
Determination of the Amino Acid Recruitment Order in Early Life by Genome-Wide Analysis of Amino Acid Usage Bias


Abstract
:1. Introduction
2. Materials and Methods
2.1. Genomic Information
2.2. Calculation of Amino Acid Usage and Genetic Codon Usage—Python Scripts
2.2.1. Amino Acid Usage
2.2.2. Genetic Codon Usage
2.3. Correlation Analysis—Corrplot R Packages
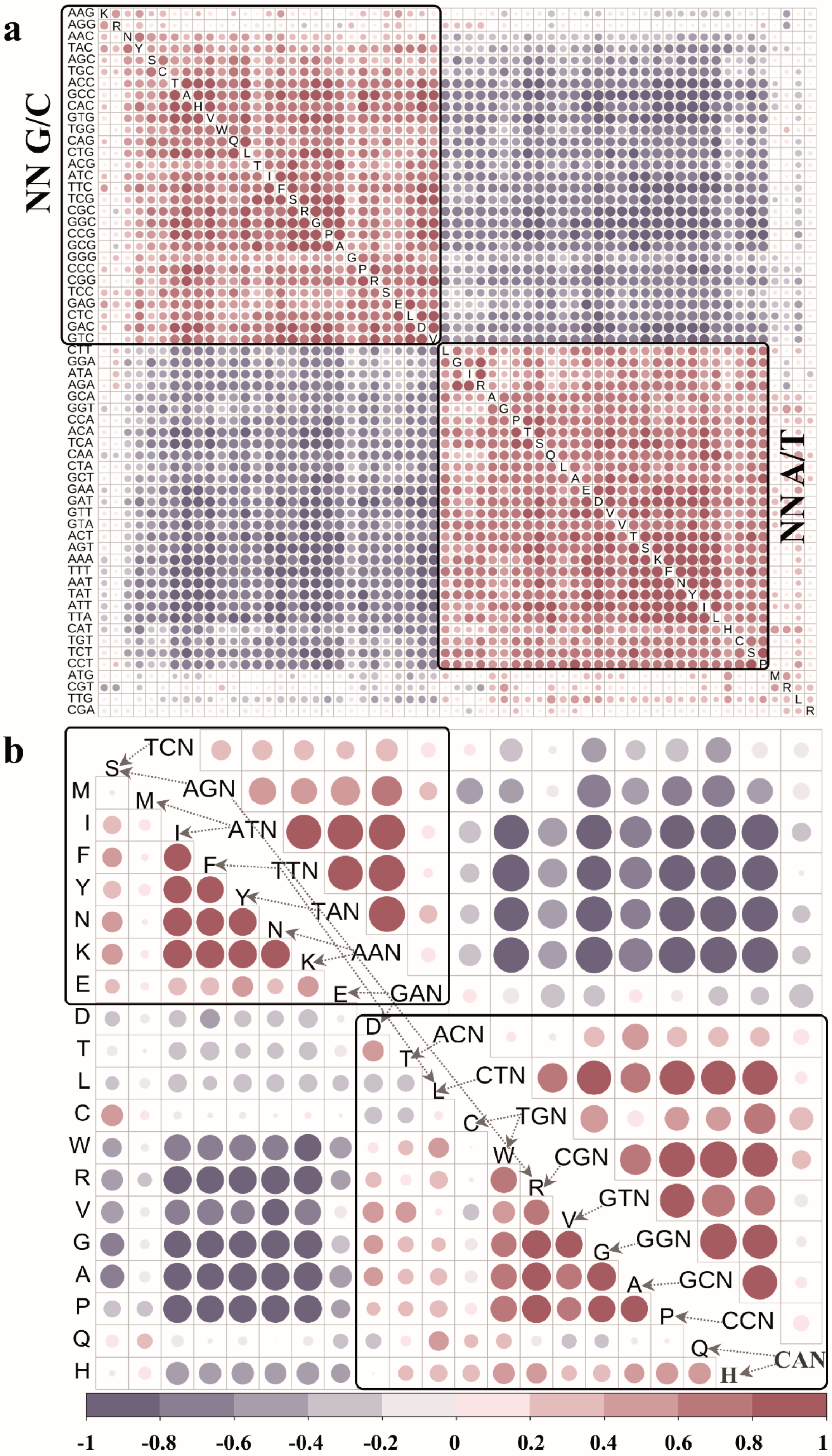
2.3.1. Usage Correlation between Amino Acids or Codons
2.3.2. Correlation of Physicochemical Properties between Amino Acids
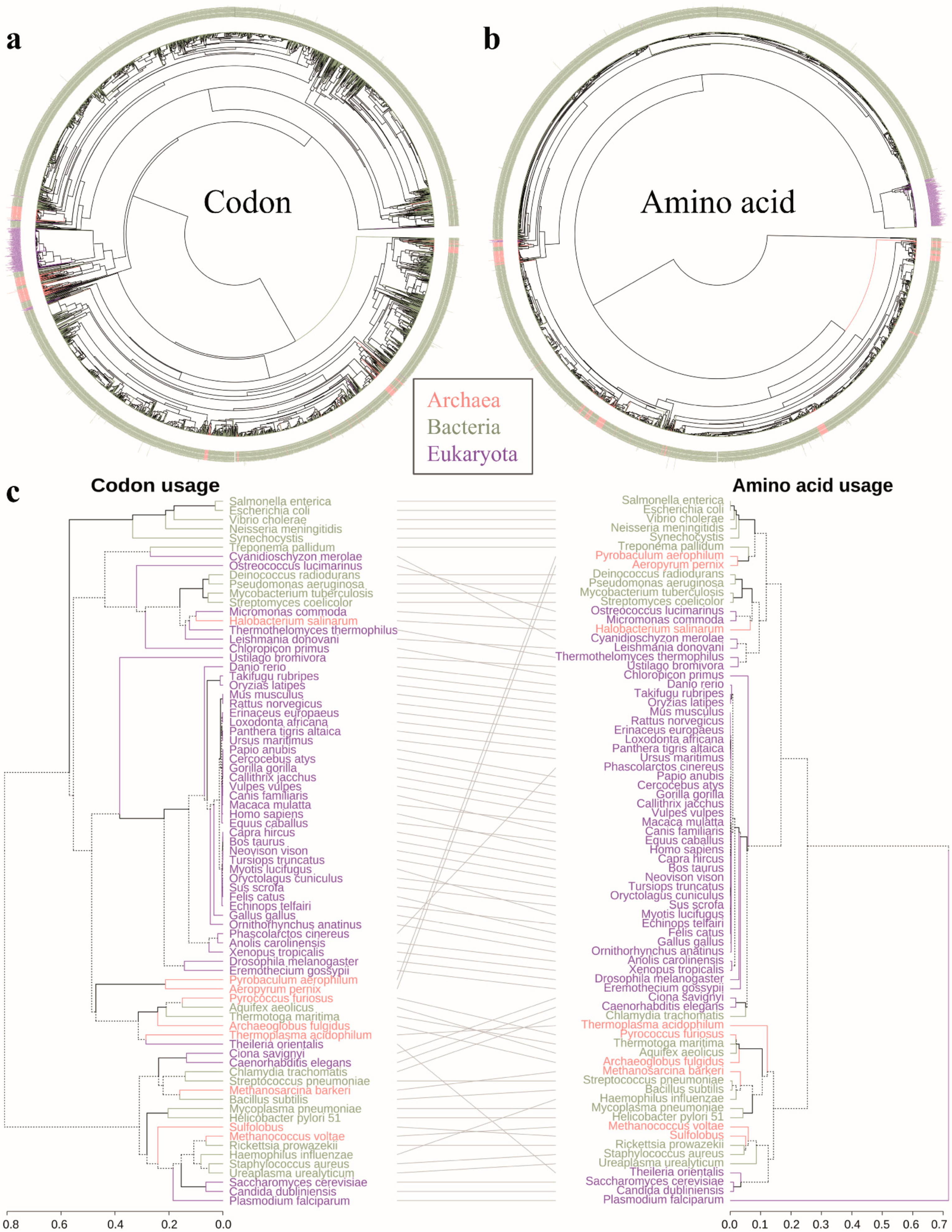
2.4. Phylogenetic Analysis
2.5. Quasi Evolutionary Time Estimation
3. Results
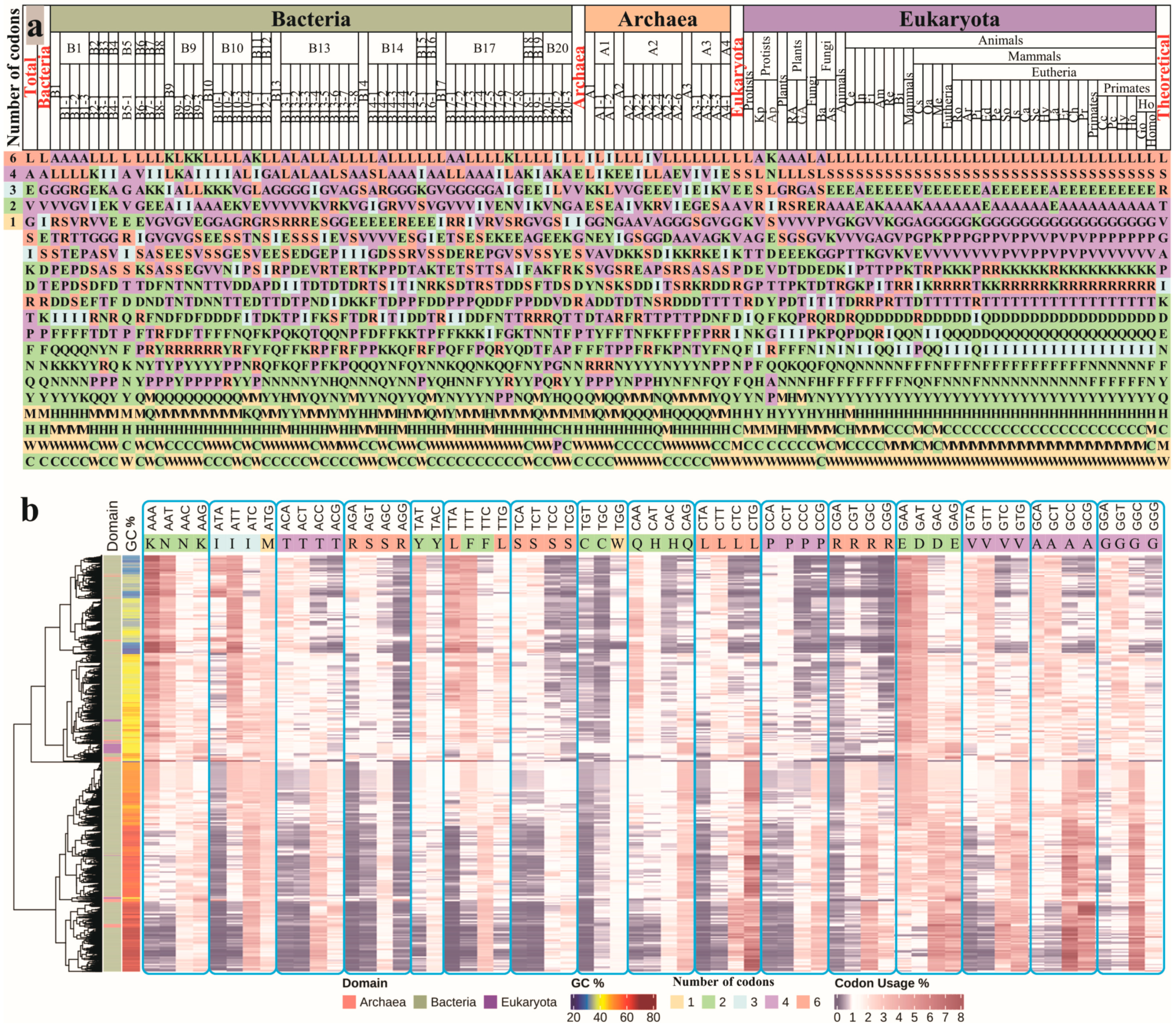
3.1. Genome-Wide Exploration of Amino Acid Usage Bias and Genetic Codon Usage Bias in the Three Domains of Life
3.1.1. Amino Acid Usage Landscape
3.1.2. Genetic Codon Usage Landscape
3.2. Chronological Order of Species Emergence Based on Codon Usage Bias
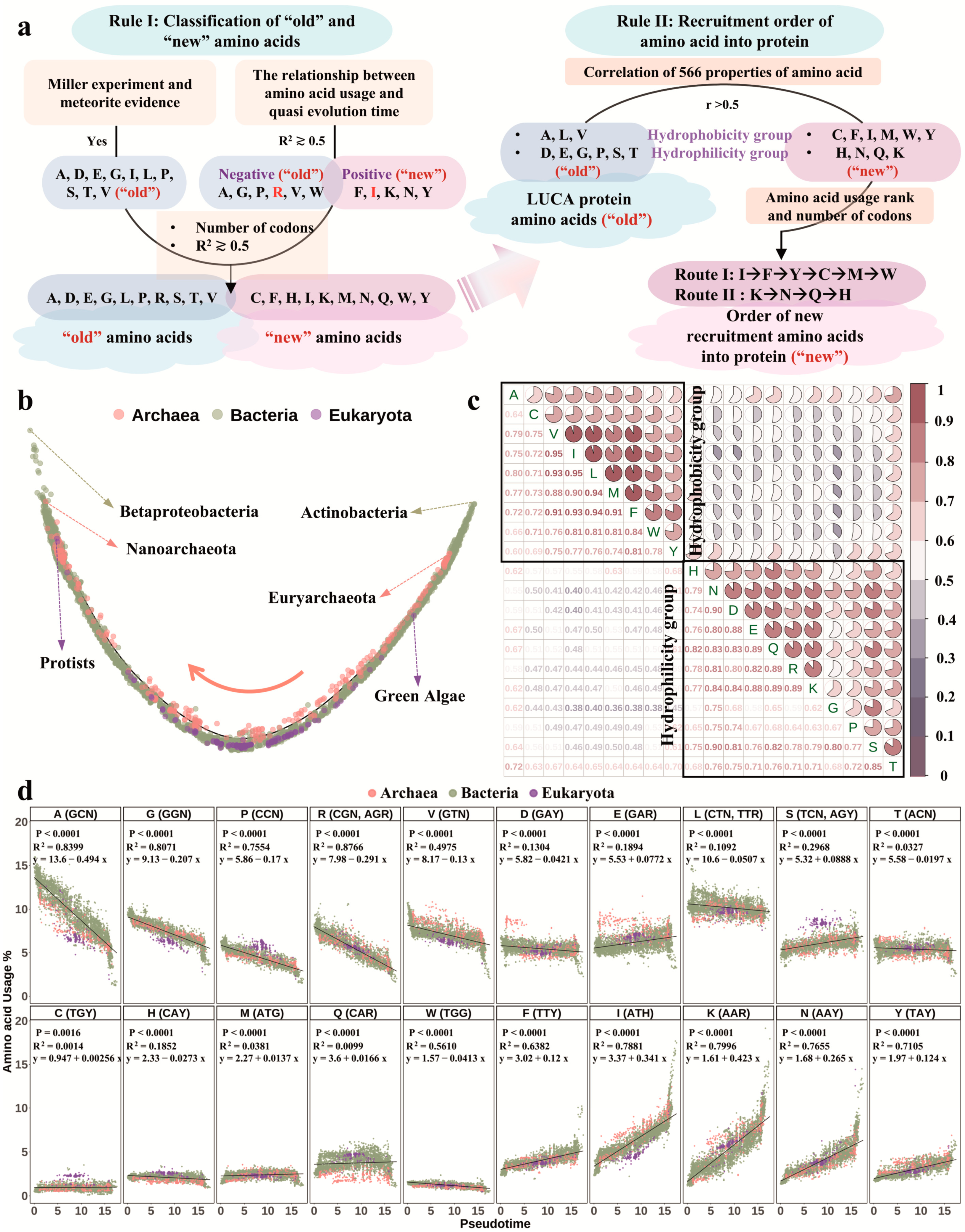
3.3. LUCA Protein Amino Acid Composition and Amino Acid Recruitment Order into Protein
3.4. Impact of Codon Selection on Amino Acid Usage and Recruitment
4. Discussion
4.1. Amino Acid Usage Bias Is Ubiquitous and Is Independent of Single Codon Usage Bias
4.2. Codon Usage Bias Is the Imprint of the Speciation Process
4.3. Recruitment of Amino Acids into Proteogenesis Is a Progressive Evolution Process
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Theobald, D.L. A formal test of the theory of universal common ancestry. Nature 2010, 465, 219–222. [Google Scholar] [CrossRef]
- Raup, D.; Valentine, J. Multiple origins of life. Proc. Natl. Acad. Sci. USA 1983, 80, 2981–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giulio, M. LUCA as well as the ancestors of archaea, bacteria and eukaryotes were progenotes: Inference from the distribution and diversity of the reading mechanism of the AUA and AUG codons in the domains of life. Biosystems 2020, 198, 104239. [Google Scholar] [CrossRef]
- Doolittle, W.F. The practice of classification and the theory of evolution, and what the demise of Charles Darwin’s tree of life hypothesis means for both of them. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 2221–2228. [Google Scholar] [CrossRef] [Green Version]
- Woese, C. The universal ancestor. Proc. Natl. Acad. Sci. USA 1998, 95, 6854–6859. [Google Scholar] [CrossRef] [Green Version]
- Wong, J. The evolution of a universal genetic code. Proc. Natl. Acad. Sci. USA 1976, 73, 2336–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifonov, E. The triplet code from first principles. J. Biomol. Struct. Dyn. 2004, 22, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Perez, M.; Jose, M.V. The evolution of proteome: From the primeval to the very dawn of LUCA. Biosystems 2019, 181, 1–10. [Google Scholar] [CrossRef]
- McDonald, J.H. Apparent trends of amino Acid gain and loss in protein evolution due to nearly neutral variation. Mol. Biol. Evol. 2006, 23, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xie, Z.; Tan, S.; Zhang, X.; Yang, S. Relationship between amino acid usage and amino acid evolution in primates. Gene 2015, 557, 182–187. [Google Scholar] [CrossRef]
- Brooks, D.; Fresco, J.; Lesk, A.; Singh, M. Evolution of amino acid frequencies in proteins over deep time: Inferred order of introduction of amino acids into the genetic code. Mol. Biol. Evol. 2002, 19, 1645–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, I.; Kondrashov, F.; Adzhubei, I.; Wolf, Y.; Koonin, E.; Kondrashov, A.; Sunyaev, S. A universal trend of amino acid gain and loss in protein evolution. Nature 2005, 433, 633–638. [Google Scholar] [CrossRef]
- Hurst, L.; Feil, E.; Rocha, E. Protein evolution: Causes of trends in amino-acid gain and loss. Nature 2006, 442, E11–E12. [Google Scholar] [CrossRef]
- Hurst, L.; Smith, N. Do essential genes evolve slowly? Curr. Biol. 1999, 9, 747–750. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, J.; Ni, F.; Dong, X.; Han, B.; Han, D.; Ji, Z.; Zhao, Y. Genome wide exploration of the origin and evolution of amino acids. BMC Evol. Biol. 2010, 10, 77. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Zhang, C.; Wang, H.; Liu, S.; Wei, W.; Guo, F. The GC Content as a Main Factor Shaping the Amino Acid Usage During Bacterial Evolution Process. Front. Microbiol. 2018, 9, 2948. [Google Scholar] [CrossRef] [PubMed]
- Wong, J. A co-evolution theory of the genetic code. Proc. Natl. Acad. Sci. USA 1975, 72, 1909–1912. [Google Scholar] [CrossRef] [Green Version]
- Trifonov, E. Consensus temporal order of amino acids and evolution of the triplet code. Gene 2000, 261, 139–151. [Google Scholar] [CrossRef]
- Osawa, S.; Muto, A.; Jukes, T.; Ohama, T. Evolutionary changes in the genetic code. Proc. Biol. Sci. 1990, 241, 19–28. [Google Scholar] [CrossRef]
- Osawa, S.; Jukes, T.H.; Watanabe, K.; Muto, A. Recent evidence for evolution of the genetic code. Microbiol. Rev. 1992, 56, 229–264. [Google Scholar] [CrossRef]
- Castresana, J.; Feldmaier-Fuchs, G.; Pääbo, S. Codon reassignment and amino acid composition in hemichordate mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 3703–3707. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Yokobori, S. tRNA Modification and Genetic Code Variations in Animal Mitochondria. J. Nucleic Acids 2011, 2011, 623095. [Google Scholar] [CrossRef]
- Simoes, J.; Bezerra, A.R.; Moura, G.R.; Araujo, H.; Gut, I.; Bayes, M.; Santos, M.A. The Fungus Candida albicans Tolerates Ambiguity at Multiple Codons. Front. Microbiol. 2016, 7, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Zhang, Z.; Du, J.; Fu, Y.; Liang, A. Large-scale mass spectrometry-based analysis of Euplotes octocarinatus supports the high frequency of +1 programmed ribosomal frameshift. Sci. Rep. 2016, 6, 33020. [Google Scholar] [CrossRef]
- Böck, A.; Forchhammer, K.; Heider, J.; Baron, C. Selenoprotein synthesis: An expansion of the genetic code. Trends Biochem. Sci. 1991, 16, 463–467. [Google Scholar] [CrossRef]
- Atkins, J.; Gesteland, R. Biochemistry. The 22nd amino acid. Science 2002, 296, 1409–1410. [Google Scholar] [CrossRef]
- Martin, W.F.; Weiss, M.C.; Neukirchen, S.; Nelson-Sathi, S.; Sousa, F.L. Physiology, phylogeny, and LUCA. Microb. Cell. 2016, 3, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muto, A.; Osawa, S. The guanine and cytosine content of genomic DNA and bacterial evolution. Proc. Natl. Acad. Sci. USA 1987, 84, 166–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, T.; Runnegar, B. Megascopic eukaryotic algae from the 2.1-billion-year-old negaunee iron-formation, Michigan. Science 1992, 257, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Granold, M.; Hajieva, P.; Toşa, M.; Irimie, F.; Moosmann, B. Modern diversification of the amino acid repertoire driven by oxygen. Proc. Natl. Acad. Sci. USA 2018, 115, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Palidwor, G.A.; Perkins, T.J.; Xia, X. A general model of codon bias due to GC mutational bias. PLoS ONE 2010, 5, e13431. [Google Scholar] [CrossRef]
- Foster, P.; Jermiin, L.; Hickey, D. Nucleotide composition bias affects amino acid content in proteins coded by animal mitochondria. J. Mol. Evol. 1997, 44, 282–288. [Google Scholar] [CrossRef] [Green Version]
- Singer, G.; Hickey, D. Nucleotide bias causes a genomewide bias in the amino acid composition of proteins. Mol. Biol. Evol. 2000, 17, 1581–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharanidharan, D.; Bhargavi, G.; Uthanumallian, K.; Gautham, N. Correlations between nucleotide frequencies and amino acid composition in 115 bacterial species. Biochem. Biophys. Res. Commun. 2004, 315, 1097–1103. [Google Scholar] [CrossRef]
- Sharp, P.; Tuohy, T.; Mosurski, K. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef]
- Hershberg, R.; Petrov, D. General rules for optimal codon choice. PLoS Genet. 2009, 5, e1000556. [Google Scholar] [CrossRef] [Green Version]
- Novoa, E.; Jungreis, I.; Jaillon, O.; Kellis, M. Elucidation of Codon Usage Signatures across the Domains of Life. Mol. Biol. Evol. 2019, 36, 2328–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlin, S.; Burge, C. Dinucleotide relative abundance extremes: A genomic signature. Trends Genet. 1995, 11, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Berezovsky, I.; Chen, W.; Choi, P.; Shakhnovich, E. Entropic stabilization of proteins and its proteomic consequences. PLoS Comput. Biol. 2005, 1, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doig, A. Frozen, but no accident—Why the 20 standard amino acids were selected. FEBS J. 2017, 284, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Bywater, R.P. On dating stages in prebiotic chemical evolution. Naturwissenschaften 2012, 99, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.B.; Hippen, A.A.; Belyeu, J.R.; Whiting, M.F.; Ridge, P.G. Missing something? Codon aversion as a new character system in phylogenetics. Cladistics 2017, 33, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Davide, A.; Maddalena, D.; Andrea, G. Codon usage bias in prokaryotic genomes and environmental adaptation. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Petrov, D.; Hartl, D. High rate of DNA loss in the Drosophila melanogaster and Drosophila virilis species groups. Mol. Biol. Evol. 1998, 15, 293–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beletskii, A.; Bhagwat, A. Transcription-induced mutations: Increase in C to T mutations in the nontranscribed strand during transcription in Escherichia coli. Proc. Natl. Acad. Sci. USA 1996, 93, 13919–13924. [Google Scholar] [CrossRef] [Green Version]
- Fryxell, K.; Zuckerkandl, E. Cytosine deamination plays a primary role in the evolution of mammalian isochores. Mol. Biol. Evol. 2000, 17, 1371–1383. [Google Scholar] [CrossRef] [Green Version]
- Lobry, J. Influence of genomic G+C content on average amino-acid composition of proteins from 59 bacterial species. Gene 1997, 205, 309–316. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Gil, M.; Zanetti, M.S.; Zoller, S.; Anisimova, M. CodonPhyML: Fast maximum likelihood phylogeny estimation under codon substitution models. Mol. Biol. Evol. 2013, 30, 1270–1280. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.B.; McKinnon, L.M.; Whiting, M.F.; Ridge, P.G. Codon use and aversion is largely phylogenetically conserved across the tree of life. Mol. Phylogenet. Evol. 2020, 144, 106697. [Google Scholar] [CrossRef]
- Williams, T.A.; Foster, P.G.; Cox, C.J.; Embley, T.M. An archaeal origin of eukaryotes supports only two primary domains of life. Nature 2013, 504, 231–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLong, E.; Pace, N. Environmental diversity of bacteria and archaea. Syst. Biol. 2001, 50, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Tautz, D.; Domazet-Loso, T. The evolutionary origin of orphan genes. Nat. Rev. Genet. 2011, 12, 692–702. [Google Scholar] [CrossRef]
- Spang, A.; Saw, J.H.; Jorgensen, S.L.; Zaremba-Niedzwiedzka, K.; Martijn, J.; Lind, A.E.; van Eijk, R.; Schleper, C.; Guy, L.; Ettema, T.J.G. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 2015, 521, 173–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, M.C.; Sousa, F.L.; Mrnjavac, N.; Neukirchen, S.; Roettger, M.; Nelson-Sathi, S.; Martin, W.F. The physiology and habitat of the last universal common ancestor. Nat. Microbiol. 2016, 1, 16116. [Google Scholar] [CrossRef]
- Akashi, H.; Gojobori, T. Metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2002, 99, 3695–3700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gospodinov, A.; Kunnev, D. Universal Codons with Enrichment from GC to AU Nucleotide Composition Reveal a Chronological Assignment from Early to Late Along with LUCA Formation. Life 2020, 10, 81. [Google Scholar] [CrossRef]
- Goldstein, R.; Pollock, D. Observations of amino acid gain and loss during protein evolution are explained by statistical bias. Mol. Biol. Evol. 2006, 23, 1444–1449. [Google Scholar] [CrossRef]
- Solis, A. Reduced alphabet of prebiotic amino acids optimally encodes the conformational space of diverse extant protein folds. BMC Evol. Biol. 2019, 19, 158. [Google Scholar] [CrossRef]
- Kimura, M.; Akanuma, S. Reconstruction and Characterization of Thermally Stable and Catalytically Active Proteins Comprising an Alphabet of ~13 Amino Acids. J. Mol. Evol. 2020, 88, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Miller, S. Which organic compounds could have occurred on the prebiotic earth? Cold Spring Harb. Symp. Quant. Biol. 1987, 52, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Rother, M.; Krzycki, J. Selenocysteine, pyrrolysine, and the unique energy metabolism of methanogenic archaea. Archaea 2010, 2010, 453642. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GC% | <45 | ≥45 | FC | GC% | <45 | ≥45 | FC |
|---|---|---|---|---|---|---|---|
| avgGC% | 36.60 | 59.10 | 1.61 | avgGC% | 36.60 | 59.10 | 1.61 |
| AAA (K) | 5.54 ± 1.93 | 1.60 ± 1.29 | 0.29 | AAT (N) | 3.70 ± 1.44 | 1.08 ± 0.68 | 0.29 |
| AAG (K) | 1.94 ± 0.81 | 2.06 ± 0.73 | 1.06 | AAC (N) | 1.57 ± 0.50 | 1.94 ± 0.33 | 1.23 |
| ATA (I) | 2.04 ± 1.61 | 0.42 ± 0.58 | 0.21 | ATT (I) | 4.19 ± 1.23 | 1.51 ± 1.11 | 0.36 |
| ATG (M) | 2.33 ± 0.32 | 2.33 ± 0.39 | 1.00 | ATC (I) | 1.68 ± 0.67 | 3.21 ± 0.74 | 1.91 |
| TTA (L) | 3.62 ± 1.60 | 0.55 ± 0.67 | 0.15 | TTT (F) | 3.45 ± 0.94 | 1.27 ± 0.90 | 0.37 |
| TTG (L) | 1.66 ± 0.64 | 1.27 ± 0.73 | 0.76 | TTC (F) | 1.20 ± 0.48 | 2.32 ± 0.64 | 1.94 |
| TAA (Stop) | 0.21 ± 0.07 | 0.10 ± 0.07 | 0.51 | TAT (Y) | 2.61 ± 0.70 | 1.10 ± 0.60 | 0.42 |
| TAG (Stop) | 0.06 ± 0.03 | 0.06 ± 0.03 | 0.90 | TAC (Y) | 1.05 ± 0.38 | 1.49 ± 0.43 | 1.42 |
| ACA (T) | 1.87 ± 0.53 | 0.55 ± 0.40 | 0.30 | ACT (T) | 1.68 ± 0.45 | 0.53 ± 0.37 | 0.31 |
| ACG (T) | 0.75 ± 0.42 | 1.70 ± 0.61 | 2.27 | ACC (T) | 1.00 ± 0.56 | 2.67 ± 0.74 | 2.66 |
| AGA (R) | 1.25 ± 0.65 | 0.26 ± 0.30 | 0.21 | AGT (S) | 1.43 ± 0.34 | 0.50 ± 0.33 | 0.35 |
| AGG (R) | 0.44 ± 0.44 | 0.37 ± 0.51 | 0.83 | AGC (S) | 0.97 ± 0.52 | 1.63 ± 0.38 | 1.68 |
| TCA (S) | 1.47 ± 0.46 | 0.47 ± 0.33 | 0.32 | TCT (S) | 1.62 ± 0.56 | 0.48 ± 0.37 | 0.30 |
| TCG (S) | 0.47 ± 0.24 | 1.41 ± 0.56 | 2.98 | TCC (S) | 0.62 ± 0.46 | 1.23 ± 0.44 | 1.99 |
| TGA (Stop) | 0.12 ± 0.22 | 0.17 ± 0.08 | 1.44 | TGT (C) | 0.64 ± 0.25 | 0.27 ± 0.18 | 0.43 |
| TGG (W) | 0.93 ± 0.28 | 1.40 ± 0.17 | 1.51 | TGC (C) | 0.36 ± 0.28 | 0.68 ± 0.19 | 1.88 |
| CAA (Q) | 2.57 ± 0.87 | 0.99 ± 0.70 | 0.39 | CAT (H) | 1.32 ± 0.31 | 0.90 ± 0.40 | 0.69 |
| CAG (Q) | 1.19 ± 0.83 | 2.71 ± 0.60 | 2.28 | CAC (H) | 0.63 ± 0.33 | 1.28 ± 0.39 | 2.03 |
| CTA (L) | 1.01 ± 0.38 | 0.34 ± 0.33 | 0.34 | CTT (L) | 1.86 ± 0.62 | 0.98 ± 0.62 | 0.53 |
| CTG (L) | 1.01 ± 1.03 | 4.95 ± 1.71 | 4.91 | CTC (L) | 0.72 ± 0.52 | 2.29 ± 1.28 | 3.19 |
| GAA (E) | 4.80 ± 0.97 | 2.70 ± 1.12 | 0.56 | GAT (D) | 3.88 ± 0.70 | 2.25 ± 0.95 | 0.58 |
| GAG (E) | 1.84 ± 0.91 | 3.12 ± 1.18 | 1.69 | GAC (D) | 1.36 ± 0.61 | 3.31 ± 1.26 | 2.44 |
| CCA (P) | 1.29 ± 0.39 | 0.53 ± 0.35 | 0.41 | CCT (P) | 1.29 ± 0.35 | 0.57 ± 0.32 | 0.44 |
| CCG (P) | 0.54 ± 0.38 | 2.46 ± 0.78 | 4.55 | CCC (P) | 0.47 ± 0.46 | 1.40 ± 0.66 | 2.94 |
| CGA (R) | 0.44 ± 0.24 | 0.41 ± 0.23 | 0.94 | CGT (R) | 1.02 ± 0.60 | 1.10 ± 0.53 | 1.08 |
| CGG (R) | 0.29 ± 0.30 | 1.39 ± 0.81 | 4.86 | CGC (R) | 0.57 ± 0.40 | 3.01 ± 1.15 | 5.26 |
| GCA (A) | 2.29 ± 0.57 | 1.36 ± 0.64 | 0.60 | GCT (A) | 2.39 ± 0.58 | 1.17 ± 0.63 | 0.49 |
| GCG (A) | 0.95 ± 0.60 | 3.95 ± 1.61 | 4.15 | GCC (A) | 1.16 ± 0.70 | 4.65 ± 1.75 | 4.00 |
| GGA (G) | 1.97 ± 0.75 | 0.88 ± 0.49 | 0.45 | GGT (G) | 2.19 ± 0.67 | 1.46 ± 0.61 | 0.67 |
| GGG (G) | 0.87 ± 0.41 | 1.39 ± 0.52 | 1.61 | GGC (G) | 1.22 ± 0.63 | 4.34 ± 1.35 | 3.56 |
| GTA (V) | 1.80 ± 0.64 | 0.67 ± 0.47 | 0.37 | GTT (V) | 2.42 ± 0.64 | 1.07 ± 0.69 | 0.44 |
| GTG (V) | 1.30 ± 0.69 | 3.11 ± 0.85 | 2.39 | GTC (V) | 0.86 ± 0.48 | 2.67 ± 1.15 | 3.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, M.; Ding, R.; Liu, Y.; Ji, Z.; Zhao, Y. Determination of the Amino Acid Recruitment Order in Early Life by Genome-Wide Analysis of Amino Acid Usage Bias. Biomolecules 2022, 12, 171. https://doi.org/10.3390/biom12020171
Zhao M, Ding R, Liu Y, Ji Z, Zhao Y. Determination of the Amino Acid Recruitment Order in Early Life by Genome-Wide Analysis of Amino Acid Usage Bias. Biomolecules. 2022; 12(2):171. https://doi.org/10.3390/biom12020171
Chicago/Turabian StyleZhao, Mingxiao, Ruofan Ding, Yan Liu, Zhiliang Ji, and Yufen Zhao. 2022. "Determination of the Amino Acid Recruitment Order in Early Life by Genome-Wide Analysis of Amino Acid Usage Bias" Biomolecules 12, no. 2: 171. https://doi.org/10.3390/biom12020171
APA StyleZhao, M., Ding, R., Liu, Y., Ji, Z., & Zhao, Y. (2022). Determination of the Amino Acid Recruitment Order in Early Life by Genome-Wide Analysis of Amino Acid Usage Bias. Biomolecules, 12(2), 171. https://doi.org/10.3390/biom12020171