
JAK2 Variant Signaling: Genetic, Hematologic and Immune Implication in Chronic Myeloproliferative Neoplasms

 ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. JAK2V617F: Genetic Implication in Signaling Pathways
3. Implications of the JAK2V617F Variant in Positive Cells and Immunothrombosis
3.1. Neutrophils
3.2. Platelets
3.3. Monocytes
3.4. T Helper and Natural Killer Lymphocytes
4. Summary and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tremblay, D.; Yacoub, A.; Hoffman, R. Overview of Myeloproliferative Neoplasms: History, Pathogenesis, Diagnostic Criteria, and Complications. Hematol. Oncol. Clin. N. Am. 2021, 35, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Chauffaille, M. Neoplasias mieloproliferativas: Revisão dos critérios diagnósticos e dos aspectos clínicos. Rev. Bras. Hematol. Hemoter. 2010, 32, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Green, A.R. Myeloproliferative neoplasms: From origins to outcomes. Hematology 2017, 1, 470–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A. The history of myeloproliferative disorders: Before and after Dameshek. Leukemia 2008, 22, 3–13. [Google Scholar] [CrossRef]
- Tefferi, A. Myeloproliferative neoplasms: A decade of discoveries and treatment advances. Am. J. Hematol. 2016, 91, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Means, R.T. JAK2 V617F and the evolving paradigm of polycythemia vera. Korean J. Hematol. 2010, 45, 90. [Google Scholar] [CrossRef] [Green Version]
- Bortolheiro, T.C.; Chiattone, C.S. Leucemia mielóide crônica: História natural e classificação. Rev. Bras. Hematol. Hemoter. 2008, 30, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Rowley, J.D. A story of swapped ends. Science 2013, 340, 1412–1413. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.; Le Beau, M.; Bloomfield, C.; Cazzola, M.; Vardiman, J. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Gisslenger, H.; Kvasnicka, H.M.; Vannucchi, A.; Guglielmelli, P.; Orazi, A.; Tefferi, A. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: Document summary and in-depth discussion. Blood Cancer J. 2018, 8, 15. [Google Scholar] [CrossRef]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, P.J.; Green, A.R. The myeloproliferative disorders. N. Engl. J. Med. 2006, 57, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, J.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef] [PubMed]
- Szuber, N.; Vallapureddy, R.; Penna, D.; Lasho, T.L.; Finke, C.; Hanson, C.A.; Ketterling, R.P.; Pardanni, A.; Gangat, N.; Tefferi, A. Myeloproliferative neoplasms in the young: Mayo Clinic experience with 361 patients age 40 years or younger. Am. J. Hematol. 2018, 93, 1474–1484. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C.N.; Koschmieder, S.; Foltz, L.; Guglielmelli, P.; Flindt, T.; Koehler, M.; Mathias, J.; Komatsu, N.; Boothroyd, R.N.; Spierer, A.; et al. The impact of myeloproliferative neoplasms (MPNs) on patient quality of life and productivity: Results from the international MPN Landmark survey. Ann. Hematol. 2017, 96, 1653–1665. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Pardanani, A. Myeloproliferative Neoplasms: A Contemporary Review. JAMA Oncol. 2015, 1, 97–105. [Google Scholar] [CrossRef]
- Meyer, S.; Levine, R.S. Molecular Pathways: Molecular Basis for Sensitivity and Resistance to JAK Kinase Inhibitors. Clin. Cancer Res. 2014, 15, 2051–2059. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Score, J.; Mannarelli, C.; Pancrazzi, A.; Biamonte, F.; Pardanani, A.; Zoi, K.; Reiter, A.; et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: An international study of 797 patients. Leukemia 2014, 28, 1804–1810. [Google Scholar] [CrossRef]
- Kralovics, R.; Stockton, D.W.; Prchal, J.T. Clonal hematopoiesis in familial polycythemia vera suggests the involvement of multiple mutational events in the early pathogenesis of the disease. Blood 2003, 102, 3793–3796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumi, E.; Harutyunyan, A.S.; Pietra, D.; Milosevic, J.D.; Casetti, I.C.; Bellini, M.; Them, N.C.C.; Cavalloni, C.; Ferretti, V.V.; Milanesi, C.; et al. CALR exon 9 mutations are somatically acquired events in familial cases of essential thrombocythemia or primary myelofibrosis. Blood 2014, 123, 2416–2419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landgren, O.; Goldin, L.R.; Kristinsson, S.Y.; Helgadottir, E.A.; Samuelsson, J.; Björkholm, M. Increased risks of polycythemia vera, essential thrombocythemia, and myelofibrosis among 24,577 first-degree relatives of 11,039 patients with myeloproliferative neoplasms in Sweden. Blood 2008, 112, 2199–2204. [Google Scholar] [CrossRef] [PubMed]
- Langabeer, S.E.; Haslam, K.; Linders, J.; Percy, M.J.; Conneally, E.; Hayat, A.; Hennessy, B.; Leahy, M.; Murphy, K.; Murray, M.; et al. Molecular heterogeneity of familial myeloproliferative neoplasms revealed by analysis of the commonly acquired JAK2, CALR and MPL mutations. Fam. Cancer. 2014, 13, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Higgs, J.R.; Sadek, I.; Neumann, P.E.; Ing, V.W.; Renault, N.K.; Berman, J.N.; Greer, W.L. Familial essential thrombocythemia with spontaneous megakaryocyte colony formation and acquired JAK2 mutations. Leukemia 2008, 22, 1551–1556. [Google Scholar] [CrossRef]
- Aljabry, M. Primary familial and congenital polycythemia; The forgotten entity. J. Appl. Hematol. 2018, 9, 39–43. [Google Scholar] [CrossRef]
- Mounier, N. Malignant hematology. Oncologie 2008, 10, 512–514. [Google Scholar] [CrossRef]
- Milosevic, J.D.; Nivarthi, H.; Gisslinger, H.; Leroy, E.; Rumi, E.; Chachoua, I.; Bagienski, K.; Kubesova, B.; Pietra, D.; Gisslinger, B.; et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 2016, 127, 325–332. [Google Scholar] [CrossRef]
- De Freitas, R.M.; da Costa Maranduba, C.M. Myeloproliferative neoplasms and the JAK/STAT signaling pathway: An overview. Rev. Bras. Hematol. Hemot. 2015, 37, 348–353. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2019, 2, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bousoik, E.; Aliabadi, H.M. Do We Know Jack2 about JAK ? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. 2018, 8, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milosevic, J.D.; Schischlik, F.; Jäger, R.; Ivanov, D.; Eisenwort, G.; Keller, A.; Schuster, M.; Hadzijusufovic, E.; Krauth, M.; Spörk, R.; et al. Overexpression of PD-L1 Correlates with JAK2-V617F Mutational Burden and Is Associated with Chromosome 9p Uniparental Disomy in MPN. Blood 2020, 136, 24. [Google Scholar] [CrossRef]
- Koschmieder, S.; Mughal, T.; Hasselbalch, H.C.; Barosi, G.; Valent, P.; Kiladjian, J.; Jeryczynski, G.; Gisslinger, H.; Jutzi, J.S.; Pahl, H.L.; et al. Myeloproliferative neoplasms and inflammation: Whether to target the malignant clone or the inflammatory process or both. Leukemia 2016, 30, 1018–1024. [Google Scholar] [CrossRef]
- Gleitz, H.; Dugourd, A.J.F.; Leimkuhler, N.B.; Snoeren, I.A.M.; Fuchs, S.N.; Menzel, S.; Ziegler, S.; Kroger, N.; Triviai, I.; Busche, G.; et al. Increased CXCL4 expression in hematopoietic cells links inflammation and progression of bone marrow fibrosis in MPN. Blood 2020, 136, 2051–2064. [Google Scholar] [CrossRef]
- Verstovsek, S.; Manshouri, T.; Pilling, D.; Bueso-Ramos, C.E.; Newberry, K.J.; Prijic, S.; Knez, L.; Bozinovic, K.; Harris, D.M.; Spaeth, E.L.; et al. Role of neoplastic monocyte-derived fibrocytes in primary myelofibrosis. J. Exp. Med. 2016, 213, 1723–1740. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.P.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005, 7, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Abdulqader, A.; Saeed, B.; Getta, H.A.; Khoshnaw, N.; Abdulqader, G.; Mohammed, A. Prevalence of JAK2 V617F, CALR, and MPL W515L Gene Mutations in Patients with Essential Thrombocythemia in Kurdistan Region of Iraq. Korean J. Clin. Lab. Sci. 2021, 53, 41–48. [Google Scholar] [CrossRef]
- Staerk, J.; Constantinescu, S.N. The JAK-STAT pathway and hematopoietic stem cells from the JAK2 V617F perspective. JAK-STAT. 2012, 1, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermouet, S.; Vilaine, M. The JAK2 46/1 haplotype: A marker of inappropriate myelomonocytic response to cytokine stimulation, leading to increased risk of inflammation, myeloid neoplasm, and impaired defense against infection? Haematologica 2011, 96, 1575–1579. [Google Scholar] [CrossRef] [PubMed]
- Olcaydu, D.; Rumi, E.; Harutyunyan, A.; Passamonti, F.; Pietra, D.; Pascutto, C.; Berg, T.; Jäger, R.; Hammond, E.; Cazzola, M.; et al. The role of the JAK2 GGCC haplotype and the TET2 gene in familial myeloproliferative neoplasms. Haematologica 2011, 96, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.V.; Cross, N.C.P. Inherited predisposition to myeloproliferative neoplasms. Ther. Adv. Hematol. 2013, 4, 237–253. [Google Scholar] [CrossRef] [Green Version]
- Tashi, T.; Swierczek, S.; Prchal, J.T. Familial MPN Predisposition. Curr. Hematol. Malig. Rep. 2017, 12, 442–447. [Google Scholar] [CrossRef]
- Koh, S.P.; Yip, S.P.; Lee, K.K.; Chan, C.C.; Lau, S.M.; Kho, C.S.; Lau, C.K.; Lin, S.Y.; Lau, Y.M.; Wong, L.G.; et al. Genetic association between germline JAK2polymorphisms and myeloproliferative neoplasms in Hong Kong Chinese population: A case—control study. BMC Genet. 2014, 15, 147. [Google Scholar] [CrossRef] [Green Version]
- Hinds, D.A.; Barnholt, K.E.; Mesa, R.A.; Kiefer, A.K.; Do, C.B.; Eriksson, N.; Mountain, J.L.; Francke, U.; Tung, J.Y.; Nguyen, H.; et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 2016, 128, 1121–1128. [Google Scholar] [CrossRef] [Green Version]
- Owen, K.L.; Brockwell, N.K.; ParkerImmune, B.S. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Marín, F.; Cuenca-Zamora, E.J.; Guijarro-Carrillo, P.J.; Teruel-Montoya, R. Emerging role of neutrophils in the thrombosis of chronic myeloproliferative neoplasms. Int. J. Mol. Sci. 2021, 22, 1143. [Google Scholar] [CrossRef]
- Landolfi, R.; Di Gennaro, L. Pathophysiology of thrombosis in myeloproliferative neoplasms. Haematologica 2011, 96, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, W.; Fidler, T.; Wang, Y.; Tang, Y.; Woods, B.; Welch, C.; Cai, B.; Silvestre-Roig, C.; Ai, D.; et al. Macrophage inflammation, erythrophagocytosis, and accelerated atherosclerosis in JAK2V617F mice. Circ. Res. 2018, 123, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Marin Oyarzún, C.P.; Heller, P.G. Platelets as mediators of thromboinflammation in chronic myeloproliferative neoplasms. Front. Immunol. 2019, 10, 1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucchi, A.M.; Guglielmelli, P. What are the current treatment approaches for patients with polycythemia vera and essential thrombocythemia? Hematology 2017, 1, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Wolach, O.; Abulafia, A.S. Can Novel Insights into the Pathogenesis of Myeloproliferative Neoplasm-Related Thrombosis Inform Novel Treatment Approaches? Hemato 2021, 2, 305–328. [Google Scholar] [CrossRef]
- Marín, C.P.; Glembotsky, A.C.; Goette, N.P.; Lev, P.R.; de Luca, G.; Baroni, M.C.; Moiraghi, B.; Castro, M.A.; Vicente, A.; Marta, R.F.; et al. Platelet Toll-Like Receptors Mediate Thromboinflammatory Responses in Patients With Essential Thrombocythemia. Front. Immunol. 2020, 11, 705. [Google Scholar] [CrossRef]
- Di Rosa, M.; Giallongo, C.; Romano, A.; Li Volti, G.; Musumeci, G.; Barbagallo, I.; Castrogiovanni, P.; Palumbo, G.A. Immunoproteasome genes are modulated in CD34+ JAK2V617F mutated cells from primary myelofibrosis patients. Int. J. Mol. Sci. 2020, 21, 2926. [Google Scholar] [CrossRef]
- Davis, Z.; Felices, M.; Lenvik, T.; Badal, S.; Walker, J.T.; Hinderlie, P.; Riley, J.L.; Vallera, D.A.; Blazar, B.R.; Miller, J.S. Low-density PD-1 expression on resting human natural killer cells is functional and upregulated after transplantation. Blood Adv. 2021, 5, 1069–1080. [Google Scholar] [CrossRef]
- Perner, F.; Perner, C.; Ernst, T.; Heidel, F.H. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef] [Green Version]
- Prestipino, A.; Emhardt, A.J.; Aumann, K.; O’Sullivan, D.; Gorantla, S.P.; Duquesne, S.; Melchinger, W.; Braun, L.; Vuckovic, S.; Boerries, M.; et al. Oncogenic JAK2V617F causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci. Transl. Med. 2019, 10, eaam7729. [Google Scholar] [CrossRef] [Green Version]
- Ginzburg, Y.Z.; Feola, M.; Zimran, E.; Varkonyi, J.; Ganz, T.; Hoffman, R. Dysregulated iron metabolism in polycythemia vera: Etiology and consequences. Leukemia 2018, 32, 2105–2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allain-Maillet, S.; Bosseboeuf, A.; Mennesson, N.; Bostoën, M.; Dufeu, L.; Choi, E.H.; Cleyrat, C.; Mansier, O.; Lippert, E.; Le Bris, J.; et al. Anti-Glucosylsphingosine Autoimmunity, JAK2V617F-Dependent Interleukin-1β and JAK2V617F-Independent Cytokines in Myeloproliferative Neoplasms. Cancers 2020, 12, 2446. [Google Scholar] [CrossRef] [PubMed]
- Hermouet, S.; Bigot-Corbel, E.; Gardie, B. Pathogenesis of Myeloproliferative Neoplasms: Role and Mechanisms of Chronic Inflammation. Mediat. Inflamm. 2015, 2015, 145293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyarzún, C.; Carestia, A.; Lev, P.R.; Glembotsky, A.C.; Castro, M.A.; Moiraghi, B.; Molinas, F.C.; Marta, R.F.; Schattner, M.; Heller, P.G. Neutrophil extracellular trap formation and circulating nucleosomes in patients with chronic myeloproliferative neoplasms. Sci. Rep. 2016, 6, 38738. [Google Scholar] [CrossRef] [Green Version]
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaan8292. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.T. Neutralize the neutrophils! Neutrophil β1/β2 integrin activation contributes to JAK2-V617F–driven thrombosis. J. Clin. Investig. 2018, 128, 4248–4250. [Google Scholar] [CrossRef]
- Gupta, N.; Edelmann, B.; Schnoeder, T.M.; Saalfeld, F.C.; Wolleschak, D.; Kliche, S.; Schraven, B.; Heidel, F.H.; Fischer, T. JAK2-V617F activates β1-integrin-mediated adhesion of granulocytes to vascular cell adhesion molecule. Leukemia 2017, 31, 1223–1226. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, B.; Gupta, N.; Schnoeder, T.M.; Oelschlegel, A.M.; Shahzad, K.; Goldschmidt, J.; Philipsen, L.; Weinert, S.; Ghosh, A.; Saalfeld, F.C.; et al. JAK2-V617F promotes venous thrombosis through β1/β2 integrin activation. J. Clin. Investig. 2018, 128, 4359–4371. [Google Scholar] [CrossRef]
- Haage, T.R.; Müller, A.J.; Arunachalam, P.; Fischer, T. Reveal the Neutrophil: Elucidating the Role of a Neutrophil-Specific JAK2-V617F Mutation. Blood 2019, 134, 2965. [Google Scholar] [CrossRef]
- Gaertner, F.; Massberg, S. Blood coagulation in immunothrombosis—At the frontline of intravascular immunity. Semin. Immunol. 2016, 28, 561–569. [Google Scholar] [CrossRef]
- Shi, C.; Yang, L.; Braun, A.; Anders, H.J. Extracellular DNA—A Danger Signal Triggering Immunothrombosis. Front. Immunol. 2020, 11, 2518. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, Z.; Long, Q.; Huang, J.; Hong, T.; Liu, W.; Lin, J. Insights into Immunothrombosis: The Interplay Among Neutrophil Extracellular Trap, von Willebrand Factor, and ADAMTS13. Front. Immunol. 2020, 11, 3116. [Google Scholar] [CrossRef] [PubMed]
- McKenna, E.; Mhaonaigh, A.U.; Wubben, R.; Dwivedi, A.; Hurley, T.; Kelly, L.A.; Stevenson, N.J.; Little, M.A.; Molloy, E.J. Neutrophils: Need for Standardized Nomenclature. Front. Immunol. 2021, 12, 1081. [Google Scholar] [CrossRef] [PubMed]
- Shaul, M.E.; Fridlender, Z.G. Cancer-related circulating and tumor-associated neutrophils—Subtypes, sources and function. FEBS J. 2018, 285, 4316–4342. [Google Scholar] [CrossRef] [PubMed]
- Giese, M.A.; Hind, L.E.; Huttenlocher, A. Neutrophil plasticity in the tumor microenvironment. Blood 2019, 133, 2159–2167. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [Green Version]
- Piccard, H.; Muschel, R.J.; Opdenakker, G. On the dual roles and polarized phenotypes of neutrophils in tumor development and progression. Crit. Rev. Oncol. Hematol. 2012, 82, 296–309. [Google Scholar] [CrossRef]
- Podaza, E.; Risnik, D. Neglected players: Tumor associated neutrophils involvement in chronic lymphocytic leukemia progression. Oncotarget 2019, 10, 1862–1863. [Google Scholar] [CrossRef]
- Castiglione, M.; Jiang, Y.P.; Mazzeo, C.; Lee, S.; Chen, J.S.; Kaushansky, K.; Yin, W.; Lin, R.Z.; Zheng, H.; Zhan, H. Endothelial JAK2V617F mutation leads to thrombosis, vasculopathy, and cardiomyopathy in a murine model of myeloproliferative neoplasm”. J. Thromb. Haemost. 2020, 18, 3359–3370. [Google Scholar] [CrossRef]
- Conran, N.; de Paula, E.V. Thromboinflammatory mechanisms in sickle cell disease—Challenging the hemostatic balance. Haematologica 2020, 105, 2380–2390. [Google Scholar] [CrossRef]
- Poisson, J.; Tanguy, M.; Davy, H.; Camara, F.; El Mdawar, M.B.; Kheloufi, M.; Dagher, T.; Devue, C.; Plessier, J.A.; Merchant, S.; et al. Erythrocyte-derived microvesicles induce arterial spasms in JAK2V617F myeloproliferative neoplasm. J. Clin. Investig. 2020, 130, 2630–2643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, M. Inflammation and cancer. Environ. Health Prev. Med. 2018, 23, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lussana, F.; Rambaldi, A. Inflammation and myeloproliferative neoplasms. J. Autoimmun. 2017, 85, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Arellano-Rodrigo, E.; Alvarez-Larra, A.; Reverter, J.C.; Colomer, D.; Villamor, N.; Bellosillo, B.; Cervantes, F. Platelet turnover, coagulation factors, and soluble markers of platelet and endothelial activation in essential thrombocythemia: Relationship with thrombosis occurrence and JAK 2 V617F allele burden. Am. J. Hematol. 2008, 84, 102–108. [Google Scholar] [CrossRef]
- Kaifie, A.; Kirschner, M.; Wolf, D.; Maintz, C.; Hänel, M.; Gattermann, N.; Gökkurt, E.; Platzbecker, U.; Hollburg, W.; Göthert, J.R.; et al. Bleeding, thrombosis, and anticoagulation in myeloproliferative neoplasms (MPN): Analysis from the German SAL-MPN-registry. J. Hematol. Oncol. 2016, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Yonal-Hindilerden, I.; Daglar-Aday, A.; Akadam-Teker, B.; Yilmaz, C.; Nalcaci, M.; Yavuz, A.S.; Dargin, D. Mutations and JAK2V617F allele burden in Philadelphia-negative myeloproliferative neoplasms. J. Blood Med. 2015, 6, 157–176. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, S.; Thompson, C.R.; Belghasem, M.E.; Bekendam, R.H.; Piasecki, A.; Leiva, O.; Ray, A.; Italiano, J.; Yang, M.; Merill-Skoloff, G.; et al. Platelet dysfunction and thrombosis in JAK2V617F-mutated primary myelofibrotic mice. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 262–272. [Google Scholar] [CrossRef]
- Greenfield, G.; McMullin, M.F.; Mills, K. Molecular pathogenesis of the myeloproliferative neoplasms. J. Hematol. Oncol. 2021, 14, 103. [Google Scholar] [CrossRef]
- Leimk€uhler, N.B.; Gleitz, H.F.E.; Ronghui, L.; Snoeren, I.A.M.; Fuchs, S.N.R.; Nagai, J.S.; Banjanin, B.; Lam, K.H.; Vogl, T.; Kuppe, C.; et al. Heterogeneous bone-marrow stromal progenitors drive myelofibrosis via a druggable alarmin axis. Cell Stem Cell. 2021, 28, 637–652. [Google Scholar] [CrossRef]
- Goette, N.P.; Lev, P.R.; Heller, P.G.; Kornblihtt, L.I.; Korin, L.; Molinas, F.C.; Marta, R.F. Monocyte IL-2Rα expression is associated with thrombosis and the JAK2V617F mutation in myeloproliferative neoplasms. Cytokine 2010, 51, 67–72. [Google Scholar] [CrossRef]
- Margraf, A.; Zarbock, A. Platelets in Inflammation and Resolution. J. Immunol. 2019, 203, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- Brostjan, C.; Oehler, R. The role of neutrophil death in chronic inflammation and cancer. Cell Death Discov. 2020, 6, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhang, S.; Wu, H.J.; Rong, X.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molitor, D.C.; Boor, P.; Buness, A.; Schneider, R.K.; Teichmann, L.L.; Körber, R.M.; Horvath, G.L.; Koschmieder, S.; Gütgemann, I. Macrophage frequency in the bone marrow correlates with morphologic subtype of myeloproliferative neoplasm. Ann. Hematol. 2021, 100, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.S.; Christensen, J.H.; Hasselbalch, H.C.; Pallisgaard, N. The JAK2 V617F mutation involves B- and T-lymphocyte lineages in a subgroup of patients with Philadelphia-chromosome negative chronic myeloproliferative disorders. Br. J. Haematol. 2007, 36, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Nicolosi, M.; Mudireddy, M.; Gangat, N.; Pardanani, A.; Hanson, C.A.; Ketterling, R.P.; Tefferi, A. Normal karyotype in myelofibrosis: Is prognostic integrity affected by the number of metaphases analyzed? Blood Cancer J. 2018, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Nicolosi, M.; Mudireddy, M.; Lasho, T.L.; Gangat, N.; Begna, K.H.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A. Revised cytogenetic risk stratification in primary myelofibrosis: Analysis based on 1002 informative patients. Leukemia 2018, 32, 1189–1199. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, A.P.; Villa-Álvarez, M.; Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; Gonzalez, S. NK Cells in the Treatment of Hematological Malignancies. J. Clin. Med. 2019, 8, 1557. [Google Scholar] [CrossRef] [Green Version]
- Arantes, A.; Leal, C.; Araújo, C.; Santos, P.; Bergamo, A.; Welner, R.S.; Tenen, D.G.; Mullally, A.; Kobayashi, S.; Magalhaes, E.; et al. Decreased Activity of NK Cells in Myeloproliferative Neoplasms. Blood 2015, 126, 1637. [Google Scholar] [CrossRef]
- Palumbo, G.A.; Stella, S.; Pennisi, M.S.; Pirosa, C.; Fermo, E.; Fabris, S.; Cattaneo, D.; Iurlo, A. The Role of New Technologies in Myeloproliferative Neoplasms. Front. Oncol. 2019, 9, 321. [Google Scholar] [CrossRef] [Green Version]
- Helbig, G. Classical Philadelphia-negative myeloproliferative neoplasms: Focus on mutations and JAK2 inhibitors. Med. Oncol. 2018, 35, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skov, V. Next Generation Sequencing in MPNs. Lessons from the Past and Prospects for Use as Predictors of Prognosis and Treatment Responses. Cancers 2021, 12, 2194. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Lasho, T.L. Genomics of myelodysplastic syndrome/myeloproliferative neoplasm overlap syndromes. Hematology 2020, 20, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Luque Paz, D.; Jouanneau-Courville, R.; Riou, J.; Ianotto, J.C.; Boyer, F.; Chauveau, A.; Renard, M.; Chomel, J.C.; Cayssials, E.; Gallego-Hernanz, M.P.; et al. Leukemic evolution ofpolycythemia vera and essential thrombocythemia: Genomic profiles predict time to transformation. Blood Adv. 2020, 4, 4887–4897. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M. From leeches to personalized medicine: Evolving concepts in the management of polycythemia vera. Haematologica 2017, 102, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moliterno, A.; Kaizer, H. Applied genomics in MPN presentation. Hematology 2020, 2020, 434–439. [Google Scholar] [CrossRef]
- Downes, C.E.J.; McClure, B.J.; Rehn, J.; Breen, J.; Bruning, J.B.; Yeung, D.T.; White, D.L. Acquired Mutations within the JAK2 Kinase Domain Confer Resistance to JAK Inhibitors in an in Vitro model of a High-Risk Acute Lymphoblastic Leukemia. Blood 2020, 136, 5–6. [Google Scholar] [CrossRef]
- Helbig, G.; Wichary, R.; Torba, K.; Kyrcz-Krzemien, S. Resolution of thrombocytopenia, but not polycythemia after ruxolitinib for polycythemia vera with detectable mutation in the exon 12 of the JAK2 gene. Med. Oncol. 2017, 34, 31. [Google Scholar] [CrossRef] [Green Version]
- Habbel, J.; Arnold, L.; Chen, Y.; Möllmann, M.; Bruderek, K.; Brandau, S.; Dührsen, U.; Hanoun, M. Inflammation-driven activation of JAK/STAT signaling reversibly accelerates acute myeloid leukemia in vitro. Blood Adv. 2020, 4, 3000–3010. [Google Scholar] [CrossRef]
- Forte, D.; Barone, M.; Palandri, F.; Catani, L. The “Vesicular Intelligence” Strategy of Blood Cancers. Genes 2021, 12, 416. [Google Scholar] [CrossRef]
- Garcia-Gisbert, N.; Fernandez-Ibarrondo, L.; Fernandez-Rodrıguez, C.; Gibert, J.; Andrade-Campos, M.; Arenillas, L.; Camacho, L.; Angona, A.; Longaron, R.; Salar, A.; et al. Circulating cell-free DNA improves the molecular characterisa- tion of Ph-negative myeloproliferative neoplasms. Br. J. Haematol. 2021, 192, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Găman, M.A.; Cozma, M.A.; Dobrică, E.C.; Cretoiu, S.M.; Găman, A.M.; Diaconu, C.C. Liquid Biopsy and Potential Liquid Biopsy-Based Biomarkers in Philadelphia-Negative Classical Myeloproliferative Neoplasms: A Systematic Review. Life 2021, 11, 677. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
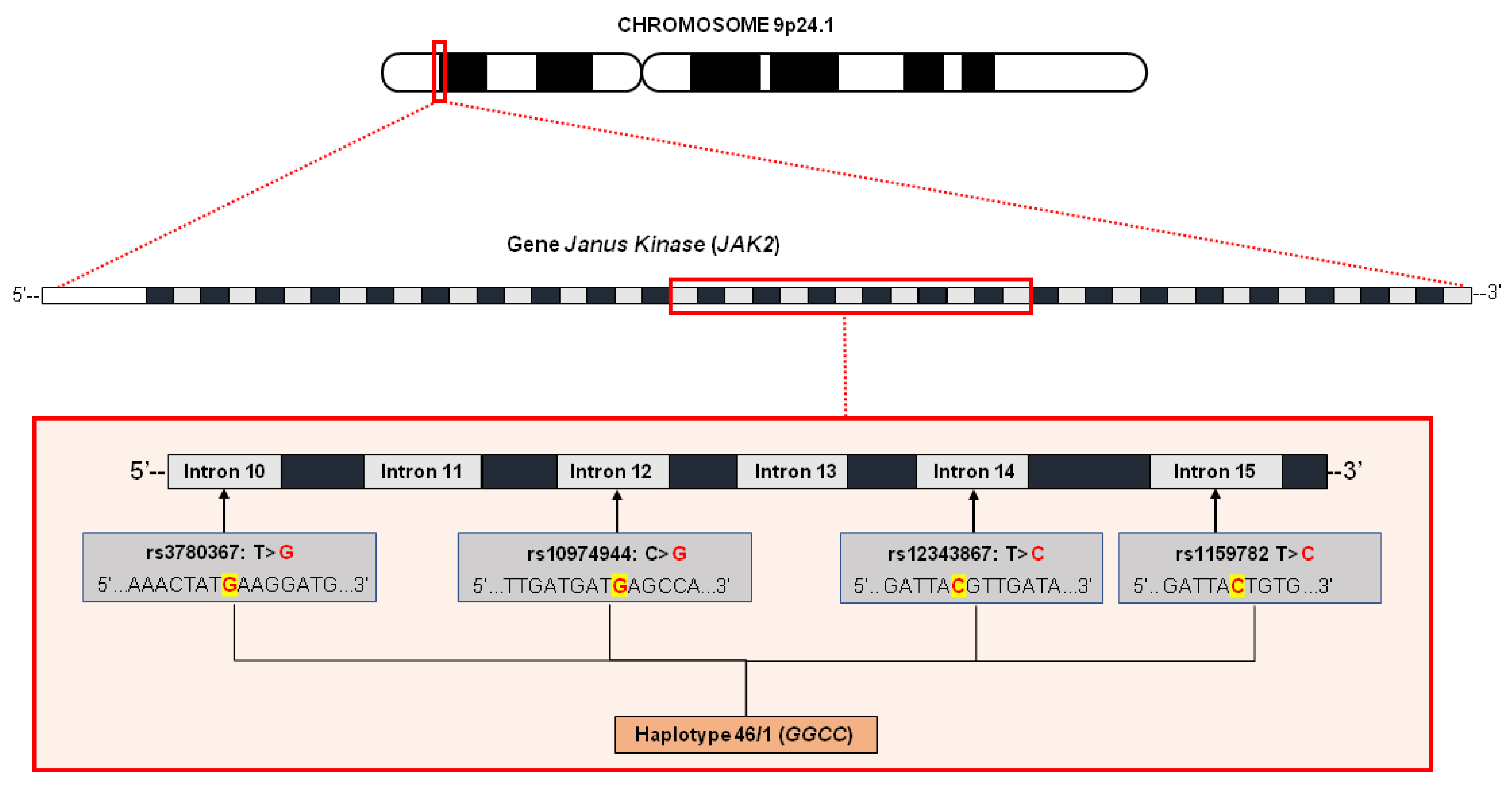{kind=link}
{kind=link}
| Neoplasm | Clinical Description | Major Diagnostic Criteria | Minor Diagnostic Criteria |
|---|---|---|---|
| PV | Exacerbated increase in erythrocyte mass (total red blood cell count). Generally, both genders are diagnosed in the 6th or 7th decade of life [11]. Annual global incidence is 0.3–1.5/100,000 and survival rate is 15 years [2]. |
|
|
| ET | Increased platelet count with megakaryocytic hyperplasia. Annual global incidence is 1.03–2.5/100,000 and diagnosis usually occurs in the 6th decade of life [15,16]. Together with PV, it presents high risks of hemorrhagic and thrombotic episodes [3,12,17,18]. |
|
|
| PMF | Indolent clinical course and has worse prognosis. Patients show increased megakaryopoiesis and extramedullary hematopoiesis [2,3,19,20] It has an annual global incidence of 1.5–2.0/100,000, and generally affects individuals over 60 to 70 years of age. | Pre-fibrotic phase:
| Pre-fibrotic phase:
|
Fibrotic phase:
| Fibrotic phase:
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, D.G.; Paes, J.; da Costa, A.G.; Malheiro, A.; Silva, G.V.; Mourão, L.P.d.S.; Tarragô, A.M. JAK2 Variant Signaling: Genetic, Hematologic and Immune Implication in Chronic Myeloproliferative Neoplasms. Biomolecules 2022, 12, 291. https://doi.org/10.3390/biom12020291
Torres DG, Paes J, da Costa AG, Malheiro A, Silva GV, Mourão LPdS, Tarragô AM. JAK2 Variant Signaling: Genetic, Hematologic and Immune Implication in Chronic Myeloproliferative Neoplasms. Biomolecules. 2022; 12(2):291. https://doi.org/10.3390/biom12020291
Chicago/Turabian StyleTorres, Dania G., Jhemerson Paes, Allyson G. da Costa, Adriana Malheiro, George V. Silva, Lucivana P. de Souza Mourão, and Andréa M. Tarragô. 2022. "JAK2 Variant Signaling: Genetic, Hematologic and Immune Implication in Chronic Myeloproliferative Neoplasms" Biomolecules 12, no. 2: 291. https://doi.org/10.3390/biom12020291
APA StyleTorres, D. G., Paes, J., da Costa, A. G., Malheiro, A., Silva, G. V., Mourão, L. P. d. S., & Tarragô, A. M. (2022). JAK2 Variant Signaling: Genetic, Hematologic and Immune Implication in Chronic Myeloproliferative Neoplasms. Biomolecules, 12(2), 291. https://doi.org/10.3390/biom12020291