
Looking at the Pathogenesis of the Rabies Lyssavirus Strain Pasteur Vaccins through a Prism of the Disorder-Based Bioinformatics


Abstract
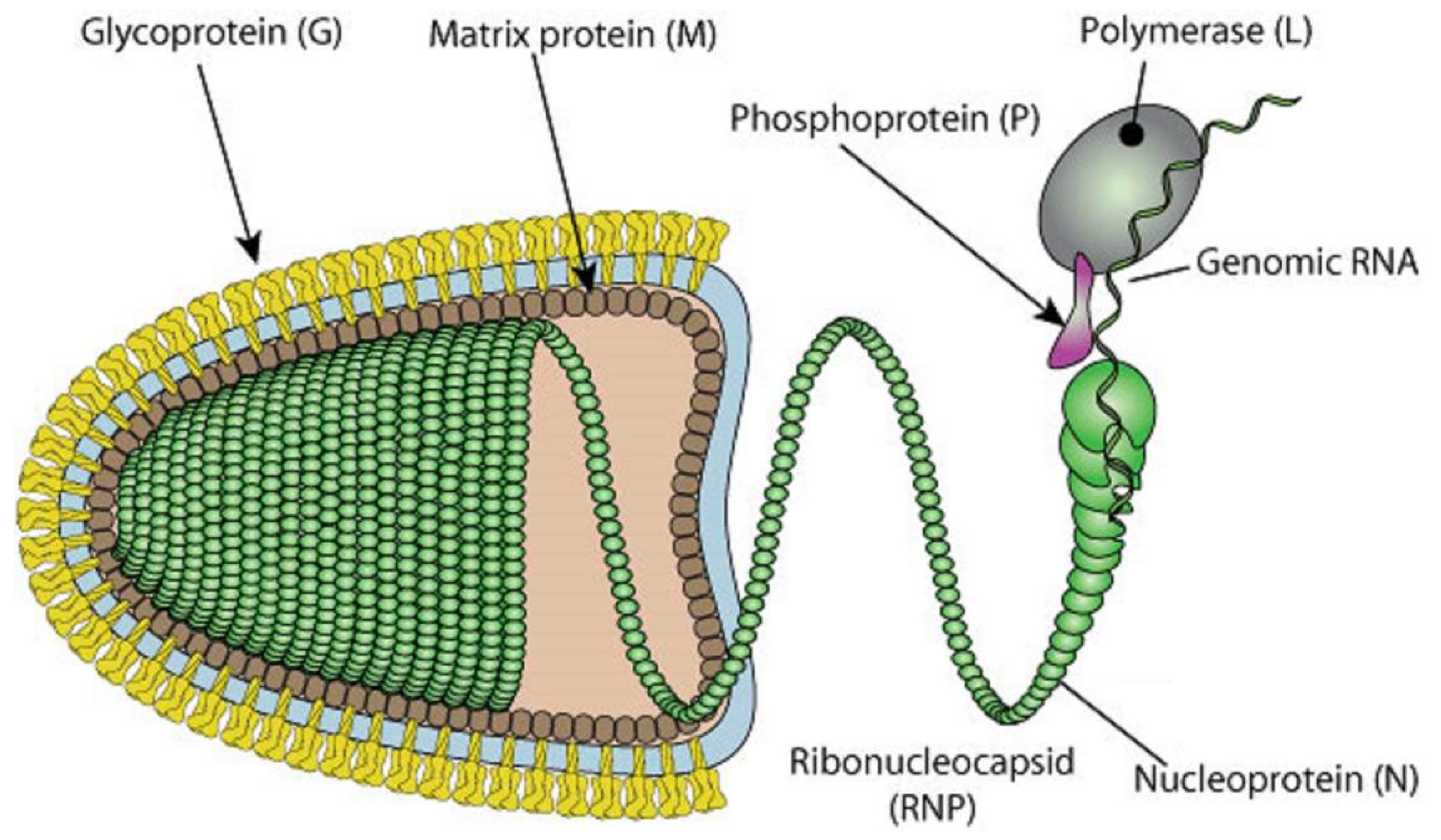
:1. Introduction
2. Materials and Methods
3. Results and Discussion
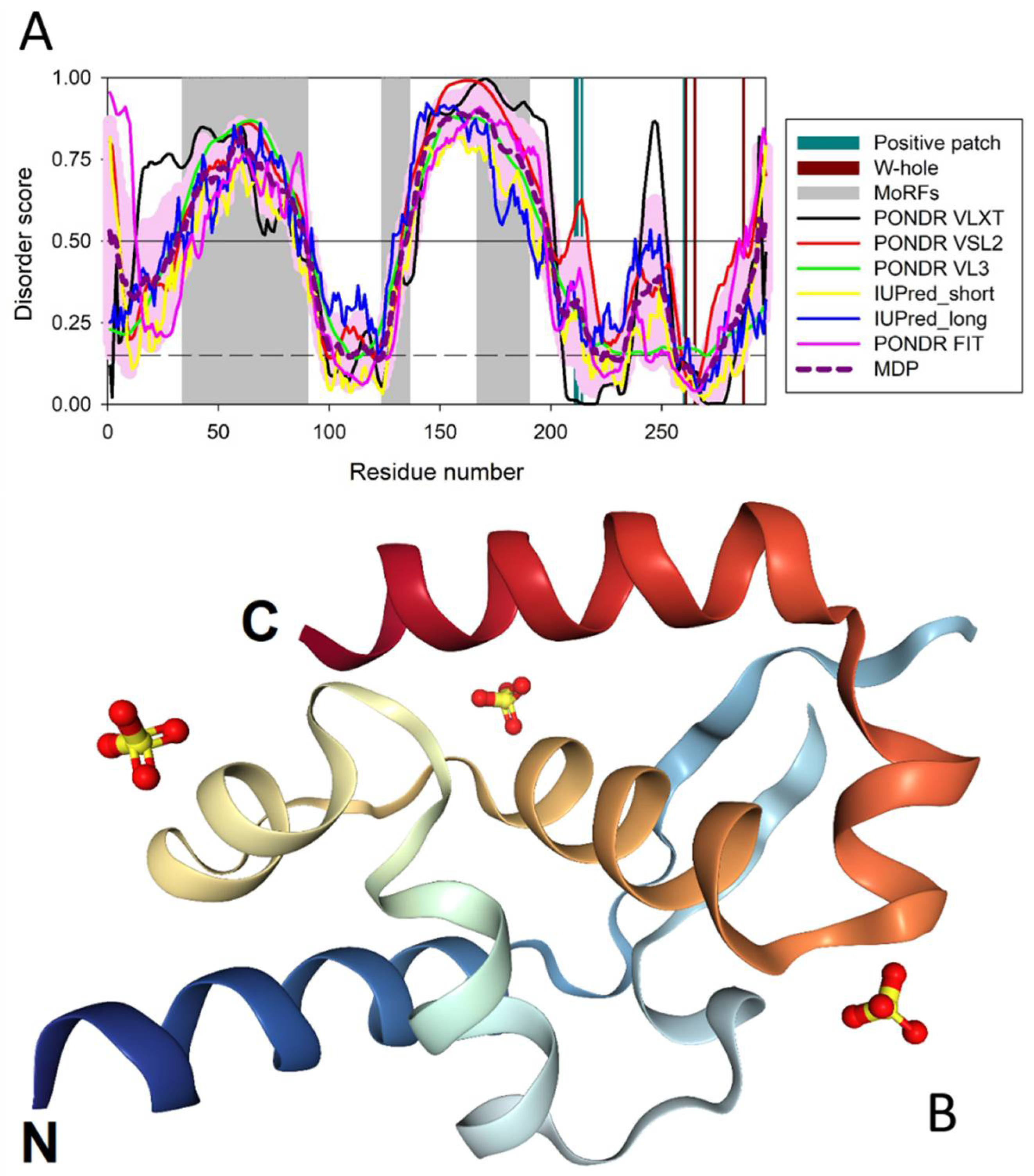
3.1. Predicted Disorder of the P-Protein and Its Suggested Functional Consequences
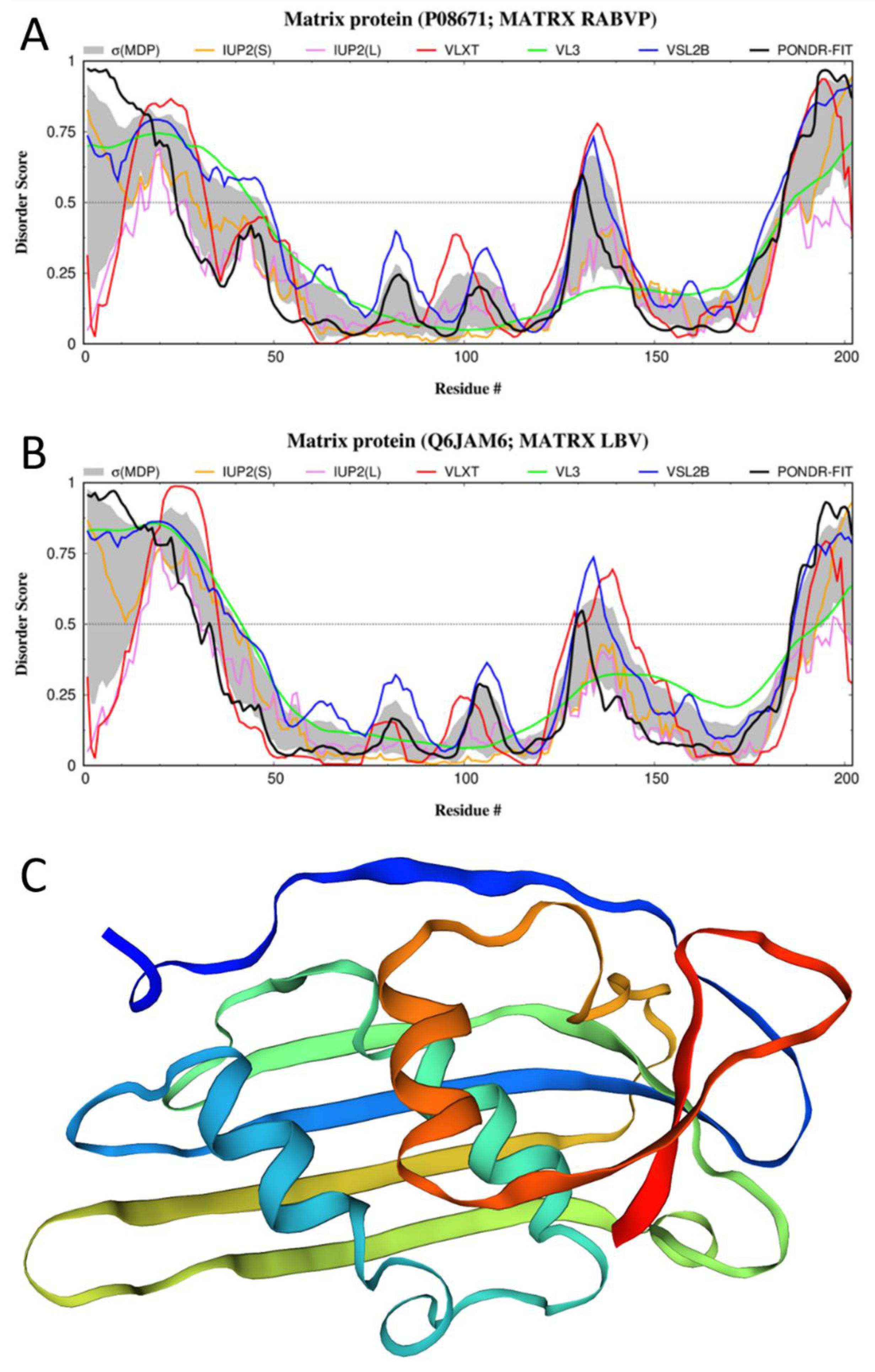
3.2. Disorder of the M-Protein and Its Suggested Functional Consequences

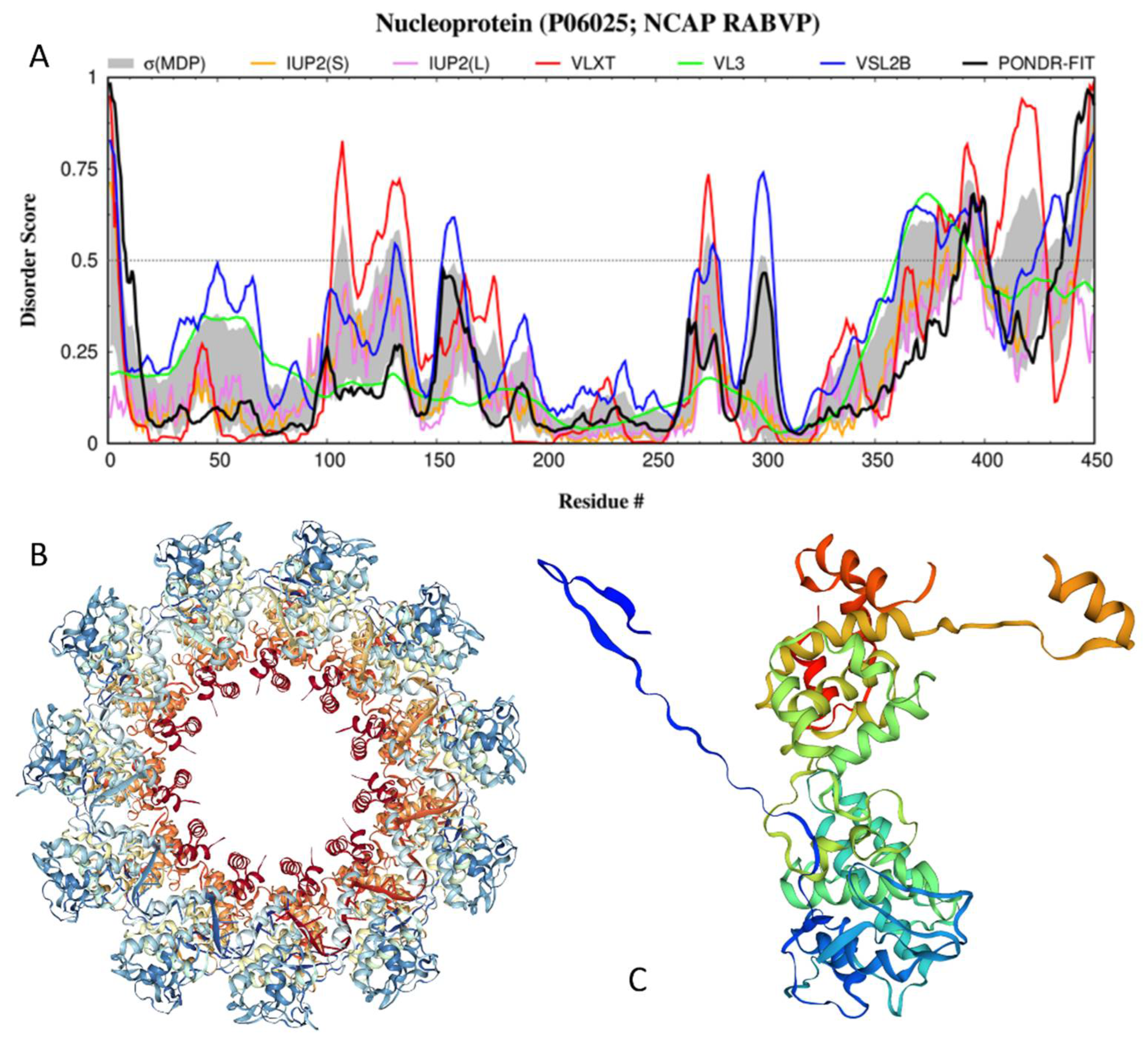
3.3. Disorder of the N-Protein and Its Suggested Functional Consequences
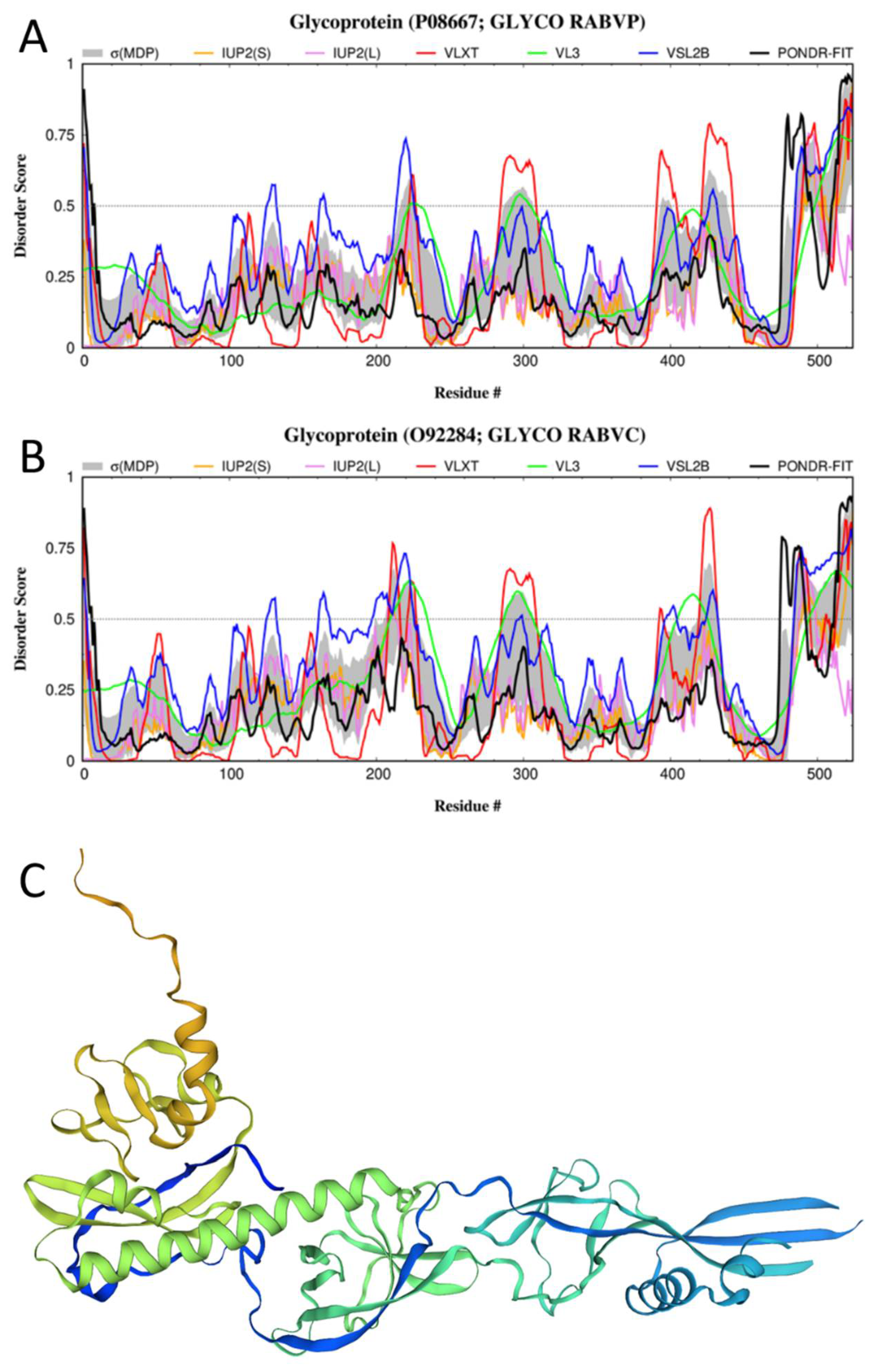
3.4. Disorder of the G-Protein and Its Suggested Functional Consequences
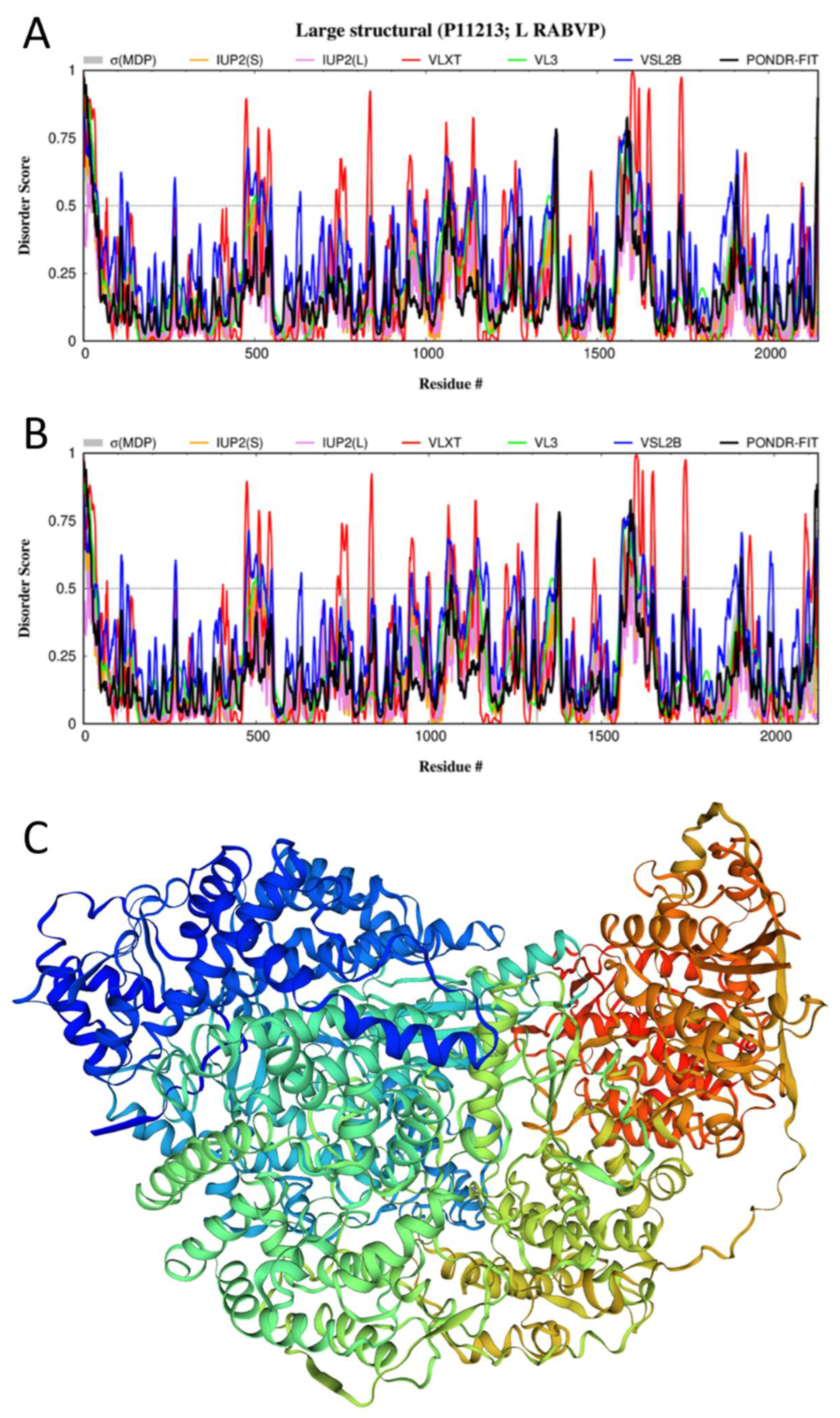
3.5. Disorder of the L-Protein and Its Suggested Functional Consequences

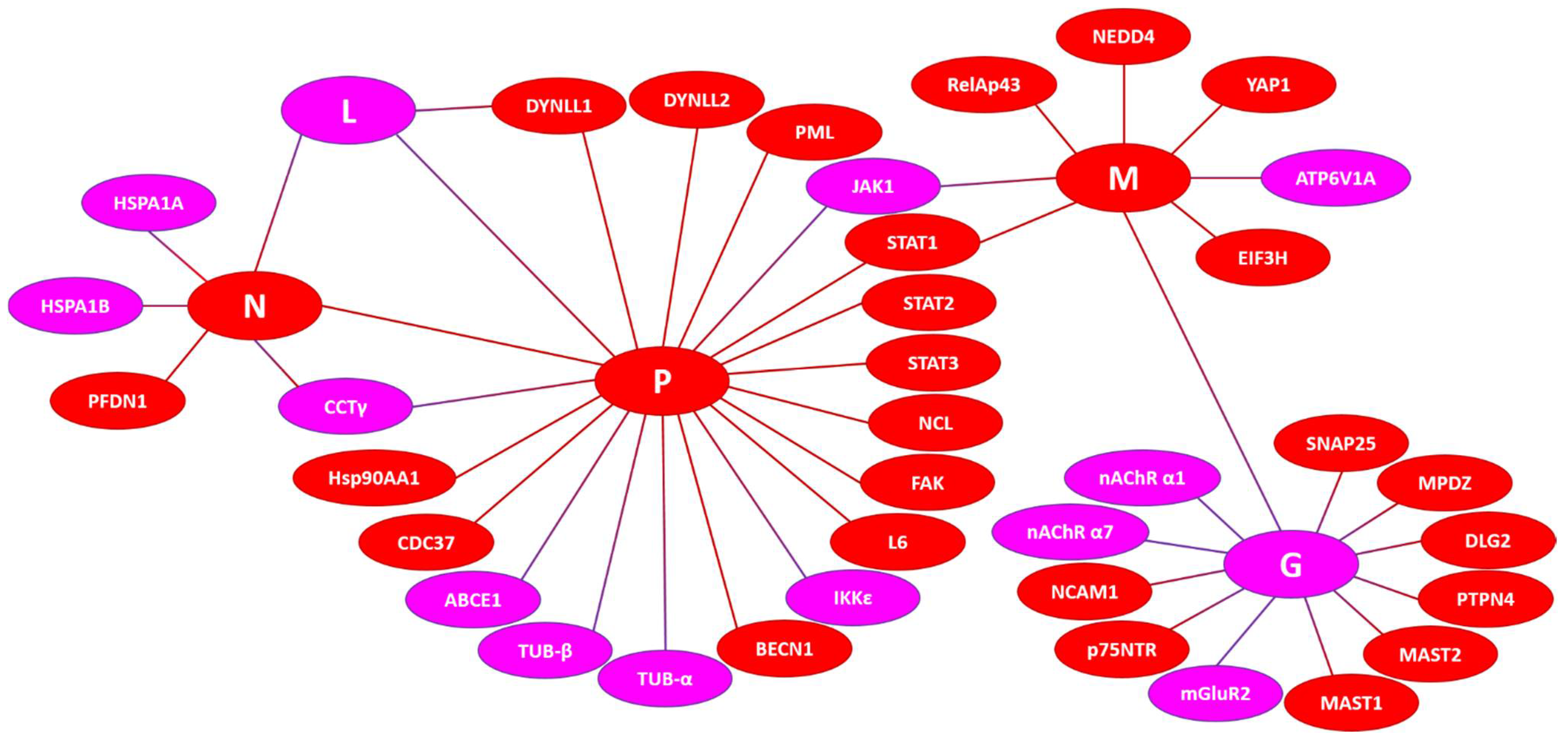
3.6. Intrinsic Disorder in Human Proteins Interacting with the RABV Proteins
3.6.1. Host Interactors of the P-Protein
3.6.2. Host Interactors of the M-Protein
3.6.3. Host Interactors of the N-Protein
3.6.4. Host Interactors of the G-Protein
3.6.5. Host Interactors of the L-Protein
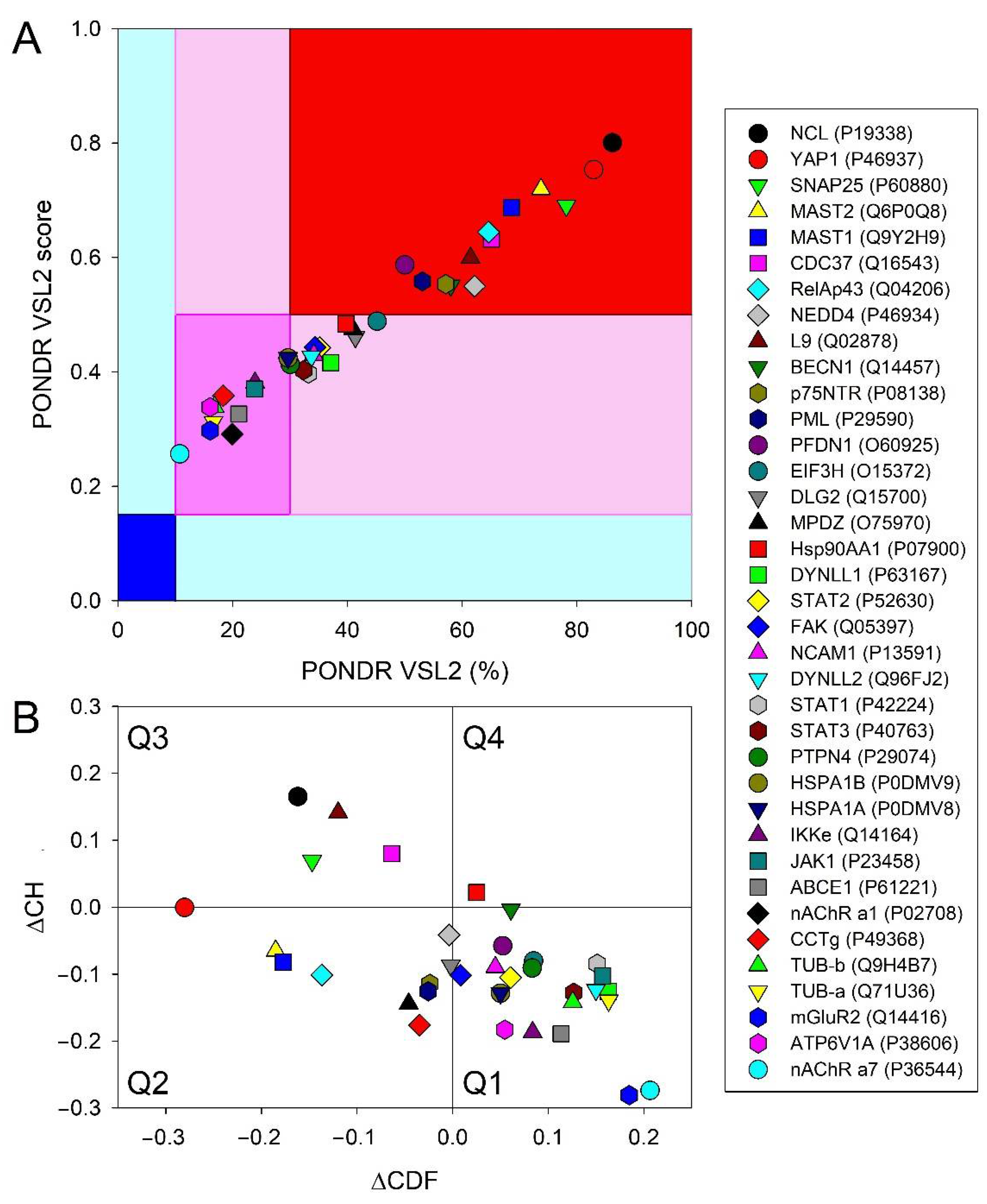
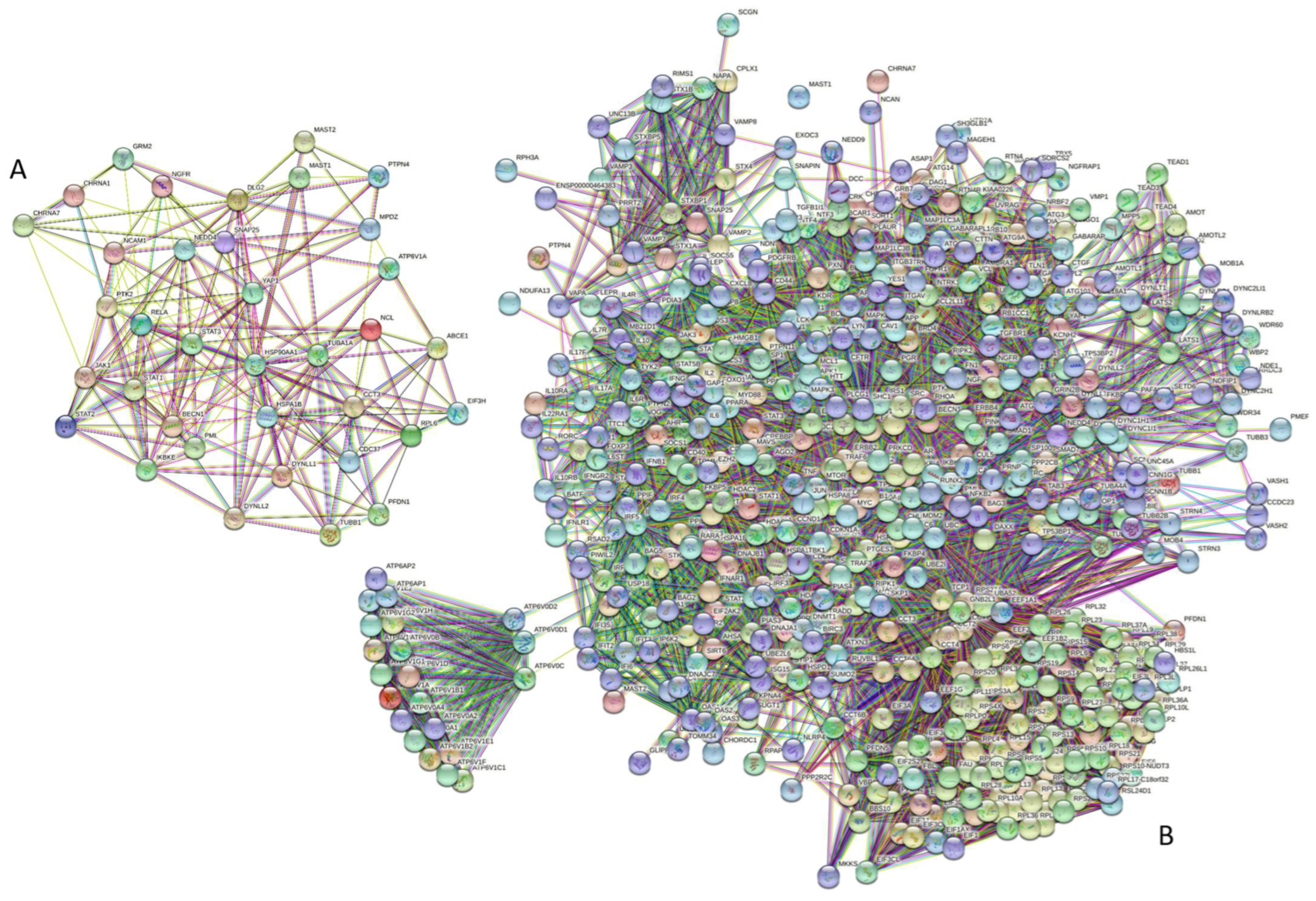
3.6.6. Prevalence of Intrinsic Disorder in Human Proteins Interacting with RABV
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rupprecht, C.E. Rhabdoviruses: Rabies Virus. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch: Galveston, TX, USA, 1996. [Google Scholar]
- Pieracci, E.G.; Pearson, C.M.; Wallace, R.M.; Blanton, J.D.; Whitehouse, E.R.; Ma, X.; Stauffer, K.; Chipman, R.B.; Olson, V. Vital Signs: Trends in Human Rabies Deaths and Exposures—United States, 1938–2018. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 524–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietzschold, B.; Li, J.; Faber, M.; Schnell, M. Concepts in the pathogenesis of rabies. Futur. Virol. 2008, 3, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gluska, S.; Zahavi, E.E.; Chein, M.; Gradus, T.; Bauer, A.; Finke, S.; Perlson, E. Rabies Virus Hijacks and Accelerates the p75NTR Retrograde Axonal Transport Machinery. PLoS Pathog. 2014, 10, e1004348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ViralZone. Lyssavirus. Available online: https://viralzone.expasy.org/resources/Rhabdoviridae_virion.jpg (accessed on 20 July 2020).
- Okada, K.; Ito, N.; Yamaoka, S.; Masatani, T.; Ebihara, H.; Goto, H.; Nakagawa, K.; Mitake, H.; Okadera, K.; Sugiyama, M. Roles of the Rabies Virus Phosphoprotein Isoforms in Pathogenesis. J. Virol. 2016, 90, 8226–8237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouznetzoff, A.; Buckle, M.; Tordo, N. Identification of a region of the rabies virus N protein involved in direct binding to the viral RNA. J. Gen. Virol. 1998, 79, 1005–1013. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, K.; Kobayashi, Y.; Ito, N.; Suzuki, Y.; Okada, K.; Makino, M.; Goto, H.; Takahashi, T.; Sugiyama, M. Molecular Function Analysis of Rabies Virus RNA Polymerase L Protein by Using an L Gene-Deficient Virus. J. Virol. 2017, 91, e00826-17. [Google Scholar] [CrossRef] [Green Version]
- Desmézières, E.; Maillard, A.P.; Gaudin, Y.; Tordo, N.; Perrin, P. Differential stability and fusion activity of Lyssavirus glycoprotein trimers. Virus Res. 2002, 91, 181–187. [Google Scholar] [CrossRef]
- Gupta, P.K.; Sharma, S.; Walunj, S.S.; Chaturvedi, V.K.; Raut, A.A.; Patial, S.; Rai, A.; Pandey, K.D.; Saini, M. Immunogenic and antigenic properties of recombinant soluble glycoprotein of rabies virus. Veter. Microbiol. 2005, 108, 207–214. [Google Scholar] [CrossRef]
- Chenik, M.; Schnell, M.; Conzelmann, K.K.; Blondel, D. Mapping the Interacting Domains between the Rabies Virus Polymerase and Phosphoprotein. J. Virol. 1998, 72, 1925–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulmanausahakul, R.; Li, J.; Schnell, M.J.; Dietzschold, B. The Glycoprotein and the Matrix Protein of Rabies Virus Affect Pathogenicity by Regulating Viral Replication and Facilitating Cell-to-Cell Spread. J. Virol. 2008, 82, 2330–2338. [Google Scholar] [CrossRef]
- Mebatsion, T.; Weiland, F.; Conzelmann, K.-K. Matrix Protein of Rabies Virus Is Responsible for the Assembly and Budding of Bullet-Shaped Particles and Interacts with the Transmembrane Spike Glycoprotein G. J. Virol. 1999, 73, 242–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chenik, M.; Chebli, K.; Blondel, D. Translation initiation at alternate in-frame AUG codons in the rabies virus phosphoprotein mRNA is mediated by a ribosomal leaky scanning mechanism. J. Virol. 1995, 69, 707–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Dong, W.; Shi, Y.; Deng, F.; Chen, X.; Wan, C.; Zhou, M.; Zhao, L.; Fu, Z.F.; Peng, G. Rabies virus phosphoprotein interacts with ribosomal protein L9 and affects rabies virus replication. Virology 2015, 488, 216–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiltzer, L.; Okada, K.; Yamaoka, S.; Larrous, F.; Kuusisto, H.V.; Sugiyama, M.; Blondel, D.; Bourhy, H.; Jans, D.; Ito, N.; et al. Interaction of Rabies Virus P-Protein with STAT Proteins is Critical to Lethal Rabies Disease. J. Infect. Dis. 2013, 209, 1744–1753. [Google Scholar] [CrossRef] [PubMed]
- Vidy, A.; El Bougrini, J.; Chelbi-Alix, M.K.; Blondel, D. The Nucleocytoplasmic Rabies Virus P Protein Counteracts Interferon Signaling by Inhibiting both Nuclear Accumulation and DNA Binding of STAT1. J. Virol. 2007, 81, 4255–4263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moseley, G.W.; Lahaye, X.; Roth, D.M.; Oksayan, S.; Filmer, R.P.; Rowe, C.L.; Blondel, D.; Jans, D. Dual modes of rabies P-protein association with microtubules: A novel strategy to suppress the antiviral response. J. Cell Sci. 2009, 122, 3652–3662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masatani, T.; Ito, N.; Ito, Y.; Nakagawa, K.; Abe, M.; Yamaoka, S.; Okadera, K.; Sugiyama, M. Importance of rabies virus nucleoprotein in viral evasion of interferon response in the brain. Microbiol. Immunol. 2013, 57, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Masatani, T.; Ito, N.; Shimizu, K.; Ito, Y.; Nakagawa, K.; Sawaki, Y.; Koyama, H.; Sugiyama, M. Rabies Virus Nucleoprotein Functions to Evade Activation of the RIG-I-Mediated Antiviral Response. J. Virol. 2010, 84, 4002–4012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogino, M.; Ito, N.; Sugiyama, M.; Ogino, T. The Rabies Virus L Protein Catalyzes mRNA Capping with GDP Polyribonucleotidyltransferase Activity. Viruses 2016, 8, 144. [Google Scholar] [CrossRef] [PubMed]
- Finke, S.; Mueller-Waldeck, R.; Conzelmann, K.-K. Rabies virus matrix protein regulates the balance of virus transcription and replication. J. Gen. Virol. 2003, 84, 1613–1621. [Google Scholar] [CrossRef]
- Ben Khalifa, Y.; Luco, S.; Besson, B.; Sonthonnax, F.; Archambaud, M.; Grimes, J.M.; Larrous, F.; Bourhy, H. The matrix protein of rabies virus binds to RelAp43 to modulate NF-κB-dependent gene expression related to innate immunity. Sci. Rep. 2016, 6, 39420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Wang, H.; Mahmood, F.; Fu, Z.F. Rabies virus glycoprotein is an important determinant for the induction of innate immune responses and the pathogenic mechanisms. Vet. Microbiol. 2013, 162, 601–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. What does it mean to be natively unfolded? Eur. J. Biochem. 2002, 269, 2–12. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta BBA Proteins Proteom. 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Dunker, A.K.; Cortese, M.; Romero, P.; Iakoucheva, L.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef]
- Uversky, V.N. Protein intrinsic disorder and structure-function continuum. Prog. Mol. Biol. Transl. Sci. 2019, 166, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genome Inform. 2000, 11, 161–171. [Google Scholar]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and Functional Analysis of Native Disorder in Proteins from the Three Kingdoms of Life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly order in protein intrinsic disorder distribution: Disorder in 3500 proteomes from viruses and the three domains of life. J. Biomol. Struct. Dyn. 2012, 30, 137–149. [Google Scholar] [CrossRef]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.J.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.N.; Kurgan, L. Exceptionally abundant exceptions: Comprehensive characterization of intrinsic disorder in all domains of life. Cell Mol. Life Sci. 2014, 72, 137–151. [Google Scholar] [CrossRef]
- Uversky, V.N. The mysterious unfoldome: Structureless, underappreciated, yet vital part of any given proteome. J. Biomed. Biotechnol. 2010, 2010, 568068. [Google Scholar] [CrossRef] [Green Version]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta BBA Proteins Proteom. 2012, 1834, 932–951. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- E Wright, P.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005, 579, 3346–3354. [Google Scholar] [CrossRef] [PubMed]
- Radivojac, P.; Iakoucheva, L.M.; Oldfield, C.J.; Obradovic, Z.; Uversky, V.N.; Dunker, A.K. Intrinsic Disorder and Functional Proteomics. Biophys. J. 2007, 92, 1439–1456. [Google Scholar] [CrossRef] [Green Version]
- Vucetic, S.; Xie, H.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Functional Anthology of Intrinsic Disorder. 2. Cellular Components, Domains, Technical Terms, Developmental Processes, and Coding Sequence Diversities Correlated with Long Disordered Regions. J. Proteome Res. 2007, 6, 1899–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Functional Anthology of Intrinsic Disorder. 3. Ligands, Post-Translational Modifications, and Diseases Associated with Intrinsically Disordered Proteins. J. Proteome Res. 2007, 6, 1917–1932. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N.; Obradovic, Z. Functional Anthology of Intrinsic Disorder. 1. Biological Processes and Functions of Proteins with Long Disordered Regions. J. Proteome Res. 2007, 6, 1882–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins in Human Diseases: Introducing the D2 Concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Markwick, P.R.L.; Oldfield, C.J.; Zhao, X.; Haynes, C.; Uversky, V.N.; Iakoucheva, L.M. Disease-Associated Mutations Disrupt Functionally Important Regions of Intrinsic Protein Disorder. PLoS Comput. Biol. 2012, 8, e1002709. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; E Villafranca, J. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. Pac. Symp. Biocomput. 1998, 3, 473–484. [Google Scholar]
- Daughdrill, G.W.; Pielak, G.J.; Uversky, V.N.; Cortese, M.S.; Dunker, A.K. Natively disordered proteins. In Handbook of Protein Folding; Buchner, J., Kiefhaber, T., Eds.; Wiley-VCH, Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; pp. 271–353. [Google Scholar]
- Uversky, V.N. Intrinsic Disorder-based Protein Interactions and their Modulators. Curr. Pharm. Des. 2013, 19, 4191–4213. [Google Scholar] [CrossRef]
- Uversky, V.N. Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J. 2015, 282, 1182–1189. [Google Scholar] [CrossRef]
- Uversky, V.N. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure–Function Continuum Concept. Int. J. Mol. Sci. 2016, 17, 1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.M.; The Consortium for Top Down Proteomics; Kelleher, N.L. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Ganti, K.; Rabionet, A.; Banks, L.; Uversky, V. Disordered Interactome of Human Papillomavirus. Curr. Pharm. Des. 2014, 20, 1274–1292. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Roman, A.; Oldfield, A.C.J.; Dunker, A.K. Protein Intrinsic Disorder and Human Papillomaviruses: Increased Amount of Disorder in E6 and E7 Oncoproteins from High Risk HPVs. J. Proteome Res. 2006, 5, 1829–1842. [Google Scholar] [CrossRef]
- Xue, B.; Mizianty, M.J.; Kurgan, L.; Uversky, V.N. Protein intrinsic disorder as a flexible armor and a weapon of HIV-1. Experientia 2011, 69, 1211–1259. [Google Scholar] [CrossRef] [PubMed]
- Dolan, P.; Roth, A.P.; Xue, B.; Sun, R.; Dunker, A.K.; Uversky, V.N.; LaCount, D.J. Intrinsic disorder mediates hepatitis C virus core-host cell protein interactions. Protein Sci. 2014, 24, 221–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Xue, B.; Dolan, P.T.; LaCount, D.J.; Kurgan, L.; Uversky, V.N. The intrinsic disorder status of the human hepatitis C virus proteome. Mol. BioSyst. 2014, 10, 1345–1363. [Google Scholar] [CrossRef]
- Meng, F.; Badierah, R.A.; Almehdar, H.A.; Redwan, E.M.; Kurgan, L.; Uversky, V.N. Unstructural biology of the dengue virus proteins. FEBS J. 2015, 282, 3368–3394. [Google Scholar] [CrossRef]
- Kumar, D.; Singh, A.; Kumar, P.; Uversky, V.N.; Rao, C.D.; Giri, R. Understanding the penetrance of intrinsic protein disorder in rotavirus proteome. Int. J. Biol. Macromol. 2019, 144, 892–908. [Google Scholar] [CrossRef]
- Whelan, J.N.; Reddy, K.D.; Uversky, V.N.; Teng, M.N. Functional correlations of respiratory syncytial virus proteins to intrinsic disorder. Mol. BioSyst. 2016, 12, 1507–1526. [Google Scholar] [CrossRef]
- Mishra, P.M.; Uversky, V.N.; Giri, R. Molecular Recognition Features in Zika Virus Proteome. J. Mol. Biol. 2017, 430, 2372–2388. [Google Scholar] [CrossRef] [PubMed]
- Giri, R.; Kumar, D.; Sharma, N.; Uversky, V.N. Intrinsically Disordered Side of the Zika Virus Proteome. Front. Cell. Infect. Microbiol. 2016, 6, 144. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kumar, A.; Yadav, R.; Uversky, V.N.; Giri, R. Deciphering the dark proteome of Chikungunya virus. Sci. Rep. 2018, 8, 5822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redwan, E.M.; AlJaddawi, A.A.; Uversky, V.N. Structural disorder in the proteome and interactome of Alkhurma virus (ALKV). Experientia 2018, 76, 577–608. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, T.; Saumya, K.U.; Kumar, P.; Sharma, N.; Gadhave, K.; Uversky, V.N.; Giri, R. Japanese encephalitis virus—Exploring the dark proteome and disorder-function paradigm. FEBS J. 2020, 287, 3751–3776. [Google Scholar] [CrossRef] [PubMed]
- Giri, R.; Bhardwaj, T.; Shegane, M.; Gehi, B.R.; Kumar, P.; Gadhave, K.; Oldfield, C.J.; Uversky, V.N. Understanding the COVID-19 via Comparative Analysis of Dark Proteomes of SARS-CoV-2, Human SARS and Bat SARS-Like Coronaviruses. Cell Mol. Life Sci. 2021, 78, 1655–1688. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Zandi, F.; Goshadrou, F.; Meyfour, A.; Vaziri, B. Rabies Infection: An Overview of Lyssavirus-Host Protein Interactions. Iran. Biomed. J. 2021, 25, 226–242. [Google Scholar] [CrossRef]
- Chen, J.; Kriwacki, R.W. Intrinsically Disordered Proteins: Structure, Function and Therapeutics. J. Mol. Biol. 2018, 430, 2275–2277. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta BBA Proteins Proteom. 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [Green Version]
- Dosztanyi, Z.; Csizmók, V.; Tompa, P.; Simon, I. The Pairwise Energy Content Estimated from Amino Acid Composition Discriminates between Folded and Intrinsically Unstructured Proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Erdős, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Simon, I.; Dosztanyi, Z. Prediction of Protein Binding Regions in Disordered Proteins. PLoS Comput. Biol. 2009, 5, e1000376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dosztanyi, Z.; Meszaros, B.; Simon, I. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayhoff, G.W.I.; Uversky, V.N. Rapid prediction and analysis of protein intrinsic disorder. Protein Sci. 2022; in press. [Google Scholar]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K.; Obradovic, Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J. Bioinform. Comput. Biol. 2005, 3, 35–60. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, A.; Sullivan, W.J., Jr.; Radivojac, P.; Dunker, A.K.; Uversky, V.N. Intrinsic disorder in pathogenic and non-pathogenic microbes: Discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol. BioSyst. 2008, 4, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xue, B.; Jones, W.T.; Rikkerink, E.; Dunker, A.K.; Uversky, V.N. A functionally required unfoldome from the plant kingdom: Intrinsically disordered N-terminal domains of GRAS proteins are involved in molecular recognition during plant development. Plant Mol. Biol. 2011, 77, 205–223. [Google Scholar] [CrossRef]
- Xue, B.; Oldfield, C.J.; Van, Y.-Y.; Dunker, A.K.; Uversky, V.N. Protein intrinsic disorder and induced pluripotent stem cells. Mol. BioSyst. 2011, 8, 134–150. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Oldfield, C.; Meng, J.; Hsu, W.-L.; Xue, B.; Uversky, V.N.; Romero, P.; Dunker, A.K. Subclassifying disordered proteins by the CH-CDF plot method. Biocomputing 2011, 128–139. [Google Scholar] [CrossRef]
- Rajagopalan, K.; Mooney, S.M.; Parekh, N.; Getzenberg, R.H.; Kulkarni, P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J. Cell. Biochem. 2011, 112, 3256–3267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztányi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D2P2: Database of disordered protein predictions. Nucleic Acids Res. 2013, 41, D508–D516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef] [Green Version]
- Walsh, I.; Martin, A.J.M.; Di Domenico, T.; Tosatto, S.C.E. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreeva, A.; Howorth, D.; Brenner, S.E.; Hubbard, T.J.; Chothia, C.; Murzin, A.G. SCOP database in 2004: Refinements integrate structure and sequence family data. Nucleic Acids Res. 2004, 32 (Suppl. S1), D226–D229. [Google Scholar] [CrossRef] [Green Version]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef]
- De Lima Morais, D.A.; Fang, H.; Rackham, O.J.L.; Wilson, D.; Pethica, R.; Chothia, C.; Gough, J. SUPERFAMILY 1.75 including a domain-centric gene ontology method. Nucleic Acids Res. 2011, 39, D427–D434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2011, 40, D261–D270. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2010, 39, D561–D568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardenberg, M.; Horvath, A.; Ambrus, V.; Fuxreiter, M.; Vendruscolo, M. Widespread occurrence of the droplet state of proteins in the human proteome. Proc. Natl. Acad. Sci. USA 2020, 117, 33254–33262. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Nevers, Q.; Albertini, A.A.; Lagaudrière-Gesbert, C.; Gaudin, Y. Negri bodies and other virus membrane-less replication compartments. Biochim. Biophys. Acta 2020, 1867, 118831. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, J.; Le Bars, R.; Lama, Z.; Scrima, N.; Lagaudrière-Gesbert, C.; Gaudin, Y.; Blondel, D. Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 2017, 8, 58. [Google Scholar] [CrossRef] [Green Version]
- Brzózka, K.; Finke, S.; Conzelmann, K.-K. Identification of the Rabies Virus Alpha/Beta Interferon Antagonist: Phosphoprotein P Interferes with Phosphorylation of Interferon Regulatory Factor 3. J. Virol. 2005, 79, 7673–7681. [Google Scholar] [CrossRef] [Green Version]
- Vidy, A.; Chelbi-Alix, M.; Blondel, D. Rabies Virus P Protein Interacts with STAT1 and Inhibits Interferon Signal Transduction Pathways. J. Virol. 2005, 79, 14411–14420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieu, K.G.; Brice, A.; Wiltzer, L.; Hirst, B.; Jans, D.A.; Blondel, D.; Moseley, G.W. The Rabies Virus Interferon Antagonist P Protein Interacts with Activated STAT3 and Inhibits Gp130 Receptor Signaling. J. Virol. 2013, 87, 8261–8265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, X.; Tang, L.; Tseggai, T.; Guo, Y.; Fu, Z.F. Wild-type rabies virus phosphoprotein is associated with viral sensitivity to type I interferon treatment. Arch. Virol. 2013, 158, 2297–2305. [Google Scholar] [CrossRef]
- Gupta, A.K.; Blondel, D.; Choudhary, S.; Banerjee, A.K. The Phosphoprotein of Rabies Virus Is Phosphorylated by a Unique Cellular Protein Kinase and Specific Isomers of Protein Kinase C. J. Virol. 2000, 74, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, J.; Watts, E.; Brice, A.M.; Metcalfe, R.D.; Rozario, A.M.; Sethi, A.; Yan, F.; Bell, T.D.M.; Griffin, M.D.W.; Moseley, G.W.; et al. Molecular Basis of Functional Effects of Phosphorylation of the C-Terminal Domain of the Rabies Virus P Protein. J. Virol. 2022, 96, e0011122. [Google Scholar] [CrossRef] [PubMed]
- Brice, A.; Whelan, D.; Ito, N.; Shimizu, K.; Wiltzer-Bach, L.; Lo, C.; Blondel, D.; Jans, D.; Bell, T.D.M.; Moseley, G.W. Quantitative Analysis of the Microtubule Interaction of Rabies Virus P3 Protein: Roles in Immune Evasion and Pathogenesis. Sci. Rep. 2016, 6, 33493. [Google Scholar] [CrossRef]
- Rowe, C.L.; Wagstaff, K.; Oksayan, S.; Glover, D.J.; Jans, D.; Moseley, G.W. Nuclear Trafficking of the Rabies Virus Interferon Antagonist P-Protein Is Regulated by an Importin-Binding Nuclear Localization Sequence in the C-Terminal Domain. PLoS ONE 2016, 11, e0150477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasdeloup, D.; Poisson, N.; Raux, H.; Gaudin, Y.; Ruigrok, R.W.; Blondel, D. Nucleocytoplasmic shuttling of the rabies virus P protein requires a nuclear localization signal and a CRM1-dependent nuclear export signal. Virology 2005, 334, 284–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blondel, D.; Regad, T.; Poisson, N.; Pavie, B.; Harper, F.; Pandolfi, P.P.; de Thé, H.; Chelbi-Alix, M.K. Rabies virus P and small P products interact directly with PML and reorganize PML nuclear bodies. Oncogene 2002, 21, 7957–7970. [Google Scholar] [CrossRef] [Green Version]
- Horvath, A.; Miskei, M.; Ambrus, V.; Vendruscolo, M.; Fuxreiter, M. Sequence-based prediction of protein binding mode landscapes. PLoS Comput. Biol. 2020, 16, e1007864. [Google Scholar] [CrossRef]
- Goh, G.K.-M.; Dunker, A.K.; Foster, J.A.; Uversky, V.N. Shell disorder analysis predicts greater resilience of the SARS-CoV-2 (COVID-19) outside the body and in body fluids. Microb. Pathog. 2020, 144, 104177. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.K.-M.; Dunker, A.K.; Foster, J.A.; Uversky, V.N. Rigidity of the Outer Shell Predicted by a Protein Intrinsic Disorder Model Sheds Light on the COVID-19 (Wuhan-2019-nCoV) Infectivity. Biomolecules 2020, 10, 331. [Google Scholar] [CrossRef] [Green Version]
- Goh, G.K.-M.; Dunker, A.K.; Foster, J.A.; Uversky, V.N. Nipah shell disorder, modes of infection, and virulence. Microb. Pathog. 2020, 141, 103976. [Google Scholar] [CrossRef]
- Goh, G.K.M.; Dunker, A.K.; Foster, J.A.; Uversky, V.N. Zika and Flavivirus Shell Disorder: Virulence and Fetal Morbidity. Biomolecules 2019, 9, 710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, G.K.M.; Dunker, A.K.; Foster, J.A.; Uversky, V.N. HIV Vaccine Mystery and Viral Shell Disorder. Biomolecules 2019, 9, 178. [Google Scholar] [CrossRef]
- Goh, G.K.-M.; Dunker, A.K.; Uversky, V.N. Correlating Flavivirus virulence and levels of intrinsic disorder in shell proteins: Protective roles vs. immune evasion. Mol. BioSyst. 2016, 12, 1881–1891. [Google Scholar] [CrossRef]
- Goh, G.K.-M.; Dunker, A.K.; Uversky, V.N. Shell disorder, immune evasion and transmission behaviors among human and animal retroviruses. Mol. BioSyst. 2015, 11, 2312–2323. [Google Scholar] [CrossRef]
- Goh, G.K.-M.; Dunker, A.K.; Uversky, V. Prediction of Intrinsic Disorder in MERS-CoV/HCoV-EMC Supports a High Oral-Fecal Transmission. PLoS Curr. 2013, 5, 24270586. [Google Scholar] [CrossRef]
- Goh, G.K.-M.; Dunker, A.K.; Uversky, V.N. Understanding Viral Transmission Behavior via Protein Intrinsic Disorder Prediction: Coronaviruses. J. Pathog. 2012, 2012, 738590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montespan, C.; Wiethoff, C.M.; Wodrich, H. A Small Viral PPxY Peptide Motif to Control Antiviral Autophagy. J. Virol. 2017, 91, e00581-17. [Google Scholar] [CrossRef] [Green Version]
- Graham, S.; Assenberg, R.; Delmas, O.; Verma, A.; Gholami, A.; Talbi, C.; Owens, R.; Stuart, D.; Grimes, J.M.; Bourhy, H. Rhabdovirus Matrix Protein Structures Reveal a Novel Mode of Self-Association. PLoS Pathog. 2008, 4, e1000251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Realegeno, S.; Niezgoda, M.; Yager, P.A.; Kumar, A.; Hoque, L.; Orciari, L.; Sambhara, S.; Olson, V.A.; Satheshkumar, P.S. An ELISA-based method for detection of rabies virus nucleoprotein-specific antibodies in human antemortem samples. PLoS ONE 2018, 13, e0207009. [Google Scholar] [CrossRef]
- Lingappa, U.F.; Wu, X.; Macieik, A.; Yu, S.F.; Atuegbu, A.; Corpuz, M.; Francis, J.; Nichols, C.; Calayag, A.; Shi, H.; et al. Host–rabies virus protein–protein interactions as druggable antiviral targets. Proc. Natl. Acad. Sci. USA 2013, 110, E861–E868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavrakis, M.; Iseni, F.; Mazzaa, C.; Schoehnac, G.; Ebelc, C.; Gentzeld, M.; Franzd, T.; Ruigrok, R.W. Isolation and Characterisation of the Rabies Virus N°-P Complex Produced in Insect Cells. Virology 2003, 305, 406–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albertini, A.A.V.; Wernimont, A.K.; Muziol, T.M.; Ravelli, R.B.G.; Clapier, C.R.; Schoehn, G.; Weissenhorn, W.; Ruigrok, R.W.H. Crystal Structure of the Rabies Virus Nucleoprotein-RNA Complex. Science 2006, 313, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Green, T.J.; Zhang, X.; Tsao, J.; Qiu, S. Conserved characteristics of the rhabdovirus nucleoprotein. Virus Res. 2007, 129, 246–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etessami, R.; Conzelmann, K.-K.; Fadai-Ghotbi, B.; Natelson, B.; Tsiang, H.; Ceccaldi, P.-E. Spread and pathogenic characteristics of a G-deficient rabies virus recombinant: An in vitro and in vivo study. J. Gen. Virol. 2000, 81, 2147–2153. [Google Scholar] [CrossRef] [PubMed]
- Nitschel, S.; Zaeck, L.M.; Potratz, M.; Nolden, T.; Kamp, V.T.; Franzke, K.; Höper, D.; Pfaff, F.; Finke, S. Point Mutations in the Glycoprotein Ectodomain of Field Rabies Viruses Mediate Cell Culture Adaptation through Improved Virus Release in a Host Cell Dependent and Independent Manner. Viruses 2021, 13, 1989. [Google Scholar] [CrossRef]
- Yang, F.; Lin, S.; Ye, F.; Yang, J.; Qi, J.; Chen, Z.; Lin, X.; Wang, J.; Yue, D.; Cheng, Y.; et al. Structural Analysis of Rabies Virus Glycoprotein Reveals pH-Dependent Conformational Changes and Interactions with a Neutralizing Antibody. Cell Host Microbe 2020, 27, 441–453.e7. [Google Scholar] [CrossRef]
- Schnell, M.J.; Conzelmann, K.-K. Polymerase Activity ofin VitroMutated Rabies Virus L Protein. Virology 1995, 214, 522–530. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, J.A.; Jenni, S.; Harrison, S.C.; Whelan, S.P.J. Structure of a rabies virus polymerase complex from electron cryo-microscopy. Proc. Natl. Acad. Sci. USA 2020, 117, 2099–2107. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Nawaz, Z.; Guo, C.; Ali, S.; Naeem, M.A.; Jamil, T.; Ahmad, W.; Siddiq, M.U.; Ahmed, S.; Idrees, M.A.; et al. Rabies Virus Exploits Cytoskeleton Network to Cause Early Disease Progression and Cellular Dysfunction. Front. Vet. Sci. 2022, 9, 889873. [Google Scholar] [CrossRef]
- Kammouni, W.; Wood, H.; Saleh, A.; Appolinario, C.M.; Fernyhough, P.; Jackson, A.C. Rabies virus phosphoprotein interacts with mitochondrial Complex I and induces mitochondrial dysfunction and oxidative stress. J. Neurovirol. 2015, 21, 370–382. [Google Scholar] [CrossRef]
- Sharma, L.K.; Lu, J.; Bai, Y. Mitochondrial Respiratory Complex I: Structure, Function and Implication in Human Diseases. Curr. Med. Chem. 2009, 16, 1266–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, Z.; Liu, R.; Shuai, L.; Wang, X.; Luo, J.; Wang, C.; Chen, W.; Wang, X.; Ge, J.; et al. Metabotropic glutamate receptor subtype 2 is a cellular receptor for rabies virus. PLoS Pathog. 2018, 14, e1007189. [Google Scholar] [CrossRef] [PubMed]
- Tuffereau, C.; Bénéjean, J.; Blondel, D.; Kieffer, B.; Flamand, A. Low-affinity nerve-growth factor receptor (P75NTR) can serve as a receptor for rabies virus. EMBO J. 1998, 17, 7250–7259. [Google Scholar] [CrossRef]
- Thoulouze, M.-I.; Lafage, M.; Schachner, M.; Hartmann, U.; Cremer, H.; Lafon, M. The Neural Cell Adhesion Molecule Is a Receptor for Rabies Virus. J. Virol. 1998, 72, 7181–7190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lentz, T.L.; Burrage, T.G.; Smith, A.L.; Crick, J.; Tignor, G.H. Is the Acetylcholine Receptor a Rabies Virus Receptor? Science 1982, 215, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Sajjanar, B.; Dhusia, K.; Saxena, S.; Joshi, V.; Bisht, D.; Thakuria, D.; Manjunathareddy, G.B.; Ramteke, P.W.; Kumar, S. Nicotinic acetylcholine receptor alpha 1(nAChRα1) subunit peptides as potential antiviral agents against rabies virus. Int. J. Biol. Macromol. 2017, 104, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Embregts, C.W.E.; Begeman, L.; Voesenek, C.J.; Martina, B.E.E.; Koopmans, M.P.G.; Kuiken, T.; GeurtsvanKessel, C.H. Street RABV Induces the Cholinergic Anti-inflammatory Pathway in Human Monocyte-Derived Macrophages by Binding to nAChr α7. Front. Immunol. 2021, 12, 622516. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.L.; Kurgan, L. Comprehensive Comparative Assessment of In-Silico Predictors of Disordered Regions. Curr. Protein Pept. Sci. 2012, 13, 6–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Necci, M.; Piovesan, D.; CAID Predictors; Curators, D.; Tosatto, S.C.E. Critical assessment of protein intrinsic disorder prediction. Nat. Methods 2021, 18, 472–481. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | UniProt Entry ID [70] | Protein Length (Residues) | Longest Disordered Region (Residues) | Percent of Disordered Residues |
|---|---|---|---|---|
| P (Phosphoprotein) | P06747 | 297 | 87 | 67.3% |
| M (Matrix protein) | P08671 | 202 | 54 | 43% |
| N (Nucleoprotein) | P06025 | 450 | 93 | 30.6% |
| G (Glycoprotein) | P08667 | 524 | 49 | 27% |
| L (Large protein) | P11213 | 2142 | 105 | 23% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhulipala, S.; Uversky, V.N. Looking at the Pathogenesis of the Rabies Lyssavirus Strain Pasteur Vaccins through a Prism of the Disorder-Based Bioinformatics. Biomolecules 2022, 12, 1436. https://doi.org/10.3390/biom12101436
Dhulipala S, Uversky VN. Looking at the Pathogenesis of the Rabies Lyssavirus Strain Pasteur Vaccins through a Prism of the Disorder-Based Bioinformatics. Biomolecules. 2022; 12(10):1436. https://doi.org/10.3390/biom12101436
Chicago/Turabian StyleDhulipala, Surya, and Vladimir N. Uversky. 2022. "Looking at the Pathogenesis of the Rabies Lyssavirus Strain Pasteur Vaccins through a Prism of the Disorder-Based Bioinformatics" Biomolecules 12, no. 10: 1436. https://doi.org/10.3390/biom12101436
APA StyleDhulipala, S., & Uversky, V. N. (2022). Looking at the Pathogenesis of the Rabies Lyssavirus Strain Pasteur Vaccins through a Prism of the Disorder-Based Bioinformatics. Biomolecules, 12(10), 1436. https://doi.org/10.3390/biom12101436