
ENT-A010, a Novel Steroid Derivative, Displays Neuroprotective Functions and Modulates Microglial Responses

 , ,
, ,  , ,
, , 
 , ,
, ,  and
and 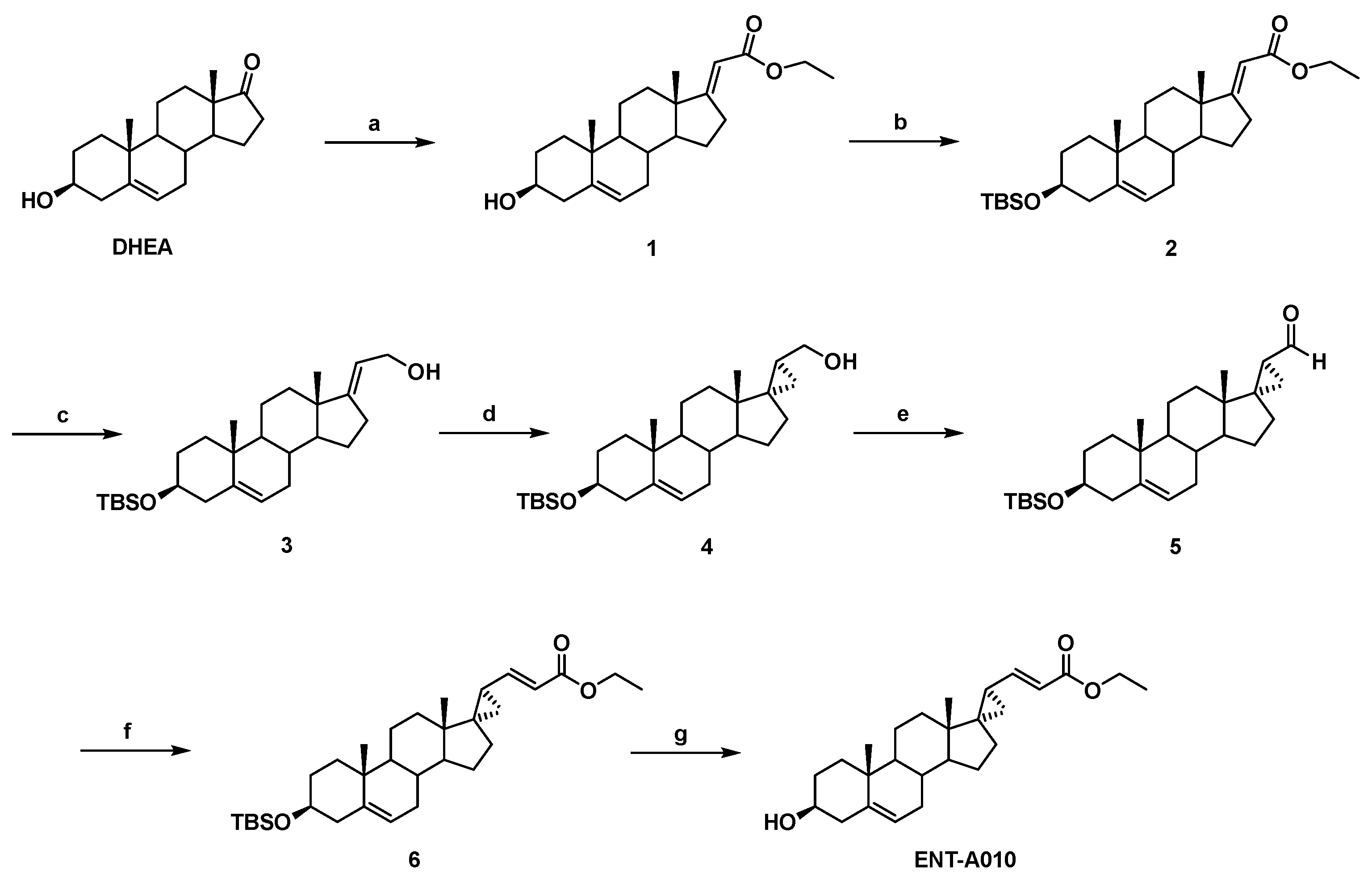{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of ENT-A010
2.1.1. Synthesis of (E)-(3β-hydroxy-5-androsten-17-ylidene)ethyl ester (1)
2.1.2. Synthesis of (E)-[3β-(t-butyldimethylsilyloxy)-5-androsten-17-ylidene]ethyl ester (2)
2.1.3. Synthesis of (E)-3β-(t-butyldimethylsilyloxy)-pregna-5,17(20)-dien-21-ol (3)
2.1.4. Synthesis of (17S,20S)-3β-(t-butyldimethylsilyloxy)-17α,20-methan-5-pregnane-21-ol (4)
2.1.5. Synthesis of (17S,20S)-3β-(t-butyldimethylsilyloxy)-17α,20-methan-5-pregnane-21-al (5)
2.1.6. Synthesis of (17S,20S)-3β-(t-butyldimethylsilyloxy)-17α,20-Methan-5,21-pregna-dien-22-ethyl ester (6)
2.1.7. Synthesis of (17S,20S)-3β-hydroxy-17α,20-methan-5,21-pregna-dien-22-ethyl ester (ENT-A010)
2.2. Mice
2.3. ENT-A010 Detection in Tissues
2.4. Cell Isolation and Culture
2.5. TUNEL Assay
2.6. CellTox Assay
2.7. Synaptophysin Detection
2.8. Phagocytosis Assay
2.9. Immunoprecipitation
2.10. Western Blotting
2.11. Immunofluorescent Staining and Confocal Microscopy
2.12. RNA Extraction and qRT-PCR
2.13. Statistical Analysis
3. Results
3.1. Synthesis of ENT-A010
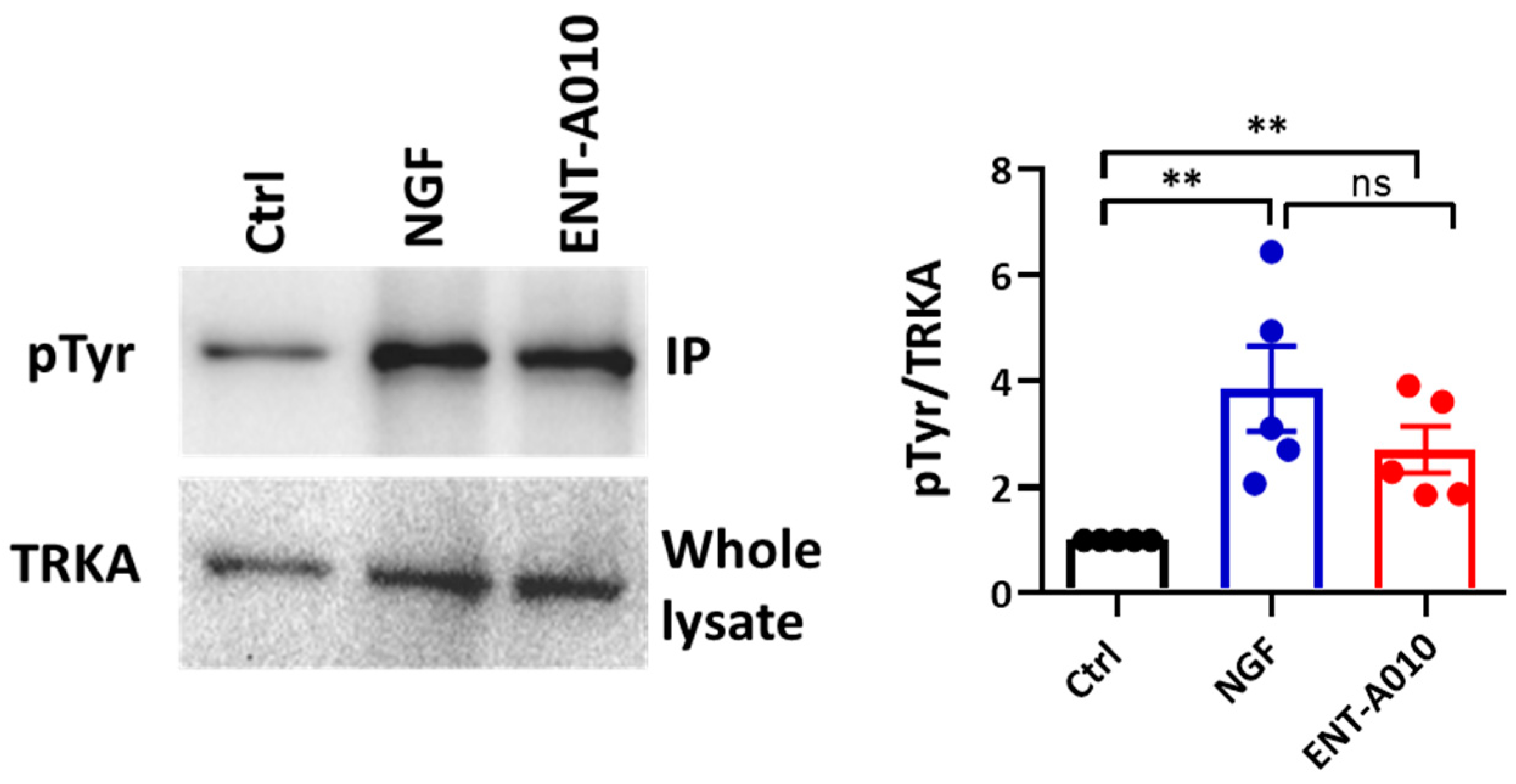
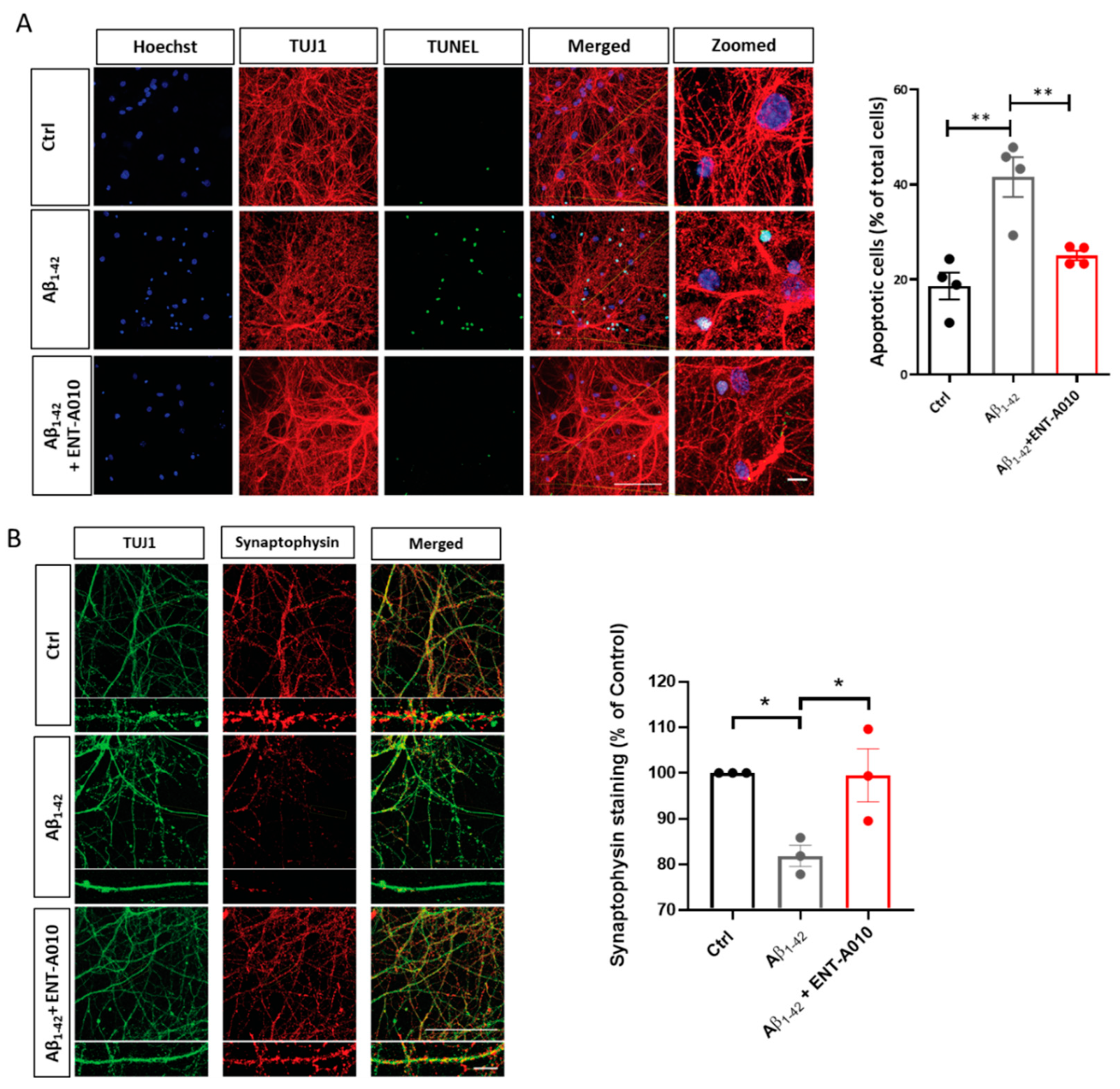
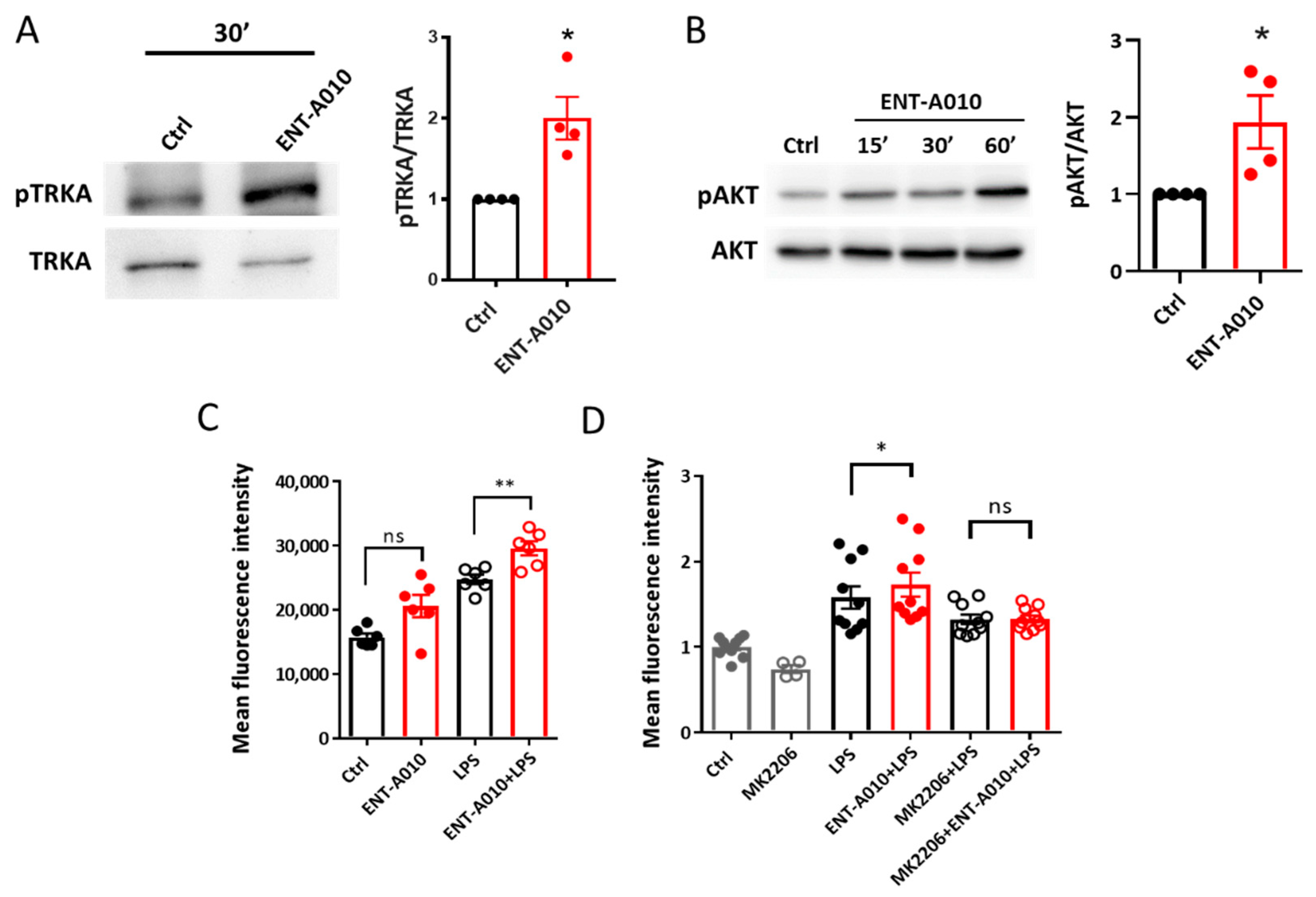
3.2. ENT-A010 Promotes Neuronal Survival in a TRKA-Dependent Manner
3.3. ENT-A010 Promotes Phagocytosis in Microglia
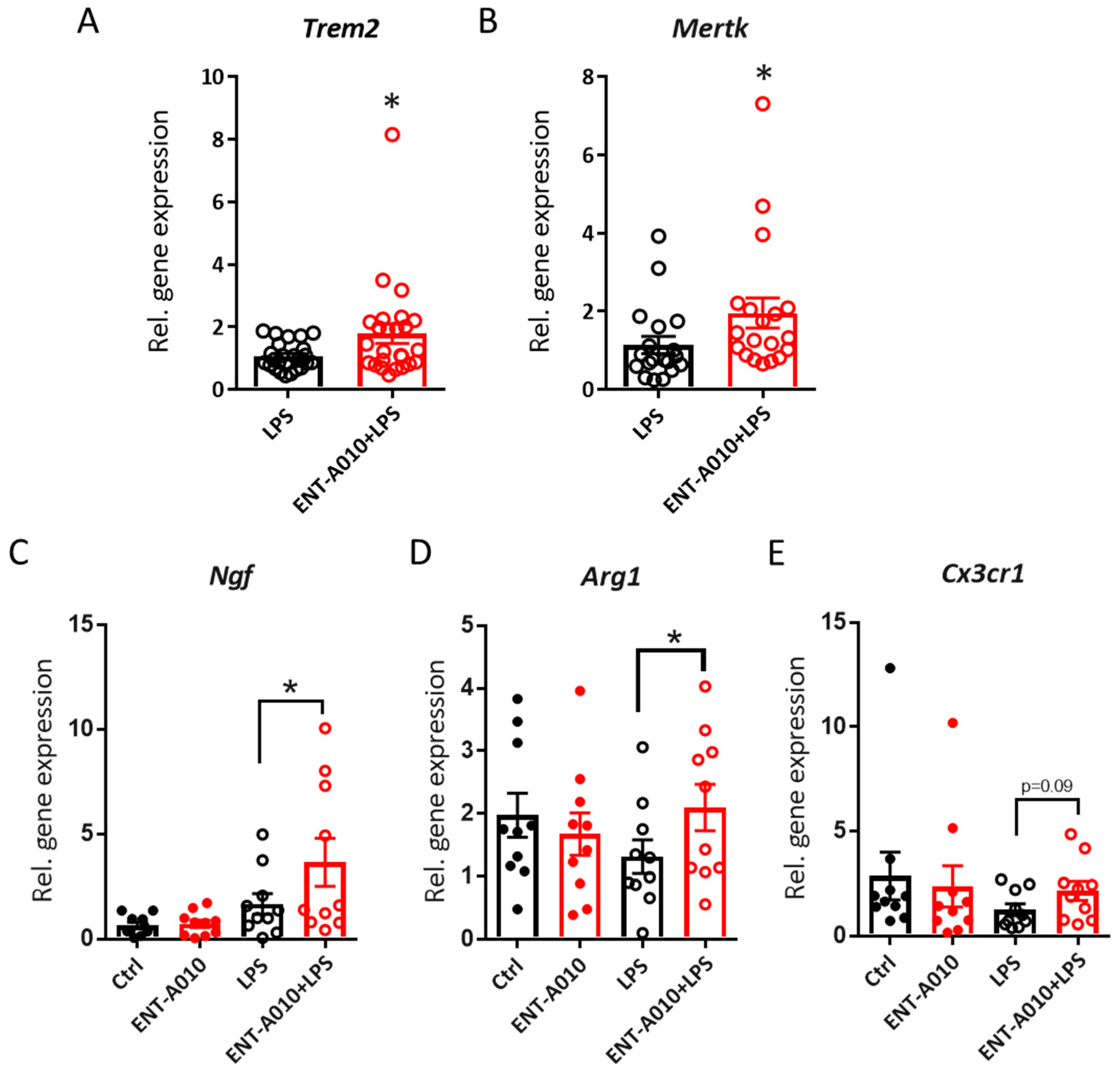
3.4. ENT-A010 Promotes a Protective Microglial Phenotype
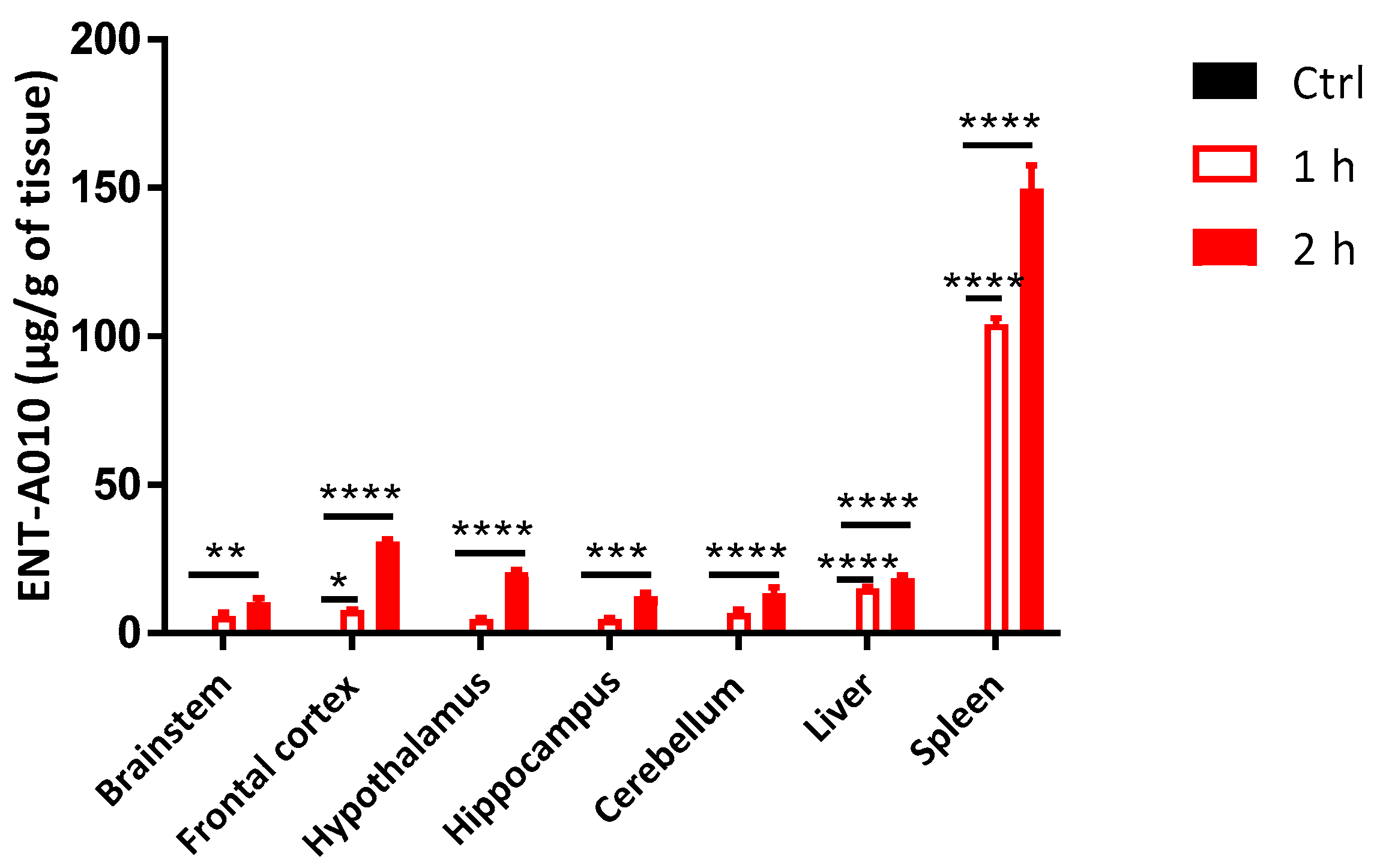
3.5. Peripherally Administered ENT-A010 Is Detected in the Brain
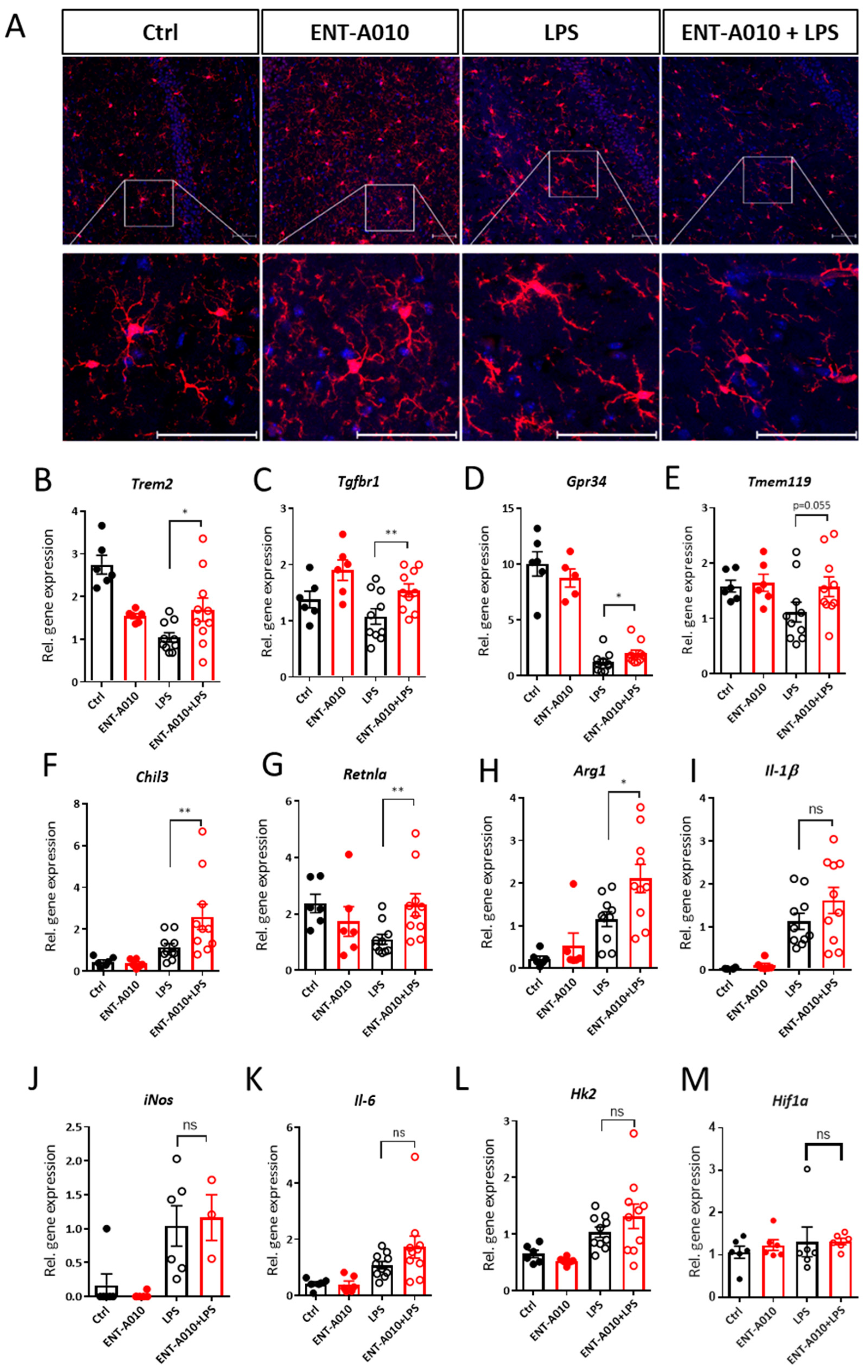
3.6. ENT-A010 Preserves the Homeostatic Phenotype of Microglia in the Hippocampus
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Adle-Biassette, H.; Milenkovic, I.; Cipriani, S.; van Scheppingen, J.; Aronica, E. Linking pathways in the developing and aging brain with neurodegeneration. Neuroscience 2014, 269, 152–172. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; El Khoury, J. Microglia in Health and Disease. Cold Spring Harb. Perspect. Biol. 2015, 8, a020560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Dissing-Olesen, L.; Stevens, B. New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 2016, 36, 128–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef]
- Niranjan, R. Recent advances in the mechanisms of neuroinflammation and their roles in neurodegeneration. Neurochem. Int. 2018, 120, 13–20. [Google Scholar] [CrossRef]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Prefontaine, P.; Plante, M.M.; Sanchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.E.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef] [Green Version]
- Sato, F.; Martinez, N.E.; Stewart, E.C.; Omura, S.; Alexander, J.S.; Tsunoda, I. “Microglial nodules” and “newly forming lesions” may be a Janus face of early MS lesions; implications from virus-induced demyelination, the Inside-Out model. BMC Neurol. 2015, 15, 219. [Google Scholar] [CrossRef] [Green Version]
- Compagnone, N.A.; Mellon, S.H. Neurosteroids: Biosynthesis and function of these novel neuromodulators. Front. Neuroendocrinol. 2000, 21, 1–56. [Google Scholar] [CrossRef]
- Yilmaz, C.; Karali, K.; Fodelianaki, G.; Gravanis, A.; Chavakis, T.; Charalampopoulos, I.; Alexaki, V.I. Neurosteroids as regulators of neuroinflammation. Front. Neuroendocrinol. 2019, 55, 100788. [Google Scholar] [CrossRef] [PubMed]
- Souza-Teodoro, L.H.; de Oliveira, C.; Walters, K.; Carvalho, L.A. Higher serum dehydroepiandrosterone sulfate protects against the onset of depression in the elderly: Findings from the English Longitudinal Study of Aging (ELSA). Psychoneuroendocrinology 2016, 64, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Yin, Y.; Xiao, C.L.; Mao, R.J.; Shi, B.H.; Jie, Y.; Wang, Z.W. Serum DHEAS levels are associated with the development of depression. Psychiat. Res. 2015, 229, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Hampl, R.; Bicikova, M. Neuroimmunomodulatory steroids in Alzheimer dementia. J. Steroid. Biochem. 2010, 119, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Aldred, S.; Mecocci, P. Decreased dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEAS) concentrations in plasma of Alzheimer’s disease (AD) patients. Arch. Gerontol. Geriat. 2010, 51, E16–E18. [Google Scholar] [CrossRef]
- Maninger, N.; Wolkowitz, O.M.; Reus, V.I.; Epel, E.S.; Mellon, S.H. Neurobiological and neuropsychiatric effects of dehydroepiandrosterone (DHEA) and DHEA sulfate (DHEAS). Front. Neuroendocrinol. 2009, 30, 65–91. [Google Scholar] [CrossRef] [Green Version]
- Du, C.G.; Khalil, M.W.; Sriram, S. Administration of dehydroepiandrosterone suppresses experimental allergic encephalomyelitis in SJL/J mice. J. Immunol. 2001, 167, 7094–7101. [Google Scholar] [CrossRef] [Green Version]
- Saijo, K.; Collier, J.G.; Li, A.C.; Katzenellenbogen, J.A.; Glass, C.K. An ADIOL-ERbeta-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell 2011, 145, 584–595. [Google Scholar] [CrossRef] [Green Version]
- Litim, N.; Morissette, M.; Di Paolo, T. Neuroactive gonadal drugs for neuroprotection in male and female models of Parkinson’s disease. Neurosci. Biobehav. Rev. 2015, 67, 79–88. [Google Scholar] [CrossRef]
- Belanger, N.; Gregoire, L.; Bedard, P.; Di Paolo, T. Estradiol and dehydroepiandrosterone potentiate levodopa-induced locomotor activity in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine monkeys. Endocrine 2003, 21, 97–101. [Google Scholar] [CrossRef]
- D’Astous, M.; Morissette, M.; Tanguay, B.; Callier, S.; Di Paolo, T. Dehydroepiandrosterone (DHEA) such as 17 beta-estradiol prevents MPTP-induced dopamine depletion in mice. Synapse 2003, 47, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Belanger, N.; Gregoire, L.; Bedard, P.J.; Di Paolo, T. DHEA improves symptomatic treatment of moderately and severely impaired MPTP monkeys. Neurobiol. Aging 2006, 27, 1684–1693. [Google Scholar] [CrossRef] [PubMed]
- Aggelakopoulou, M.; Kourepini, E.; Paschalidis, N.; Simoes, D.C.; Kalavrizioti, D.; Dimisianos, N.; Papathanasopoulos, P.; Mouzaki, A.; Panoutsakopoulou, V. ERbeta-Dependent Direct Suppression of Human and Murine Th17 Cells and Treatment of Established Central Nervous System Autoimmunity by a Neurosteroid. J. Immunol. 2016, 197, 2598–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Klein, G.M.; Sun, P.; Buchan, A.M. Dehydroepiandrosterone (DHEA) reduces neuronal injury in a rat model of global cerebral ischemia. Brain Res. 2001, 888, 263–266. [Google Scholar] [CrossRef]
- Charalampopoulos, L.; Tsatsanis, C.; Dermitzaki, E.; Alexaki, V.I.; Castanas, E.; Margioris, A.N.; Gravanis, A. Dehydroepiandrosterone and allopregnanolone protect sympathoadrenal medulla cells against apoptosis via antiapoptotic Bcl-2 proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 8209–8214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charalampopoulos, I.; Alexaki, V.I.; Lazaridis, I.; Dermitzaki, E.; Avlonitis, N.; Tsatsanis, C.; Calogeropoulou, T.; Margioris, A.N.; Castanas, E.; Gravanis, A. G protein-associated, specific membrane binding sites mediate the neuroprotective effect of dehydroepiandrosterone. FASEB J. 2006, 20, 577–579. [Google Scholar] [CrossRef]
- Charalampopoulos, I.; Alexaki, V.I.; Tsatsanis, C.; Minas, V.; Dermitzaki, E.; Lasaridis, I.; Vardouli, L.; Stournaras, C.; Margioris, A.N.; Castanas, E.; et al. Neurosteroids as endogenous inhibitors of neuronal cell apoptosis in aging. Neuroendocr. Immune Crosstalk 2006, 1088, 139–152. [Google Scholar] [CrossRef]
- Lazaridis, I.; Charalampopoulos, I.; Alexaki, V.I.; Avlonitis, N.; Pediaditakis, I.; Efstathopoulos, P.; Calogeropoulou, T.; Castanas, E.; Gravanis, A. Neurosteroid Dehydroepiandrosterone Interacts with Nerve Growth Factor (NGF) Receptors, Preventing Neuronal Apoptosis. PLoS Biol. 2011, 9, e1001051. [Google Scholar] [CrossRef]
- Gravanis, A.; Calogeropoulou, T.; Panoutsakopoulou, V.; Thermos, K.; Neophytou, C.; Charalampopoulos, I. Neurosteroids and Microneurotrophins Signal Through NGF Receptors to Induce Prosurvival Signaling in Neuronal Cells. Sci. Signal. 2012, 5, pt8. [Google Scholar] [CrossRef] [Green Version]
- Charalampopoulos, I.; Remboutsika, E.; Margioris, A.N.; Gravanis, A. Neurosteroids as modulators of neurogenesis and neuronal survival. Trends Endocrin. Met. 2008, 19, 300–307. [Google Scholar] [CrossRef]
- Du, C.G.; Guan, Q.N.; Khalil, M.W.; Sriram, S. Stimulation of Th2 response by high doses of dehydroepiandrosterone in KLH-primed splenocytes. Exp. Biol. Med. 2001, 226, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Kipper-Galperin, M.; Galilly, R.; Danenberg, H.D.; Brenner, T. Dehydroepiandrosterone selectively inhibits production of tumor necrosis factor alpha and interleukin-6 [correction of interlukin-6] in astrocytes. Int. J. Dev. Neurosci. 1999, 17, 765–775. [Google Scholar] [CrossRef]
- Straub, R.H.; Konecna, L.; Hrach, S.; Rothe, G.; Kreutz, M.; Scholmerich, J.; Falk, W.; Lang, B. Serum dehydroepiandrosterone (DHEA) and DHEA sulfate are negatively correlated with serum interleukin-6 (IL-6), and DHEA inhibits IL-6 secretion from mononuclear cells in man in vitro: Possible link between endocrinosenescence and immunosenescence. J. Clin. Endocrinol. Metab. 1998, 83, 2012–2017. [Google Scholar] [CrossRef] [PubMed]
- Hoyk, Z.; Parducz, A.; Garcia-Segura, L.M. Dehydroepiandrosterone regulates astroglia reaction to denervation of olfactory glomeruli. Glia 2004, 48, 207–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazeldine, J.; Arlt, W.; Lord, J.M. Dehydroepiandrosterone as a regulator of immune cell function. J. Steroid. Biochem. 2010, 120, 127–136. [Google Scholar] [CrossRef]
- Ziogas, A.; Maekawa, T.; Wiessner, J.R.; Le, T.T.; Sprott, D.; Troullinaki, M.; Neuwirth, A.; Anastasopoulou, V.; Grossklaus, S.; Chung, K.J.; et al. DHEA Inhibits Leukocyte Recruitment through Regulation of the Integrin Antagonist DEL-1. J. Immunol. 2020, 204, 1214–1224. [Google Scholar] [CrossRef]
- Barger, S.W.; Chavis, J.A.; Drew, P.D. Dehydroepiandrosterone inhibits microglial nitric oxide production in a stimulus-specific manner. J. Neurosci. Res. 2000, 62, 503–509. [Google Scholar] [CrossRef]
- Alexaki, V.I.; Fodelianaki, G.; Neuwirth, A.; Mund, C.; Kourgiantaki, A.; Ieronimaki, E.; Lyroni, K.; Troullinaki, M.; Fujii, C.; Kanczkowski, W.; et al. DHEA inhibits acute microglia-mediated inflammation through activation of the TrkA-Akt1/2-CREB-Jmjd3 pathway. Mol. Psychiatry 2018, 23, 1410–1420. [Google Scholar] [CrossRef]
- Calogeropoulou, T.; Avlonitis, N.; Minas, V.; Alexi, X.; Pantzou, A.; Charalampopoulos, I.; Zervou, M.; Vergou, V.; Katsanou, E.S.; Lazaridis, I.; et al. Novel dehydroepiandrosterone derivatives with antiapoptotic, neuroprotective activity. J. Med. Chem. 2009, 52, 6569–6587. [Google Scholar] [CrossRef]
- Pediaditakis, I.; Efstathopoulos, P.; Prousis, K.C.; Zervou, M.; Arevalo, J.C.; Alexaki, V.I.; Nikoletopoulou, V.; Karagianni, E.; Potamitis, C.; Tavernarakis, N.; et al. Selective and differential interactions of BNN27, a novel C17-spiroepoxy steroid derivative, with TrkA receptors, regulating neuronal survival and differentiation. Neuropharmacology 2016, 111, 266–282. [Google Scholar] [CrossRef] [Green Version]
- Pediaditakis, I.; Kourgiantaki, A.; Prousis, K.C.; Potamitis, C.; Xanthopoulos, K.P.; Zervou, M.; Calogeropoulou, T.; Charalampopoulos, I.; Gravanis, A. BNN27, a 17-Spiroepoxy Steroid Derivative, Interacts With and Activates p75 Neurotrophin Receptor, Rescuing Cerebellar Granule Neurons from Apoptosis. Front. Pharmacol. 2016, 7, 512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzi, C.; Tiberi, A.; Giustizieri, M.; Marrone, M.C.; Gobbo, F.; Carucci, N.M.; Meli, G.; Arisi, I.; D’Onofrio, M.; Marinelli, S.; et al. NGF steers microglia toward a neuroprotective phenotype. Glia 2018, 66, 1395–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fodelianaki, G.; Lansing, F.; Bhattarai, P.; Troullinaki, M.; Zeballos, M.A.; Charalampopoulos, I.; Gravanis, A.; Mirtschink, P.; Chavakis, T.; Alexaki, V.I. Nerve Growth Factor modulates LPS—Induced microglial glycolysis and inflammatory responses. Exp. Cell Res. 2019, 377, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Prencipe, G.; Minnone, G.; Strippoli, R.; De Pasquale, L.; Petrini, S.; Caiello, I.; Manni, L.; De Benedetti, F.; Bracci-Laudiero, L. Nerve growth factor downregulates inflammatory response in human monocytes through TrkA. J. Immunol. 2014, 192, 3345–3354. [Google Scholar] [CrossRef] [PubMed]
- Aloe, L.; Rocco, M.L.; Bianchi, P.; Manni, L. Nerve growth factor: From the early discoveries to the potential clinical use. J. Transl. Med. 2012, 10, 239. [Google Scholar] [CrossRef] [Green Version]
- Talele, T.T. The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem. 2016, 59, 8712–8756. [Google Scholar] [CrossRef]
- Gomes, A.R.; Varela, C.L.; Tavares-da-Silva, E.J.; Roleira, F.M.F. Epoxide containing molecules: A good or a bad drug design approach. Eur. J. Med. Chem 2020, 201, 112327. [Google Scholar] [CrossRef]
- Albuquerque, C.; Joseph, D.J.; Choudhury, P.; MacDermott, A.B. Dissection, plating, and maintenance of dorsal root ganglion neurons for monoculture and for coculture with dorsal horn neurons. Cold Spring Harb. Protoc. 2009, 2009, pdb.prot5275. [Google Scholar] [CrossRef]
- Seibenhener, M.L.; Wooten, M.W. Isolation and culture of hippocampal neurons from prenatal mice. J. Vis. Exp. 2012, 65, e3634. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Poteet, E.; Xie, L.; Liu, R.; Wen, Y.; Yang, S.H. Regulation of matrix metalloproteinase 2 by oligomeric amyloid beta protein. Brain Res. 2011, 1387, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellissier, H. Recent developments in asymmetric cyclopropanation. Tetrahedron 2008, 64, 7041–7095. [Google Scholar] [CrossRef]
- Ruit, K.G.; Elliott, J.L.; Osborne, P.A.; Yan, Q.; Snider, W.D. Selective dependence of mammalian dorsal root ganglion neurons on nerve growth factor during embryonic development. Neuron 1992, 8, 573–587. [Google Scholar] [CrossRef]
- Alexaki, V.I.; Charalampopoulos, I.; Kampa, M.; Vassalou, H.; Theodoropoulos, P.; Stathopoulos, E.N.; Hatzoglou, A.; Gravanis, A.; Castanas, E. Estrogen exerts neuroprotective effects via membrane estrogen receptors and rapid Akt/NOS activation. FASEB J. 2004, 18, 1594–1596. [Google Scholar] [CrossRef] [Green Version]
- Reifert, J.; Hartung-Cranston, D.; Feinstein, S.C. Amyloid beta-mediated cell death of cultured hippocampal neurons reveals extensive Tau fragmentation without increased full-length tau phosphorylation. J. Biol. Chem. 2011, 286, 20797–20811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid beta-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Pina-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for beta-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e1027. [Google Scholar] [CrossRef] [Green Version]
- Lemke, G. How macrophages deal with death. Nat. Rev. Immunol. 2019, 19, 539–549. [Google Scholar] [CrossRef]
- Savage, J.C.; Jay, T.; Goduni, E.; Quigley, C.; Mariani, M.M.; Malm, T.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Nuclear receptors license phagocytosis by trem2+ myeloid cells in mouse models of Alzheimer’s disease. J. Neurosci. 2015, 35, 6532–6543. [Google Scholar] [CrossRef] [Green Version]
- Alexaki, V. The Impact of Obesity on Microglial Function: Immune, Metabolic and Endocrine Perspectives. Cells 2021, 10, 1584. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, L.; Heinen, Y.; van Dam, A.M.; Lucassen, P.J.; Korosi, A. Microglial Priming and Alzheimer’s Disease: A Possible Role for (Early) Immune Challenges and Epigenetics? Front. Hum. Neurosci. 2016, 10, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivest, S. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 2009, 9, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Lana, D.; Ugolini, F.; Wenk, G.L.; Giovannini, M.G.; Zecchi-Orlandini, S.; Nosi, D. Microglial distribution, branching, and clearance activity in aged rat hippocampus are affected by astrocyte meshwork integrity: Evidence of a novel cell-cell interglial interaction. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 4007–4020. [Google Scholar] [CrossRef] [PubMed]
- Fulle, L.; Offermann, N.; Hansen, J.N.; Breithausen, B.; Erazo, A.B.; Schanz, O.; Radau, L.; Gondorf, F.; Knopper, K.; Alferink, J.; et al. CCL17 exerts a neuroimmune modulatory function and is expressed in hippocampal neurons. Glia 2018, 66, 2246–2261. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chetelat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Longo, F.M.; Massa, S.M. Small-molecule modulation of neurotrophin receptors: A strategy for the treatment of neurological disease. Nat. Rev. Drug Discov. 2013, 12, 507–525. [Google Scholar] [CrossRef]
- Allen, S.J.; Dawbarn, D. Clinical relevance of the neurotrophins and their receptors. Clin. Sci. 2006, 110, 175–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardridge, W.M. Neurotrophins, neuroprotection and the blood-brain barrier. Curr. Opin. Investig. Drugs 2002, 3, 1753–1757. [Google Scholar] [PubMed]
- Thorne, R.G.; Frey, W.H., 2nd. Delivery of neurotrophic factors to the central nervous system: Pharmacokinetic considerations. Clin. Pharmacokinet. 2001, 40, 907–946. [Google Scholar] [CrossRef] [PubMed]
- Rocco, M.L.; Soligo, M.; Manni, L.; Aloe, L. Nerve Growth Factor: Early Studies and Recent Clinical Trials. Curr. Neuropharmacol. 2018, 16, 1455–1465. [Google Scholar] [CrossRef]
- Petty, B.G.; Cornblath, D.R.; Adornato, B.T.; Chaudhry, V.; Flexner, C.; Wachsman, M.; Sinicropi, D.; Burton, L.E.; Peroutka, S.J. The effect of systemically administered recombinant human nerve growth factor in healthy human subjects. Ann. Neurol. 1994, 36, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Eriksdotter Jonhagen, M.; Nordberg, A.; Amberla, K.; Backman, L.; Ebendal, T.; Meyerson, B.; Olson, L.; Seiger, Å.; Shigeta, M.; Theodorsson, E.; et al. Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 1998, 9, 246–257. [Google Scholar] [CrossRef]
- Simmons, D.A.; Belichenko, N.P.; Yang, T.; Condon, C.; Monbureau, M.; Shamloo, M.; Jing, D.; Massa, S.M.; Longo, F.M. A small molecule TrkB ligand reduces motor impairment and neuropathology in R6/2 and BACHD mouse models of Huntington’s disease. J. Neurosci. 2013, 33, 18712–18727. [Google Scholar] [CrossRef]
- Tep, C.; Lim, T.H.; Ko, P.O.; Getahun, S.; Ryu, J.C.; Goettl, V.M.; Massa, S.M.; Basso, M.; Longo, F.M.; Yoon, S.O. Oral administration of a small molecule targeted to block proNGF binding to p75 promotes myelin sparing and functional recovery after spinal cord injury. J. Neurosci. 2013, 33, 397–410. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Meeker, R.B.; Massa, S.M.; Longo, F.M. Modulation of the p75 neurotrophin receptor suppresses age-related basal forebrain cholinergic neuron degeneration. Sci. Rep. 2019, 9, 5273. [Google Scholar] [CrossRef] [Green Version]
- Tsoka, P.; Matsumoto, H.; Maidana, D.E.; Kataoka, K.; Naoumidi, I.; Gravanis, A.; Vavvas, D.G.; Tsilimbaris, M.K. Effects of BNN27, a novel C17-spiroepoxy steroid derivative, on experimental retinal detachment-induced photoreceptor cell death. Sci. Rep. 2018, 8, 10661. [Google Scholar] [CrossRef] [Green Version]
- Iban-Arias, R.; Lisa, S.; Mastrodimou, N.; Kokona, D.; Koulakis, E.; Iordanidou, P.; Kouvarakis, A.; Fothiadaki, M.; Papadogkonaki, S.; Sotiriou, A.; et al. The Synthetic Microneurotrophin BNN27 Affects Retinal Function in Rats With Streptozotocin-Induced Diabetes. Diabetes 2018, 67, 321–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetto, G.; Charalampopoulos, I.; Gravanis, A.; Karagogeos, D. The novel synthetic microneurotrophin BNN27 protects mature oligodendrocytes against cuprizone-induced death, through the NGF receptor TrkA. Glia 2017, 65, 1376–1394. [Google Scholar] [CrossRef] [PubMed]
- Glajch, K.E.; Ferraiuolo, L.; Mueller, K.A.; Stopford, M.J.; Prabhkar, V.; Gravanis, A.; Shaw, P.J.; Sadri-Vakili, G. MicroNeurotrophins Improve Survival in Motor Neuron-Astrocyte Co-Cultures but Do Not Improve Disease Phenotypes in a Mutant SOD1 Mouse Model of Amyotrophic Lateral Sclerosis. PLoS ONE 2016, 11, e0164103. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e569. [Google Scholar] [CrossRef] [Green Version]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Ulland, T.K.; Colonna, M. TREM2—A key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Sousa, C.; Golebiewska, A.; Poovathingal, S.K.; Kaoma, T.; Pires-Afonso, Y.; Martina, S.; Coowar, D.; Azuaje, F.; Skupin, A.; Balling, R.; et al. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. 2018, 19, e46171. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yilmaz, C.; Rogdakis, T.; Latorrata, A.; Thanou, E.; Karadima, E.; Papadimitriou, E.; Siapi, E.; Li, K.W.; Katsila, T.; Calogeropoulou, T.; et al. ENT-A010, a Novel Steroid Derivative, Displays Neuroprotective Functions and Modulates Microglial Responses. Biomolecules 2022, 12, 424. https://doi.org/10.3390/biom12030424
Yilmaz C, Rogdakis T, Latorrata A, Thanou E, Karadima E, Papadimitriou E, Siapi E, Li KW, Katsila T, Calogeropoulou T, et al. ENT-A010, a Novel Steroid Derivative, Displays Neuroprotective Functions and Modulates Microglial Responses. Biomolecules. 2022; 12(3):424. https://doi.org/10.3390/biom12030424
Chicago/Turabian StyleYilmaz, Canelif, Thanasis Rogdakis, Alessia Latorrata, Evangelia Thanou, Eleftheria Karadima, Eleni Papadimitriou, Eleni Siapi, Ka Wan Li, Theodora Katsila, Theodora Calogeropoulou, and et al. 2022. "ENT-A010, a Novel Steroid Derivative, Displays Neuroprotective Functions and Modulates Microglial Responses" Biomolecules 12, no. 3: 424. https://doi.org/10.3390/biom12030424
APA StyleYilmaz, C., Rogdakis, T., Latorrata, A., Thanou, E., Karadima, E., Papadimitriou, E., Siapi, E., Li, K. W., Katsila, T., Calogeropoulou, T., Charalampopoulos, I., & Alexaki, V. I. (2022). ENT-A010, a Novel Steroid Derivative, Displays Neuroprotective Functions and Modulates Microglial Responses. Biomolecules, 12(3), 424. https://doi.org/10.3390/biom12030424