyons, C.; Sankar, S.; Svendrovski, A.; Baral, S.; Oh, J. Spinal Cord Atrophy in Multiple Sclerosis: A Systematic Review and Meta-Analysis. J. Neuroimaging 2018, 28, 556–586. [Google Scholar] [CrossRef] [PubMed]
- Moccia, M.; Ruggieri, S.; Ianniello, A.; Toosy, A.; Pozzilli, C.; Ciccarelli, O. Advances in spinal cord imaging in multiple sclerosis. Ther. Adv. Neurol. Disord. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Rudick, R.; Antel, J.; Confavreux, C.; Cutter, G.; Ellison, G.; Fischer, J.; Lublin, F.; Miller, A.; Petkau, J.; Rao, S.; et al. Recommendations from the national multiple sclerosis society clinical outcomes assessment task force. Ann. Neurol. 1997, 42, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.S.; Rudick, R.A.; Cutter, G.R.; Reingold, S.C.; National MS Society Clinical Outcomes Assessment Task Force. The Multiple Sclerosis Functional Composite Measure (MSFC): An integrated approach to MS clinical outcome assessment. National MS Society Clinical Outcomes Assessment Task Force. Mult. Scler. J. 1999, 5, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Cadavid, D.; Cohen, J.A.; Freedman, M.S.; Goldman, M.D.; Hartung, H.-P.; Havrdova, E.K.; Jeffery, D.; Kapoor, R.; Miller, A.; Sellebjerg, F.; et al. The EDSS-Plus, an improved endpoint for disability progression in secondary progressive multiple sclerosis. Mult. Scler. J. 2016, 23, 94–105. [Google Scholar] [CrossRef]
- Inojosa, H.; Schriefer, D.; Ziemssen, T. Clinical outcome measures in multiple sclerosis: A review. Autoimmun. Rev. 2020, 19, 102512. [Google Scholar] [CrossRef]
- Bin Sawad, A.; Seoane-Vazquez, E.; Rodriguez-Monguio, R.; Turkistani, F. Evaluation of the Expanded Disability Status Scale and the Multiple Sclerosis Functional Composite as clinical endpoints in multiple sclerosis clinical trials: Quantitative meta-analyses. Curr. Med. Res. Opin. 2016, 32, 1969–1974. [Google Scholar] [CrossRef]
- Bosma, L.; Kragt, J.; Brieva, L.; Khaleeli, Z.; Montalban, X.; Polman, C.; Thompson, A.; Tintoré, M.; Uitdehaag, B.; Thompson, A. The search for responsive clinical endpoints in primary progressive multiple sclerosis. Mult. Scler. J. 2009, 15, 715–720. [Google Scholar] [CrossRef]
- Wolinsky, J.S.; Montalban, X.; Hauser, S.L.; Giovannoni, G.; Vermersch, P.; Bernasconi, C.; Deol-Bhullar, G.; Garren, H.; Chin, P.; Belachew, S.; et al. Evaluation of no evidence of progression or active disease (NEPAD) in patients with primary progressive multiple sclerosis in the ORATORIO trial. Ann. Neurol. 2018, 84, 527–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegen, H.; Bsteh, G.; Berger, T. ‘No evidence of disease activity’—Is it an appropriate surrogate in multiple sclerosis? Eur. J. Neurol. 2018, 25, 1107-e101. [Google Scholar] [CrossRef]
- Goldberg, R.; Gore, J.M.; Barton, B.; Gurwitz, J. Individual and Composite Study Endpoints: Separating the Wheat from the Chaff. Am. J. Med. 2014, 127, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Lorscheider, J.; Buzzard, K.; Jokubaitis, V.; Spelman, T.; Havrdova, E.; Horakova, D.; Trojano, M.; Izquierdo, G.; Girard, M.; Duquette, P.; et al. Defining secondary progressive multiple sclerosis. Brain 2016, 139, 2395–2405. [Google Scholar] [CrossRef] [Green Version]
- Krajnc, N.; Bsteh, G.; Berger, T. Clinical and Paraclinical Biomarkers and the Hitches to Assess Conversion to Secondary Progressive Multiple Sclerosis: A Systematic Review. Front. Neurol. 2021, 12, 666868. [Google Scholar] [CrossRef]
- Hohol, M.J.; Orav, E.J.; Weiner, H.L. Disease steps in multiple sclerosis: A longitudinal study comparing disease steps and EDSS to evaluate disease progression. Mult. Scler. J. 1999, 5, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Ravnborg, M.; Blinkenberg, M.; Sellebjerg, F.; Ballegaard, M.; Larsen, S.H.; Sørensen, P.S. Responsiveness of the Multiple Sclerosis Impairment Scale in comparison with the Expanded Disability Status Scale. Mult. Scler. J. 2005, 11, 81–84. [Google Scholar] [CrossRef] [PubMed]
- University of California SFMSET; Cree BAC; Hollenbach, J.A.; Bove, R.; Kirkish, G.; Sacco, S.; Caverzasi, E.; Bischof, A.; Gundel, T.; Zhu, A.H.; et al. Silent progression in disease activity-free relapsing multiple sclerosis. Ann. Neurol. 2019, 85, 653–666. [Google Scholar]
- Benedict, R.H.; Pol, J.; Yasin, F.; Hojnacki, D.; Kolb, C.; Eckert, S.; Tacca, B.; Drake, A.; Wojcik, C.; Morrow, S.A.; et al. Recovery of cognitive function after relapse in multiple sclerosis. Mult. Scler. J. 2020, 27, 71–78. [Google Scholar] [CrossRef]
- Kalinowski, A.; Cutter, G.; Bozinov, N.; Hinman, J.A.; Hittle, M.; Motl, R.; Odden, M.; Nelson, L.M. The timed 25-foot walk in a large cohort of multiple sclerosis patients. Mult. Scler. J. 2021, 13524585211017013. [Google Scholar] [CrossRef]
- Goldman, M.D.; Motl, R.W.; Scagnelli, J.; Pula, J.H.; Sosnoff, J.J.; Cadavid, D. Clinically meaningful performance benchmarks in MS: Timed 25-Foot Walk and the real world. Neurology 2013, 81, 1856–1863. [Google Scholar] [CrossRef]
- Ferraro, D.; Guicciardi, C.; DE Biasi, S.; Pinti, M.; Bedin, R.; Camera, V.; Vitetta, F.; Nasi, M.; Meletti, S.; Sola, P. Plasma neurofilaments correlate with disability in progressive multiple sclerosis patients. Acta Neurol. Scand. 2019, 141, 16–21. [Google Scholar] [CrossRef]
- Ebers, G.C.; Heigenhauser, L.; Daumer, M.; Lederer, C.; Noseworthy, J.H. Disability as an outcome in MS clinical trials. Neurology 2008, 71, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.W.; Mostert, J.; Repovic, P.; Bowen, J.D.; Uitdehaag, B.; Cutter, G. Reliability of Outcome Measures in Clinical Trials in Secondary Progressive Multiple Sclerosis. Neurology 2020, 96, e111–e120. [Google Scholar] [CrossRef]
- Kalincik, T.; Sormani, M.P.; Tur, C. Has the Time Come to Revisit Our Standard Measures of Disability Progression in Multiple Sclerosis? Neurology 2021, 96, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Tourbah, A.; Lebrun-Frenay, C.; Edan, G.; Clanet, M.; Papeix, C.; Vukusic, S.; De Sèze, J.; Debouverie, M.; Gout, O.; Clavelou, P.; et al. MD1003 (high-dose biotin) for the treatment of progressive multiple sclerosis: A randomised, double-blind, placebo-controlled study. Mult. Scler. J. 2016, 22, 1719–1731. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Blumhardt, L.D. Disability outcome measures in therapeutic trials of relapsing-remitting multiple sclerosis: Effects of heterogeneity of disease course in placebo cohorts. J. Neurol. Neurosurg. Psychiatry 2000, 68, 450–457. [Google Scholar] [CrossRef] [Green Version]
- Kalincik, T.; Cutter, G.; Spelman, T.; Jokubaitis, V.; Havrdova, E.K.; Horáková, D.; Trojano, M.; Izquierdo, G.; Girard, M.; Duquette, P.; et al. Defining reliable disability outcomes in multiple sclerosis. Brain 2015, 138, 3287–3298. [Google Scholar] [CrossRef] [PubMed]
- Ontaneda, D.; Cohen, J.A.; Amato, M.P. Clinical outcome measures for progressive MS trials. Mult. Scler. J. 2017, 23, 1627–1635. [Google Scholar] [CrossRef] [Green Version]
- Kappos, L.; Wolinsky, J.S.; Giovannoni, G.; Arnold, D.L.; Wang, Q.; Bernasconi, C.; Model, F.; Koendgen, H.; Manfrini, M.; Belachew, S.; et al. Contribution of Relapse-Independent Progression vs Relapse-Associated Worsening to Overall Confirmed Disability Accumulation in Typical Relapsing Multiple Sclerosis in a Pooled Analysis of 2 Randomized Clinical Trials. JAMA Neurol. 2020, 77, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Saidha, S.; Al-Louzi, O.; Ratchford, J.N.; Bhargava, P.; Oh, J.; Newsome, S.D.; Prince, J.L.; Pham, D.; Roy, S.; Van Zijl, P.; et al. Optical coherence tomography reflects brain atrophy in multiple sclerosis: A four-year study. Ann. Neurol. 2015, 78, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Saidha, S.; Sotirchos, E.S.; Oh, J.; Syc, S.B.; Seigo, M.A.; Shiee, N.; Eckstein, C.; Durbin, M.K.; Oakley, J.D.; Meyer, S.A.; et al. Relationships between retinal axonal and neuronal measures and global central nervous system pathology in multiple sclerosis. JAMA Neurol. 2013, 70, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Bsteh, G.; Hegen, H.; Teuchner, B.; Amprosi, M.; Berek, K.; Ladstätter, F.; Wurth, S.; Auer, M.; Di Pauli, F.; Deisenhammer, F.; et al. Peripapillary retinal nerve fibre layer as measured by optical coherence tomography is a prognostic biomarker not only for physical but also for cognitive disability progression in multiple sclerosis. Mult. Scler. J. 2017, 25, 196–203. [Google Scholar] [CrossRef]
- Martínez-Lapiscina, E.H.; Arnow, S.; Wilson, J.A.; Saidha, S.; Preiningerova, J.L.; Oberwahrenbrock, T.; Brandt, A.U.; Pablo, L.E.; Guerrieri, S.; González-Suárez, I.; et al. Retinal thickness measured with optical coherence tomography and risk of disability worsening in multiple sclerosis: A cohort study. Lancet Neurol. 2016, 15, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Bsteh, G.; Hegen, H.; Teuchner, B.; Berek, K.; Wurth, S.; Auer, M.; Di Pauli, F.; Deisenhammer, F.; Berger, T. Peripapillary retinal nerve fibre layer thinning rate as a biomarker discriminating stable and progressing relapsing–remitting multiple sclerosis. Eur. J. Neurol. 2019, 26, 865–871. [Google Scholar] [CrossRef]
- Bsteh, G.; Berek, K.; Hegen, H.; Altmann, P.; Wurth, S.; Auer, M.; Zinganell, A.; Di Pauli, F.; Rommer, P.; Leutmezer, F.; et al. Macular ganglion cell–inner plexiform layer thinning as a biomarker of disability progression in relapsing multiple sclerosis. Mult. Scler. J. 2020, 27, 684–694. [Google Scholar] [CrossRef]
- Bowd, C.; Zangwill, L.M.; Weinreb, R.N.; Medeiros, F.A.; Belghith, A. Estimating Optical Coherence Tomography Structural Measurement Floors to Improve Detection of Progression in Advanced Glaucoma. Am. J. Ophthalmol. 2016, 175, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Disanto, G.; Barro, C.; Benkert, P.; Naegelin, Y.; Schädelin, S.; Giardiello, A.; Zecca, C.; Blennow, K.; Zetterberg, H.; Leppert, D.; et al. Serum Neurofilament light: A biomarker of neuronal damage in multiple sclerosis. Ann. Neurol. 2017, 81, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Novakova, L.; Zetterberg, H.; Sundström, P.; Axelsson, M.; Khademi, M.; Gunnarsson, M.; Malmeström, C.; Svenningsson, A.; Olsson, T.; Piehl, F.; et al. Monitoring disease activity in multiple sclerosis using serum neurofilament light protein. Neurology 2017, 89, 2230–2237. [Google Scholar] [CrossRef] [Green Version]
- Damasceno, A.; Dias-Carneiro, R.; Moraes, A.; Boldrini, V.O.; Quintiliano, R.P.S.; Silva, V.A.D.P.G.D.; Farias, A.; Brandão, C.O.; Damasceno, B.P.; dos Santos, L.M.B.; et al. Clinical and MRI correlates of CSF neurofilament light chain levels in relapsing and progressive MS. Mult. Scler. Relat. Disord. 2019, 30, 149–153. [Google Scholar] [CrossRef]
- Barro, C.; Benkert, P.; Disanto, G.; Tsagkas, C.; Amann, M.; Naegelin, Y.; Leppert, D.; Gobbi, C.; Granziera, C.; Yaldizli, Ö.; et al. Serum neurofilament as a predictor of disease worsening and brain and spinal cord atrophy in multiple sclerosis. Brain 2018, 141, 2382–2391. [Google Scholar] [CrossRef]
- Kuhle, J.; Kropshofer, H.; Haering, D.A.; Kundu, U.; Meinert, R.; Barro, C.; Dahlke, F.; Tomic, D.; Leppert, D.; Kappos, L. Blood neurofilament light chain as a biomarker of MS disease activity and treatment response. Neurology 2019, 92, e1007–e1015. [Google Scholar] [CrossRef]
- Sellebjerg, F.; Royen, L.; Sørensen, P.S.; Oturai, A.B.; Jensen, P.E.H. Prognostic value of cerebrospinal fluid neurofilament light chain and chitinase-3-like-1 in newly diagnosed patients with multiple sclerosis. Mult. Scler. J. 2018, 25, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Smith, K.E.; Allegretta, M.; Arnold, D.L.; Carroll, W.; Comabella, M.; Furlan, R.; Harp, C.; Kuhle, J.; Leppert, D.; et al. Serum neurofilament light as a biomarker in progressive multiple sclerosis. Neurology 2020, 95, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Altmann, D.R.; Jasperse, B.; Barkhof, F.; Beckmann, K.; Filippi, M.; Kappos, L.D.; Molyneux, P.; Polman, C.H.; Pozzilli, C.; Thompson, A.; et al. Sample sizes for brain atrophy outcomes in trials for secondary progressive multiple sclerosis. Neurology 2008, 72, 595–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delcoigne, B.; Manouchehrinia, A.; Barro, C.; Benkert, P.; Michalak, Z.; Kappos, L.; Leppert, D.; Tsai, J.A.; Plavina, T.; Kieseier, B.C.; et al. Blood neurofilament light levels segregate treatment effects in multiple sclerosis. Neurology 2020, 94, e1201–e1212. [Google Scholar] [CrossRef] [Green Version]
- Kosa, P.; Ghazali, D.; Tanigawa, M.; Barbour, C.; Cortese, I.; Kelley, W.; Snyder, B.; Ohayon, J.; Fenton, K.; Lehky, T.; et al. Development of a Sensitive Outcome for Economical Drug Screening for Progressive Multiple Sclerosis Treatment. Front. Neurol. 2016, 7, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, V.; Sharma, H.; Afroz, N.; Callan, A.; Medin, J. Patient-reported outcomes in multiple sclerosis: A systematic comparison of available measures. Eur. J. Neurol. 2017, 24, 1099–1107. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Study Name | Study Design | Inclusion Criteria | Intervention and Comparator | Study Duration | Primary Outcome Measure | Definition of Progression | Sustained Progression | Secondary Outcome Measures |
|---|---|---|---|---|---|---|---|---|
| Primary progressive multiple sclerosis | ||||||||
| ORATORIO | Randomized, double-blind, placebo-controlled | Age 18–55 years EDSS 3.0–6.5 with pyramidal FS ≥ 2 Duration of MS symptoms of <15 years (EDSS > 5.0) or <10 years (EDSS ≤ 5.0) OCB in the CSF | Ocrelizumab (n = 488) vs. placebo (n = 244) | 120 weeks | EDSS | Increase in EDSS score for ≥1.0 point (EDSS ≤ 5.5) or ≥0.5 point (EDSS ≥ 6.0) | 12 weeks | 24 week CDP, change in T25FW from baseline to week 120 MRI: change in the total volume of brain lesions (T2-weighted) at week 120, change in whole brain volume from week 24 to week 120 PROMs: change in the SF-36 Physical component |
| INFORMS | Randomized, double-blind, placebo-controlled | Age 25–65 years EDSS 3.5–6.0 with pyramidal FS ≥ 2 Increase in EDSS for ≥0.5 point in the past 2 years T25FW < 30 s Disease duration 2–10 years | Fingolimod (n = 336) vs. placebo (n = 487) | 36 months | EDSS-Plus | Increase in EDSS score for ≥1.0 point (EDSS ≤ 5.0) or ≥0.5 point (EDSS ≥ 5.5) or increase of ≥20% in the T25FW or 9HPT | 3 months | Time to 3 month CDP MRI: brain volume change, number of Gd-positive lesions, number of T2 lesions PROMs: PRIMUS, EQ-5D, UFIS, MSWS-12 |
| OLYMPUS | Randomized, double-blind, placebo-controlled | Age 18–65 years EDSS 2.0–6.5 with pyramidal FS ≥ 2 OCB in CSF Disease duration ≥1 year | Rituximab (n = 292) vs. placebo (n = 147) | 96 weeks | EDSS | Increase in EDSS score for ≥1.0 point (EDSS ≤ 5.5) or ≥0.5 point (EDSS ≥ 6.0) | 12 weeks | Time to 24 week CDP, change in MSFC Pharmacodynamic assessments of B-cells counts and immunoglobulin levels MRI: number of Gd-enhancing lesions, change in volume of T2 lesions, change in brain volume |
| PROMiSE | Randomized, double-blind, placebo-controlled | Age 30–65 years EDSS 3.0–6.5 with pyramidal FS ≥ 2 Evidence of myelopathy for at least 6 months before the screening | Glatiramer acetate (n = 627) vs. placebo (n = 316) | 36 months | EDSS | Increase in EDSS score for ≥1.0 point (EDSS ≤ 5.0) or ≥0.5 point (EDSS ≥ 5.5) | 3 months | Proportion of progression-free patients, change in EDSS and MSFC MRI: number and volume of T2 lesions, number of Gd-enhancing lesions, brain volume loss |
| Secondary progressive multiple sclerosis | ||||||||
| SPECTRIMS | Randomized, double-blind, placebo-controlled | Age 18–55 years EDSS 3.0–6.5 ≥2 relapses or increase in EDSS for ≥1.0 point in the previous 2 years | Interferon 1a (n = 360) vs. placebo (n = 358) | 36 months | EDSS | Increase in EDSS score for ≥1.0 point (EDSS ≤ 5.5) or ≥0.5 point (EDSS ≥ 6.0) | 3 months | Time to becoming wheelchair-bound (EDSS ≥ 7.0), ARR, time to first relapse, proportion of patients with moderate/severe relapses MRI: volume of T2 lesions, number of newly active lesions, change in brain volume |
| Interferon 1a study | Randomized, double-blind, placebo-controlled | Age 18–55 years EDSS 1.0–3.5 ≥2 relapses in the prior 3 years No relapse up to 2 months before randomization Disease duration ≥1 year | Interferon 1a (n = 158) vs. placebo (n = 143) | 104 weeks | EDSS | Increase in EDSS score for ≥1.0 point | 6 months | Number of relapses MRI: number and volume of Gd-enhancing lesions, T2 lesion volume |
| IMPACT | Randomized, double-blind, placebo-controlled | Age 18–60 years EDSS 3.5–6.5 | Interferon β 1a (n = 217) vs. placebo (n = 219) | 24 months | MSFC | Decrease in the mean and median MSFC Z-scores | 3 months | Change in EDSS, ARR MRI: number of new or enlarging T2 lesions and number of Gd-enhancing lesions PROMs: BDI, MSQLI |
| Cladribine study | Randomized, double-blind, placebo-controlled | Clinically definite secondary progressive MS for ≥2 years | Cladribine (n = 24) vs. placebo (n = 24) | 2 years | EDSS, SNRS | The change in both measurements was calculated every month1 | MRI: number and volume of T2-lesions CSF: total protein and immunoglobulin concentration, oligoclonal bands | |
| MIMS | Randomized, double-blind, placebo-controlled | Age 18–55 years EDSS 3.0–6.0 Increase in EDSS for ≥1.0 point during 18 months before enrolment (progressive relapsing-remitting or secondary progressive MS) No relapse up to 8 weeks before randomization | Mitoxantrone (n = 124) vs. placebo (n = 64) | 24 months | Composite score2 | Only a change between baseline and 24 months in each measurement of the composite score was calculated | Proportion of patients with deterioration of ≥1.0 EDSS point, proportion of patients with such deterioration confirmed after 3 and 6 months, time to first EDSS deterioration, time to first relapse, ARR, proportion of patients without relapse, number of days in hospital, use of wheelchair assistance MRI: number and volume of Gd-enhancing, and nonenhancing T1 and T2 lesions PROMs: quality of life (Stanford health assessment questionnaire) | |
| ASCEND | Randomized, double-blind, placebo-controlled | Age 18–58 years EDSS 3.0–6.5 with disability progression during the year before enrolment MSSS ≥ 4 No relapse up to 3 months before randomization | Natalizumab (n = 439) vs. placebo (n = 448) | 96 weeks | EDSS-Plus | Increase in EDSS score for ≥1.0 point (EDSS ≤ 5.5) or ≥0.5 point (EDSS ≥ 6.0) or increase of ≥20% in the T25FW or 9HPT | 3 months | Proportion of patients with improvement in T25FW, proportion of patients with disability progression, measured by individual physical EDSS FS scores MRI: change in whole brain volume between week 24 and 96 PROMs: change in patient-reported ambulatory status (MSWS-12), manual ability (ABILHAND questionnaire), quality of life (MSIS-29 physical score)3 |
| EXPAND | Randomized, double-blind, placebo-controlled | Age 18–60 years EDSS 3.0–6.5 progression during 2 years before enrolment No relapse up to 3 months before randomization | Siponimod (n = 1105) vs. placebo (n = 546) | Protocol amended after occurrence of 374 CDP events and after ≥95% of patients had been assigned to treatment for ≥12 months | EDSS | Increase in EDSS score for ≥1.0 points (EDSS ≤ 5.0) or increase of ≥0.5 (EDSS ≥ 5.5) | 3 months | Time to 3 and 6 month confirmed worsening of T25FW, ARR, time to first relapse, proportion of relapse-free patients MRI: number of new or enlarging T2 lesions, change in T2 lesion volume, number of Gd-enhancing lesions, brain volume change PROMs: change in score on the MSWS-12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krajnc, N.; Berger, T.; Bsteh, G. Measuring Treatment Response in Progressive Multiple Sclerosis—Considerations for Adapting to an Era of Multiple Treatment Options. Biomolecules 2021, 11, 1342. https://doi.org/10.3390/biom11091342
Krajnc N, Berger T, Bsteh G. Measuring Treatment Response in Progressive Multiple Sclerosis—Considerations for Adapting to an Era of Multiple Treatment Options. Biomolecules. 2021; 11(9):1342. https://doi.org/10.3390/biom11091342
Chicago/Turabian StyleKrajnc, Nik, Thomas Berger, and Gabriel Bsteh. 2021. "Measuring Treatment Response in Progressive Multiple Sclerosis—Considerations for Adapting to an Era of Multiple Treatment Options" Biomolecules 11, no. 9: 1342. https://doi.org/10.3390/biom11091342
APA StyleKrajnc, N., Berger, T., & Bsteh, G. (2021). Measuring Treatment Response in Progressive Multiple Sclerosis—Considerations for Adapting to an Era of Multiple Treatment Options. Biomolecules, 11(9), 1342. https://doi.org/10.3390/biom11091342