
Roles of PI3K/AKT/mTOR Axis in Arteriovenous Fistula

 , , ,
, , ,  and
and 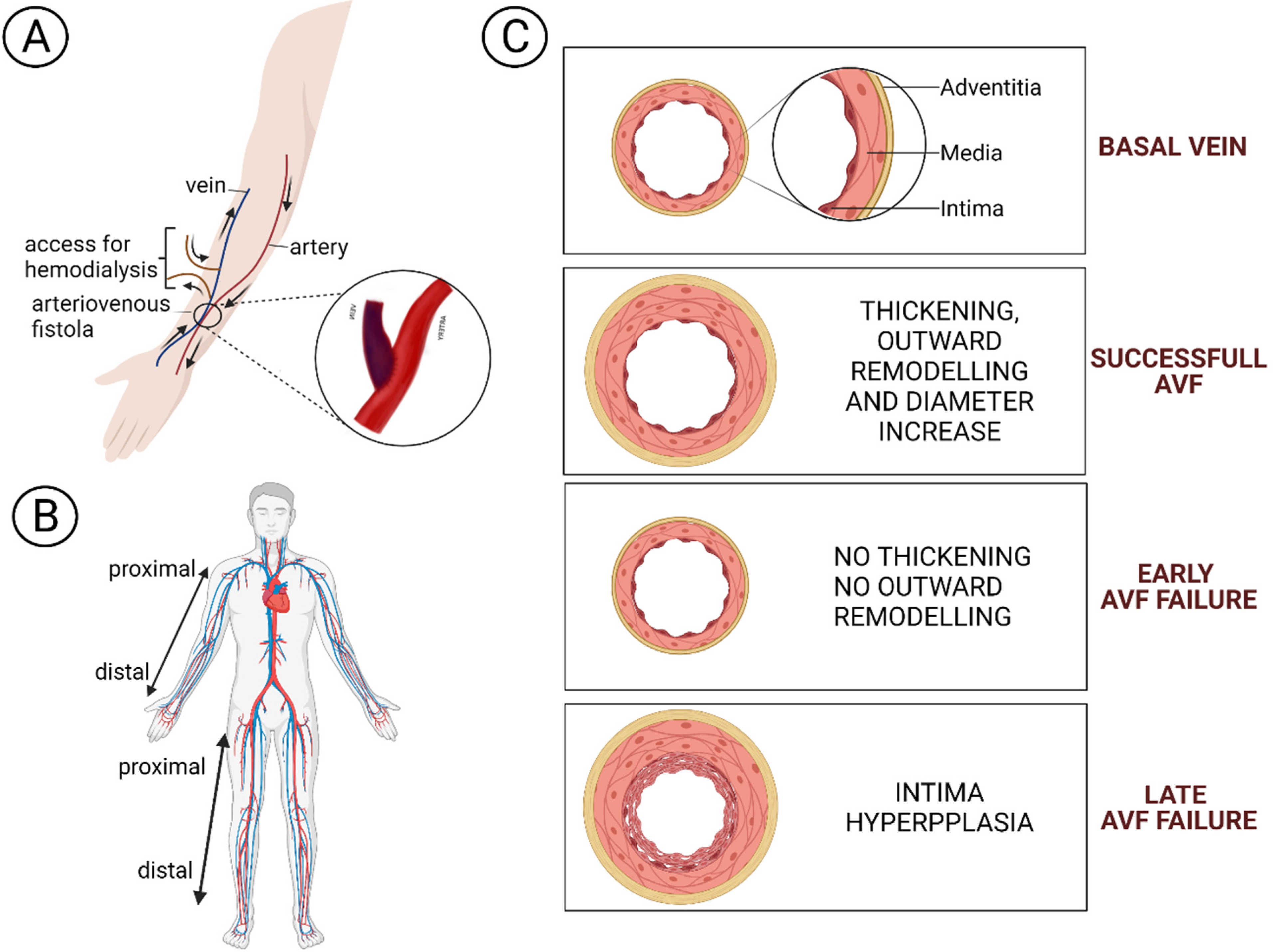{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Arteriovenous Fistula
2. Phosphoinositides
3. PI3K/AKT/mTOR Axis
4. PI3K/Akt/mTOR Pathway in AVF
5. Conclusions
Funding
Conflicts of Interest
References
- Deng, Y.; Li, N.; Wu, Y.; Wang, M.; Yang, S.; Zheng, Y.; Deng, X.; Xiang, D.; Zhu, Y.; Xu, P.; et al. Global, Regional, and National Burden of Diabetes-Related Chronic Kidney Disease from 1990 to 2019. Front. Endocrinol. 2021, 12, 672350. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.; Mone, P.; Jankauskas, S.S.; Gambardella, J.; Santulli, G. Chronic kidney disease: Definition, updated epidemiology, staging, and mechanisms of increased cardiovascular risk. J. Clin. Hypertens. 2021, 23, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Crews, D.C.; Bello, A.K.; Saadi, G. Burden, access, and disparities in kidney disease. J. Nephrol. 2019, 32, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Jager, K.J.; Fraser, S.D.S. The ascending rank of chronic kidney disease in the global burden of disease study. Nephrol. Dial. Transpl. 2017, 32, ii121–ii128. [Google Scholar] [CrossRef] [PubMed]
- Gorecka, J.; Fereydooni, A.; Gonzalez, L.; Lee, S.R.; Liu, S.; Ono, S.; Xu, J.; Liu, J.; Taniguchi, R.; Matsubara, Y.; et al. Molecular Targets for Improving Arteriovenous Fistula Maturation and Patency. Vasc. Investig. Ther. 2019, 2, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Ravani, P.; Quinn, R.; Oliver, M.; Robinson, B.; Pisoni, R.; Pannu, N.; MacRae, J.; Manns, B.; Hemmelgarn, B.; James, M.; et al. Examining the Association between Hemodialysis Access Type and Mortality: The Role of Access Complications. Clin. J. Am. Soc. Nephrol. 2017, 12, 955–964. [Google Scholar] [CrossRef] [Green Version]
- Gameiro, J.; Ibeas, J. Factors affecting arteriovenous fistula dysfunction: A narrative review. J. Vasc. Access. 2020, 21, 134–147. [Google Scholar] [CrossRef]
- Lopes, J.R.A.; Marques, A.L.B.; Correa, J.A. Randomised clinical study of the impact of routine preoperative Doppler ultrasound for the outcome of autologous arteriovenous fistulas for haemodialysis. J. Vasc. Access. 2021, 22, 107–114. [Google Scholar] [CrossRef]
- Matsubara, Y.; Kiwan, G.; Fereydooni, A.; Langford, J.; Dardik, A. Distinct subsets of T cells and macrophages impact venous remodeling during arteriovenous fistula maturation. JVS Vasc. Sci. 2020, 1, 207–218. [Google Scholar] [CrossRef]
- Remuzzi, A.; Bozzetto, M. Biological and Physical Factors Involved in the Maturation of Arteriovenous Fistula for Hemodialysis. Cardiovasc. Eng. Technol. 2017, 8, 273–279. [Google Scholar] [CrossRef]
- Paszkowiak, J.J.; Dardik, A. Arterial wall shear stress: Observations from the bench to the bedside. Vasc. Endovasc. Surg. 2003, 37, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Mauro, R.; Rocchi, C.; Vasuri, F.; Pini, A.; Croci Chiocchini, A.L.; Ciavarella, C.; La Manna, G.; Pasquinelli, G.; Faggioli, G.; Gargiulo, M. Tissue Ki67 proliferative index expression and pathological changes in hemodialysis arteriovenous fistulae: Preliminary single-center results. J. Vasc. Access. 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Zonnebeld, N.; Huberts, W.; van Loon, M.M.; Delhaas, T.; Tordoir, J.H.M. Natural Vascular Remodelling After Arteriovenous Fistula Creation in Dialysis Patients With and Without Previous Ipsilateral Vascular Access. Eur. J. Vasc. Endovasc. Surg. 2020, 59, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Roy-Chaudhury, P.; Spergel, L.M.; Besarab, A.; Asif, A.; Ravani, P. Biology of arteriovenous fistula failure. J. Nephrol. 2007, 20, 150–163. [Google Scholar] [PubMed]
- Oliver, M.J. The Science of Fistula Maturation. J. Am. Soc. Nephrol. 2018, 29, 2607–2609. [Google Scholar] [CrossRef] [Green Version]
- Shih, Y.-C.; Chen, P.-Y.; Ko, T.-M.; Huang, P.-H.; Ma, H.; Tarng, D.-C. MMP-9 Deletion Attenuates Arteriovenous Fistula Neointima through Reduced Perioperative Vascular Inflammation. Int. J. Mol. Sci. 2021, 22, 5448. [Google Scholar] [CrossRef]
- Yao, J.; Zhao, X.; Tan, F.; Cao, X.; Guo, S.; Li, X.; Huang, Z.; Diabakte, K.; Wang, L.; Liu, M.; et al. Early modulation of macrophage ROS-PPARgamma-NF-kappaB signalling by sonodynamic therapy attenuates neointimal hyperplasia in rabbits. Sci. Rep. 2020, 10, 11638. [Google Scholar] [CrossRef]
- Wimmer, R.A.; Leopoldi, A.; Aichinger, M.; Kerjaschki, D.; Penninger, J.M. Generation of blood vessel organoids from human pluripotent stem cells. Nat. Protoc. 2019, 14, 3082–3100. [Google Scholar] [CrossRef]
- Valente, S.; Ciavarella, C.; Hernandez-Aguilera, A.; Salvador, F.A.; Buzzi, M.; Joven, J.; Pasquinelli, G. Phenotypic, morphological, and metabolic characterization of vascular-spheres from human vascular mesenchymal stem cells. Microsc. Res. Tech. 2021, 10, 23918. [Google Scholar] [CrossRef]
- Hokin, M.R.; Hokin, L.E. Enzyme secretion and the incorporation of P32 into phospholipides of pancreas slices. J. Biol. Chem. 1953, 203, 967–977. [Google Scholar] [CrossRef]
- Lete, M.G.; Tripathi, A.; Chandran, V.; Bankaitis, V.A.; McDermott, M.I. Lipid transfer proteins and instructive regulation of lipid kinase activities: Implications for inositol lipid signaling and disease. Adv. Biol. Regul. 2020, 78, 100740. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Jian, Y.; Sun, X.; Yang, C.; Gao, Z.; Zhang, Z.; Liu, X.; Li, Y.; Xu, J.; Jing, Y.; et al. Negative regulation of phosphatidylinositol 3-phosphate levels in early-to-late endosome conversion. J. Cell. Biol. 2016, 212, 181–198. [Google Scholar] [CrossRef] [Green Version]
- Gozzelino, L.; De Santis, M.C.; Gulluni, F.; Hirsch, E.; Martini, M. PI(3,4)P2 Signaling in Cancer and Metabolism. Front. Oncol. 2020, 10, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamann, B.L.; Blind, R.D. Nuclear phosphoinositide regulation of chromatin. J. Cell. Physiol. 2018, 233, 107–123. [Google Scholar] [CrossRef] [PubMed]
- Sztacho, M.; Sobol, M.; Balaban, C.; Escudeiro Lopes, S.E.; Hozak, P. Nuclear phosphoinositides and phase separation: Important players in nuclear compartmentalization. Adv. Biol. Regul. 2019, 71, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Stijf-Bultsma, Y.; Sommer, L.; Tauber, M.; Baalbaki, M.; Giardoglou, P.; Jones, D.R.; Gelato, K.A.; van Pelt, J.; Shah, Z.; Rahnamoun, H.; et al. The basal transcription complex component TAF3 transduces changes in nuclear phosphoinositides into transcriptional output. Mol. Cell. 2015, 58, 453–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiume, R.; Stijf-Bultsma, Y.; Shah, Z.H.; Keune, W.J.; Jones, D.R.; Jude, J.G.; Divecha, N. PIP4K and the role of nuclear phosphoinositides in tumour suppression. Biochim. Biophys. Acta 2015, 1851, 898–910. [Google Scholar] [CrossRef]
- Shah, Z.H.; Jones, D.R.; Sommer, L.; Foulger, R.; Bultsma, Y.; D’Santos, C.; Divecha, N. Nuclear phosphoinositides and their impact on nuclear functions. FEBS J. 2013, 280, 6295–6310. [Google Scholar] [CrossRef]
- Hammond, G.R.; Balla, T. Polyphosphoinositide binding domains: Key to inositol lipid biology. Biochim. Biophys. Acta 2015, 1851, 746–758. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, B.D.; Goncalves, M.D.; Cantley, L.C. Insulin-PI3K signalling: An evolutionarily insulated metabolic driver of cancer. Nat. Rev. Endocrinol. 2020, 16, 276–283. [Google Scholar] [CrossRef]
- Kang, D.S.; Yang, Y.R.; Lee, C.; Park, B.; Park, K.I.; Seo, J.K.; Seo, Y.K.; Cho, H.; Lucio, C.; Suh, P.G. Netrin-1/DCC-mediated PLCgamma1 activation is required for axon guidance and brain structure development. EMBO Rep. 2018, 19, 4250. [Google Scholar] [CrossRef] [PubMed]
- Barker, C.J.; Leibiger, I.B.; Leibiger, B.; Berggren, P.O. Phosphorylated inositol compounds in beta -cell stimulus-response coupling. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E1113–E1122. [Google Scholar] [CrossRef] [PubMed]
- Tronchere, H.; Buj-Bello, A.; Mandel, J.L.; Payrastre, B. Implication of phosphoinositide phosphatases in genetic diseases: The case of myotubularin. Cell. Mol. Life Sci. 2003, 60, 2084–2099. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, P.T.; Stephens, L.R. PI3K signalling in inflammation. Biochim. Biophys. Acta 2015, 1851, 882–897. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, P.T.; Anderson, K.E.; Davidson, K.; Stephens, L.R. Signalling through Class I PI3Ks in mammalian cells. Biochem. Soc. Trans. 2006, 34 Pt 5, 647–662. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, D.R.; Whitman, M.; Schaffhausen, B.; Pallas, D.C.; White, M.; Cantley, L.; Roberts, T.M. Common elements in growth factor stimulation and oncogenic transformation: 85 kd phosphoprotein and phosphatidylinositol kinase activity. Cell 1987, 50, 1021–1029. [Google Scholar] [CrossRef]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef]
- Okkenhaug, K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu. Rev. Immunol. 2013, 31, 675–704. [Google Scholar] [CrossRef] [Green Version]
- Gulluni, F.; De Santis, M.C.; Margaria, J.P.; Martini, M.; Hirsch, E. Class II PI3K Functions in Cell Biology and Disease. Trends Cell Biol. 2019, 29, 339–359. [Google Scholar] [CrossRef]
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell. Biol. 2019, 20, 515–534. [Google Scholar] [CrossRef]
- Braccini, L.; Ciraolo, E.; Campa, C.C.; Perino, A.; Longo, D.L.; Tibolla, G.; Pregnolato, M.; Cao, Y.; Tassone, B.; Damilano, F.; et al. PI3K-C2gamma is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat. Commun. 2015, 6, 7400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backer, J.M. The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem. J. 2016, 473, 2251–2271. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Biswas, S.K.; McNulty, P.H.; Kozak, M.; Jun, J.Y.; Segar, L. PDGF-induced vascular smooth muscle cell proliferation is associated with dysregulation of insulin receptor substrates. Am. J. Physiol. Cell. Physiol. 2011, 300, C1375–C1385. [Google Scholar] [CrossRef] [Green Version]
- Simone, S.; Loverre, A.; Cariello, M.; Divella, C.; Castellano, G.; Gesualdo, L.; Pertosa, G.; Grandaliano, G. Arteriovenous fistula stenosis in hemodialysis patients is characterized by an increased adventitial fibrosis. J. Nephrol. 2014, 27, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lee, F.Y.; Bhalla, K.N.; Wu, J. Potent inhibition of platelet-derived growth factor-induced responses in vascular smooth muscle cells by BMS-354825 (dasatinib). Mol. Pharmacol. 2006, 69, 1527–1533. [Google Scholar] [CrossRef]
- Yang, H.H.; Xu, Y.X.; Chen, J.Y.; Harn, H.J.; Chiou, T.W. N-Butylidenephthalide Inhibits the Phenotypic Switch of VSMCs through Activation of AMPK and Prevents Stenosis in an Arteriovenous Fistula Rat Model. Int. J. Mol. Sci. 2020, 21, 7403. [Google Scholar] [CrossRef]
- Shih, Y.C.; Wu, C.C.; Wang, S.C.; Liou, J.Y.; Huang, P.H.; Tarng, D.C. Oral Charcoal Adsorbents Attenuate Neointima Formation of Arteriovenous Fistulas. Toxins 2020, 12, 237. [Google Scholar] [CrossRef] [Green Version]
- Abizaid, A.; Costa, M.A.; Blanchard, D.; Albertal, M.; Eltchaninoff, H.; Guagliumi, G.; Geert-Jan, L.; Abizaid, A.S.; Sousa, A.G.; Wuelfert, E.; et al. Sirolimus-eluting stents inhibit neointimal hyperplasia in diabetic patients. Insights from the RAVEL Trial. Eur. Heart J. 2004, 25, 107–112. [Google Scholar] [CrossRef] [Green Version]
- Jadlowiec, C.C.; Feigel, A.; Yang, C.; Feinstein, A.J.; Kim, S.T.; Collins, M.J.; Kondo, Y.; Muto, A.; Dardik, A. Reduced adult endothelial cell EphB4 function promotes venous remodeling. Am. J. Physiol. Cell. Physiol. 2013, 304, C627–C635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protack, C.D.; Foster, T.R.; Hashimoto, T.; Yamamoto, K.; Lee, M.Y.; Kraehling, J.R.; Bai, H.; Hu, H.; Isaji, T.; Santana, J.M.; et al. Eph-B4 regulates adaptive venous remodeling to improve arteriovenous fistula patency. Sci. Rep. 2017, 7, 15386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Fereydooni, A.; Isaji, T.; Gorecka, J.; Liu, S.; Hu, H.; Ono, S.; Alozie, M.; Lee, S.R.; Taniguchi, R.; et al. Inhibition of the Akt1-mTORC1 Axis Alters Venous Remodeling to Improve Arteriovenous Fistula Patency. Sci. Rep. 2019, 9, 11046. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; Borok, Z. TGF-beta-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef]
- Postlethwaite, A.E.; Shigemitsu, H.; Kanangat, S. Cellular origins of fibroblasts: Possible implications for organ fibrosis in systemic sclerosis. Curr. Opin. Rheumatol. 2004, 16, 733–738. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Kaygin, M.A.; Halici, U.; Aydin, A.; Dag, O.; Binici, D.N.; Limandal, H.K.; Arslan, U.; Kiymaz, A.; Kahraman, N.; Calik, E.S.; et al. The relationship between arteriovenous fistula success and inflammation. Ren. Fail. 2013, 35, 1085–1088. [Google Scholar] [CrossRef]
- Schofer, J.; Schluter, M.; Gershlick, A.H.; Wijns, W.; Garcia, E.; Schampaert, E.; Breithardt, G.; Investigators, E.S. Sirolimus-eluting stents for treatment of patients with long atherosclerotic lesions in small coronary arteries: Double-blind, randomised controlled trial (E-SIRIUS). Lancet 2003, 362, 1093–1099. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ratti, S.; Mauro, R.; Rocchi, C.; Mongiorgi, S.; Ramazzotti, G.; Gargiulo, M.; Manzoli, L.; Cocco, L.; Fiume, R. Roles of PI3K/AKT/mTOR Axis in Arteriovenous Fistula. Biomolecules 2022, 12, 350. https://doi.org/10.3390/biom12030350
Ratti S, Mauro R, Rocchi C, Mongiorgi S, Ramazzotti G, Gargiulo M, Manzoli L, Cocco L, Fiume R. Roles of PI3K/AKT/mTOR Axis in Arteriovenous Fistula. Biomolecules. 2022; 12(3):350. https://doi.org/10.3390/biom12030350
Chicago/Turabian StyleRatti, Stefano, Raffaella Mauro, Cristina Rocchi, Sara Mongiorgi, Giulia Ramazzotti, Mauro Gargiulo, Lucia Manzoli, Lucio Cocco, and Roberta Fiume. 2022. "Roles of PI3K/AKT/mTOR Axis in Arteriovenous Fistula" Biomolecules 12, no. 3: 350. https://doi.org/10.3390/biom12030350
APA StyleRatti, S., Mauro, R., Rocchi, C., Mongiorgi, S., Ramazzotti, G., Gargiulo, M., Manzoli, L., Cocco, L., & Fiume, R. (2022). Roles of PI3K/AKT/mTOR Axis in Arteriovenous Fistula. Biomolecules, 12(3), 350. https://doi.org/10.3390/biom12030350