

Common Mechanism of Activated Catalysis in P-loop Fold Nucleoside Triphosphatases—United in Diversity
 , and
, and 
Abstract
:
1. Introduction

- (1)
- In most analysed structures, a short H-bond (<2.7 Å) connects AspWB and [Ser/Thr]K+1; this bond is extremely short (2.4–2.5 Å) in those structures that contain NDP:AlF4− as a TS analogue; see Figure 1E,F.
- (2)
- In TS-like structures of P-loop NTPases of all classes, except those of the TRAFAC class, the Wcat-coordinating “catalytic” Glu or Asp residue provides a proton pathway from Wcat to the nearest ligand of Mg2+.
- (3)
- The distance between neighboring ligands in the coordination shell of Mg2+ is 2.9–3.0 Å, which implies the possibility of proton exchange between all of them.
- (i)
- The twisting γ-phosphate by stimulator(s) should affect the properties of Mg2+ ligands including [S/T]K+1; we suggest that the functional pKa of [S/T]K+1 becomes lower than that of AspWB, and the proton relocates from [S/T]K+1 to AspWB.
- (ii)
- The remaining proton vacancy at the anionic [Ser/Thr]K+1 alkoxide is refilled by a proton that comes from Wcat (or a sugar moiety in some kinases), after which the nascent nucleophilic anion attacks γ-phosphate.
- (iii)
2. Materials and Methods
3. Results
3.1. Generic Designation of Structure Elements in P-loop Fold NTPases
3.2. Catalytically Relevant Amino Acids, Stimulatory Patterns, and Activation Mechanisms in Different Classes of P-loop NTPases
3.2.1. Coordination of the Mg-triphosphate Moiety

3.2.2. Variability of Catalytically Relevant Amino Acids, Activating Partners, Stimulatory Patterns, and Coordination of Wcat




3.2.3. Interim Summary on the Common Structural Traits of P-loop NTPases
- (i)
- In all inspected structures for which TS-like structures are available, some of the amino acid residues that immediately follow AspWB (at the D+1–D+5 positions of the WB-crest) are involved in catalytic interactions with Wcat (Figure 1, Figure 2, Figure 3, Figure 4, Figure 5 and Figure 6 and Figure S5, Table 1 and Table S1).
- (ii)
- (iii)
3.2.4. Water Molecules in the Vicinity of the Mg2+ Coordination Shell

3.3. Global Structural Analysis of Hydrogen Bonding between the Walker A and Walker B Motifs in the Whole Set of P-loop NTPases with Bound Mg-NTP Complexes or Their Analogs
3.3.1. The H-Bond between AspWB and [Ser/Thr]K+1 Is Shorter in the Presence of Transition State Analogs

3.3.2. The TS-Analog AlF4− Makes More Bonds within the Catalytic Site than MgF3− or AlF3
4. Discussion
4.1. Structure Comparison of P-loop NTPases
4.1.1. Constriction of the Catalytic Site in the Transition State
4.1.2. Coordination of Wcat and Auxiliary Interactions
4.1.3. Summary on Novel Common Structural Traits of P-loop NTPases
- (i)
- (ii)
- In agreement with previous data (see, e.g., [39,159]), the activating partner usually binds to the P-loop domain by making new H-bonds and salt bridges with the residues of the WB-crest; see Figure 1C,D and Figure S5. In all inspected structures for which TS-like structures are available, some of the amino acid residues that immediately follow AspWB (at the D+1–D+5 positions of the WB-crest) are involved in catalytic interactions with Wcat (Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure S5, Table 1 and Table S1). The energy of binding of the activation partner seems to be used for pushing Wcat towards the PG atom of γ-phosphate, constricting the catalytic site, and inserting the stimulatory moiety (see Supplementary File S2 for a detailed consideration).
- (iii)
- In the companion article [30], we provide evidence that the common trait of all inspected stimulators is their mechanistic interaction with the oxygen atom(s) of γ-phosphate, which may cause its rotation by 30–40°.
- (iv)
- Comparing the structures with bound analogues of ATP/GTP and TS, respectively, we noticed that the binding of ADP:AlF4− as a TS-analogue results in greater constriction of catalytic sites than the binding of ATP or GTP. The constriction manifests itself in the shorter distances between AspWB and [Ser/Thr]K+1, which are as short as 2.5 Å on average (Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6 and Figure 8A). The distances between HNK-3 and the analogues of γ-phosphate are also shorter in the structures with ADP:AlF4− bound; see Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6 and the companion article [30].
- (v)
4.2. ATP and GTP Hydrolysis by P-loop NTPases
4.2.1. Background on the Catalysis by P-loop NTPases
- (1)
- (2)
- (3)
- (4)
- (5)
4.2.2. Catalytic Factors in P-loop NTPases: Destabilization of the O3B–PG Bond
4.2.3. Catalytic Factors in P-loop NTPases: Planarization of γ-phosphate
4.2.4. Catalytic Factors in P-loop NTPases: Electrostatic Compensation of the Negative Charge of the Phosphate Groups
4.2.5. Catalytic Factors in P-loop NTPases: Does the [Ser/Thr]K+1–AspWB Pair Accept a Proton from Wcat?

- (1)
- We discovered that the “catalytic” Glu/Asp residues connect Wcat with ligands of Mg2+ in positions #3 or #6 in TS-like structures of P-loop NTPases of various classes (except the TRAFAC class); see Figure 1F, Figure 4, Figure 5, Figure 6 and Figure 7A. Notably, the six ligands of Mg2+ form a regular octahedron with edges 2.9–3.0 Å long, so that the ligands #3 and #6 are on a H+-transfer distance from [Ser/Thr]K+1 (Figure 10). The short H-bond between [Ser/Thr]K+1 and AspWB completes the proton-conducting pathway that connects Wcat with AspWB in all families of P-loop NTPases except the TRAFAC class. The proton pathways from Wcat to AspWB, which resemble proton translocation systems of PRC and BR (cf Figure 9), are shown by the red dashed lines for P-loop NTPases of different classes in Figure 4, Figure 5, Figure 6 and Figure 7 and by dashed arrows in Figure 10.
- (2)
- Mutations of AspWB to Asn, while retarding dramatically the activated hydrolysis, had no effect on the NTP binding [185,189,190,191,192,193]. In the case of E. coli F1-ATPase, the mutation even increased the affinity for ATP [191]. The AspWB to Asn mutation mimics the charge state of a protonated AspWB. Hence, the protonation of AspWB is unlikely to distort the catalytic pocket and be the cause of universal catalytic incompetence of the AspWB to Asn mutants. We attribute this incompetence to the inability of AsnWB to trap a proton from [Ser/Thr]K+1.Figure 10. Schematic presentation of tentative proton routes along the edges of the octahedral coordination shell of Mg2+ ion. The Mg2+ ligand #3 is assumed to be a water molecule. Proton entry points via ligand #6 or ligand #3 are shown with red and orange arrows, respectively. Protonic connection between [S/T]K+1 and AspWB are shown by magenta and light blue arrows, the route from [S/T]K+1 to O2B of β-phosphate is shown as a dark blue arrow. The movement of the O1G atom as a result of γ-phosphate twist is shown by purple arrows.Figure 10. Schematic presentation of tentative proton routes along the edges of the octahedral coordination shell of Mg2+ ion. The Mg2+ ligand #3 is assumed to be a water molecule. Proton entry points via ligand #6 or ligand #3 are shown with red and orange arrows, respectively. Protonic connection between [S/T]K+1 and AspWB are shown by magenta and light blue arrows, the route from [S/T]K+1 to O2B of β-phosphate is shown as a dark blue arrow. The movement of the O1G atom as a result of γ-phosphate twist is shown by purple arrows.
![Biomolecules 12 01346 g010]()
- (3)
- The pKa of an aspartate residue in water is about 4.0, much lower than that of water (14.0). However, unlike the “catalytic” Glu/Asp residues or γ-phosphate surrounded by charged residues (Figure 1E,F, Figure 3, Figure 4, Figure 5 and Figure 6), AspWB is in a nonpolar environment and its functional pK is likely to be high when the catalytic site is closed. AspWB is in the middle of an αβα sandwich, on the interface between the β-pleated sheet and the α1-helix; such interfaces are stabilized by hydrophobic interactions [169]. In addition, AspWB is preceded by four hydrophobic residues of the Walker B motif (Figure 1); the adjacent β strands, as well as the α1-helix, also contain many hydrophobic residues; see the sequence alignments in [9,10,12,188].Our data show that the relative SASA of AspWB drops below 6% in the presence of TS-analogues (Figure 8B). Upon constriction of the catalytic pocket and expulsion of eventually present water molecules, the hydrophobic environment should elevate the proton affinity of the H-bonded AspWB, as it happens with similarly H-bonded Asp96, which has a functional pKa of ~12.0 in a hydrophobic environment of the ground-state BR. Figure 9C shows that the structure of the Ser186K+1–Asp454WB pair of myosin overlaps nicely with the Thr46–Asp96 pair of BR.
- (4)
- Unfortunately, we are not aware of non-commercial software for reliable calculating the absolute pKa values in proteins. Hence, we used the PROPKA web server at https://www.ddl.unimi.it/vegaol/propka.htm (accessed on 14 August 2022). The server uses the PROPKA v. 2.0 half-empiric software that allows to assess pKa changes in response to ligand binding in the catalytic center [66].We applied PROPKA to the bovine ATP synthase, an extremely well-studied enzyme with a plethora of structures available [251]. The three catalytic centers of this ring-shaped enzyme work in turn, according to the so-called binding change mechanism [252]. Therefore, the catalytic pockets are usually open to varying extents. Using mixtures of nucleotides and their analogues, Walker and co-workers managed to obtain several structures where different centers in the same structure are as if in different stages of the catalytic cycle [253,254,255].We calculated the changes in pKa values of Asp256WB and the catalytic Glu188 (see Figure 6A) in response to opening/closing of the catalytic pockets and in the presence of different ligands. The values given in Supplementary Table S3 show that the estimated pKa of Asp256WB varies from around 9.0 (closed or constricted site with a nucleotide bound) to around 2.5 (empty site or a site with Pi bound). Interestingly, an intermediate pKa value of around 5.5 was obtained for a site containing ADP and Pi, which is believed to be half-opened [43]. Hence, the pKa of AspWB can increase by 7 units upon closing of the catalytic pocket. The estimated pKa values of Glu188 are much lower, usually around 5.0 when a nucleotide is bound (Table S3).
- (5)
- In contrast, the pK of [Ser/Thr]K+1, which is about 13.0 in water, is likely to be reduced when [Ser/Thr]K+1 serves as a Mg2+ ligand. Coordination of a Zn2+ ion by a serine side chain is known to decrease the pK value of the latter up to 5.5 yielding a serine anion (alkoxide) at neutral pH; see [52] and the references therein. The impact of a Mg2+ ion should be weaker; still, within a closed/constricted catalytic site, the low dielectric permittivity would enhance electrostatic interactions. [Ser/Thr]K+1 is the most deeply buried of the Mg2+ ligands (see Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6 and Figure 10), and thus it should be the most sensitive to electrostatic effects. As a result, the functional pK value of [Ser/Thr]K+1 may dramatically decrease upon constriction of the catalytic site.
- (6)
- The shortening of H-bonds found in the TS-like structures (Figure 8A) enables estimation of the difference in functional pK values of [Ser/Thr]K+1 and AspWB in the TS. It is well established, based on ample experimental evidence, that hydrogen bonds “generally shorten as ΔpKa, the difference in the donor and acceptor pKa values, decreases” (quoted from [142]). Specifically, Herschlag and colleagues observed, on various systems, that the ΔpKa decreases linearly from 20 to 0 with the decrease in the O—H••••O distance from 2.9 Å to 2.4 Å, with a slope of 0.02 Å/pKa unit, [142,256]. In NDP:AlF4--containing structures with constricted catalytic site, the length of the H-bonds between [Ser/Thr]K+1 and AspWB varies around 2.5 Å (Figure 1E,F, Figure 3, Figure 4A,B, Figure 5C,D, Figure 6A and Figure 8A), which corresponds to ΔpK < 3.0 and indicates a low-barrier hydrogen bond [142,256]. Hence, [Ser/Thr]K+1 and AspWB may have comparably high proton affinities in a constricted catalytic site.
- (7)
- Limbach and colleagues combined low-temperature UV-Vis and 1H/13C NMR spectroscopy (UVNMR) to study the effect of solvent polarity on the proton equilibrium between phenols and carboxylic acids [257,258,259], which system can be viewed as a model of the [Ser/Thr]K+1—AspWB H-bonded pair. These authors have shown that proton relocates from the hydroxy group to the carboxyl with decrease in polarity.
- (8)
- In the octahedral coordination shell of Mg2+, the O1G atom of γ-phosphate is the ligand opposite to [Ser/Thr]K+1 (Figure 10). Therefore, the stimulator-induced rotation of γ-phosphate, by moving O1G in any direction (as shown by dashed purple arrows in Figure 10), would inevitably increase the distance between O1G and the hydroxyl of [Ser/Thr]K+1. Pulling away the negatively charged O1G will increase the cumulative positive charge at [Ser/Thr]K+1 prompting the relocation of its proton to AspWB, e.g., in response to a thermal fluctuation [259] (Figure 11A).
- (9)
- We suggest that the resulting Mg2+-coordinated Ser/Thr anion (alkoxide), used as a proton acceptor from water by many enzymes [52,260,261,262], withdraws the proton from Wcat (or the sugar moiety in some kinases) via proton pathways shown in Figure 4, Figure 5, Figure 6, Figure 7A, Figure 10 and Figure 11B. This proton transfer should be additionally driven by strong local electric field (see Supplementary File S3 on the uneven electric field distribution in the catalytic sites of P-loop NTPases). The resulting state where both [Ser/Thr]K+1 and AspWB are protonated corresponds to the ground state of the Thr46-Asp96 pair in the BR (see the light-green structures in Figure 9B,C).
- (10)
- The formed anionic nucleophile (e.g., OH−cat), while stabilized and polarized by its ligands, is attracted by the electrophilic PG atom (Figure 11B). The proton affinity of the anionic nucleophile decreases as it gets closer to PG, so that proton return from the [Ser/Thr]K+1—AspWB couple becomes increasingly unfavorable, eventually satisfying the Eigen’s condition for proton transfer and making it complete.


4.2.6. Catalytic Factors in P-loop NTPases: Charge Compensation at β-phosphate
4.2.7. Evidence for Transient Protonation of AspWB from Infrared Spectroscopy Data
4.2.8. Re-Assignment of Functions in and around the Walker A and Walker B Motifs
4.2.9. Minimal Mechanistic Model of NTP Hydrolysis by P-loop NTPases

- (A)
- A Mg-NTP complex binds to the Walker A and Walker B motifs; the binding energy is used to bring the NTP molecule into an elongated conformation with eclipsed β- and γ-phosphates and to surround the triphosphate chain by positively charged groups that are provided by the P-loop, WB-crest and, only in TRAFAC NTPases, Switch I loop. The accompanying protein conformational changes can power useful mechanical work. The catalytic site is further stabilized by the H-bond between AspWB and [Ser/Thr]K+1. The H-bond length is about 2.6–2.7 Å (Figure 8A); in this state AspWB is negatively charged.
- (B)
- An exergonic interaction between the activating partner (another protein domain and/or an RNA/DNA molecule) and the WB-crest (i) shields and constricts the catalytic site, (ii) moves the WB-crest residues closer to the γ-phosphate, and (iii) inserts the stimulator(s) next to the phosphate chain. The constriction of the catalytic site shortens the H-bond between [Ser/Thr]K+1 and AspWB to 2.4–2.5 Å, turning AspWB into a potent proton trap. In most cases, (one of) the stimulator(s) links the O2A and O3G atoms of the triphosphate (Figure 1E,F, Figure 3B–D, Figure 4A–D, Figure 5A–C and Figure 6A) and twists γ-phosphate counter clockwise; the rotated γ-phosphate is stabilized by a new H-bond between O2G and HNK-3.In other cases, the stimulators drag only γ-phosphate and, supposedly, twists it in some direction; see Figure 3A, Figure 5D and Figure 6B–D. The interaction of stimulators with γ-phosphate (i) increases the electrophilicity of the PG atom, (ii) weakens the O3B–PG bond, (iii) promotes the transition of γ-phosphate to a more planar conformation, and (iv) inevitably affects the coordination of the Mg2+ ion by displacing the O1G atom. The increase of local positive charge at [Ser/Thr]K+1—after O1G is moved aside by the stimulator—promotes the relocation of proton from [Ser/Thr]K+1 to AspWB.
- (C)
- The anionic [Ser/Thr]K+1 alkoxide withdraws a proton from the polarized Wcat molecule via intermediate proton carriers. Here, we depicted the simplest proton route as envisioned for TRAFAC NTPases (see Figure 7D and Figure 11D–F). More complex proton routes via Wcat-coordinating Glu/Asp residues, as found in other classes of NTPases, are indicated by red dashed lines in Figure 4, Figure 5 and Figure 6 and Figure 7A and differently shaded red arrows in Figure 10 and Figure 11A–C.
- (D)
- The resulting OH—cat, stabilized/polarized by its ligands, attacks the PG atom. Although the simplified diagram in Figure 12D shows only one stabilizing interaction of OH—cat with the HN group of the WB-crest residue, several ligands are usually involved in the stabilization; see Figure 1E,F, Figure 3, Figure 4, Figure 5 and Figure 6. During this step, the proton stays on AspWB. The formation of a covalent bond between OH—cat and PG increases the planarization of γ-phosphate; its oxygen atoms repel the β-phosphate oxygen atoms, resulting in a lengthening of the O3B–PG bond. With the inversion of γ-phosphate, increase in the O3B–PG distance, and γ-phosphate moving away from β-phosphate, HNK−3 detaches from γ-phosphate and, together with LysWA, Mg2+ and the stimulator, neutralizes the negative charge appearing on the O3B atom, thereby lowering the activation barrier. In addition, the negative charge on β-phosphate attracts a proton from [Ser/Thr]K+1.
- (E)
- (F)
- The H-bond between β- and γ-phosphate gradually dissociates as H2PO42− leaves the catalytic site. The departure of H2PO42− is an exergonic reaction that may be coupled to conformational changes, detachment of the activating partner from the WB-crest, and useful mechanical work.
5. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walker, J.E.; Saraste, M.; Runswick, M.J.; Gay, N.J. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982, 1, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Saraste, M.; Sibbald, P.R.; Wittinghofer, A. The P-loop—A common motif in ATP- and GTP-binding proteins. Trends Biochem. Sci. 1990, 15, 430–434. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Koonin, E.V. Helicases: Amino acid sequence comparisons and structure-function relationships. Curr. Opin. Struc. Biol. 1993, 3, 419–429. [Google Scholar] [CrossRef]
- Smith, C.A.; Rayment, I. Active site comparisons highlight structural similarities between myosin and other P-loop proteins. Biophys. J. 1996, 70, 1590–1602. [Google Scholar] [CrossRef]
- Sprang, S.R. G protein mechanisms: Insights from structural analysis. Annu. Rev. Biochem. 1997, 66, 639–678. [Google Scholar] [CrossRef]
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome. Res. 1999, 9, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Wolf, Y.I.; Aravind, L. Protein fold recognition using sequence profiles and its application in structural genomics. Adv. Protein Chem. 2000, 54, 245–275. [Google Scholar] [PubMed]
- Muneyuki, E.; Noji, H.; Amano, T.; Masaike, T.; Yoshida, M. FOF1-ATP synthase: General structural features of ‘ATP-engine’ and a problem on free energy transduction. Biochim. Biophys. Acta 2000, 1458, 467–481. [Google Scholar] [CrossRef]
- Leipe, D.D.; Koonin, E.V.; Aravind, L. Evolution and classification of P-loop kinases and related proteins. J. Mol. Biol. 2003, 333, 781–815. [Google Scholar] [CrossRef]
- Leipe, D.D.; Wolf, Y.I.; Koonin, E.V.; Aravind, L. Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol. 2002, 317, 41–72. [Google Scholar] [CrossRef] [Green Version]
- Anantharaman, V.; Aravind, L.; Koonin, E.V. Emergence of diverse biochemical activities in evolutionarily conserved structural scaffolds of proteins. Curr. Opin. Chem. Biol. 2003, 7, 12–20. [Google Scholar] [CrossRef]
- Iyer, L.M.; Leipe, D.D.; Koonin, E.V.; Aravind, L. Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 2004, 146, 11–31. [Google Scholar] [CrossRef]
- Ammelburg, M.; Frickey, T.; Lupas, A.N. Classification of AAA+ proteins. J. Struct. Biol. 2006, 156, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, N.D.; Berger, J.M. Structural frameworks for considering microbial protein- and nucleic acid-dependent motor ATPases. Mol. Microbiol. 2008, 69, 1071–1090. [Google Scholar] [CrossRef] [PubMed]
- Wittinghofer, A.; Vetter, I.R. Structure-function relationships of the G domain, a canonical switch motif. Annu. Rev. Biochem. 2011, 80, 943–971. [Google Scholar] [CrossRef] [PubMed]
- Wendler, P.; Ciniawsky, S.; Kock, M.; Kube, S. Structure and function of the AAA+ nucleotide binding pocket. Biochim. Biophys. Acta 2012, 1823, 2–14. [Google Scholar] [CrossRef]
- Burroughs, A.M.; Aravind, L. The Origin and Evolution of Release Factors: Implications for Translation Termination, Ribosome Rescue, and Quality Control Pathways. Int. J. Mol. Sci. 2019, 20, 1981. [Google Scholar] [CrossRef]
- Longo, L.M.; Jablonska, J.; Vyas, P.; Kanade, M.; Kolodny, R.; Ben-Tal, N.; Tawfik, D.S. On the emergence of P-Loop NTPase and Rossmann enzymes from a Beta-Alpha-Beta ancestral fragment. Elife 2020, 9, e64415. [Google Scholar] [CrossRef]
- Krishnan, A.; Burroughs, A.M.; Iyer, L.M.; Aravind, L. Comprehensive classification of ABC ATPases and their functional radiation in nucleoprotein dynamics and biological conflict systems. Nucleic Acids Res. 2020, 48, 10045–10075. [Google Scholar] [CrossRef]
- La Cour, T.F.; Nyborg, J.; Thirup, S.; Clark, B.F. Structural details of the binding of guanosine diphosphate to elongation factor Tu from E. coli as studied by X-ray crystallography. EMBO J. 1985, 4, 2385–2388. [Google Scholar] [CrossRef]
- Jurnak, F. Induction of elongation factor Tu-GDP crystal polymorphism by polyethylene glycol contaminants. J. Mol. Biol. 1985, 185, 215–217. [Google Scholar] [CrossRef]
- Hingorani, V.N.; Ho, Y.K. A structural model for the alpha-subunit of transducin. Implications of its role as a molecular switch in the visual signal transduction mechanism. FEBS Lett. 1987, 220, 15–22. [Google Scholar] [CrossRef]
- Pai, E.F.; Krengel, U.; Petsko, G.A.; Goody, R.S.; Kabsch, W.; Wittinghofer, A. Refined crystal structure of the triphosphate conformation of H-Ras p21 at 1.35 A resolution: Implications for the mechanism of GTP hydrolysis. EMBO J. 1990, 9, 2351–2359. [Google Scholar] [CrossRef] [PubMed]
- Kanade, M.; Chakraborty, S.; Shelke, S.S.; Gayathri, P. A Distinct Motif in a Prokaryotic Small Ras-Like GTPase Highlights Unifying Features of Walker B Motifs in P-Loop NTPases. J. Mol. Biol. 2020, 432, 5544–5564. [Google Scholar] [CrossRef]
- Matte, A.; Tari, L.W.; Delbaere, L.T. How do kinases transfer phosphoryl groups? Structure 1998, 6, 413–419. [Google Scholar] [CrossRef]
- Vetter, I.R.; Wittinghofer, A. Nucleoside triphosphate-binding proteins: Different scaffolds to achieve phosphoryl transfer. Q. Rev. Biophys. 1999, 32, 1–56. [Google Scholar] [CrossRef]
- Allin, C.; Gerwert, K. Ras catalyzes GTP hydrolysis by shifting negative charges from gamma- to beta-phosphate as revealed by time-resolved FTIR difference spectroscopy. Biochemistry 2001, 40, 3037–3046. [Google Scholar] [CrossRef]
- Kotting, C.; Gerwert, K. Time-resolved FTIR studies provide activation free energy, activation enthalpy and activation entropy for GTPase reactions. Chem. Phys. 2004, 307, 227–232. [Google Scholar] [CrossRef]
- Shalaeva, D.N.; Cherepanov, D.A.; Galperin, M.Y.; Golovin, A.V.; Mulkidjanian, A.Y. Evolution of cation binding in the active sites of P-loop nucleoside triphosphatases in relation to the basic catalytic mechanism. Elife 2018, 7, e37373. [Google Scholar] [CrossRef]
- Kozlova, M.I.; Shalaeva, D.N.; Dibrova, D.V.; Mulkidjanian, A.Y. Patterns of hydrolysis initiation in P-loop fold nucleoside triphosphatases. Biomolecules 2022, 12, 1345. [Google Scholar] [CrossRef]
- Coleman, D.E.; Berghuis, A.M.; Lee, E.; Linder, M.E.; Gilman, A.G.; Sprang, S.R. Structures of active conformations of Gi alpha 1 and the mechanism of GTP hydrolysis. Science 1994, 265, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Sondek, J.; Lambright, D.G.; Noel, J.P.; Hamm, H.E.; Sigler, P.B. GTPase mechanism of Gproteins from the 1.7-A crystal structure of transducin alpha-GDP-AIF4. Nature 1994, 372, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Scheffzek, K.; Ahmadian, M.R.; Wittinghofer, A. GTPase-activating proteins: Helping hands to complement an active site. Trends Biochem. Sci. 1998, 23, 257–262. [Google Scholar] [CrossRef]
- Ogura, T.; Whiteheart, S.W.; Wilkinson, A.J. Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. J. Struct. Biol. 2004, 146, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Scrima, A.; Wittinghofer, A. Dimerisation-dependent GTPase reaction of MnmE: How potassium acts as GTPase-activating element. EMBO J. 2006, 25, 2940–2951. [Google Scholar] [CrossRef] [PubMed]
- Ash, M.-R.; Maher, M.J.; Guss, J.M.; Jormakka, M. The cation-dependent G-proteins: In a class of their own. FEBS Lett. 2012, 586, 2218–2224. [Google Scholar] [CrossRef]
- Jin, Y.; Molt, R.W., Jr.; Blackburn, G.M. Metal fluorides: Tools for structural and computational analysis of phosphoryl transfer enzymes. Top. Curr. Chem. 2017, 375, 36. [Google Scholar] [CrossRef]
- Gasper, R.; Wittinghofer, F. The Ras switch in structural and historical perspective. Biol. Chem. 2019, 401, 143–163. [Google Scholar] [CrossRef]
- Vetter, I.R.; Wittinghofer, A. Signal transduction—The guanine nucleotide-binding switch in three dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef]
- Nam, K.; Pu, J.; Karplus, M. Trapping the ATP binding state leads to a detailed understanding of the F1-ATPase mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, 17851–17856. [Google Scholar] [CrossRef] [Green Version]
- Llinas, P.; Isabet, T.; Song, L.; Ropars, V.; Zong, B.; Benisty, H.; Sirigu, S.; Morris, C.; Kikuti, C.; Safer, D.; et al. How actin initiates the motor activity of myosin. Dev. Cell 2015, 33, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Wittinghofer, A. Signaling mechanistics: Aluminum fluoride for molecule of the year. Curr. Biol. 1997, 7, R682–R685. [Google Scholar] [CrossRef]
- Menz, R.I.; Walker, J.E.; Leslie, A.G. Structure of bovine mitochondrial F1-ATPase with nucleotide bound to all three catalytic sites: Implications for the mechanism of rotary catalysis. Cell 2001, 106, 331–341. [Google Scholar] [CrossRef]
- Graham, D.L.; Lowe, P.N.; Grime, G.W.; Marsh, M.; Rittinger, K.; Smerdon, S.J.; Gamblin, S.J.; Eccleston, J.F. MgF3− as a transition state analog of phosphoryl transfer. Chem. Biol. 2002, 9, 375–381. [Google Scholar] [CrossRef]
- Davies, D.R.; Hol, W.G. The power of vanadate in crystallographic investigations of phosphoryl transfer enzymes. FEBS Lett. 2004, 577, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Richards, N.G.; Waltho, J.P.; Blackburn, G.M. Metal fluorides as analogues for studies on phosphoryl transfer enzymes. Angew. Chem. Int. Ed. Engl. 2017, 56, 4110–4128. [Google Scholar] [CrossRef]
- Lacabanne, D.; Wiegand, T.; Wili, N.; Kozlova, M.I.; Cadalbert, R.; Klose, D.; Mulkidjanian, A.Y.; Meier, B.H.; Bockmann, A. ATP Analogues for Structural Investigations: Case Studies of a DnaB Helicase and an ABC Transporter. Molecules 2020, 25, 5268. [Google Scholar] [CrossRef]
- Blackburn, G.M.; Cherfils, J.; Moss, G.P.; Richards, N.G.J.; Waltho, J.P.; Williams, N.H.; Wittinghofer, A. How to name atoms in phosphates, polyphosphates, their derivatives and mimics, and transition state analogues for enzyme-catalysed phosphoryl transfer reactions (IUPAC Recommendations 2016). Pure Appl. Chem. 2017, 89, 653–675. [Google Scholar] [CrossRef]
- Knowles, J.R. Enzyme-catalyzed phosphoryl transfer reactions. Annu. Rev. Biochem. 1980, 49, 877–919. [Google Scholar] [CrossRef]
- Westheimer, F.H. Why Nature Chose Phosphates. Science 1987, 235, 1173–1178. [Google Scholar] [CrossRef]
- Shabarova, Z.A.; Bogdanov, A.A. Advanced Organic Chemistry of Nucleic Acids; VCH: Weinheim, Germany, 1994. [Google Scholar]
- Cleland, W.W.; Hengge, A.C. Enzymatic mechanisms of phosphate and sulfate transfer. Chem. Rev. 2006, 106, 3252–3278. [Google Scholar] [CrossRef] [PubMed]
- Bowler, M.W.; GCliff, M.J.; Waltho, J.P.; Blackburn, G.M. Why did Nature select phosphate for its dominant roles in biology? New J. Chem. 2010, 34, 784–794. [Google Scholar] [CrossRef]
- Lassila, J.K.; Zalatan, J.G.; Herschlag, D. Biological phosphoryl-transfer reactions: Understanding mechanism and catalysis. Annu. Rev. Biochem. 2011, 80, 669–702. [Google Scholar] [CrossRef] [PubMed]
- Kiani, F.A.; Fischer, S. Comparing the catalytic strategy of ATP hydrolysis in biomolecular motors. Phys. Chem. Chem. Phys. 2016, 18, 20219–20233. [Google Scholar] [CrossRef] [PubMed]
- Law, Y.S.; Utt, A.; Tan, Y.B.; Zheng, J.; Wang, S.; Chen, M.W.; Griffin, P.R.; Merits, A.; Luo, D. Structural insights into RNA recognition by the Chikungunya virus nsP2 helicase. Proc. Natl. Acad. Sci. USA 2019, 116, 9558–9567. [Google Scholar] [CrossRef]
- Langen, R.; Schweins, T.; Warshel, A. On the mechanism of guanosine triphosphate hydrolysis in ras p21 proteins. Biochemistry 1992, 31, 8691–8696. [Google Scholar] [CrossRef]
- Schweins, T.; Langen, R.; Warshel, A. Why Have Mutagenesis Studies Not Located the General Base in Ras P21. Nat. Struct. Biol. 1994, 1, 476–484. [Google Scholar] [CrossRef]
- Schweins, T.; Geyer, M.; Scheffzek, K.; Warshel, A.; Kalbitzer, H.R.; Wittinghofer, A. Substrate-assisted catalysis as a mechanism for GTP hydrolysis of p21ras and other GTP-binding proteins. Nat. Struct. Biol. 1995, 2, 36–44. [Google Scholar] [CrossRef]
- Braig, K.; Menz, R.I.; Montgomery, M.G.; Leslie, A.G.; Walker, J.E. Structure of bovine mitochondrial F1-ATPase inhibited by Mg2+ ADP and aluminium fluoride. Structure 2000, 8, 567–573. [Google Scholar] [CrossRef]
- Maegley, K.A.; Admiraal, S.J.; Herschlag, D. Ras-catalyzed hydrolysis of GTP: A new perspective from model studies. Proc. Natl. Acad. Sci. USA 1996, 93, 8160–8166. [Google Scholar] [CrossRef] [Green Version]
- Glennon, T.M.; Villa, J.; Warshel, A. How does GAP catalyze the GTPase reaction of Ras?: A computer simulation study. Biochemistry 2000, 39, 9641–9651. [Google Scholar] [CrossRef] [PubMed]
- Kotting, C.; Kallenbach, A.; Suveyzdis, Y.; Wittinghofer, A.; Gerwert, K. The GAP arginine finger movement into the catalytic site of Ras increases the activation entropy. Proc. Natl. Acad. Sci. USA 2008, 105, 6260–6265. [Google Scholar] [CrossRef] [PubMed]
- Mondal, D.; Warshel, A. EF-Tu and EF-G are activated by allosteric effects. Proc. Natl. Acad. Sci. USA 2018, 115, 3386–3391. [Google Scholar] [CrossRef] [PubMed]
- Shrake, A.; Rupley, J.A. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J. Mol. Biol. 1973, 79, 351–371. [Google Scholar] [CrossRef]
- Bas, D.C.; Rogers, D.M.; Jensen, J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins 2008, 73, 765–783. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodova, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koca, J.; Rose, A.S. Mol* Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System; Version 1.7.2.1; Schrödinger, LLC.: New York, NY, USA, 2010. [Google Scholar]
- Mueller, M.P.; Goody, R.S. Review: Ras GTPases and myosin: Qualitative conservation and quantitative diversification in signal and energy transduction. Biopolymers 2016, 105, 422–430. [Google Scholar] [CrossRef]
- Chappie, J.S.; Acharya, S.; Leonard, M.; Schmid, S.L.; Dyda, F. G domain dimerization controls dynamin’s assembly-stimulated GTPase activity. Nature 2010, 465, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Gara, S.K.; Mishra, S.; Prakash, B. Analysis of GTPases carrying hydrophobic amino acid substitutions in lieu of the catalytic glutamine: Implications for GTP hydrolysis. Proteins 2005, 59, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Fasano, O.; De Vendittis, E.; Parmeggiani, A. Hydrolysis of GTP by elongation factor Tu can be induced by monovalent cations in the absence of other effectors. J. Biol. Chem. 1982, 257, 3145–3150. [Google Scholar] [CrossRef]
- Maracci, C.; Peske, F.; Dannies, E.; Pohl, C.; Rodnina, M.V. Ribosome-induced tuning of GTP hydrolysis by a translational GTPase. Proc. Natl. Acad. Sci. USA 2014, 111, 14418–14423. [Google Scholar] [CrossRef]
- Chinali, G.; Parmeggiani, A. The coupling with polypeptide synthesis of the GTPase activity dependent on elongation factor G. J. Biol. Chem. 1980, 255, 7455–7459. [Google Scholar] [CrossRef]
- Ivell, R.; Sander, G.; Parmeggiani, A. Modulation by monovalent and divalent cations of the guanosine-5′-triphosphatase activity dependent on elongation factor Tu. Biochemistry 1981, 20, 6852–6859. [Google Scholar] [CrossRef] [PubMed]
- Parmeggiani, A.; Sander, G. Properties and regulation of the GTPase activities of elongation factors Tu and G, and of initiation factor 2. Mol. Cell Biochem. 1981, 35, 129–158. [Google Scholar] [CrossRef] [PubMed]
- Voigt, J.; Sander, G.; Nagel, K.; Parmeggiani, A. Effect of NH4+ and K+ on the activity of the ribosomal subunits in the EF-G- and EF-T-dependent GTR hydrolysis. Biochem. Biophys. Res. Commun. 1974, 57, 1279–1286. [Google Scholar] [CrossRef]
- Mulkidjanian, A.Y.; Bychkov, A.Y.; Dibrova, D.V.; Galperin, M.Y.; Koonin, E.V. Origin of first cells at terrestrial, anoxic geothermal fields. Proc. Natl. Acad. Sci. USA 2012, 109, E821–E830. [Google Scholar] [CrossRef]
- Kuhle, B.; Ficner, R. A monovalent cation acts as structural and catalytic cofactor in translational GTPases. EMBO J. 2014, 33, 2547–2563. [Google Scholar] [CrossRef]
- Voorhees, R.M.; Schmeing, T.M.; Kelley, A.C.; Ramakrishnan, V. The mechanism for activation of GTP hydrolysis on the ribosome. Science 2010, 330, 835–838. [Google Scholar] [CrossRef] [Green Version]
- Byrnes, L.J.; Singh, A.; Szeto, K.; Benvin, N.M.; O’Donnell, J.P.; Zipfel, W.R.; Sondermann, H. Structural basis for conformational switching and GTP loading of the large G protein atlastin. EMBO J. 2013, 32, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, M.; Willard, F.S.; Kimple, A.J.; Turnbull, A.P.; Ball, L.J.; Schoch, G.A.; Gileadi, C.; Fedorov, O.Y.; Dowler, E.F.; Higman, V.A.; et al. Structural diversity in the RGS domain and its interaction with heterotrimeric G protein alpha-subunits. Proc. Natl. Acad. Sci. USA 2008, 105, 6457–6462. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.J.; Walker, J.E. Homology between human bladder carcinoma oncogene product and mitochondrial ATP-synthase. Nature 1983, 301, 262–264. [Google Scholar] [CrossRef]
- Sprang, S.R. G proteins, effectors and GAPs: Structure and mechanism. Curr. Opin. Struc. Biol. 1997, 7, 849–856. [Google Scholar] [CrossRef]
- Wittinghofer, A. GTP and ATP hydrolysis in biology. Biopolymers 2016, 105, 419–421. [Google Scholar] [CrossRef]
- Scheffzek, K.; Ahmadian, M.R.; Kabsch, W.; Wiesmuller, L.; Lautwein, A.; Schmitz, F.; Wittinghofer, A. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 1997, 277, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.J.; Smith, C.A.; Thoden, J.B.; Smith, R.; Sutoh, K.; Holden, H.M.; Rayment, I. X-ray structures of the myosin motor domain of Dictyostelium discoideum complexed with MgADP•BeFx and MgADP•AlF4−. Biochemistry 1995, 34, 8960–8972. [Google Scholar] [CrossRef] [PubMed]
- Geeves, M.A. Review: The ATPase mechanism of myosin and actomyosin. Biopolymers 2016, 105, 483–491. [Google Scholar] [CrossRef]
- Cross, R.A. Review: Mechanochemistry of the kinesin-1 ATPase. Biopolymers 2016, 105, 476–482. [Google Scholar] [CrossRef]
- O’Donnell, J.P.; Byrnes, L.J.; Cooley, R.B.; Sondermann, H. A hereditary spastic paraplegia-associated atlastin variant exhibits defective allosteric coupling in the catalytic core. J. Biol. Chem. 2018, 293, 687–700. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.A.; Rayment, I. X-ray structure of the magnesium(II).ADP•vanadate complex of the Dictyostelium discoideum myosin motor domain to 1.9 A resolution. Biochemistry 1996, 35, 5404–5417. [Google Scholar] [CrossRef]
- Bange, G.; Sinning, I. SIMIBI twins in protein targeting and localization. Nat. Struct. Mol. Biol. 2013, 20, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Lutkenhaus, J. The ParA/MinD family puts things in their place. Trends Microbiol. 2012, 20, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Ataide, S.F.; Schmitz, N.; Shen, K.; Ke, A.; Shan, S.O.; Doudna, J.A.; Ban, N. The crystal structure of the signal recognition particle in complex with its receptor. Science 2011, 331, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Moser, J.; Lange, C.; Krausze, J.; Rebelein, J.; Schubert, W.D.; Ribbe, M.W.; Heinz, D.W.; Jahn, D. Structure of ADP-aluminium fluoride-stabilized protochlorophyllide oxidoreductase complex. Proc. Natl. Acad. Sci. USA 2013, 110, 2094–2098. [Google Scholar] [CrossRef]
- Focia, P.J.; Gawronski-Salerno, J.; Coon, J.S.t.; Freymann, D.M. Structure of a GDP:AlF4− complex of the SRP GTPases Ffh and FtsY, and identification of a peripheral nucleotide interaction site. J. Mol. Biol. 2006, 360, 631–643. [Google Scholar] [CrossRef]
- Ostermann, N.; Segura-Pena, D.; Meier, C.; Veit, T.; Monnerjahn, C.; Konrad, M.; Lavie, A. Structures of human thymidylate kinase in complex with prodrugs: Implications for the structure-based design of novel compounds. Biochemistry 2003, 42, 2568–2577. [Google Scholar] [CrossRef]
- Poyraz, O.; Brunner, K.; Lohkamp, B.; Axelsson, H.; Hammarstrom, L.G.; Schnell, R.; Schneider, G. Crystal structures of the kinase domain of the sulfate-activating complex in Mycobacterium tuberculosis. PLoS ONE 2015, 10, e0121494. [Google Scholar] [CrossRef]
- Cheek, S.; Ginalski, K.; Zhang, H.; Grishin, N.V. A comprehensive update of the sequence and structure classification of kinases. BMC Struct. Biol. 2005, 5, 6. [Google Scholar] [CrossRef]
- Kenyon, C.P.; Roth, R.L.; van der Westhuyzen, C.W.; Parkinson, C.J. Conserved phosphoryl transfer mechanisms within kinase families and the role of the C8 proton of ATP in the activation of phosphoryl transfer. BMC Res. Notes 2012, 5, 131. [Google Scholar] [CrossRef] [Green Version]
- Lansdon, E.B.; Segel, I.H.; Fisher, A.J. Ligand-induced structural changes in adenosine 5′-phosphosulfate kinase from Penicillium chrysogenum. Biochemistry 2002, 41, 13672–13680. [Google Scholar] [CrossRef] [PubMed]
- Kerns, S.J.; Agafonov, R.V.; Cho, Y.J.; Pontiggia, F.; Otten, R.; Pachov, D.V.; Kutter, S.; Phung, L.A.; Murphy, P.N.; Thai, V.; et al. The energy landscape of adenylate kinase during catalysis. Nat. Struct. Mol. Biol. 2015, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Makarova, K.S.; Koonin, E.V.; Aravind, L. Comparative genomics of the FtsK-HerA superfamily of pumping ATPases: Implications for the origins of chromosome segregation, cell division and viral capsid packaging. Nucleic Acids Res. 2004, 32, 5260–5279. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Iyer, L.M.; Leipe, D.D.; Koonin, E.V. A novel family of P-loop NTPases with an unusual phyletic distribution and transmembrane segments inserted within the NTPase domain. Genome Biol. 2004, 5, R30. [Google Scholar] [CrossRef]
- Lupas, A.N.; Martin, J. AAA proteins. Curr. Opin. Struct. Biol. 2002, 12, 746–753. [Google Scholar] [CrossRef]
- Miller, J.M.; Enemark, E.J. Fundamental Characteristics of AAA+ Protein Family Structure and Function. Archaea 2016, 2016, 9294307. [Google Scholar] [CrossRef]
- Yu, R.C.; Hanson, P.I.; Jahn, R.; Brunger, A.T. Structure of the ATP-dependent oligomerization domain of N-ethylmaleimide sensitive factor complexed with ATP. Nat. Struct. Biol. 1998, 5, 803–811. [Google Scholar] [CrossRef]
- Singleton, M.R.; Dillingham, M.S.; Wigley, D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007, 76, 23–50. [Google Scholar] [CrossRef]
- Sengoku, T.; Nureki, O.; Nakamura, A.; Satoru, K.I.; Yokoyama, S. Structural basis for RNA unwinding by the DEAD-box protein Drosophila vasa. Cell 2006, 125, 287–300. [Google Scholar] [CrossRef]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef]
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC transporters: The power to change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Kerr, I.D. Sequence analysis of twin ATP binding cassette proteins involved in translational control, antibiotic resistance, and ribonuclease L inhibition. Biochem. Biophys. Res. Commun. 2004, 315, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P. Rustless translation. Biol. Chem. 2012, 393, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Oldham, M.L.; Chen, J. Snapshots of the maltose transporter during ATP hydrolysis. Proc. Natl. Acad. Sci. USA 2011, 108, 15152–15156. [Google Scholar] [CrossRef]
- Gai, D.; Zhao, R.; Li, D.; Finkielstein, C.V.; Chen, X.S. Mechanisms of conformational change for a replicative hexameric helicase of SV40 large tumor antigen. Cell 2004, 119, 47–60. [Google Scholar] [CrossRef]
- Gu, M.; Rice, C.M. The Spring alpha-Helix Coordinates Multiple Modes of HCV (Hepatitis C Virus) NS3 Helicase Action. J. Biol. Chem. 2016, 291, 14499–14509. [Google Scholar] [CrossRef]
- Dibrova, D.V.; Konovalov, K.A.; Perekhvatov, V.V.; Skulachev, K.V.; Mulkidjanian, A.Y. COGcollator: A web server for analysis of distant relationships between homologous protein families. Biol. Direct. 2017, 12, 29. [Google Scholar] [CrossRef]
- Lawson, M.R.; Ma, W.; Bellecourt, M.J.; Artsimovitch, I.; Martin, A.; Landick, R.; Schulten, K.; Berger, J.M. Mechanism for the Regulated Control of Bacterial Transcription Termination by a Universal Adaptor Protein. Mol. Cell 2018, 71, 911–922.e914. [Google Scholar] [CrossRef]
- Dibrova, D.V.; Galperin, M.Y.; Koonin, E.V.; Mulkidjanian, A.Y. Ancient systems of sodium/potassium homeostasis as predecessors of membrane bioenergetics. Biochemistry (Moscow) 2015, 80, 495–516. [Google Scholar] [CrossRef]
- Malaer, A.A.; Wili, N.; Volker, L.A.; Kozlova, M.I.; Cadalbert, R.; Dapp, A.; Weber, M.E.; Zehnder, J.; Jeschke, G.; Eckert, H.; et al. Spectroscopic glimpses of the transition state of ATP hydrolysis trapped in a bacterial DnaB helicase. Nat. Commun. 2021, 12, 5293. [Google Scholar] [CrossRef]
- Abe, J.; Hiyama, T.B.; Mukaiyama, A.; Son, S.; Mori, T.; Saito, S.; Osako, M.; Wolanin, J.; Yamashita, E.; Kondo, T.; et al. Circadian rhythms. Atomic-scale origins of slowness in the cyanobacterial circadian clock. Science 2015, 349, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Singleton, M.R.; Sawaya, M.R.; Ellenberger, T.; Wigley, D.B. Crystal structure of T7 gene 4 ring helicase indicates a mechanism for sequential hydrolysis of nucleotides. Cell 2000, 101, 589–600. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, H.; Pavletich, N.P. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature 2008, 453, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; He, Y.; Wu, Y.; Luo, Y. Asp302 determines potassium dependence of a RadA recombinase from Methanococcus voltae. J. Mol. Biol. 2006, 360, 537–547. [Google Scholar] [CrossRef]
- Itsathitphaisarn, O.; Wing, R.A.; Eliason, W.K.; Wang, J.; Steitz, T.A. The hexameric helicase DnaB adopts a nonplanar conformation during translocation. Cell 2012, 151, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Yamaichi, Y.; Niki, H. Active segregation by the Bacillus subtilis partitioning system in Escherichia coli. Proc. Natl. Acad. Sci. USA 2000, 97, 14656–14661. [Google Scholar] [CrossRef] [PubMed]
- Marsin, S.; Adam, Y.; Cargemel, C.; Andreani, J.; Baconnais, S.; Legrand, P.; Li de la Sierra-Gallay, I.; Humbert, A.; Aumont-Nicaise, M.; Velours, C.; et al. Study of the DnaB:DciA interplay reveals insights into the primary mode of loading of the bacterial replicative helicase. Nucleic Acids Res 2021, 49, 6569–6586. [Google Scholar] [CrossRef]
- Li, Y.; He, Y.; Luo, Y. Conservation of a conformational switch in RadA recombinase from Methanococcus maripaludis. Acta Cryst. D Biol. Cryst. 2009, 65, 602–610. [Google Scholar] [CrossRef]
- Kamerlin, S.C.; Sharma, P.K.; Prasad, R.B.; Warshel, A. Why nature really chose phosphate. Q. Rev. Biophys. 2013, 46, 1–132. [Google Scholar] [CrossRef]
- Fothergill, M.; Goodman, M.F.; Petrushka, J.; Warshel, A. Structure-energy analysis of the role of metal ions in phosphodiester bond hydrolysis by DNA polymerase I. J. Amer. Chem. Soc. 1995, 117, 11619–11627. [Google Scholar] [CrossRef]
- Da Silva, J.F.; Williams, R.J.P. The Biological Chemistry of the Elements: The Inorganic Chemistry of Life; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Berta, D.; Buigues, P.J.; Badaoui, M.; Rosta, E. Cations in motion: QM/MM studies of the dynamic and electrostatic roles of H+ and Mg2+ ions in enzyme reactions. Curr. Opin. Struct. Biol. 2020, 61, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, T.P. How enzymes harness highly unfavorable proton transfer reactions. Protein Sci. 2021, 30, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Ehebauer, M.T.; Pukala, T.; Marsh, M.; Blundell, T.L.; Venkitaraman, A.R.; Abell, C.; Hyvonen, M. Using a fragment-based approach to target protein-protein interactions. Chembiochem 2013, 14, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Miyano, N.; Baba, S.; Liao, J.; Kawamura, T.; Tsuda, C.; Takeda, A.; Yamamoto, M.; Kumasaka, T.; Kataoka, T.; et al. Molecular Mechanism for Conformational Dynamics of Ras.GTP Elucidated from In-Situ Structural Transition in Crystal. Sci. Rep. 2016, 6, 25931. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Grosseruschkamp, F.; Stephan, S.; Cui, Q.; Kotting, C.; Xia, F.; Gerwert, K. Specific Substates of Ras To Interact with GAPs and Effectors: Revealed by Theoretical Simulations and FTIR Experiments. J. Phys. Chem. Lett. 2018, 9, 1312–1317. [Google Scholar] [CrossRef]
- Johansen, J.S.; Kavaliauskas, D.; Pfeil, S.H.; Blaise, M.; Cooperman, B.S.; Goldman, Y.E.; Thirup, S.S.; Knudsen, C.R.E. E. coli elongation factor Tu bound to a GTP analogue displays an open conformation equivalent to the GDP-bound form. Nucleic Acids Res. 2018, 46, 8641–8650. [Google Scholar] [CrossRef]
- Bauer, C.B.; Holden, H.M.; Thoden, J.B.; Smith, R.; Rayment, I. X-ray structures of the apo and MgATP-bound states of Dictyostelium discoideum myosin motor domain. J. Biol. Chem. 2000, 275, 38494–38499. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, L. Unraveling the structural and chemical features of biological short hydrogen bonds. Chem. Sci. 2019, 10, 7734–7745. [Google Scholar] [CrossRef]
- Herschlag, D.; Pinney, M.M. Hydrogen Bonds: Simple after All? Biochemistry 2018, 57, 3338–3352. [Google Scholar] [CrossRef]
- Rittinger, K.; Walker, P.A.; Eccleston, J.F.; Smerdon, S.J.; Gamblin, S.J. Structure at 1.65 A of RhoA and its GTPase-activating protein in complex with a transition-state analogue. Nature 1997, 389, 758–762. [Google Scholar] [CrossRef]
- Yang, X.; Chen, C.; Tian, H.; Chi, H.; Mu, Z.; Zhang, T.; Yang, K.; Zhao, Q.; Liu, X.; Wang, Z.; et al. Mechanism of ATP hydrolysis by the Zika virus helicase. Faseb J. 2018, 32, 5250–5257. [Google Scholar] [CrossRef] [PubMed]
- Mulkidjanian, A.Y.; Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Evolutionary primacy of sodium bioenergetics. Biol. Direct 2008, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Dibrova, D.V.; Cherepanov, D.A.; Galperin, M.Y.; Skulachev, V.P.; Mulkidjanian, A.Y. Evolution of cytochrome bc complexes: From membrane-anchored dehydrogenases of ancient bacteria to triggers of apoptosis in vertebrates. Biochim. Biophys. Acta 2013, 1827, 1407–1427. [Google Scholar] [CrossRef] [PubMed]
- Dibrova, D.V.; Shalaeva, D.N.; Galperin, M.Y.; Mulkidjanian, A.Y. Emergence of cytochrome bc complexes in the context of photosynthesis. Physiol. Plant 2017, 161, 150–170. [Google Scholar] [CrossRef]
- Shalaeva, D.N.; Cherepanov, D.A.; Galperin, M.Y.; Vriend, G.; Mulkidjanian, A.Y. G protein-coupled receptors of class A harness the energy of membrane potential to increase their sensitivity and selectivity. Biochim. Biophys. Acta Biomembr. 2019, 1861, 183051. [Google Scholar] [CrossRef] [PubMed]
- Shalaeva, D.N.; Galperin, M.Y.; Mulkidjanian, A.Y. Eukaryotic G protein-coupled receptors as descendants of prokaryotic sodium-translocating rhodopsins. Biol. Direct 2015, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Lawson, J.D.; Pate, E.; Rayment, I.; Yount, R.G. Molecular dynamics analysis of structural factors influencing back door Pi release in myosin. Biophys. J. 2004, 86, 3794–3803. [Google Scholar] [CrossRef]
- Zhang, N.; Buck, M. Formation of MgF3−-dependent complexes between an AAA(+) ATPase and sigma(54.). FEBS Open Bio. 2012, 2, 89–92. [Google Scholar] [CrossRef]
- Chaney, M.; Grande, R.; Wigneshweraraj, S.R.; Cannon, W.; Casaz, P.; Gallegos, M.T.; Schumacher, J.; Jones, S.; Elderkin, S.; Dago, A.E.; et al. Binding of transcriptional activators to sigma 54 in the presence of the transition state analogue ADP-aluminum fluoride: Insights into activator mechanochemical action. Genes Dev. 2001, 15, 2282–2294. [Google Scholar] [CrossRef]
- Higashijima, T.; Ferguson, K.M.; Sternweis, P.C.; Ross, E.M.; Smigel, M.D.; Gilman, A.G. The effect of activating ligands on the intrinsic fluorescence of guanine nucleotide-binding regulatory proteins. J. Biol. Chem. 1987, 262, 752–756. [Google Scholar] [CrossRef]
- Gremer, L.; Gilsbach, B.; Ahmadian, M.R.; Wittinghofer, A. Fluoride complexes of oncogenic Ras mutants to study the Ras-RasGap interaction. Biol. Chem. 2008, 389, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.; Ahmadian, M.R.; Goody, R.S.; Wittinghofer, A. Formation of a transition-state analogue of the Ras GTPase reaction by Ras-GDP, tetrafluoroaluminate, and GTPase-activating proteins. Science 1996, 273, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, P.P.; Daumke, O.; Suveyzdis, Y.; Kotting, C.; Gerwert, K.; Wittinghofer, A. Insight into catalysis of a unique GTPase reaction by a combined biochemical and FTIR approach. J. Mol. Biol. 2007, 367, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Baxter, N.J.; Blackburn, G.M.; Marston, J.P.; Hounslow, A.M.; Cliff, M.J.; Bermel, W.; Williams, N.H.; Hollfelder, F.; Wemmer, D.E.; Waltho, J.P. Anionic charge is prioritized over geometry in aluminum and magnesium fluoride transition state analogues of phosphoryl transfer enzymes. J. Am. Chem. Soc. 2008, 130, 3952–3958. [Google Scholar] [CrossRef]
- Majumdar, S.; Acharya, A.; Prakash, B. Structural plasticity mediates distinct GAP-dependent GTP hydrolysis mechanisms in Rab33 and Rab5. FEBS J. 2017, 284, 4358–4375. [Google Scholar] [CrossRef]
- Nassar, N.; Hoffman, G.R.; Manor, D.; Clardy, J.C.; Cerione, R.A. Structures of Cdc42 bound to the active and catalytically compromised forms of Cdc42GAP. Nat. Struct. Biol. 1998, 5, 1047–1052. [Google Scholar] [CrossRef]
- Klahn, M.; Rosta, E.; Warshel, A. On the mechanism of hydrolysis of phosphate monoesters dianions in solutions and proteins. J. Am. Chem. Soc. 2006, 128, 15310–15323. [Google Scholar] [CrossRef]
- Williams, N.H. Magnesium ion catalyzed ATP hydrolysis. JACS 2000, 122, 12023–12024. [Google Scholar] [CrossRef]
- Li, G.; Zhang, X.C. GTP hydrolysis mechanism of Ras-like GTPases. J. Mol. Biol. 2004, 340, 921–932. [Google Scholar] [CrossRef]
- Rudack, T.; Xia, F.; Schlitter, J.; Kotting, C.; Gerwert, K. The role of magnesium for geometry and charge in GTP hydrolysis, revealed by quantum mechanics/molecular mechanics simulations. Biophys. J. 2012, 103, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.C.; Sun, S.; Chandler, D.; Oster, G. The conformational states of Mg.ATP in water. Eur. Biophys. J. 2004, 33, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.F. AMBER force-field parameters for guanosine triphosphate and its imido and methylene analogs. LJ Comput. Chem. 1993, 14, 995–1005. [Google Scholar] [CrossRef]
- Delbaere, L.T.; Sudom, A.M.; Prasad, L.; Leduc, Y.; Goldie, H. Structure/function studies of phosphoryl transfer by phosphoenolpyruvate carboxykinase. Biochim. Biophys. Acta 2004, 1697, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Rudack, T.; Xia, F.; Schlitter, J.; Kotting, C.; Gerwert, K. Ras and GTPase-activating protein (GAP) drive GTP into a precatalytic state as revealed by combining FTIR and biomolecular simulations. Proc. Natl. Acad. Sci. USA 2012, 109, 15295–15300. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Sukal, S.; Deng, H.; Leyh, T.S.; Callender, R. Vibrational structure of GDP and GTP bound to RAS: An isotope-edited FTIR study. Biochemistry 2001, 40, 4035–4043. [Google Scholar] [CrossRef]
- Finkelstein, A.; Ptitsyn, B. Protein Physics; Academic Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Shutes, A.; Der, C.J. Real-time in vitro measurement of intrinsic and Ras GAP-mediated GTP hydrolysis. Methods Enzym. 2006, 407, 9–22. [Google Scholar]
- delToro, D.; Ortiz, D.; Ordyan, M.; Pajak, J.; Sippy, J.; Catala, A.; Oh, C.S.; Vu, A.; Arya, G.; Smith, D.E.; et al. Functional Dissection of a Viral DNA Packaging Machine’s Walker B Motif. J. Mol. Biol. 2019, 431, 4455–4474. [Google Scholar] [CrossRef]
- Voigts-Hoffmann, F.; Schmitz, N.; Shen, K.; Shan, S.O.; Ataide, S.F.; Ban, N. The structural basis of FtsY recruitment and GTPase activation by SRP RNA. Mol. Cell 2013, 52, 643–654. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, S.; Wu, Z.; Li, X.; Wang, W.L.; Zhu, Y.; Stoilova-McPhie, S.; Lu, Y.; Finley, D.; Mao, Y. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 2019, 565, 49–55. [Google Scholar] [CrossRef]
- Lu, K.Y.; Chen, W.F.; Rety, S.; Liu, N.N.; Wu, W.Q.; Dai, Y.X.; Li, D.; Ma, H.Y.; Dou, S.X.; Xi, X.G. Insights into the structural and mechanistic basis of multifunctional S. cerevisiae Pif1p helicase. Nucleic Acids Res. 2018, 46, 1486–1500. [Google Scholar] [CrossRef]
- Schweins, T.; Warshel, A. Mechanistic analysis of the observed linear free energy relationships in p21ras and related systems. Biochemistry 1996, 35, 14232–14243. [Google Scholar] [CrossRef] [PubMed]
- Allin, C.; Ahmadian, M.R.; Wittinghofer, A.; Gerwert, K. Monitoring the GAP catalyzed H-Ras GTPase reaction at atomic resolution in real time. Proc. Natl. Acad. Sci. USA 2001, 98, 7754–7759. [Google Scholar] [CrossRef] [PubMed]
- Valiev, M.; Kawai, R.; Adams, J.A.; Weare, J.H. The role of the putative catalytic base in the phosphoryl transfer reaction in a protein kinase: First-principles calculations. J. Am. Chem. Soc. 2003, 125, 9926–9927. [Google Scholar] [CrossRef]
- Valiev, M.; Yang, J.; Adams, J.A.; Taylor, S.S.; Weare, J.H. Phosphorylation reaction in cAPK protein kinase-free energy quantum mechanical/molecular mechanics simulations. J. Phys. Chem. B 2007, 111, 13455–13464. [Google Scholar] [CrossRef]
- Cheng, H.; Sukal, S.; Callender, R.; Leyh, T.S. γ-phosphate protonation and pH-dependent unfolding of the Ras•GTP•Mg2+ complex: A vibrational spectroscopy study. J. Biol. Chem. 2001, 276, 9931–9935. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.; Guldenhaupt, J.; Schartner, J.; Gerwert, K.; Kotting, C. The protonation states of GTP and GppNHp in Ras proteins. J. Biol. Chem. 2018, 293, 3871–3879. [Google Scholar] [CrossRef]
- Eigen, M.; Kruse, W. Über den Einfluß von Wasserstoffbrücken-Struktur und elektrostatischer Wechselwirkung auf die Geschwindigkeit protolytischer Reaktionen. Z. Naturforsch. B 1963, 18, 857–865. [Google Scholar] [CrossRef]
- Eigen, M. Proton transfer, acid-base catalysis, and enzymatic hydrolysis. Part I: Elementary processes. Angew. Chem. Int. Ed. Engl. 1964, 3, 1–19. [Google Scholar] [CrossRef]
- Molt, R.W., Jr.; Pellegrini, E.; Jin, Y. A GAP-GTPase-GDP-Pi Intermediate Crystal Structure Analyzed by DFT Shows GTP Hydrolysis Involves Serial Proton Transfers. Chemistry 2019, 25, 8484–8488. [Google Scholar] [CrossRef]
- Frick, D.N.; Rypma, R.S.; Lam, A.M.; Frenz, C.M. Electrostatic analysis of the hepatitis C virus NS3 helicase reveals both active and allosteric site locations. Nucleic Acids Res. 2004, 32, 5519–5528. [Google Scholar] [CrossRef]
- Frick, D.N. The hepatitis C virus NS3 protein: A model RNA helicase and potential drug target. Curr. Issues Mol. Biol. 2007, 9, 1–20. [Google Scholar] [PubMed]
- Alexov, E.G.; Gunner, M.R. Incorporating protein conformational flexibility into the calculation of pH-dependent protein properties. Biophys. J. 1997, 72, 2075–2093. [Google Scholar] [CrossRef]
- Gu, M.; Rice, C.M. Three conformational snapshots of the hepatitis C virus NS3 helicase reveal a ratchet translocation mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Leipe, D.D.; Aravind, L.; Grishin, N.V.; Koonin, E.V. The bacterial replicative helicase DnaB evolved from a RecA duplication. Genome Res. 2000, 10, 5–16. [Google Scholar]
- Senior, A.E.; al-Shawi, M.K. Further examination of seventeen mutations in Escherichia coli F1-ATPase beta-subunit. J. Biol. Chem. 1992, 267, 21471–21478. [Google Scholar] [CrossRef]
- van der Wolk, J.P.; Klose, M.; de Wit, J.G.; den Blaauwen, T.; Freudl, R.; Driessen, A.J. Identification of the magnesium-binding domain of the high-affinity ATP-binding site of the Bacillus subtilis and Escherichia coli SecA protein. J. Biol. Chem. 1995, 270, 18975–18982. [Google Scholar] [CrossRef]
- Lobau, S.; Weber, J.; Wilke-Mounts, S.; Senior, A.E. F1-ATPase, roles of three catalytic site residues. J. Biol. Chem. 1997, 272, 3648–3656. [Google Scholar] [CrossRef]
- Frelet, A.; Klein, M. Insight in eukaryotic ABC transporter function by mutation analysis. FEBS Lett. 2006, 580, 1064–1084. [Google Scholar] [CrossRef]
- John, J.; Rensland, H.; Schlichting, I.; Vetter, I.; Borasio, G.D.; Goody, R.S.; Wittinghofer, A. Kinetic and structural analysis of the Mg2+-binding site of the guanine nucleotide-binding protein p21H-ras. J. Biol. Chem. 1993, 268, 923–929. [Google Scholar] [CrossRef]
- Frick, D.N.; Banik, S.; Rypma, R.S. Role of divalent metal cations in ATP hydrolysis catalyzed by the hepatitis C virus NS3 helicase: Magnesium provides a bridge for ATP to fuel unwinding. J. Mol. Biol. 2007, 365, 1017–1032. [Google Scholar] [CrossRef]
- Yamashita, T.; Unno, H.; Mori, Y.; Tani, H.; Moriishi, K.; Takamizawa, A.; Agoh, M.; Tsukihara, T.; Matsuura, Y. Crystal structure of the catalytic domain of Japanese encephalitis virus NS3 helicase/nucleoside triphosphatase at a resolution of 1.8 A. Virology 2008, 373, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Scavetta, R.; Chang, X.B. Interaction between the bound Mg.ATP and the Walker A serine residue in NBD2 of multidrug resistance-associated protein MRP1 plays a crucial role for the ATP-dependent leukotriene C4 transport. Biochemistry 2008, 47, 8456–8464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zundel, G. Proton polarizability of hydrogen bonds: Infrared methods, relevance to electrochemical and biological systems. Methods Enzym. 1986, 127, 439–455. [Google Scholar]
- Brzezinski, B.; Zuńdel, G. The role of water and proton-transfer processes in hydrogen-bonded chains with large proton polarizability. Faraday Discuss. 1996, 103, 363–370. [Google Scholar] [CrossRef]
- Kaulen, A.D. Electrogenic processes and protein conformational changes accompanying the bacteriorhodopsin photocycle. Biochim. Biophys. Acta 2000, 1460, 204–219. [Google Scholar] [CrossRef]
- Heberle, J. Proton transfer reactions across bacteriorhodopsin and along the membrane. Biochim. Biophys. Acta 2000, 1458, 135–147. [Google Scholar] [CrossRef]
- Decoursey, T.E. Voltage-gated proton channels and other proton transfer pathways. Physiol. Rev. 2003, 83, 475–579. [Google Scholar] [CrossRef]
- Wraight, C.A. Chance and design—proton transfer in water, channels and bioenergetic proteins. Biochim. Biophys. Acta 2006, 1757, 886–912. [Google Scholar] [CrossRef]
- Mulkidjanian, A.Y. Proton in the well and through the desolvation barrier. Biochim. Biophys. Acta 2006, 1757, 415–427. [Google Scholar] [CrossRef]
- Mulkidjanian, A.Y.; Heberle, J.; Cherepanov, D.A. Protons @ interfaces: Implications for biological energy conversion. Biochim. Biophys. Acta 2006, 1757, 913–930. [Google Scholar] [CrossRef]
- Schubert, L.; Langner, P.; Ehrenberg, D.; Lorenz-Fonfria, V.A.; Heberle, J. Protein conformational changes and protonation dynamics probed by a single shot using quantum-cascade-laser-based IR spectroscopy. J. Chem. Phys. 156 2022, 204201. [Google Scholar] [CrossRef] [PubMed]
- Kaur, D.; Khaniya, U.; Zhang, Y.; Gunner, M.R. Protein Motifs for Proton Transfers That Build the Transmembrane Proton Gradient. Front. Chem. 2021, 9, 660954. [Google Scholar] [CrossRef] [PubMed]
- Pines, D.; Nibbering, E.T.J.; Pines, E. Monitoring the Microscopic Molecular Mechanisms of Proton Transfer in Acid-base Reactions in Aqueous Solutions. Isr. J. Chem. 2015, 55, 1240–1251. [Google Scholar] [CrossRef]
- Borshchevskiy, V.; Kovalev, K.; Round, E.; Efremov, R.; Astashkin, R.; Bourenkov, G.; Bratanov, D.; Balandin, T.; Chizhov, I.; Baeken, C.; et al. True-atomic-resolution insights into the structure and functional role of linear chains and low-barrier hydrogen bonds in proteins. Nat. Struct. Mol. Biol. 2022, 29, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.S. Light-driven proton transfers and proton transport by microbial rhodopsins—A biophysical perspective. Biochim Biophys. Acta Biomembr. 2022, 1864, 183867. [Google Scholar] [CrossRef] [PubMed]
- de Grotthuss, C.J.T. Mémoire Sur la Décomposition de L’Eau ET Des Corps Qu’Elle Tient en Dissolution à L‘Aide de L‘éLectricité Galvanique; Rome, Italy, 1805. [Google Scholar]
- Marx, D. Proton transfer 200 years after von Grotthuss: Insights from ab initio simulations. Chemphyschem 2006, 7, 1848–1870. [Google Scholar] [CrossRef] [PubMed]
- Ceriotti, M.; Cuny, J.; Parrinello, M.; Manolopoulos, D.E. Nuclear quantum effects and hydrogen bond fluctuations in water. Proc. Natl. Acad. Sci. USA 2013, 110, 15591–15596. [Google Scholar] [CrossRef]
- Kaur, D.; Zhang, Y.; Reiss, K.M.; Mandal, M.; Brudvig, G.W.; Batista, V.S.; Gunner, M.R. Proton exit pathways surrounding the oxygen evolving complex of photosystem II. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148446. [Google Scholar] [CrossRef]
- Nadolny, C.; Zundel, G. Fourier transform infrared spectroscopic studies of proton transfer processes and the dissociation of Zn2+-bound water in alcohol dehydrogenases. Eur. J. Biochem. 1997, 247, 914–919. [Google Scholar] [CrossRef]
- Axelrod, H.L.; Abresch, E.C.; Paddock, M.L.; Okamura, M.Y.; Feher, G. Determination of the binding sites of the proton transfer inhibitors Cd2+ and Zn2+ in bacterial reaction centers. Proc. Natl. Acad. Sci. USA 2000, 97, 1542–1547. [Google Scholar] [CrossRef]
- Stowell, M.H.; McPhillips, T.M.; Rees, D.C.; Soltis, S.M.; Abresch, E.; Feher, G. Light-induced structural changes in photosynthetic reaction center: Implications for mechanism of electron-proton transfer. Science 1997, 276, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Epilogue: From energetic abstraction to biochemical mechanism. Symp. Soc. Gen. Microbiol. 1977, 27, 383–423. [Google Scholar]
- Paddock, M.L.; Rongey, S.H.; Feher, G.; Okamura, M.Y. Pathway of proton transfer in bacterial reaction centers: Replacement of glutamic acid 212 in the L subunit by glutamine inhibits quinone (secondary acceptor) turnover. Proc. Natl. Acad. Sci. USA 1989, 86, 6602–6606. [Google Scholar] [CrossRef] [PubMed]
- Drachev, L.; Mamedov, M.; Mulkidjanian, A.Y.; Semenov, A.Y.; Shinkarev, V.; Verkhovsky, M. Electrogenesis associated with proton transfer in the reaction center protein of the purple bacterium Rhodobacter sphaeroides. FEBS Lett. 1990, 259, 324–326. [Google Scholar] [CrossRef] [Green Version]
- Hienerwadel, R.; Grzybek, S.; Fogel, C.; Kreutz, W.; Okamura, M.Y.; Paddock, M.L.; Breton, J.; Nabedryk, E.; Mantele, W. Protonation of Glu L212 following QB− formation in the photosynthetic reaction center of Rhodobacter sphaeroides: Evidence from time-resolved infrared spectroscopy. Biochemistry 1995, 34, 2832–2843. [Google Scholar] [CrossRef]
- Nabedryk, E.; Breton, J.; Hienerwadel, R.; Fogel, C.; Mantele, W.; Paddock, M.L.; Okamura, M.Y. Fourier transforms infrared difference spectroscopy of secondary quinone acceptor photoreduction in proton transfer mutants of Rhodobacter sphaeroides. Biochemistry 1995, 34, 14722–14732. [Google Scholar] [CrossRef]
- Mulkidjanian, A.Y. Conformationally controlled pK-switching in membrane proteins: One more mechanism specific to the enzyme catalysis? FEBS Lett. 1999, 463, 199–204. [Google Scholar] [CrossRef]
- Paddock, M.L.; McPherson, P.H.; Feher, G.; Okamura, M.Y. Pathway of proton transfer in bacterial reaction centers: Replacement of serine-L223 by alanine inhibits electron and proton transfers associated with reduction of quinone to dihydroquinone. Proc. Natl. Acad. Sci. USA 1990, 87, 6803–6807. [Google Scholar] [CrossRef]
- Paddock, M.L.; Rongey, S.H.; McPherson, P.H.; Juth, A.; Feher, G.; Okamura, M.Y. Pathway of proton transfer in bacterial reaction centers: Role of aspartate-L213 in proton transfers associated with reduction of quinoneto dihydroquinone. Biochemistry 1994, 33, 734–745. [Google Scholar] [CrossRef]
- Gopta, O.A.; Cherepanov, D.A.; Junge, W.; Mulkidjanian, A.Y. Proton transfer from the bulk to the bound ubiquinone QB of the reaction center in chromatophores of Rhodobacter sphaeroides: Retarded conveyance by neutral water. Proc. Natl. Acad. Sci. USA 1999, 96, 13159–13164. [Google Scholar] [CrossRef]
- Paddock, M.L.; Feher, G.; Okamura, M.Y. Proton transfer pathways and mechanism in bacterial reaction centers. FEBS Lett. 2003, 555, 45–50. [Google Scholar] [CrossRef]
- Cherepanov, D.A.; Bibikov, S.I.; Bibikova, M.V.; Bloch, D.A.; Drachev, L.A.; Gopta, O.A.; Oesterhelt, D.; Semenov, A.Y.; Mulkidjanian, A.Y. Reduction and protonation of the secondary quinone acceptor of Rhodobacter sphaeroides photosynthetic reaction center: Kinetic model based on a comparison of wild-type chromatophores with mutants carrying Arg-->Ile substitution at sites 207 and 217 in the L-subunit. Biochim. Biophys. Acta 2000, 1459, 10–34. [Google Scholar] [PubMed]
- Mulkidjanian, A.Y.; Kozlova, M.A.; Cherepanov, D.A. Ubiquinone reduction in the photosynthetic reaction centre of Rhodobacter sphaeroides: Interplay between electron transfer, proton binding and flips of the quinone ring. Biochem. Soc. Trans. 2005, 33, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Klare, J.P.; Bordignon, E.; Engelhard, M.; Steinhoff, H.J. Sensory rhodopsin II and bacteriorhodopsin: Light activated helix F movement. Photochem Photobiol Sci 2004, 3, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Lanyi, J.K. Proton transfers in the bacteriorhodopsin photocycle. Biochim. Et Biophys. Acta Bioenerg. 2006, 1757, 1012–1018. [Google Scholar] [CrossRef]
- Nogly, P.; Weinert, T.; James, D.; Carbajo, S.; Ozerov, D.; Furrer, A.; Gashi, D.; Borin, V.; Skopintsev, P.; Jaeger, K.; et al. Retinal isomerization in bacteriorhodopsin captured by a femtosecond x-ray laser. Science 2018, 361, eaat0094. [Google Scholar] [CrossRef]
- Schobert, B.; Brown, L.S.; Lanyi, J.K. Crystallographic structures of the M and N intermediates of bacteriorhodopsin: Assembly of a hydrogen-bonded chain of water molecules between Asp-96 and the retinal Schiff base. J. Mol. Biol. 2003, 330, 553–570. [Google Scholar] [CrossRef]
- Marti, T.; Otto, H.; Mogi, T.; Rosselet, S.J.; Heyn, M.P.; Khorana, H.G. Bacteriorhodopsin mutants containing single substitutions of serine or threonine residues are all active in proton translocation. J. Biol. Chem. 1991, 266, 6919–6927. [Google Scholar] [CrossRef]
- Steinhoff, H.J.; Mollaaghababa, R.; Altenbach, C.; Hideg, K.; Krebs, M.; Khorana, H.G.; Hubbell, W.L. Time-resolved detection of structural changes during the photocycle of spin-labeled bacteriorhodopsin. Science 1994, 266, 105–107. [Google Scholar] [CrossRef]
- Luecke, H.; Schobert, B.; Cartailler, J.P.; Richter, H.T.; Rosengarth, A.; Needleman, R.; Lanyi, J.K. Coupling photoisomerization of retinal to directional transport in bacteriorhodopsin. J. Mol. Biol. 2000, 300, 1237–1255. [Google Scholar] [CrossRef]
- Rink, T.; Pfeiffer, M.; Oesterhelt, D.; Gerwert, K.; Steinhoff, H.J. Unraveling photoexcited conformational changes of bacteriorhodopsin by time resolved electron paramagnetic resonance spectroscopy. Biophys. J. 2000, 78, 1519–1530. [Google Scholar] [CrossRef]
- Heberle, J.; Fitter, J.; Sass, H.J.; Buldt, G. Bacteriorhodopsin: The functional details of a molecular machine are being resolved. Biophys. Chem. 2000, 85, 229–248. [Google Scholar] [CrossRef]
- Lazaratos, M.; Siemers, M.; Brown, L.S.; Bondar, A.N. Conserved hydrogen-bond motifs of membrane transporters and receptors. Biochim. Biophys. Acta 2022, 1864, 183896. [Google Scholar] [CrossRef] [PubMed]
- Zscherp, C.; Schlesinger, R.; Tittor, J.; Oesterhelt, D.; Heberle, J. In situ determination of transient pKa changes of internal amino acids of bacteriorhodopsin by using time-resolved attenuated total reflection Fourier-transform infrared spectroscopy. Proc. Natl. Acad. Sci. USA 1999, 96, 5498–5503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Varo, G.; Klinger, A.L.; Czajkowsky, D.M.; Braiman, M.S.; Needleman, R.; Lanyi, J.K. Proton transfer from Asp-96 to the bacteriorhodopsin Schiff base is caused by a decrease of the pKa of Asp-96 which follows a protein backbone conformational change. Biochemistry 1993, 32, 1981–1990. [Google Scholar] [CrossRef]
- Drachev, L.A.; Kaulen, A.D.; Khitrina, L.V.; Skulachev, V.P. Fast stages of photoelectric processes in biological membranes. I. Bacteriorhodopsin. Eur. J. Biochem. 1981, 117, 461–470. [Google Scholar] [CrossRef]
- Drachev, L.; Kaulen, A.; Skulachev, V. Correlation of photochemical cycle, H+ release and uptake, and electric events in bacteriorhodopsin. FEBS Lett. 1984, 178, 331–335. [Google Scholar] [CrossRef]
- Brzezinski, P.; Paddock, M.L.; Okamura, M.Y.; Feher, G. Light-induced electrogenic events associated with proton uptake upon forming QB- in bacterial wild-type and mutant reaction centers. Biochim. Biophys. Acta 1997, 1321, 149–156. [Google Scholar] [CrossRef]
- Drachev, L.; Kaurov, B.; Mamedov, M.; Mulkidjanian, A.Y.; Semenov, A.Y.; Shinkarev, V.; Skulachev, V.; Verkhovsky, M. Flash-induced electrogenic events in the photosynthetic reaction center and bc1 complexes of Rhodobacter sphaeroides chromatophores. Biochim. Biophys. Acta. 1989, 973, 189–197. [Google Scholar] [CrossRef]
- Kozlova, M.A.; Juhnke, H.D.; Cherepanov, D.A.; Lancaster, C.R.; Mulkidjanian, A.Y. Proton transfer in the photosynthetic reaction center of Blastochloris viridis. FEBS Lett. 2008, 582, 238–242. [Google Scholar] [CrossRef]
- Engelhard, M.; Gerwert, K.; Hess, B.; Kreutz, W.; Siebert, F. Light-driven protonation changes of internal aspartic acids of bacteriorhodopsin: An investigation by static and time-resolved infrared difference spectroscopy using [4-13C]aspartic acid labeled purple membrane. Biochemistry 1985, 24, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Wegener, A.A.; Chizhov, I.; Engelhard, M.; Steinhoff, H.J. Time-resolved detection of transient movement of helix F in spin-labelled pharaonis sensory rhodopsin II. J. Mol. Biol. 2000, 301, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Warshel, A.; Russell, S.T. Calculations of electrostatic interactions in biological systems and in solutions. Q. Rev. Biophys. 1984, 17, 283–422. [Google Scholar] [CrossRef] [PubMed]
- Bozdaganyan, M.E.; Lokhmatikov, A.V.; Voskoboynikova, N.; Cherepanov, D.A.; Steinhoff, H.J.; Shaitan, K.V.; Mulkidjanian, A.Y. Proton leakage across lipid bilayers: Oxygen atoms of phospholipid ester linkers align water molecules into transmembrane water wires. Biochim. Biophys. Acta 2019, 1860, 439–451. [Google Scholar] [CrossRef]
- Harris, T.K.; Turner, G.J. Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life 2002, 53, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Walker, J. ATP synthesis by rotary catalysis. Angew. Chem. Int. Ed. Engl. 1998, 37, 2309–2319. [Google Scholar] [CrossRef]
- Boyer, P.D. The ATP synthase—A splendid molecular machine. Annu. Rev. Biochem. 1997, 66, 717–749. [Google Scholar] [CrossRef]
- Kagawa, R.; Montgomery, M.G.; Braig, K.; Leslie, A.G.; Walker, J.E. The structure of bovine F1-ATPase inhibited by ADP and beryllium fluoride. EMBO J. 2004, 23, 2734–2744. [Google Scholar] [CrossRef]
- Bowler, M.W.; Montgomery, M.G.; Leslie, A.G.; Walker, J.E. Ground state structure of F1-ATPase from bovine heart mitochondria at 1.9 A resolution. J. Biol. Chem. 2007, 282, 14238–14242. [Google Scholar] [CrossRef]
- Menz, R.I.; Leslie, A.G.; Walker, J.E. The structure and nucleotide occupancy of bovine mitochondrial F1-ATPase are not influenced by crystallisation at high concentrations of nucleotide. FEBS Lett. 2001, 494, 11–14. [Google Scholar] [CrossRef]
- Sigala, P.A.; Ruben, E.A.; Liu, C.W.; Piccoli, P.M.; Hohenstein, E.G.; Martinez, T.J.; Schultz, A.J.; Herschlag, D. Determination of Hydrogen Bond Structure in Water versus Aprotic Environments To Test the Relationship Between Length and Stability. J. Am. Chem. Soc. 2015, 137, 5730–5740. [Google Scholar] [CrossRef] [PubMed]
- Koeppe, B.; Tolstoy, P.M.; Limbach, H.H. Reaction pathways of proton transfer in hydrogen-bonded phenol-carboxylate complexes explored by combined UV-vis and NMR spectroscopy. J. Am. Chem. Soc. 2011, 133, 7897–7908. [Google Scholar] [CrossRef] [PubMed]
- Koeppe, B.; Guo, J.; Tolstoy, P.M.; Denisov, G.S.; Limbach, H.H. Solvent and H/D isotope effects on the proton transfer pathways in heteroconjugated hydrogen-bonded phenol-carboxylic acid anions observed by combined UV-vis and NMR spectroscopy. J. Am. Chem. Soc. 2013, 135, 7553–7566. [Google Scholar] [CrossRef] [PubMed]
- Pylaeva, S.; Allolio, C.; Koeppe, B.; Denisov, G.S.; Limbach, H.H.; Sebastiani, D.; Tolstoy, P.M. Proton transfer in a short hydrogen bond caused by solvation shell fluctuations: An ab initio MD and NMR/UV study of an (OHO)(-) bonded system. Phys. Chem. Chem. Phys. 2015, 17, 4634–4644. [Google Scholar] [CrossRef] [PubMed]
- Fersht, A. Enzyme Structure and Mechanism; W. H. Freeman & Co: New York, NY, USA, 1985. [Google Scholar]
- Jencks, W.P. Catalysis in Chemistry and Enzymology; Courier Corporation: Chelmsford, MA, USA, 1987. [Google Scholar]
- Uversky, V.N. The intrinsic disorder alphabet. III. Dual personality of serine. Intrinsically Disord. Proteins 2015, 3, e1027032. [Google Scholar] [CrossRef] [PubMed]
- Dehghani-Tafti, S.; Levdikov, V.; Antson, A.A.; Bax, B.; Sanders, C.M. Structural and functional analysis of the nucleotide and DNA binding activities of the human PIF1 helicase. Nucleic Acids Res. 2019, 47, 3208–3222. [Google Scholar] [CrossRef]
- Dai, Y.X.; Chen, W.F.; Liu, N.N.; Teng, F.Y.; Guo, H.L.; Hou, X.M.; Dou, S.X.; Rety, S.; Xi, X.G. Structural and functional studies of SF1B Pif1 from Thermus oshimai reveal dimerization-induced helicase inhibition. Nucleic Acids Res. 2021, 49, 4129–4143. [Google Scholar] [CrossRef]
- Coureux, P.D.; Sweeney, H.L.; Houdusse, A. Three myosin V structures delineate essential features of chemo-mechanical transduction. EMBO J. 2004, 23, 4527–4537. [Google Scholar] [CrossRef]
- Lin, Y.; Lu, S.; Zhang, J.; Zheng, Y. Structure of an inactive conformation of GTP-bound RhoA GTPase. Structure 2021, 29, 553–563.e555. [Google Scholar] [CrossRef]
- Kumawat, A.; Chakrabarty, S.; Kulkarni, K. Nucleotide Dependent Switching in Rho GTPase: Conformational Heterogeneity and Competing Molecular Interactions. Sci. Rep. 2017, 7, 45829. [Google Scholar] [CrossRef]
- Pasqualato, S.; Cherfils, J. Crystallographic evidence for substrate-assisted GTP hydrolysis by a small GTP binding protein. Structure 2005, 13, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Kotting, C.; Gerwert, K. Proteins in action monitored by time-resolved FTIR spectroscopy. Chemphyschem 2005, 6, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Barth, A.; Zscherp, C. What vibrations tell us about proteins. Q. Rev. Biophys. 2002, 35, 369–430. [Google Scholar] [CrossRef] [PubMed]
- Parke, C.L.; Wojcik, E.J.; Kim, S.; Worthylake, D.K. ATP hydrolysis in Eg5 kinesin involves a catalytic two-water mechanism. J. Biol. Chem. 2010, 285, 5859–5867. [Google Scholar] [CrossRef] [Green Version]
- Wojcik, E.J.; Dalrymple, N.A.; Alford, S.R.; Walker, R.A.; Kim, S. Disparity in allosteric interactions of monastrol with Eg5 in the presence of ADP and ATP: A difference FT-IR investigation. Biochemistry 2004, 43, 9939–9949. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Sardana, V.; Xu, B.; Homnick, C.; Halczenko, W.; Buser, C.A.; Schaber, M.; Hartman, G.D.; Huber, H.E.; Kuo, L.C. Inhibition of a mitotic motor protein: Where, how, and conformational consequences. J. Mol. Biol. 2004, 335, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Cochran, J.C.; Gatial, J.E., 3rd; Kapoor, T.M.; Gilbert, S.P. Monastrol inhibition of the mitotic kinesin Eg5. J. Biol. Chem. 2005, 280, 12658–12667. [Google Scholar] [CrossRef]
- Jun, B.; Kim, S. Real-time structural transitions are coupled to chemical steps in ATP hydrolysis by Eg5 kinesin. J. Biol. Chem. 2010, 285, 11073–11077. [Google Scholar] [CrossRef]
- Xu, R.; Yang, Y.; Zheng, X. Unique structural features of the adenylate kinase hCINAP/AK6 and its multifaceted functions in carcinogenesis and tumor progression. FEBS Lett. 2021, 595, 2071–2084. [Google Scholar] [CrossRef]
- Drakou, C.E.; Malekkou, A.; Hayes, J.M.; Lederer, C.W.; Leonidas, D.D.; Oikonomakos, N.G.; Lamond, A.I.; Santama, N.; Zographos, S.E. hCINAP is an atypical mammalian nuclear adenylate kinase with an ATPase motif: Structural and functional studies. Proteins 2012, 80, 206–220. [Google Scholar] [CrossRef]
- Knihtila, R.; Holzapfel, G.; Weiss, K.; Meilleur, F.; Mattos, C. Neutron Crystal Structure of RAS GTPase Puts in Question the Protonation State of the GTP gamma-Phosphate. J. Biol. Chem. 2015, 290, 31025–31036. [Google Scholar] [CrossRef] [PubMed]
- Leipe, D.D.; Koonin, E.V.; Aravind, L. STAND, a class of P-loop NTPases including animal and plant regulators of programmed cell death: Multiple, complex domain architectures, unusual phyletic patterns, and evolution by horizontal gene transfer. J. Mol. Biol. 2004, 343, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Rayment, I.; Holden, H.M. The three-dimensional structure of a molecular motor. Trends Biochem. Sci. 1994, 19, 129–134. [Google Scholar] [CrossRef]
- Smith, C.A.; Rayment, I. X-ray structure of the magnesium(II)-pyrophosphate complex of the truncated head of Dictyostelium discoideum myosin to 2.7 A resolution. Biochemistry 1995, 34, 8973–8981. [Google Scholar] [CrossRef]
- Rayment, I.; Smith, C.; Yount, R.G. The active site of myosin. Annu. Rev. Physiol. 1996, 58, 671–702. [Google Scholar] [CrossRef]
- Gulick, A.M.; Bauer, C.B.; Thoden, J.B.; Rayment, I. X-ray structures of the MgADP, MgATPgammaS, and MgAMPPNP complexes of the Dictyostelium discoideum myosin motor domain. Biochemistry 1997, 36, 11619–11628. [Google Scholar] [CrossRef]
- Fedorov, R.; Bohl, M.; Tsiavaliaris, G.; Hartmann, F.K.; Taft, M.H.; Baruch, P.; Brenner, B.; Martin, R.; Knolker, H.J.; Gutzeit, H.O.; et al. The mechanism of pentabromopseudilin inhibition of myosin motor activity. Nat. Struct. Mol. Biol. 2009, 16, 80–88. [Google Scholar] [CrossRef]
- Frye, J.J.; Klenchin, V.A.; Bagshaw, C.R.; Rayment, I. Insights into the importance of hydrogen bonding in the gamma-phosphate binding pocket of myosin: Structural and functional studies of serine 236. Biochemistry 2010, 49, 4897–4907. [Google Scholar] [CrossRef]
- Shalaeva, D.N.; Cherepanov, D.A.; Galperin, M.Y.; Mulkidjanian, A.Y. Comparative analysis of active sites in P-loop nucleoside triphosphatases suggests an ancestral activation mechanism. BioRxiv 2018. [Google Scholar] [CrossRef]
- Cleland, W.W. Low-barrier hydrogen bonds and low fractionation factor bases in enzymatic reactions. Biochemistry 1992, 31, 317–319. [Google Scholar] [CrossRef]
- Frey, P.A.; Whitt, S.A.; Tobin, J.B. A low-barrier hydrogen bond in the catalytic triad of serine proteases. Science 1994, 264, 1927–1930. [Google Scholar] [CrossRef] [PubMed]
- Perrin, C.L.; Nielson, J.B. “Strong” hydrogen bonds in chemistry and biology. Annu. Rev. Phys. Chem. 1997, 48, 511–544. [Google Scholar] [CrossRef] [PubMed]
- Perrin, C.L. Are short, low-barrier hydrogen bonds unusually strong? Acc. Chem. Res. 2010, 43, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Wahlgren, W.Y.; Pal, G.; Kardos, J.; Porrogi, P.; Szenthe, B.; Patthy, A.; Graf, L.; Katona, G. The catalytic aspartate is protonated in the Michaelis complex formed between trypsin and an in vitro evolved substrate-like inhibitor: A refined mechanism of serine protease action. J. Biol. Chem. 2011, 286, 3587–3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Agarwal, P.K.; Waddell, M.B.; Mittag, T.; Serpersu, E.H.; Cuneo, M.J. Low-Barrier and Canonical Hydrogen Bonds Modulate Activity and Specificity of a Catalytic Triad. Angew. Chem. Int. Ed. Engl. 2019, 58, 16260–16266. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Agarwal, P.K.; Cuneo, M.J. On the Case of the Misplaced Hydrogens. Chembiochem 2021, 22, 288–297. [Google Scholar] [CrossRef]
- Nagy, G.N.; Suardiaz, R.; Lopata, A.; Ozohanics, O.; Vekey, K.; Brooks, B.R.; Leveles, I.; Toth, J.; Vertessy, B.G.; Rosta, E. Structural Characterization of Arginine Fingers: Identification of an Arginine Finger for the Pyrophosphatase dUTPases. J. Am. Chem. Soc. 2016, 138, 15035–15045. [Google Scholar] [CrossRef]
- Grigorenko, B.; Polyakov, I.; Nemukhin, A. Mechanisms of ATP to cAMP Conversion Catalyzed by the Mammalian Adenylyl Cyclase: A Role of Magnesium Coordination Shells and Proton Wires. J. Phys. Chem. B 2020, 124, 451–460. [Google Scholar] [CrossRef]
- Lopata, A.; Jambrina, P.G.; Sharma, P.K.; Brooks, B.R.; Toth, J.; Vertessy, B.G.; Rosta, E. Mutations decouple proton transfer from phosphate cleavage in the dutpase catalytic reaction. ACS Catal. 2015, 5, 3225–3237. [Google Scholar] [CrossRef]
- Cleland, W.W.; Frey, P.A.; Gerlt, J.A. The low barrier hydrogen bond in enzymatic catalysis. J. Biol. Chem. 1998, 273, 25529–25532. [Google Scholar] [CrossRef]
- Ngo, P.D.; Mansoorabadi, S.O.; Frey, P.A. Serine Protease Catalysis: A Computational Study of Tetrahedral Intermediates and Inhibitory Adducts. J. Phys. Chem. B 2016, 120, 7353–7359. [Google Scholar] [CrossRef] [PubMed]
- Reikhardt, B.A.; Shabanov, P.D. Catalytic Subunit of PKA as a Prototype of the Eukaryotic Protein Kinase Family. Biochemistry (Moscow) 2020, 85, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Endicott, J.A.; Noble, M.E.; Johnson, L.N. The structural basis for control of eukaryotic protein kinases. Annu. Rev. Biochem. 2012, 81, 587–613. [Google Scholar] [CrossRef]
- Kottke, T.; Lorenz-Fonfria, V.A.; Heberle, J. The Grateful Infrared: Sequential Protein Structural Changes Resolved by Infrared Difference Spectroscopy. J. Phys. Chem. B 2017, 121, 335–350. [Google Scholar] [CrossRef] [PubMed]

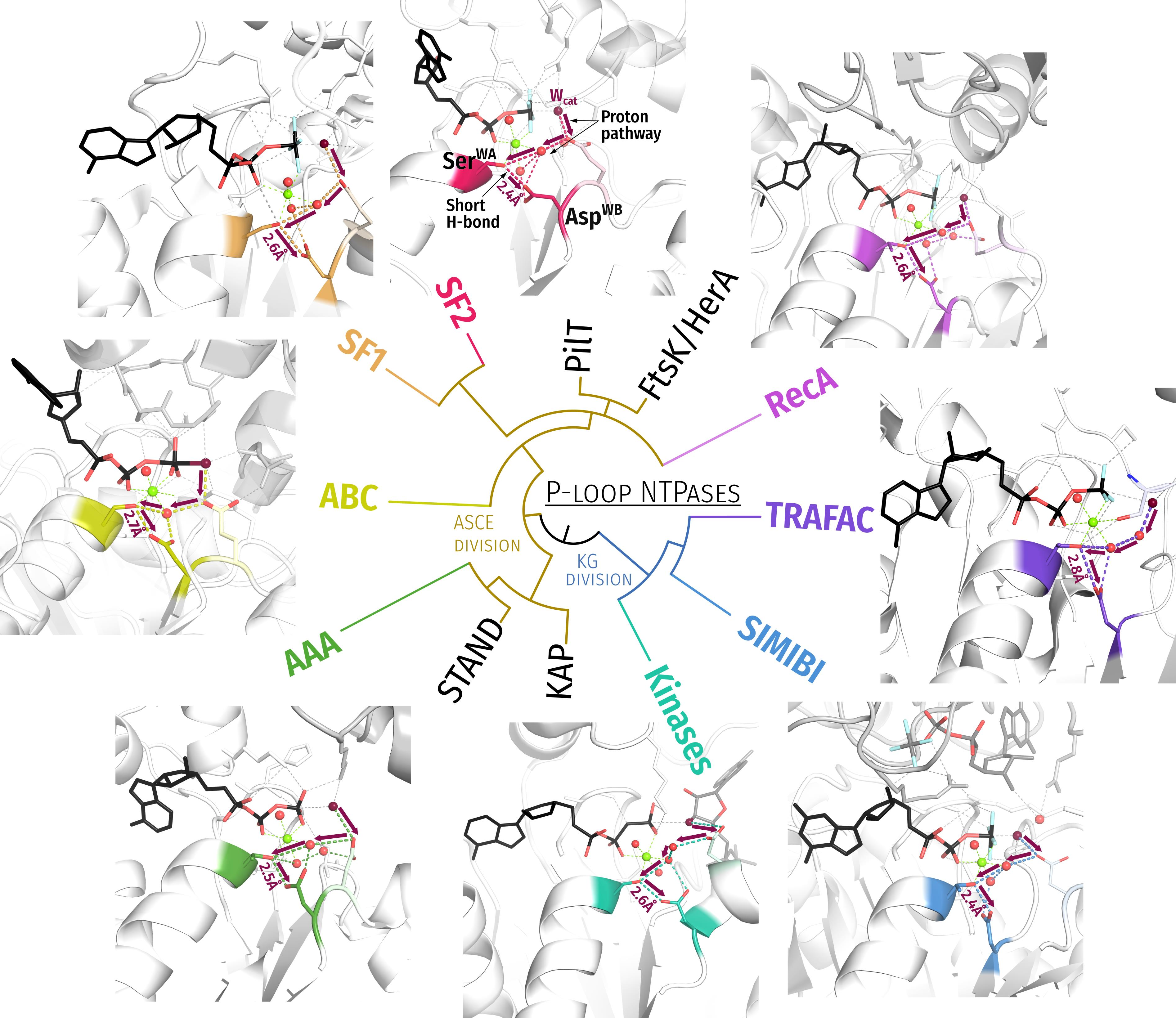{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P-loop Class, Activation Mechanism | Representative Protein Structure | Site ID in Table S1 | Figure in the Text | PDB Entry ID, Resolution | NTP or NTP Analog | Walker A Motif (K-3 and K+1 Residues) | Walker B Asp/Glu | Stimulatory Moieties | Coordination of Wcat a | |
|---|---|---|---|---|---|---|---|---|---|---|
| Kinase-GTPase Division | ||||||||||
| TRAFAC Interaction with the activating partner or dimerization in the presence of the activating partner leads to the stabilization of the Switch I loop and insertion of diverse stimulatory moieties into the catalytic site | RhoA | 3508 | 1C,E | 1OW3, 1.8 Å | GDP:MgF3− | Ala15 | Thr19 | Asp59 | Arg85†-NH2 (AG) | Gln63D+4-OE1, Thr37SwI-CO |
| MnmE | 236 | 2A | 2GJ8, 1.7 Å | GDP:AlF4− | Asn226 | Ser230 | Asp270 | K+ ion (AG) | Thr251SwI-CO, Gly249T−2-HN, Thr250T−1-HN, Gly273D+3-HN, Gly273D+3-CO* | |
| Dynamin | 3550 | 3A | 2X2E, 2.0 Å | GDP:AlF4− | Ser41 | Ser45 | Asp136 | K+ or Na+ ion (G) | Thr65SwI-CO, Gly139D+3-HN, Gly139D+3-CO*, Gln40K−4-OE1* | |
| Atlastin | 3661 | 3B | 6B9F, 1.9 Å | GDP:AlF4− | Arg77 | Ser81 | Asp146 | Arg77K−3-NH2 (AG) | Gly149D+3-HN, Thr120SwI-CO, Gly149D+3-CO*, Asp152D+6-OD2* | |
| Gα3 | 3545 | 3C | 2ODE, 1.9 Å | GDP:AlF4− | Glu43 | Ser47 | Asp200 | Arg178T−3-NH1 (AG) | Thr181SwI-CO, Gln204D+4-OE1, Gly203D+3-HN | |
| Myosin II | 42 | 3D | 1VOM, 1.9 Å | ADP-VO43− | Gly182 | Thr186 | Asp454 | Asn233S−4-ND2 (AG) | Ser237SwI-CO, Ser236S−1-OG, Gly457D+3-HN, Gly457D+3-CO, Arg238S+1*-NH1,Glu459D+5*-OE1 | |
| SIMIBI Monomeric domains dimerize and provide activating Lys or Arg fingers for each other in response to the interaction with the activating partner | GET3 | 64 | 4A | 2YNM, 2.1 Å | ADP:AlF4− | Gly39 | Ser43 | Asp151 | Lys37†-NZ (AG) | Asp66WB+1-OD2, Asp155†-OD1, Lys68WB+1-NZ, Gly154D+3-HN, Lys37†-HN* |
| Signal recognition particle | 3522 | 4B | 2CNW, 2.39 Å | GDP:AlF4− | Gly108 | Thr112 | Asp187 | Arg138-NH1 (AG) | Gly190D+3-HN, Asp135WB+1-OD2, GTP†-O3′*, Gly190D+3-CO*, Glu284†-OE1* | |
| Kinases Rearrangement of the lid domain upon binding of the second substrate leads to the insertion of Arg/Lys fingers into the catalytic site | Thymidylate kinase | 1207 | 4C | 1NN5, 1.5 Å | ANP | Arg16 | Ser20 | Asp96 | Arg16K−3-NH1 (AG?), Arg97D+1-NH2 (G) | The second substrate is coordinated by Arg45WB+1-NH2, Arg-97D+1-NE, Glu149Lid-OE1, Pro43-CO* |
| Adenosine 5′-phosphosulfate kinase | 1490 | 4D | 4BZX, 1.7 Å | ANP | Gly453 | Ser457 | Asp478 | Lys562Lid-NZ (G) | The second substrate is coordinated by Asp41D+2-OD1, Lys562Lid-NZ, Arg483D+5-NH2, Arg497-NH1 | |
| Adenylate kinase | xxx0 | S8B | 3SR0, 1.6 Å | ADP:AlF4− | Gly10 | Gly14 | Asp81 | Arg124Lid-NH1 (AG) Arg124Lid-NH2 (G) Arg150Lid-NH1 (G) Arg161Lid-NH1(G) | The second substrate (phosphate acceptor) is coordinated by Arg150Lid-NH2, Arg85D+4-NH1, Arg85D+4-NH2, Arg36WB+1-NH1, Arg36WB+1-NH2 | |
| P-loop class, activation mechanism | Representative protein structure | Site ID in Table S1 | Figure in the text | PDB entry ID | NTP or NTP analogue | Walker A motif (K-3 and and K+1 residues) | Walker B motif, [Asp/Glu]WB | Stimulatory moieties | Coordination of Wcata | |
| ASCE Division | ||||||||||
| AAA+/SF3 ATPases In a hexamer, the binding/hydrolysis of ATP in one subunit causes conformational changes activating the adjacent subunit; the activation involves class-specific helical domain | N-ethylmaleimide sensitive factor | 359 | 5A | 1NSF, 1.9 Å | ATP | His546 | Thr550 | Asp603 | Lys708‡-NZ (AG) Lys631†-NZ (G) | Asp604D+1-OD2, Lys631†-NZ, Ser647WB−1-OG |
| SV40 large T antigen helicase (SF3) | 372 | 5B | 1SVM, 1.9 Å | ATP | Asp429 | Thr433 | Glu473 | Lys418†-NZ (AG), Arg540†-NH2 (G) | Asp474E+1-OD1, Asn529WB−1-OD1, Arg498†-NH1, Arg540†-NH2 | |
| Helicases SF1/2: Rearrangement of the C-terminal domain upon DNA or RNA binding leads to the insertion of an Arg finger into the nucleotide-binding domain | Chikungunya virus nsP2 helicase (SF1) | N/A | 1D, 1F | 6JIM, 2.0 Å | ADP:AlF4− | Gly189 | Ser193 | Asp252 | Arg312‡-NH2 (AG), Arg416‡-NH1 (G), Arg416‡-NH2 (G), | Glu253D+1-OE2,Gln283D+1-OE2, Gly384‡-HN |
| HCV NS3 helicase (SF2) | 136 | 5C | 3KQL, 2.5 Å | ADP:AlF4− | Gly207 | Ser211 | Asp290 | Arg467‡-NH2 (AG), Arg467‡ -NH1 (G), Arg467‡ -NH2 (G), Arg464‡ -NH1 (G), Arg464‡ -NH2 (G), | Glu291D+1-OE1,Gln460‡-OE1, Arg464‡-NH2,Gly417‡-NH, Ala323-NHWB+1* | |
| Multifunctional helicase Pif1p (SF1) | 233 | 11B | 5O6B, 2.0Å | ADP:AlF4− | Gly261 | Ser265 | Asp-341 | Arg417‡-NH2 (AG) | Glu342D+1-OE1,Gln381WB−1-OE1, Arg734‡-NH1, Gly709‡-HN | |
| ABC ATPases: Monomeric domains dimerize and provide activating LSGGQ motifs for each other in response to the substrate binding | Maltose transporter | 154 | 5D | 3PUW, 2.2 Å | ADP:AlF4− | Gly39 | Ser43 | Asp158 | Ser135†-OG (G), Gly137†-HN (G) | Gln82WB+1-NE2,Glu159D+1-OE1, Glu159D+1-OE2,Asn163†-CO, His192 WB−1-NE2 |
| F1/RecA-like: In an oligomer, ATP binding/hydrolysis in one subunit causes conformational changes that activate the adjacent subunit by inserting Arg/Lys fingers | F1-ATPase | 18 | 6A | 1H8E, 2.0 Å | ADP:AlF4− | Gly159 | Thr163 | Asp256 | Arg373†-NH1 (AG), Arg189WB−1-NH1 (G), Arg189WB−1-NH2 (G), | Glu188WB−1-OE1, Arg260D+4-NH2, Ser344†-CO |
| Replicative helicase DnaB | N/A | 6B | 6T66, 3.9 Å | GDP-AlF4− | Ser231 | Thr235 | Asp340 | Arg439†-NE (G), Lys437†-NZ (G) | Glu259WB+1**, Tyr341D+1** Gln381WB−1**Arg439† | |
| Circadian clock protein KaiC | 683 | 6C | 4TL7, 1.9 Å | ATP | Gly49 | Thr53 | Asp145 | Lys224†-NZ (G) Arg226†-NH2 (G) | Glu183WB−1-OE1**, Ser146D+1**, Phe199†-CO | |
| Recombinase RadA | 1376 | 6D | 3EW9, 2.4 Å | ANP | Gly108 | Thr112 | Asp211 | K+-503 (G), K+-504 (G) | Glu151WB+1**, Ser212D+1** Gln257WB−1**, | |
| RecA | 100 | N/A | 3CMX, 3.4 Å | ADP:AlF4− | Ser69 | Thr73 | Asp144 | Lys248†-NZ (G) Lys250†-NZ (G) | Glu96D+1**, Phe216**†-CO, Gln194**D−1, | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozlova, M.I.; Shalaeva, D.N.; Dibrova, D.V.; Mulkidjanian, A.Y. Common Mechanism of Activated Catalysis in P-loop Fold Nucleoside Triphosphatases—United in Diversity. Biomolecules 2022, 12, 1346. https://doi.org/10.3390/biom12101346
Kozlova MI, Shalaeva DN, Dibrova DV, Mulkidjanian AY. Common Mechanism of Activated Catalysis in P-loop Fold Nucleoside Triphosphatases—United in Diversity. Biomolecules. 2022; 12(10):1346. https://doi.org/10.3390/biom12101346
Chicago/Turabian StyleKozlova, Maria I., Daria N. Shalaeva, Daria V. Dibrova, and Armen Y. Mulkidjanian. 2022. "Common Mechanism of Activated Catalysis in P-loop Fold Nucleoside Triphosphatases—United in Diversity" Biomolecules 12, no. 10: 1346. https://doi.org/10.3390/biom12101346
APA StyleKozlova, M. I., Shalaeva, D. N., Dibrova, D. V., & Mulkidjanian, A. Y. (2022). Common Mechanism of Activated Catalysis in P-loop Fold Nucleoside Triphosphatases—United in Diversity. Biomolecules, 12(10), 1346. https://doi.org/10.3390/biom12101346