
Exploring the Interactome of Cytochrome P450 2E1 in Human Liver Microsomes with Chemical Crosslinking Mass Spectrometry

 ,
, 
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Protein Expression and Purification
2.3. Pooled Human Liver Microsomes
2.4. Characterization of the Content of Protein and Cytochromes P450 in HLM
2.5. Modification of CYP2E1 with BPM and BPS
2.6. Incorporation of CYP2E1 into HLM
2.7. Probing the Catalytic Activity of BPM- and BPS-Modified CYP2E1
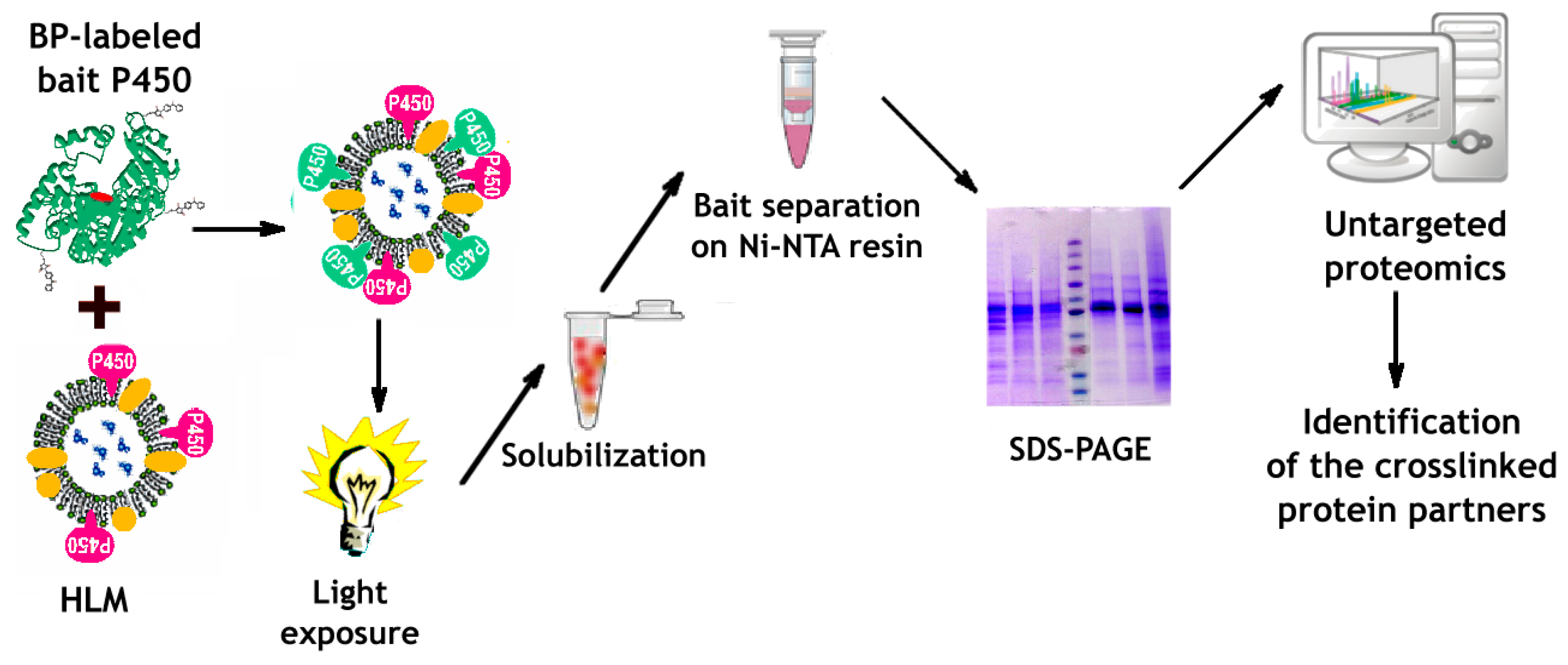
2.8. Crosslinking of the Bait Protein and Its Subsequent Isolation from HLM
2.9. Untargeted Proteomics Assays
3. Results
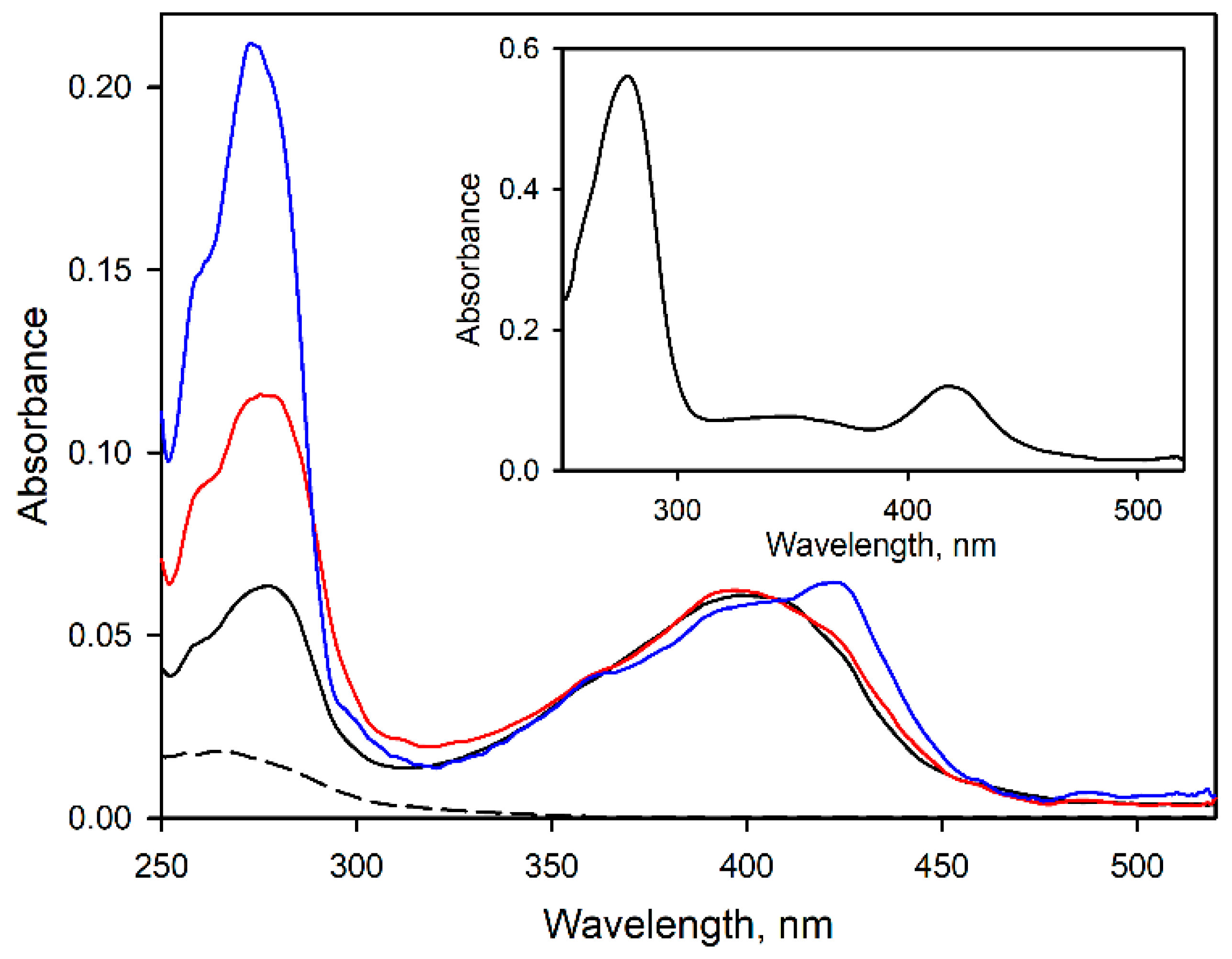
3.1. Modification of CYP2E1 with Benzophenone Derivatives
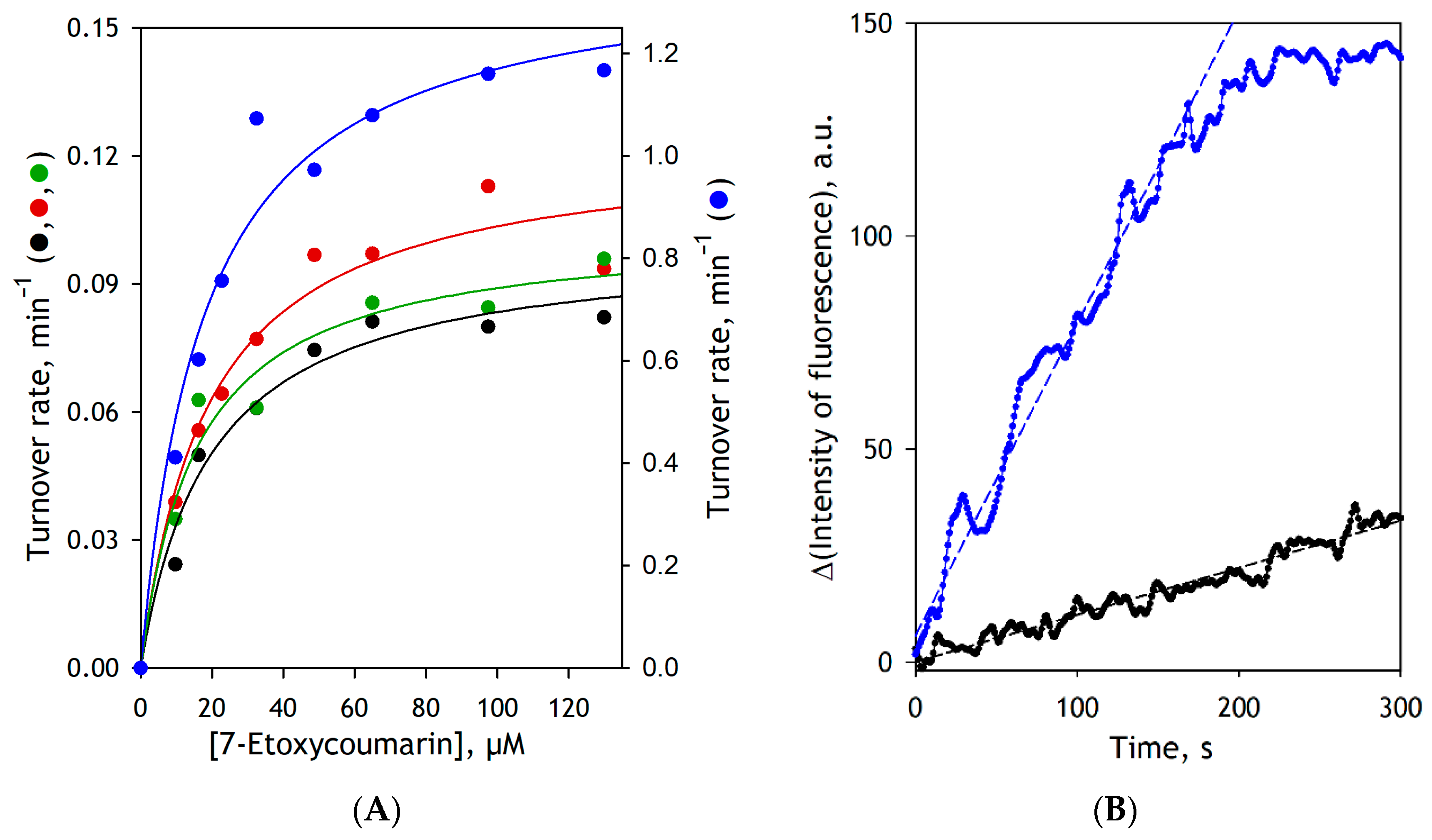
3.2. Effect of Modifications of CYP2E1 by BPM and BPS on the Functional Properties of the Enzyme
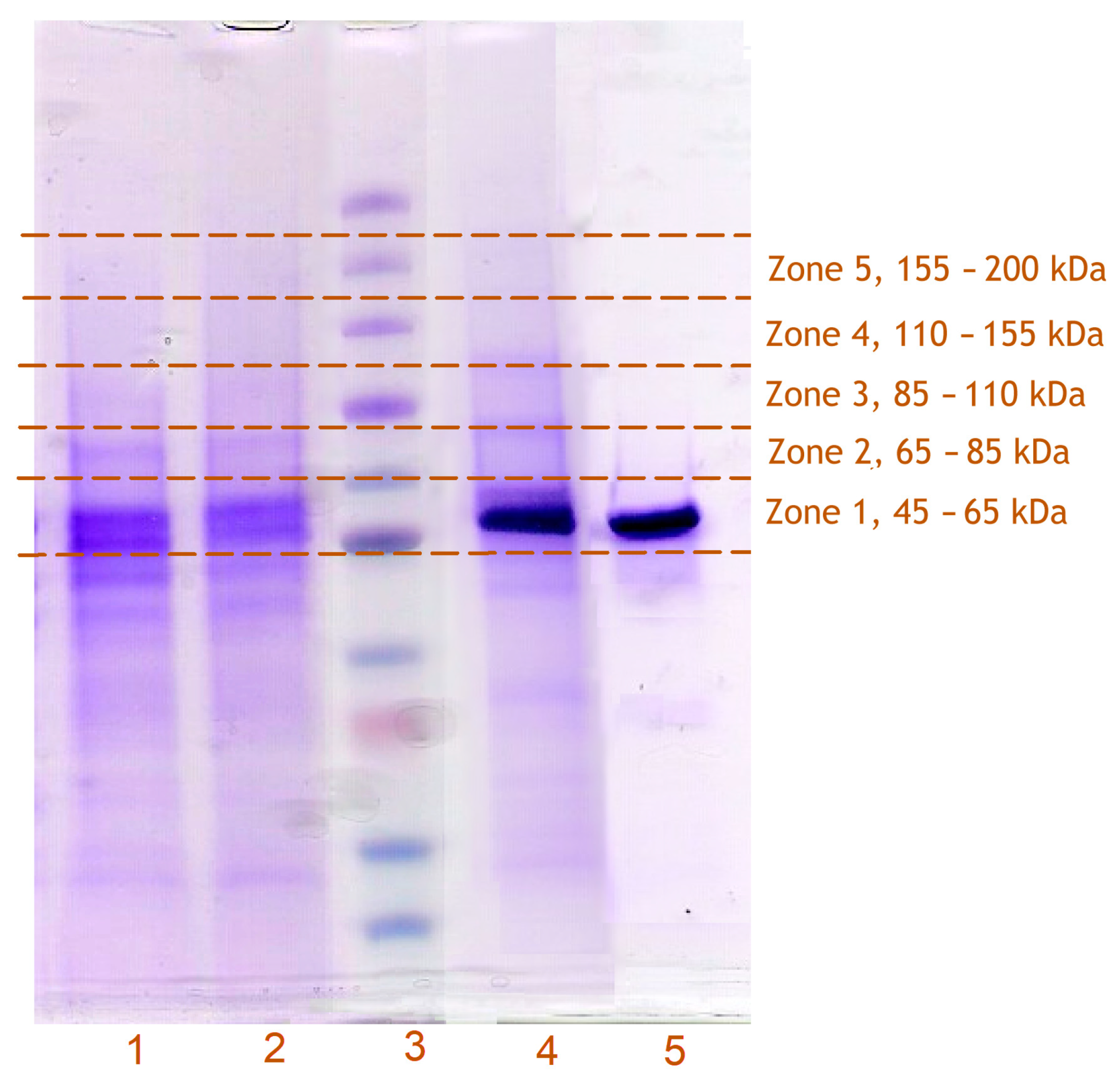
3.3. Incorporation of Modified CYP2E1 into HLM, Its Photo-Activated Crosslinking, and Subsequent Isolation from the Membranes
3.4. Identification of CYP2E1-Crosslinked Proteins with Untargeted Proteomics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rendic, S.; Guengerich, F.P. Update Information on Drug Metabolism Systems-2009, Part II. Summary of Information on the Effects of Diseases and Environmental Factors on Human Cytochrome P450 (CYP) Enzymes and Transporters. Curr. Drug Metab. 2010, 11, 4–84. [Google Scholar] [CrossRef]
- Wienkers, L.C.; Heath, T.G. Predicting in vivo drug interactions from in vitro drug discovery data. Nat. Rev. Drug Discov. 2005, 4, 825–833. [Google Scholar] [CrossRef]
- Gao, N.; Tian, X.; Fang, Y.; Zhou, J.; Zhang, H.F.; Wen, Q.; Jia, L.J.; Gao, J.; Sun, B.; Wei, J.Y.; et al. Gene polymorphisms and contents of cytochrome P450s have only limited effects on metabolic activities in human liver microsomes. Eur. J. Pharm. Sci. 2016, 92, 86–97. [Google Scholar] [CrossRef]
- Zhang, H.F.; Wang, H.H.; Gao, N.; Wei, J.Y.; Tian, X.; Zhao, Y.; Fang, Y.; Zhou, J.; Wen, Q.; Gao, J.; et al. Physiological content and intrinsic Activities of 10 cytochrome P450 isoforms in human normal liver microsomes. J. Pharm. Exp. Ther. 2016, 358, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Gao, J.; Wang, T.; Tian, X.; Gao, N.; Zhou, J.; Zhang, H.F.; Wen, Q.; Jin, H.; Xing, Y.R.; et al. Intraindividual Variation and Correlation of Cytochrome P450 Activities in Human Liver Microsomes. Mol. Pharmaceut. 2018, 15, 5312–5318. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Gao, N.; Tian, X.; Zhou, J.; Zhang, H.F.; Gao, J.; He, X.P.; Wen, Q.; Jia, L.J.; Jin, H.; et al. Effect of P450 Oxidoreductase Polymorphisms on the Metabolic Activities of Ten Cytochrome P450s Varied by Polymorphic CYP Genotypes in Human Liver Microsomes. Cell Physiol. Biochem. 2018, 47, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Volpe, D.A. Interindividual Variability in Drug Metabolizing Enzymes. Curr. Drug Metab. 2019, 20, 1041–1043. [Google Scholar] [CrossRef]
- Davydov, D.R. Molecular organization of the microsomal oxidative system: A new connotation for an old term. Biochem. Mosc.-Suppl. Ser. B-Biomed. Chem. 2016, 10, 10–21. [Google Scholar] [CrossRef]
- Davydov, D.R.; Prasad, B. Assembling the P450 puzzle: On the sources of non-additivity in drug metabolism. Thrends Pharmacol. Sci. 2021, 42, 988–997. [Google Scholar] [CrossRef]
- Reed, J.; Backes, W. The functional effects of physical interactions involving cytochromes P450: Putative mechanisms of action and the extent of these effects in biological membranes. Drug Metab Rev. 2016, 48, 453–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, J.R.; Backes, W.L. Physical studies of P450-P450 interactions: Predicting quaternary structures of P450 complexes in membranes from their X-ray crystal structures. Front. Pharmacol. 2017, 8, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, C.S.; Klein, K.; Zanger, U.M. Membrane Associated Progesterone Receptors: Promiscuous Proteins with Pleiotropic Functions-Focus on Interactions with Cytochromes P450. Front. Pharmacol. 2017, 8, 159. [Google Scholar] [CrossRef] [Green Version]
- Cederbaum, A.I. CYP2E1-Biochemical and toxicological aspects and role in alcohol-induced liver injury. Mount Sinai J. Med. 2006, 73, 657–672. [Google Scholar]
- Cederbaum, A.I. Ethanol-related cytotoxicity catalyzed by CYP2E1-dependent generation of reactive oxygen intermediates in transduced HepG2 cells. Biofactors 1998, 8, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.-N.; Anderson, G.D. Pharmacokinetic and Pharmacodynamic Drug Interactions with Ethanol (Alcohol). Clin. Pharmacokinet. 2014, 53, 1115–1136. [Google Scholar] [CrossRef]
- Jang, G.R.; Harris, R.Z. Drug interactions involving ethanol and alcoholic beverages. Expert Opin. Drug Metab. Toxicol. 2007, 3, 719–731. [Google Scholar] [CrossRef]
- Neuvonen, P.J.; Penttila, O.; Roos, M.; Tirkkonen, J. Effect of long-term alcohol consumption on the half-life of tetracycline and doxycycline in man. Int. J. Clini. Pharm. Biopharm. 1976, 14, 303–307. [Google Scholar]
- Sellman, R.; Kanto, J.; Raijola, E.; Pekkarinen, A. Human and animal study of elimination from plasma and metabolism of diazepam after chronic alcohol intake. Acta Pharmacol. Tox. 1975, 36, 33–38. [Google Scholar] [CrossRef]
- Sellman, R.; Pekkarinen, A.; Kangas, L.; Raijola, E. Reduced concentrations of plasma diazepam in chronic-alcoholic patients following an oral-administration of diazepam. Acta Pharmacol. Tox. 1975, 36, 25–32. [Google Scholar] [CrossRef]
- Sandor, P.; Sellers, E.M.; Dumbrell, M.; Khouw, V. Effect of short-term and long-term alcohol-use on phenytoin kinetics in chronic-alcoholics. Clin. Pharmacol. Ther. 1981, 30, 390–397. [Google Scholar] [CrossRef]
- Kater, R.M.H.; Roggin, G.; Tobon, F.; Zieve, P.; Iber, F.L. Increased rate of clearance of drugs from circulation of alcoholics. Am. J. Med. Sci. 1969, 258, 35–39. [Google Scholar] [CrossRef]
- Dangi, B.; Davydova, N.Y.; Maldonado, M.A.; Ahire, D.; Prasad, B.; Davydov, D.R. Probing functional interactions between cytochromes P450 with principal component analysis of substrate saturation profiles and targeted proteomics. Arch. Biochem. Biophys. 2021, 708, 108937. [Google Scholar] [CrossRef]
- Davydova, N.Y.; Dangi, B.; Maldonado, M.A.; Vavilov, N.E.; Zgoda, V.G.; Davydov, D.R. Toward a systems approach to cytochrome P450 ensemble: Interactions of CYP2E1 with other P450 species and their impact on CYP1A2. Biochem. J. 2019, 476, 3661–3685. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.M.; Olaoye, O.O. Recent Advances in Chemical Biology Using Benzophenones and Diazirines as Radical Precursors. Molecules 2020, 25, 2285. [Google Scholar] [CrossRef]
- Spatzenegger, M.; Liu, H.; Wang, Q.M.; Debarber, A.; Koop, D.R.; Halpert, J.R. Analysis of differential substrate selectivities of CYP2B6 and CYP2E1 by site-directed mutagenesis and molecular modeling. J. Pharm. Exp. Ther. 2003, 304, 477–487. [Google Scholar] [CrossRef]
- Dangi, B.; Davydova, N.Y.; Maldonado, M.A.; Abbasi, A.; Vavilov, N.E.; Zgoda, V.G.; Davydov, D.R. Effects of alcohol-induced increase in CYP2E1 content in human liver microsomes on the activity and cooperativity of CYP3A4. Arch. Biochem. Biophys. 2021, 698, 108677. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Davydov, D.R.; Davydova, N.Y.; Rodgers, J.T.; Rushmore, T.H.; Jones, J.P. Toward a systems approach to the human cytochrome P450 ensemble: Interactions between CYP2D6 and CYP2E1 and their functional consequences. Biochem. J. 2017, 474, 3523–3542. [Google Scholar] [CrossRef]
- Davydov, D.R.; Davydova, N.Y.; Tsalkova, T.N.; Halpert, J.R. Effect of glutathione on homo- and heterotropic cooperativity in cytochrome P450 3A4. Arch. Biochem. Biophys. 2008, 471, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Petushkova, N.A.; Zgoda, V.G.; Pyatnitskiy, M.A.; Larina, O.V.; Teryaeva, N.B.; Potapov, A.A.; Lisitsa, A.V. Post-translational modifications of FDA-approved plasma biomarkers in glioblastoma samples. PLoS ONE 2017, 12, e0177427. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Tomas, H.; Havli, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef]
- MaxQuant. Available online: https://maxquant.org/ (accessed on 21 December 2021).
- Balhara, A.; Basit, A.; Argikar, U.A.; Dumouchel, J.L.; Singh, S.; Prasad, B. Comparative proteomics analysis of the post-mitochondrial supernatant fraction of human lens-free whole eye and liver. Drug Metab. Disp. 2021, DMD-AR-2020-000297. [Google Scholar] [CrossRef]
- Ekins, S.; VandenBranden, M.; Ring, B.J.; Wrighton, S.A. Examination of purported probes of human CYP2B6. Pharmacogenetics 1997, 7, 165–179. [Google Scholar] [CrossRef]
- Donato, M.T.; Jimenez, N.; Castell, J.V.; Gomez-Lechon, M.J. Fluorescence-based assays for screening nine cytochrome P450 (P450) activities in intact cells expressing individual human P450 enzymes. Drug Metab. Disp. 2004, 32, 699–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davydov, D.R.; Davydova, N.Y.; Sineva, E.V.; Halpert, J.R. Interactions among cytochromes P450 in microsomal membranes: Oligomerization of cytochromes P450 3A4, 3A5 and 2E1 and its functional consequences. J. Biol.Chem. 2015, 453, 219–230. [Google Scholar]
- Lewis, D.F.V.; Hlavica, P. Interactions between redox partners in various cytochrome P450 systems: Functional and structural aspects. Biochim. Biophys. Acta 2000, 1460, 353–374. [Google Scholar] [CrossRef] [Green Version]
- Monk, B.C.; Tomasiak, T.M.; Keniya, M.V.; Huschmann, F.U.; Tyndall, J.D.A.; O’Connell, J.D., III.; Cannon, R.D.; McDonald, J.G.; Rodriguez, A.; Finer-Moore, J.S.; et al. Architecture of a single membrane spanning cytochrome P450 suggests constraints that orient the catalytic domain relative to a bilayer. Proc. Natl. Acad. Sci. USA 2014, 111, 3865–3870. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.A.; Cosme, J.; Sridhar, V.; Johnson, E.F.; McRee, D.E. Mammalian microsomal cytochrome P450 monooxygenase: Structural adaptations for membrane binding and functional diversity. Mol. Cell 2000, 5, 121–131. [Google Scholar] [CrossRef]
- Liu, J.; Tawa, G.J.; Wallqvist, A. Identifying Cytochrome P450 Functional Networks and Their Allosteric Regulatory Elements. PLoS ONE 2013, 8, e81980. [Google Scholar] [CrossRef] [PubMed]
- Baylon, J.L.; Lenov, I.L.; Sligar, S.G.; Tajkhorshid, E. Characterizing the Membrane-Bound State of Cytochrome P450 3A4: Structure, Depth of Insertion, and Orientation. J. Am. Chem. Soc. 2013, 135, 8542–8551. [Google Scholar] [CrossRef]
- Berka, K.; Paloncyova, M.; Anzenbacher, P.; Otyepka, M. Behavior of Human Cytochromes P450 on Lipid Membranes. J. Phys. Chem. B 2013, 117, 11556–11564. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, V.; Balali-Mood, K.; Sansom, M.S.P.; Wade, R.C. Structure and Dynamics of the Membrane-Bound Cytochrome P450 2C9. PLoS Comput. Biol. 2011, 7, e1002152. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, G.; Nandekar, P.P.; Bruce, N.J.; Wade, R.C. Differing Membrane Interactions of Two Highly Similar Drug-Metabolizing Cytochrome P450 Isoforms: CYP 2C9 and CYP 2C19. Int. J. Mol. Sci. 2019, 20, 4328. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, G.; Nandekar, P.P.; Camp, T.J.; Bruce, N.J.; Gregory, M.C.; Sligar, S.G.; Wade, R.C. Influence of Transmembrane Helix Mutations on Cytochrome P450-Membrane Interactions and Function. Biophys. J. 2019, 116, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, G.; Nandekar, P.P.; Mukherjee, G.; Bruce, N.J.; Wade, R.C. The Effect of Force-Field Parameters on Cytochrome P450-Membrane Interactions: Structure and Dynamics. Sci. Rep. 2020, 10, 7284. [Google Scholar] [CrossRef]
- Gomes, A.M.; Winter, S.; Klein, K.; Turpeinen, M.; Schaeffeler, E.; Schwab, M.; Zanger, U.M. Pharmacogenomics of human liver cytochrome P450 oxidoreductase: Multifactorial analysis and impact on microsomal drug oxidation. Pharmacogenomics 2009, 10, 579–599. [Google Scholar] [CrossRef] [Green Version]
- Connick, J.P.; Reed, J.R.; Cawley, G.F.; Backes, W.L. Heteromeric complex formation between human cytochrome P450 CYP1A1 and heme oxygenase-1. Biochem. J. 2021, 478, 377–388. [Google Scholar] [CrossRef]
- Connick, J.P.; Reed, J.R.; Cawley, G.F.; Backes, W.L. Heme oxygenase-1 affects cytochrome P450 function through the formation of heteromeric complexes: Interactions between CYP1A2 and heme oxygenase-1. J. Biol. Chem. 2021, 296. [Google Scholar] [CrossRef]
- Esteves, F.; Urban, P.; Rueff, J.; Truan, G.; Kranendonk, M. Interaction Modes of Microsomal Cytochrome P450s with Its Reductase and the Role of Substrate Binding. Int. J. Mol. Sci. 2020, 21, 6669. [Google Scholar] [CrossRef] [PubMed]
- Kandel, S.E.; Lampe, J.N. Role of Protein-Protein Interactions in Cytochrome P450-Mediated Drug Metabolism and Toxicity. Chem. Res. Toxicol. 2014, 27, 1474–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubner, N.R.; Kozar-Konieczna, A.; Szoltysek-Boldys, I.; Slodczyk-Mankowska, E.; Goniewicz, J.; Sobczak, A.; Jacob, P.; Benowitz, N.L.; Goniewicz, M.L. Cessation of alcohol consumption decreases rate of nicotine metabolism in male alcohol-dependent smokers. Drug Alcohol Depen. 2016, 163, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnert, M.; Kovacs, E.J.; Molina, P.E.; Relja, B. Modulation of Inflammation by Alcohol Exposure. Mediat. Inflamm. 2014, 2014, 283756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, W.B. Sjogren-Larsson syndrome: Molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol. Genet. Metab. 2007, 90, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Michaels, S.; Wang, M.Z. The Revised Human Liver Cytochrome P450 “Pie”: Absolute Protein Quantification of CYP4F and CYP3A Enzymes Using Targeted Quantitative Proteomics. Drug Metab. Disp. 2014, 42, 1241–1251. [Google Scholar] [CrossRef] [Green Version]
- Uehara, S.; Murayama, N.; Nakanishi, Y.; Nakamura, C.; Hashizume, T.; Zeldin, D.C.; Yamazaki, H.; Uno, Y. Immunochemical quantification of cynomolgus CYP2J2, CYP4A and CYP4F enzymes in liver and small intestine. Xenobiotica 2015, 45, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Ishii, Y.; Iwanaga, M.; Nishimura, Y.; Takeda, S.; Ikushiro, S.I.; Nagata, K.; Yamazoe, Y.; Mackenzie, P.I.; Yamada, H. Protein-protein interactions between rat hepatic Cytochromes P450 (P450s) and UDP-glucuronosyltransferases (UGTs): Evidence for the functionally active UGT in P450-UGT complex. Drug Metab. Pharmacok. 2007, 22, 367–376. [Google Scholar] [CrossRef]
- Hu, D.G.; Hulin, J.U.A.; Nair, P.C.; Haines, A.Z.; McKinnon, R.A.; Mackenzie, P.I.; Meech, R. The UGTome: The expanding diversity of UDP glycosyltransferases and its impact on small molecule metabolism. Pharmacol. Ther. 2019, 204. [Google Scholar] [CrossRef]
- Radominska-Pandya, A.; Ouzzine, M.; Fournel-Gigleux, S.; Magdalou, J. Structure of UDP-glucuronosyltransferases in membranes. In Phase II Conjugation Enzymes and Transport Systems; Sies, H., Packer, L., Eds.; Methods in Enzymology; Elsevier Academic Press Inc.: San Diego, CA, USA, 2005; Volume 400, pp. 116–147. [Google Scholar]
- Miyauchi, Y.; Kimura, S.; Kimura, A.; Kurohara, K.; Hirota, Y.; Fujimoto, K.; Mackenzie, P.I.; Tanaka, Y.; Ishii, Y. Investigation of the Endoplasmic Reticulum Localization of UDP-Glucuronosyltransferase 2B7 with Systematic Deletion Mutants. Mol. Pharm. 2019, 95, 551–562. [Google Scholar] [CrossRef] [Green Version]
- Ishii, Y.; Egoshi, N.; Miyauchi, Y.; Yamada, H. Mutual modulation of UDP-glucuronosyltransferases and cytochrome P450 3A4: The domains involved in the functional and physical interactions. Drug Metab. Pharmacok. 2017, 32, S21. [Google Scholar] [CrossRef]
- Miyauchi, Y.; Ishii, Y.; Nagata, K.; Yamazoe, Y.; Mackenzie, P.I.; Yamada, H. Suppression of cytochrome P450 3A4 activity by UDP-glucuronosyltransferase (UGT) 2B7: The role of charged residue(s) in the cytosolic tail of UGT2B7. Drug Metab. Rev. 2015, 47, 34. [Google Scholar]
- Takeda, S.; Ishii, Y.; Iwanaga, M.; Nurrochmad, A.; Ito, Y.; Mackenzie, P.I.; Nagata, K.; Yamazoe, Y.; Oguri, K.; Yamada, H. Interaction of Cytochrome P450 3A4 and UDP-Glucuronosyltransferase 2B7: Evidence for Protein-Protein Association and Possible Involvement of CYP3A4 J-Helix in the Interaction. Mol. Pharm. 2009, 75, 956–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyauchi, Y.; Tanaka, Y.; Nagata, K.; Yamazoe, Y.; Mackenzie, P.I.; Yamada, H.; Ishii, Y. UDP-Glucuronosyltransferase (UGT)-mediated attenuations of cytochrome P450 3A4 activity: UGT isoform-dependent mechanism of suppression. Brit. J. Pharmacol. 2020, 177, 1077–1089. [Google Scholar] [CrossRef]
- Fremont, J.J.; Wang, R.W.; King, C.D. Coimmunoprecipitation of UDP-glucuronosyltransferase isoforms and cytochrome P450 3A4. Mol. Pharm. 2005, 67, 260–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, Y.; Koba, H.; Kinoshita, K.; Oizaki, T.; Iwamoto, Y.; Takeda, S.; Miyauchi, Y.; Nishimura, Y.; Egoshi, N.; Taura, F.; et al. Alteration of the Function of the UDP-Glucuronosyltransferase 1A Subfamily by Cytochrome P450 3A4: Different Susceptibility for UGT Isoforms and UGT1A1/7 Variants. Drug Metab. Disp. 2014, 42, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Taura, K.; Naito, E.; Ishii, Y.; Mori, M.A.; Oguri, K.; Yamada, H. Cytochrome P450 1A1 (CYP1A1) inhibitor alpha-naphthoflavone interferes with UDP-glucuronosyltransferase (UGT) activity in intact but not in permeabilized hepatic microsomes from 3-methylcholanthrene-treated rats: Possible involvement of UGT-P450 interactions. Biol. Pharm. Bull. 2004, 27, 56–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, R.; Itoh, T. Extensive Protein-Protein Interactions Involving UDP-glucuronosyltransferase (UGT) 2B7 in Human Liver Microsomes. Drug Metab. Pharmacok. 2014, 29, 259–265. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microsomal Preparation | KM, µM | Vmax, s−1 a |
|---|---|---|
| SS(2E1) | 19.1 ± 5.2 | 0.099 ± 0.019 |
| SS(CPR+b5)2E1 | 17.5 ± 3.7 | 0.122 ± 0.018 |
| SS(CPR+b5)2E1-BPM | 16.2 ± 0.7 | 0.104 ± 0.005 |
| SS(CPR+b5)2E1-BPS | 20.3 ± 3.3 | 1.51 ± 0.42 |
| Protein Group | Gene Name | Number of Hits |
|---|---|---|
| Cytochromes P450 | CYP2A6 and CYP2A7 | 4 |
| CYP2C8 | 5 | |
| CYP2C9 and CYP2C19 | 5 | |
| CYP3A4 | 4 | |
| CYP4A11 and CYP4A22 | 4 | |
| UDP-glucuronosyltransferases | UGT1A1 | 4 |
| UGT1A4; UGT1A5 | 4 | |
| UGT1A6 | 6 | |
| UGT1A9, UGT1A8, UGT1A7, and UGT1A10 | 6 | |
| UGT2B17 | 6 | |
| UGT2B4 | 6 | |
| UGT2B7 | 6 | |
| Other proteins of microsomal membrane | ALDH3A2 | 4 |
| EPHX1 | 4 | |
| ERO1L | 5 | |
| FMO3 | 4 | |
| RPN2 | 4 |
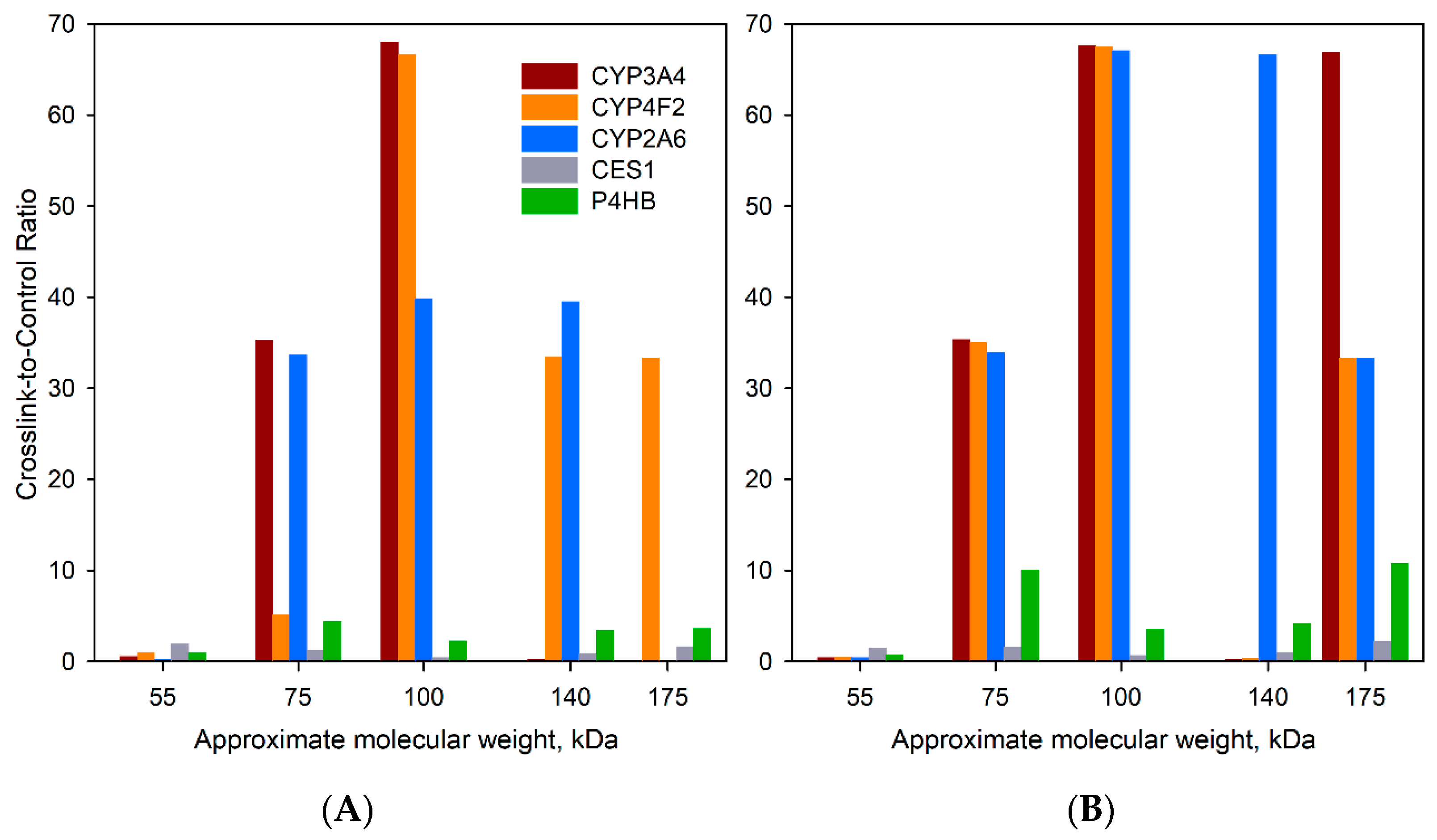
| Protein | Crosslinking with BPM | Crosslinking with BPS | Total Number of Positives | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HLM(LFJ), Assay 1 | HLM(LFJ), Assay 2 | HLM(LBA) | HLM(LFJ), Assay1 | HLM(LFJ), Assay 2 | HLM(LBA) | ||||||||||||||||||||||||||
| 55 a | 75 | 100 | 140 | 180 | 55 | 75 | 100 | 140 | 180 | 55 | 75 | 100 | 140 | 180 | 55 | 75 | 100 | 140 | 180 | 55 | 75 | 100 | 140 | 180 | 55 | 75 | 100 | 140 | 180 | ||
| CYP2A6 b | 0.3 | 0.7 | ∝ | 0.5 | 0 | 0.2 | 0.3 | 2 | ∝ | 0 | 0.1 | ∝ | 18 | 18 | 0 | 0.7 | 1.1 | ∝ | 0 | 0 | 0.5 | 0.6 | 1.2 | ∝ | 0 | 0.1 | ∝ | ∝ | ∝ | ∝ | 6 |
| CYP2C8 | 0.7 | 1.2 | ∝ | 0.8 | 0.2 | 1.3 | 1.4 | 2.8 | 0.3 | 0 | 0.4 | ∝ | 6.6 | 0 | 0 | 1.4 | 1.2 | ∝ | 0 | 0 | 0.5 | 1.5 | 3.1 | 0.1 | 8.9 | 0.1 | ∝ | 4.7 | 3 | 8.3 | 4 |
| CYP2C9 c | 1.2 | 0.5 | ∝ | 0.8 | 0.1 | 0.9 | 1.3 | 2.6 | 0.8 | 0 | 0.6 | 13 | 4.7 | 3.2 | 0 | 1.1 | 1.3 | ∝ | 0 | 0 | 0.4 | 2.9 | 4.4 | 1.6 | ∝ | 0.3 | 25 | 6.8 | 2 | 4.6 | 3 |
| CYP3A4 | 0.7 | 2.1 | ∝ | 0.4 | 0 | 0.4 | 3.7 | 4 | 0.4 | 0 | 0.5 | ∝ | ∝ | 0 | 0 | 1 | 1.9 | ∝ | 0 | 0.7 | 0.2 | 4.2 | 3 | 0.7 | ∝ | 0 | ∝ | ∝ | 0 | ∝ | 5 |
| CYP4A11 d | 0.7 | 1.2 | ∝ | 0.9 | 0 | 0.5 | 6.9 | 5 | 0 | 0 | 0.6 | ∝ | 0 | 0 | 0 | 1 | 0.6 | ∝ | 0 | 0 | 0.3 | 7.8 | 6 | 0.3 | 0 | 0.2 | ∝ | 2.4 | 0 | 0 | 4 |
| CYP4F2 | 0.6 | 1 | ∝ | ∝ | 0 | 0.9 | 1.3 | ∝ | 0.2 | ∝ | 1.2 | 13 | 0 | 0 | 0 | 1 | 0.3 | ∝ | 0 | 0 | 0.3 | 5 | ∝ | 1.2 | ∝ | 1.2 | ∝ | 0 | 0 | 0 | 5 |
| CYB5A | ∝ | 0.9 | 0 | 0 | 0 | ∝ | 0.9 | ∝ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ∝ | 0.8 | 0 | 0 | 0 | ∝ | 0.6 | ∝ | ∝ | ∝ | 0 | 0 | 0 | 0 | 0 | 4 |
| CPR | 3.9 | 1.2 | 0.2 | 3.3 | 0 | 0.7 | ∝ | 0.1 | 0.1 | 0 | 1.7 | 11 | 0 | 0 | 0 | 11 | 1.1 | 0.3 | 0 | 0 | 3.5 | ∝ | 0.1 | 0.1 | ∝ | 19 | 0.8 | 129 | 0.0 | ∝ | 2 |
| UGT1A1 | 1 | 0.7 | ∝ | 0.5 | 0 | 1.5 | 0.3 | 6.4 | 0.4 | 0 | 1 | 0 | 0 | 0 | 0 | 0.5 | 2.5 | ∝ | 0 | 0 | 1 | 0.6 | 6.3 | 0.4 | ∝ | 0 | ∝ | 0 | 0 | 0 | 4 |
| UGT 1A4 e | 0.8 | 0.6 | ∝ | 0.6 | ∝ | 1 | 0.3 | 3.9 | 1.2 | 0 | 1 | 0 | 0 | 0 | 0 | 0.5 | 1.4 | ∝ | 0 | ∝ | 0.6 | 0.9 | 5.2 | 1.6 | ∝ | 0.5 | ∝ | 0 | 0 | 0 | 4 |
| UGT 1A6 | 0.6 | 1.7 | ∝ | 1.3 | ∝ | 0.7 | 0.9 | 3 | 1.3 | ∝ | 0.7 | ∝ | 0 | ∝ | 0 | 0.7 | 1.4 | ∝ | 0 | 0 | 0.5 | 0.6 | 2.7 | 2.8 | ∝ | 0.6 | ∝ | ∝ | ∝ | ∝ | 6 |
| UGT 1A9 f | 0.6 | ∝ | ∝ | ∝ | 0 | 0.7 | ∝ | ∝ | ∝ | 0 | 0.5 | ∝ | ∝ | 1 | 0 | 0.6 | ∝ | ∝ | 0 | 0 | 0.3 | ∝ | ∝ | ∝ | 0 | 0.3 | ∝ | ∝ | 1 | ∝ | 6 |
| UGT 2B10 | 0.2 | 3.1 | ∝ | 0 | 0 | 0.2 | ∝ | 2 | ∝ | 0 | 0.6 | ∝ | 0 | 0 | 0 | 1.1 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | ∝ | 0 | ∝ | 0 | 0 | 0 | 5 |
| UGT 2B15 | 0.6 | 0.9 | ∝ | 2.1 | ∝ | 0.7 | ∝ | 2.9 | 1.1 | 0 | 0.6 | ∝ | 0 | 0 | 0 | 1 | 1.5 | ∝ | 0 | 0 | 0.2 | ∝ | 5.3 | 1.4 | ∝ | 0 | 0 | 0 | 0 | 0 | 5 |
| UGT 2B17 | 0.8 | 1 | ∝ | 0.6 | 1.5 | 0.6 | 0.4 | 7 | 0.7 | 14 | 0.5 | ∝ | 8.3 | ∝ | 0 | 1 | 1.5 | ∝ | 0 | 0 | 0.4 | 0.4 | 3.4 | 1.6 | 36 | 0 | ∝ | 6.8 | ∝ | ∝ | 5 |
| UGT 2B4 | 0.7 | 0.9 | ∝ | 0.9 | 2 | 0.5 | 5 | 4.4 | 1.9 | ∝ | 0.7 | ∝ | 1.7 | ∝ | 0 | 1 | 0.9 | ∝ | 0 | 0 | 0.2 | 12 | 3 | 4.9 | ∝ | 0.4 | ∝ | 0 | ∝ | 0 | 6 |
| UGT 2B7 | 0.6 | 0.9 | ∝ | 1 | 1.2 | 0.5 | 1.2 | 2.3 | 0.8 | 3.1 | 0.7 | ∝ | ∝ | 7.6 | 0 | 0.6 | 1 | ∝ | 0 | 4 | 0.2 | 3.3 | 2.3 | 1.4 | 3.3 | 0.1 | ∝ | ∝ | 8.4 | ∝ | 4 |
| ALDH3A2 | 0.4 | 0.6 | ∝ | ∝ | 0 | 0.3 | ∝ | 5 | 3.4 | 0 | 0 | 0 | 0 | 0 | 0 | 0.8 | 0.8 | ∝ | 0 | 0 | 0.2 | ∝ | 6.5 | 2.3 | ∝ | 0 | 0 | 0 | 0 | 0 | 4 |
| EPHX1 | 0.2 | 0.6 | ∝ | 0.6 | 1.5 | 0.7 | ∝ | 1 | 0.4 | 0.8 | 0.2 | ∝ | 1.6 | 0.8 | ∝ | 0.2 | 0.4 | ∝ | 0.4 | 2.7 | 0.9 | ∝ | 0.8 | 1.2 | 0.7 | 0.2 | ∝ | 0.7 | 0.9 | ∝ | 6 |
| ERO1L | 0.8 | 1 | 0 | ∝ | 0 | 0.7 | ∝ | 0 | ∝ | 0 | 0 | 0 | 0 | 0 | 0 | 0.7 | 1.8 | ∝ | 0 | 0 | 0.4 | ∝ | ∝ | ∝ | ∝ | 0 | 0 | 0 | 0 | 0 | 4 |
| RPN2 | 0.7 | 1.3 | ∝ | ∝ | ∝ | 0.4 | ∝ | ∝ | ∝ | ∝ | 0.8 | ∝ | ∝ | ∝ | ∝ | 0.9 | 1.8 | ∝ | ∝ | ∝ | 0.7 | ∝ | ∝ | ∝ | ∝ | 0.4 | ∝ | ∝ | ∝ | ∝ | 6 |
| CES1 | 1.4 | 1.6 | 0.4 | 1.1 | 1.4 | 1.2 | 0.8 | 0.3 | 0.9 | 3.3 | 3.5 | 1.2 | 0.7 | 0.6 | 0.1 | 1.1 | 1.5 | 0.4 | 1.4 | 1.9 | 1.2 | 0.5 | 0.5 | 1.2 | 3 | 2.1 | 2.7 | 1.1 | 0.4 | 1.7 | 0 |
| P4HB | 1.1 | 1.5 | 0.2 | 5.7 | 6.4 | 1.1 | 1 | 0.3 | 3.8 | 1.9 | 0.8 | 11 | 6.3 | 0.8 | 2.6 | 0.8 | 1.6 | 0.3 | 6.4 | 13 | 0.9 | 0.5 | 0.6 | 5.5 | 4.6 | 0.5 | 28 | 9.7 | 0.8 | 15 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davydov, D.R.; Dangi, B.; Yue, G.; Ahire, D.S.; Prasad, B.; Zgoda, V.G. Exploring the Interactome of Cytochrome P450 2E1 in Human Liver Microsomes with Chemical Crosslinking Mass Spectrometry. Biomolecules 2022, 12, 185. https://doi.org/10.3390/biom12020185
Davydov DR, Dangi B, Yue G, Ahire DS, Prasad B, Zgoda VG. Exploring the Interactome of Cytochrome P450 2E1 in Human Liver Microsomes with Chemical Crosslinking Mass Spectrometry. Biomolecules. 2022; 12(2):185. https://doi.org/10.3390/biom12020185
Chicago/Turabian StyleDavydov, Dmitri R., Bikash Dangi, Guihua Yue, Deepak S. Ahire, Bhagwat Prasad, and Victor G. Zgoda. 2022. "Exploring the Interactome of Cytochrome P450 2E1 in Human Liver Microsomes with Chemical Crosslinking Mass Spectrometry" Biomolecules 12, no. 2: 185. https://doi.org/10.3390/biom12020185
APA StyleDavydov, D. R., Dangi, B., Yue, G., Ahire, D. S., Prasad, B., & Zgoda, V. G. (2022). Exploring the Interactome of Cytochrome P450 2E1 in Human Liver Microsomes with Chemical Crosslinking Mass Spectrometry. Biomolecules, 12(2), 185. https://doi.org/10.3390/biom12020185