
Metabolic Changes Induced by Cerebral Ischemia, the Effect of Ischemic Preconditioning, and Hyperhomocysteinemia

 , , ,
, , ,
Abstract
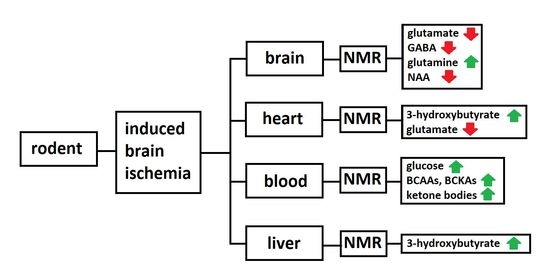
:
1. Introduction
1.1. General Description of Ischemic/Reperfusion (IR) Animal Models
1.2. Metabolomic Approach
2. Metabolomic Alterations
2.1. Brain Tissues after Cerebral Ischemia Studied by the 1H NMR
2.2. Circulating Metabolites in Animal Models of Cerebral Ischemia
2.3. Possible 1H NMR Biomarkers of the Cerebral Ischemia
2.4. 1H NMR Metabolomics Approach in the Ischemic Preconditioning (IPC)
2.5. 1H NMR Approach in Animal Models of Hyperhomocysteinemia and Cerebral Ischemia with Induced Hyperhomocysteinemia
3. Challenges to Clinical Translation and Future Perspectives
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bonita, R. Epidemiology of Stroke. Lancet 1992, 339, 342–344. [Google Scholar] [CrossRef]
- Pulsinelli, W.A.; Brierley, J.B. A New Model of Bilateral Hemispheric Ischemia in the Unanesthetized Rat. Stroke 1979, 10, 267–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traystman, R.J. Animal Models of Focal and Global Cerebral Ischemia. ILAR J. 2003, 44, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S. NMR Metabolomics: A Look Ahead. J. Magn. Reson. 2019, 306, 155–161. [Google Scholar] [CrossRef]
- Tognarelli, J.M.; Dawood, M.; Shariff, M.I.F.; Grover, V.P.B.; Crossey, M.M.E.; Cox, I.J.; Taylor-Robinson, S.D.; McPhail, M.J.W. Magnetic Resonance Spectroscopy: Principles and Techniques: Lessons for Clinicians. J. Clin. Exp. Hepatol. 2015, 5, 320. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.Z. Magic Angle Spinning NMR Metabolomics. Metab. Open Access 2016, 6, 1000e147. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.W.; Rothman, S.M. The Role of Glutamate Neurotoxicity in Hypoxic-Ischemic Neuronal Death. Annu. Rev. Neurosci. 1990, 13, 171–182. [Google Scholar] [CrossRef]
- Nishizawa, Y. Glutamate Release and Neuronal Damage in Ischemia. Life Sci. 2001, 69, 369–381. [Google Scholar] [CrossRef]
- Pomytkin, I.; Krasil’nikova, I.; Bakaeva, Z.; Surin, A.; Pinelis, V. Excitotoxic Glutamate Causes Neuronal Insulin Resistance by Inhibiting Insulin Receptor/Akt/MTOR Pathway. Mol. Brain 2019, 12, 112. [Google Scholar] [CrossRef]
- Rose, C.F.; Verkhratsky, A.; Parpura, V. Astrocyte Glutamine Synthetase: Pivotal in Health and Disease. Biochem. Soc. Trans. 2013, 41, 1518–1524. [Google Scholar] [CrossRef]
- Anlauf, E.; Derouiche, A. Glutamine Synthetase as an Astrocytic Marker: Its Cell Type and Vesicle Localization. Front. Endocrinol. 2013, 4, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranovicova, E.; Kalenska, D.; Grendar, M.; Lehotsky, J. Metabolomic Recovery as a Result of Ischemic Preconditioning Was More Pronounced in Hippocampus than in Cortex That Appeared More Sensitive to Metabolomic Blood Components. Metabolites 2021, 11, 516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wang, W.; Huang, J.; Liu, X.; Zhang, H.; Zhang, N. Metabolomic Investigation of Regional Brain Tissue Dysfunctions Induced by Global Cerebral Ischemia. BMC Neurosci. 2016, 17, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.-L.; Xia, H.-H.; Yang, Y.-J.; Zheng, H.; Zhao, L.-C.; Chen, Y.-C.; Zhuge, Q.-C.; Xia, N.-Z.; Gao, H.-C.; Chen, W.-J. Metabolic Alterations in the Rat Cerebellum Following Acute Middle Cerebral Artery Occlusion, as Determined by 1H NMR Spectroscopy. Mol. Med. Rep. 2018, 17, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Ruan, L.; Wang, Y.; Chen, S.; Zhao, T.; Huang, Q.; Hu, Z.; Xia, N.; Liu, J.; Chen, W.; Zhang, Y.; et al. Metabolite Changes in the Ipsilateral and Contralateral Cerebral Hemispheres in Rats with Middle Cerebral Artery Occlusion. Neural Regen. Res. 2017, 12, 931. [Google Scholar] [CrossRef]
- Huang, Q.; Li, C.; Xia, N.; Zhao, L.; Wang, D.; Yang, Y.; Gao, H. Neurochemical Changes in Unilateral Cerebral Hemisphere during the Subacute Stage of Focal Cerebral Ischemia–reperfusion in Rats: An Ex Vivo 1H Magnetic Resonance Spectroscopy Study. Brain Res. 2018, 1684, 67–74. [Google Scholar] [CrossRef]
- Kovalenko, T.; Osadchenko, I.; Nikonenko, A.; Lushnikova, I.; Voronin, K.; Nikonenko, I.; Muller, D.; Skibo, G. Ischemia-Induced Modifications in Hippocampal CA1 Stratum Radiatum Excitatory Synapses. Hippocampus 2006, 16, 814–825. [Google Scholar] [CrossRef]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Newsholme, P.; Curi, R.; Pithon Curi, T.C.; Murphy, C.J.; Garcia, C.; Pires de Melo, M. Glutamine Metabolism by Lymphocytes, Macrophages, and Neutrophils: Its Importance in Health and Disease. J. Nutr. Biochem. 1999, 10, 316–324. [Google Scholar] [CrossRef]
- Denes, A.; Vidyasagar, R.; Feng, J.; Narvainen, J.; McColl, B.W.; Kauppinen, R.A.; Allan, S.M. Proliferating Resident Microglia after Focal Cerebral Ischaemia in Mice. J. Cereb. Blood Flow Metab. 2007, 27, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Yan, T.; Chopp, M.; Chen, J. Experimental Animal Models and Inflammatory Cellular Changes in Cerebral Ischemic and Hemorrhagic Stroke. Neurosci. Bull. 2015, 31, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, P.J.; O’Connell, M.T.; Al-Rawi, P.G.; Kett-White, C.R.; Gupta, A.K.; Maskell, L.B.; Pickard, J.D.; Kirkpatrick, P.J. Increases in GABA Concentrations during Cerebral Ischaemia: A Microdialysis Study of Extracellular Amino Acids. J. Neurol. Neurosurg. Psychiatry 2002, 72, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Zhou, X.; He, J.; Xie, Z.; Xia, S.; Lu, G. The Roles of GABA in Ischemia–reperfusion Injury in the Central Nervous System and Peripheral Organs. Oxid. Med. Cell. Longev. 2019, 2019, 4028394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herring, B.E.; Silm, K.; Edwards, R.H.; Nicoll, R.A. Is Aspartate an Excitatory Neurotransmitter? J. Neurosci. 2015, 35, 10168. [Google Scholar] [CrossRef]
- Phillis, J.W.; Smith-Barbour, M.; Perkins, L.M.; O’Regan, M.H. Characterization of Glutamate, Aspartate, and GABA Release from Ischemic Rat Cerebral Cortex. Brain Res. Bull. 1994, 34, 457–466. [Google Scholar] [CrossRef]
- Phillis, J.W.; O’Regan, M.H. Mechanisms of Glutamate and Aspartate Release in the Ischemic Rat Cerebral Cortex. Brain Res. 1996, 730, 150–164. [Google Scholar] [CrossRef]
- Demougeot, C.; Garnier, P.; Mossiat, C.; Bertrand, N.; Giroud, M.; Beley, A.; Marie, C. N-Acetylaspartate, a Marker of Both Cellular Dysfunction and Neuronal Loss: Its Relevance to Studies of Acute Brain Injury. J. Neurochem. 2001, 77, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Schuff, N.; Meyerhoff, D.J.; Mueller, S.; Chao, L.; Sacrey, D.T.; Laxer, K.; Weiner, M.W. N-Acetylaspartate as a Marker of Neuronal Injury in Neurodegenerative Disease. Adv. Exp. Med. Biol. 2006, 576, 241–363. [Google Scholar]
- Wesley, U.V.; Bhute, V.J.; Hatcher, J.F.; Palecek, S.P.; Dempsey, R.J. Local and Systemic Metabolic Alterations in Brain, Plasma, and Liver of Rats in Response to Aging and Ischemic Stroke, as Detected by Nuclear Magnetic Resonance (NMR) Spectroscopy. Neurochem. Int. 2019, 127, 113–124. [Google Scholar] [CrossRef]
- Jin, X.; Wang, R.; Wang, H.; Long, C.; Wang, H. Brain Protection against Ischemic Stroke Using Choline as a New Molecular Bypass Treatment. Acta Pharmacol. Sin. 2015, 36, 1416–1425. [Google Scholar] [CrossRef] [Green Version]
- Borges, A.A.; El-Batah, P.N.; Yamashita, L.F.; Santana, A.d.S.; Lopes, A.C.; Freymuller-Haapalainen, E.; Coimbra, C.G.; Sinigaglia-Coimbra, R. Neuroprotective Effect of Oral Choline Administration after Global Brain Ischemia in Rats. Nutr. Neurosci. 2015, 18, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Fernstrom, J.D. Branched-Chain Amino Acids and Brain Function. J. Nutr. 2005, 135, 1539S–1546S. [Google Scholar] [CrossRef] [PubMed]
- Kasparová, S.; Brezová, V.; Valko, M.; Horecký, J.; Mlynárik, V.; Liptaj, T.; Vancová, O.; Ulicná, O.; Dobrota, D. Study of the Oxidative Stress in a Rat Model of Chronic Brain Hypoperfusion. Neurochem. Int. 2005, 46, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Baranovicova, E.; Kalenska, D.; Tomascova, A.; Holubcikova, S.; Lehotsky, J. Time-Related Metabolomics Study in the Rat Plasma after Global Cerebral Ischemia and Reperfusion: Effect of Ischemic Preconditioning. IUBMB Life 2020, 72, 2010–2023. [Google Scholar] [CrossRef] [PubMed]
- Baranovicova, E.; Grendar, M.; Kalenska, D.; Tomascova, A.; Cierny, D.; Lehotsky, J. NMR Metabolomic Study of Blood Plasma in Ischemic and Ischemically Preconditioned Rats: An Increased Level of Ketone Bodies and Decreased Content of Glycolytic Products 24 h after Global Cerebral Ischemia. J. Physiol. Biochem. 2018, 74, 417–429. [Google Scholar] [CrossRef]
- Baranovicova, E.; Kalenska, D.; Tomascova, A.; Lehotsky, J. Metabolomic Study of Altered Energy Metabolism during Global Forebrain Ischemia and Ischemic Precoditioning in Blood Plasma in Homocysteine Treated Rats. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2018, 69, 901–909. [Google Scholar] [CrossRef]
- Ma, S.; Zhao, H.; Ji, X.; Luo, Y. Peripheral to Central: Organ Interactions in Stroke Pathophysiology. Exp. Neurol. 2015, 272, 41–49. [Google Scholar] [CrossRef]
- Koch, K.; Berressem, D.; Konietzka, J.; Thinnes, A.; Eckert, G.P.; Klein, J. Hepatic Ketogenesis Induced by Middle Cerebral Artery Occlusion in Mice. J. Am. Heart Assoc. 2017, 6, e005556. [Google Scholar] [CrossRef]
- Nagana Gowda, G.A.; Gowda, Y.N.; Raftery, D. Expanding the Limits of Human Blood Metabolite Quantitation Using NMR Spectroscopy. Anal. Chem. 2015, 87, 706–715. [Google Scholar] [CrossRef] [Green Version]
- White, H.; Venkatesh, B. Clinical Review: Ketones and Brain Injury. Crit. Care Lond. Engl. 2011, 15, 219. [Google Scholar] [CrossRef] [Green Version]
- Svart, M.; Gormsen, L.C.; Hansen, J.; Zeidler, D.; Gejl, M.; Vang, K.; Aanerud, J.; Moeller, N. Regional Cerebral Effects of Ketone Body Infusion with 3-Hydroxybutyrate in Humans: Reduced Glucose Uptake, Unchanged Oxygen Consumption and Increased Blood Flow by Positron Emission Tomography. A Randomized, Controlled Trial. PLoS ONE 2018, 13, e0190556. [Google Scholar] [CrossRef] [PubMed]
- Prins, M.L. Cerebral Metabolic Adaptation and Ketone Metabolism after Brain Injury. J. Cereb. Blood Flow Metab. 2008, 28, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, D.R.; Ribeiro, L.C.; Hagenn, M.; Siqueira, I.R.; Araújo, E.; Torres, I.L.S.; Gottfried, C.; Netto, C.A.; Gonçalves, C.-A. Ketogenic Diet Increases Glutathione Peroxidase Activity in Rat Hippocampus. Neurochem. Res. 2003, 28, 1793–1797. [Google Scholar] [CrossRef] [PubMed]
- Gasior, M.; Rogawski, M.A.; Hartman, A.L. Neuroprotective and Disease-Modifying Effects of the Ketogenic Diet. Behav. Pharmacol. 2006, 17, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Yudkoff, M.; Daikhin, Y.; Nissim, I.; Lazarow, A.; Nissim, I. Ketogenic Diet, Amino Acid Metabolism, and Seizure Control. J. Neurosci. Res. 2001, 66, 931–940. [Google Scholar] [CrossRef]
- LaManna, J.C.; Salem, N.; Puchowicz, M.; Erokwu, B.; Koppaka, S.; Flask, C.; Lee, Z. KETONES SUPPRESS BRAIN GLUCOSE CONSUMPTION. Adv. Exp. Med. Biol. 2009, 645, 301. [Google Scholar] [CrossRef] [Green Version]
- Hasselbalch, S.G.; Madsen, P.L.; Hageman, L.P.; Olsen, K.S.; Justesen, N.; Holm, S.; Paulson, O.B. Changes in Cerebral Blood Flow and Carbohydrate Metabolism during Acute Hyperketonemia. Am. J. Physiol. 1996, 270, E746–E751. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.S.; Pulido, O.M.; Mueller, R.W.; McGuire, P.F. Molecular and Immunochemical Characterization of the Ionotropic Glutamate Receptors in the Rat Heart. Brain Res. Bull. 1998, 46, 429–434. [Google Scholar] [CrossRef]
- Gill, S.S.; Pulido, O.M.; Mueller, R.W.; McGuire, P.F. Immunochemical Localization of the Metabotropic Glutamate Receptors in the Rat Heart. Brain Res. Bull. 1999, 48, 143–146. [Google Scholar] [CrossRef]
- Xia, J.; Broadhurst, D.I.; Wilson, M.; Wishart, D.S. Translational Biomarker Discovery in Clinical Metabolomics: An Introductory Tutorial. Metabolomics Off. J. Metab. Soc. 2013, 9, 280–299. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Zhang, H.; Liang, X.; Dai, Y.; Liu, L.; Tan, K.; Ma, R.; Luo, J.; Ding, Y.; Ke, C. Application of Metabolomics to the Discovery of Biomarkers for Ischemic Stroke in the Murine Model: A Comparison with the Clinical Results. Mol. Neurobiol. 2021, 58, 6415–6426. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Li, M.; Xu, P.; Gu, T.; Ma, T.; Gu, S. (1)H NMR-Based Metabolomics Exploring Biomarkers in Rat Cerebrospinal Fluid after Cerebral Ischemia/Reperfusion. Mol. Biosyst. 2013, 9, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Yokota, H.; Jover, T.; Cappuccio, I.; Calderone, A.; Simionescu, M.; Bennett, M.V.L.; Zukin, R.S. Ischemic Preconditioning: Neuronal Survival in the Face of Caspase-3 Activation. J. Neurosci. 2004, 24, 2750–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.-H.; Yang, J.-T.; Lin, J.-R.; Hu, C.-J.; Chou, W.-H.; Lin, C.-P.; Chi, N.-F. Protective Effects of Ischemic Preconditioning against Neuronal Apoptosis and Dendritic Injury in the Hippocampus Are Age-Dependent. J. Neurochem. 2020, 155, 430–447. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-Y.; Chang, Y.-C.; Ho, C.-J.; Huang, C.-C. Ischemic Preconditioning Reduces Neurovascular Damage after Hypoxia-Ischemia via the Cellular Inhibitor of Apoptosis 1 in Neonatal Brain. Stroke 2013, 44, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, D.F.d.C.; Fontes, B.; Shimazaki, J.K.; Heimbecker, A.M.C.; Jacysyn, J.d.F.; Rasslan, S.; Montero, E.F.d.S.; Utiyama, E.M. Ischemic Preconditioning Modifies Mortality and Inflammatory Response. Acta Cir. Bras. 2016, 31, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Han, R.; Zhou, B. Metabolic Reprogramming: Strategy for Ischemic Stroke Treatment by Ischemic Preconditioning. Biology 2021, 10, 424. [Google Scholar] [CrossRef]
- Yong, M.; Kaste, M. Dynamic of Hyperglycemia as a Predictor of Stroke Outcome in the ECASS-II Trial. Stroke 2008, 39, 2749–2755. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.-M.; Lee, Y.-S.; Bae, H.-J.; Kang, D.-W. Homocysteine as a Predictor of Early Neurological Deterioration in Acute Ischemic Stroke. Stroke 2014, 45, 871–873. [Google Scholar] [CrossRef]
- Lehotský, J.; Burda, J.; Danielisová, V.; Gottlieb, M.; Kaplán, P.; Saniová, B. Ischemic Tolerance: The Mechanisms of Neuroprotective Strategy. Anat. Rec. 2009, 292, 2002–2012. [Google Scholar] [CrossRef]
- Petras, M.; Tatarkova, Z.; Kovalska, M.; Mokra, D.; Dobrota, D.; Lehotsky, J.; Drgova, A. Hyperhomocysteinemia as a Risk Factor for the Neuronal System Disorders. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2014, 65, 15–23. [Google Scholar]
- Williams, S.R.; Yang, Q.; Chen, F.; Liu, X.; Keene, K.L.; Jacques, P.; Chen, W.-M.; Weinstein, G.; Hsu, F.-C.; Beiser, A.; et al. Genome-Wide Meta-Analysis of Homocysteine and Methionine Metabolism Identifies Five One Carbon Metabolism Loci and a Novel Association of ALDH1L1 with Ischemic Stroke. PLoS Genet. 2014, 10, e1004214. [Google Scholar] [CrossRef]
- Kalhan, S.C.; Marczewski, S.E. Methionine, Homocysteine, One Carbon Metabolism and Fetal Growth. Rev. Endocr. Metab. Disord. 2012, 13, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Lehotský, J.; Tothová, B.; Kovalská, M.; Dobrota, D.; Beňová, A.; Kalenská, D.; Kaplán, P. Role of Homocysteine in the Ischemic Stroke and Development of Ischemic Tolerance. Front. Neurosci. 2016, 10, 538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, P.; Tatarkova, Z.; Sivonova, M.K.; Racay, P.; Lehotsky, J. Homocysteine and Mitochondria in Cardiovascular and Cerebrovascular Systems. Int. J. Mol. Sci. 2020, 21, 7698. [Google Scholar] [CrossRef]
- Büdy, B.; O’Neill, R.; DiBello, P.M.; Sengupta, S.; Jacobsen, D.W. Homocysteine Transport by Human Aortic Endothelial Cells: Identification and Properties of Import Systems. Arch. Biochem. Biophys. 2006, 446, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Yang, F.; Brailoiu, E.; Jakubowski, H.; Dun, N.J.; Schafer, A.I.; Yang, X.; Durante, W.; Wang, H. Differential Regulation of Homocysteine Transport in Vascular Endothelial and Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1976–1983. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Huang, Y.; Hu, Q.; Ma, L. Hyperhomocysteinemia Stimulates Hepatic Glucose Output and PEPCK Expression. Acta Biochim. Biophys. Sin. 2009, 41, 1027–1032. [Google Scholar] [CrossRef] [Green Version]
- Tessari, P.; Kiwanuka, E.; Coracina, A.; Zaramella, M.; Vettore, M.; Valerio, A.; Garibotto, G. Insulin in Methionine and Homocysteine Kinetics in Healthy Humans: Plasma vs. Intracellular Models. Am. J. Physiol.-Endocrinol. Metab. 2005, 288, E1270–E1276. [Google Scholar] [CrossRef] [Green Version]
- Kovalska, M.; Baranovicova, E.; Kalenska, D.; Tomascova, A.; Adamkov, M.; Kovalska, L.; Lehotsky, J. Methionine Diet Evoked Hyperhomocysteinemia Causes Hippocampal Alterations, Metabolomics Plasma Changes and Behavioral Pattern in Wild Type Rats. Int. J. Mol. Sci. 2021, 22, 4961. [Google Scholar] [CrossRef]
- Kovalska, M.; Hnilicova, P.; Kalenska, D.; Tothova, B.; Adamkov, M.; Lehotsky, J. Effect of Methionine Diet on Metabolic and Histopathological Changes of Rat Hippocampus. Int. J. Mol. Sci. 2019, 20, 6234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, J.L.; Choi, I.-Y.; Brooks, W.M. Probing Astrocyte Metabolism in Vivo: Proton Magnetic Resonance Spectroscopy in the Injured and Aging Brain. Front. Aging Neurosci. 2015, 7, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehotsky, J.; Kovalska, M.; Baranovicova, E.; Hnilicova, P.; Kalenska, D.; Kaplan, P. Ischemic Brain Injury in Hyperhomocysteinemia. In Cerebral Ischemia [Internet]; Pluta, R., Ed.; Exon Publications: Brisbane, Australia, 2021; Chapter 5. [Google Scholar]
- Kovalska, M.; Hnilicova, P.; Kalenska, D.; Tomascova, A.; Adamkov, M.; Lehotsky, J. Effect of Methionine Diet on Time-Related Metabolic and Histopathological Changes of Rat Hippocampus in the Model of Global Brain Ischemia. Biomolecules 2020, 10, 1128. [Google Scholar] [CrossRef] [PubMed]
- Role of Methionine on Epigenetic Modification of DNA Methylation and Gene Expression in Animals-ScienceDirect. Available online: https://www.sciencedirect.com/science/article/pii/S240565451730094X?via%3Dihub (accessed on 7 March 2022).
- Wu, X.; Zhang, L.; Miao, Y.; Yang, J.; Wang, X.; Wang, C.; Feng, J.; Wang, L. Homocysteine Causes Vascular Endothelial Dysfunction by Disrupting Endoplasmic Reticulum Redox Homeostasis. Redox Biol. 2019, 20, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Soares, D.P.; Law, M. Magnetic Resonance Spectroscopy of the Brain: Review of Metabolites and Clinical Applications. Clin. Radiol. 2009, 64, 12–21. [Google Scholar] [CrossRef]
- Sajja, B.R.; Wolinsky, J.S.; Narayana, P.A. Proton Magnetic Resonance Spectroscopy in Multiple Sclerosis. Neuroimaging Clin. N. Am. 2009, 19, 45–58. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer Disease: Postmortem Neuropathologic Correlates of Antemortem 1H MR Spectroscopy Metabolite Measurements1 | Radiology. Available online: https://pubs.rsna.org/doi/10.1148/radiol.2481071590 (accessed on 7 March 2022).
- Unschuld, P.G.; Edden, R.A.E.; Carass, A.; Liu, X.; Shanahan, M.; Wang, X.; Oishi, K.; Brandt, J.; Bassett, S.S.; Redgrave, G.W.; et al. Brain Metabolite Alterations and Cognitive Dysfunction in Early Huntington’s Disease. Mov. Disord. 2012, 27, 895–902. [Google Scholar] [CrossRef] [Green Version]
- Clinical Proton MR Spectroscopy in Central Nervous System Disorders | Radiology. Available online: https://pubs.rsna.org/doi/10.1148/radiol.13130531 (accessed on 7 March 2022).
- Faghihi, R.; Zeinali-Rafsanjani, B.; Mosleh-Shirazi, M.-A.; Saeedi-Moghadam, M.; Lotfi, M.; Jalli, R.; Iravani, V. Magnetic Resonance Spectroscopy and Its Clinical Applications: A Review. J. Med. Imaging Radiat. Sci. 2017, 48, 233–253. [Google Scholar] [CrossRef] [Green Version]
- Perła-Kaján, J.; Jakubowski, H. Dysregulation of Epigenetic Mechanisms of Gene Expression in the Pathologies of Hyperhomocysteinemia. Int. J. Mol. Sci. 2019, 20, 3140. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W.; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Stanga, S.; Caretto, A.; Boido, M.; Vercelli, A. Mitochondrial Dysfunctions: A Red Thread across Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 3719. [Google Scholar] [CrossRef] [PubMed]
- Timkova, V.; Tatarkova, Z.; Lehotsky, J.; Racay, P.; Dobrota, D.; Kaplan, P. Effects of Mild Hyperhomocysteinemia on Electron Transport Chain Complexes, Oxidative Stress, and Protein Expression in Rat Cardiac Mitochondria. Mol. Cell. Biochem. 2016, 411, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Tatarkova, Z.; Bencurova, M.; Lehotsky, J.; Racay, P.; Kmetova Sivonova, M.; Dobrota, D.; Kaplan, P. Effect of Hyperhomocysteinemia on Rat Cardiac Sarcoplasmic Reticulum. Mol. Cell. Biochem. 2022, 477, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Tang, H.; Nicholson, J.; Lindon, J. Use of H-1 NMR-Determined Diffusion Coefficients to Characterize Lipoprotein Fractions in Human Blood Plasma. Magn. Reson. Chem. 2002, 40, S83–S88. [Google Scholar] [CrossRef]
- Kovalska, M.; Tothova, B.; Kovalska, L.; Tatarkova, Z.; Kalenska, D.; Tomascova, A.; Adamkov, M.; Lehotsky, J. Association of Induced Hyperhomocysteinemia with Alzheimer’s Disease-Like Neurodegeneration in Rat Cortical Neurons After Global Ischemia–reperfusion Injury. Neurochem. Res. 2018, 43, 1766–1778. [Google Scholar] [CrossRef]
- Zaragozá, R. Transport of Amino Acids Across the Blood–brain Barrier. Front. Physiol. 2020, 11, 973. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Reper-Fusion | Increase | Decrease | References | |||
|---|---|---|---|---|---|---|
| 4VO | rats | 24 h | cortex | glutamine, isoleucine, valine, phenylalanine, fumarate | glutamate, GABA, NAA, choline | [12] |
| 24 h | hippocampus | glutamate, GABA, ascorbate, inosine, NAA, choline, myo-inositol | [12] | |||
| 3 h | blood plasma | glucose, 3-hydroxybutyrate, acetoacetate, BCAAs, BCKAs, phenylalanine | lactate, pyruvate, alanine, citrate, glutamine, lipoproteins, tyrosine, lysine | [34] | ||
| 24 h | blood plasma | glucose, 3-hydroxybutyrate, acetoacetate, acetone, BCAAs, BCKAs, phenylalanine | lactate, pyruvate, alanine, citrate, glutamine, lipoproteins, citrate | [34,35] | ||
| 72 h | blood plasma | glucose | citrate | [34] | ||
| 24 h | heart | 3-hydroxybutyrate | glutamate | [12] | ||
| 2VO | mice female | 1 h | cortex | leucine, isoleucine, valine, alanine, lysine, glutamine, succinate, myo-inositol, GABA | glutamate, aspartate, taurine, NAA | [13] |
| 1 h | hippocampus | leucine, isoleucine, valine, alanine, GABA, tyrosine | glutamate, aspartate | [13] | ||
| mice male | 1 h | cortex | leucine, isoleucine, valine, alanine, lysine, GABA, glutamine, tyrosine | glutamate, aspartate, NAA, | [13] | |
| 1 h | hippocampus | isoleucine, valine. alanine, GABA, glutamine | glutamate | [13] | ||
| MCAO | rat | 24 h | left ischemic cerebral hemisphere | glycine, GABA, alanine, choline | myo-inositol, NAA, aspartate, glutamate, creatine | [14] |
| 24 h | right cerebellum | GABA, aspartate, glutamine, choline | glutamate, succinate, creatine | [14] | ||
| MCAO | rat | 1 h | ipsilateral (ischemic) hemisphere | alanine, GABA, choline, glycine | NAA, glutamate, creatine | [15] |
| 3 h | ipsilateral (ischemic) hemisphere | alanine, GABA, choline, glycine | NAA, glutamate, aspartate, creatine | [15] | ||
| 9 h | ipsilateral (ischemic) hemisphere | alanine, glycine | NAA, glutamate, aspartate, creatine | [15] | ||
| 24 h | ipsilateral (ischemic) hemisphere | alanine, GABA, choline, glycine | NAA, glutamate, aspartate, creatine | [15] | ||
| rat | 1 h | contralateral hemisphere | alanine, aspartate | GABA, glutamate | [15] | |
| 3 h | contralateral hemisphere | aspartate, glycine | [15] | |||
| 9 h | contralateral hemisphere | aspartate | [15] | |||
| 24 h | contralateral hemisphere | alanine, aspartate, choline | creatine | [15] | ||
| pMCAO | rat | 7 days | left brain extracts | glutamine | NAA, GABA, glutamate, succinate | [16] |
| tMCAO | rat | 2 h–7 days | left brain extracts | NAA, GABA, glutamate, succinate | [16] | |
| MCAO | rat 3 months | day 2 | brain | lysine, tryptophan, glycine, fumarate | NAA | [29] |
| rat 12 months | day 2 | brain | NAA | [29] | ||
| rat 3 months | day 2 | liver | [29] | |||
| rat 12 months | day 2 | liver | choline, 3-hydroxybutyrate, taurine | tyrosine, tryptophan, serine, alanine, histamine | [29] | |
| rat 3 months | day 2 | blood plasma | lysine | tyrosine, choline, citrate | [29] | |
| rat 12 months | day 2 | blood plasma | lysine, isoleucine | tyrosine, citrate, proline, threonine | [29] | |
| 4VO | rat-hHcy | 24 h | blood plasma | lactate, leucine, isoleucine, valine, acetone, 3-hydroxybutyrate, phenylalanine, creatine | glucose, pyruvate, citrate, lipoproteins | [36] |
| Reperfusion | Medium | Metabolites Discriminating Ischemic Rats against Controls with AUC Value Obtained after Discrimination | References |
|---|---|---|---|
| 24 h | cortex | fumarate 1, choline 0.998, phenylalanine 0.997, valine 0.995, isoleucine 0.925, glutamine 0.923, alanine 0.842, GABA 0.809 | [12] |
| 24 h | hippocampus | glutamate 1, GABA 1, choline 1, myo-inositol 0.978, ascorbate 0.963, inosine 0.919, NAA 0.918, creatine 0.894, succinate 0.878 | [12] |
| 3 h | blood plasma | alanine, phenylalanine, acetoacetate, all in combination AUC = 1 | [34] |
| 24 h | blood plasma | 3-hydroxybutyrate, acetoacetate, leucine, pyruvate, choline, all in combination AUC = 1 | [35] |
| 24 h | blood plasma | alanine, isoleucine, leucine acetoacetate, pyruvate, all in combination AUC = 1 | [34] |
| 72 h | blood plasma | acetoacetate, lysine, 3-hydroxybutyrate, glucose, citrate, all in combination AUC = 0.96 | [34] |
| 24 h | heart | glutamate 0.898 | [12] |
| 24 h | blood plasma, hHcy rats | citrate, 3-hydroxybutyrate, creatine, glucose, all in combination AUC = 1 | [36] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baranovicova, E.; Hnilicova, P.; Kalenska, D.; Kaplan, P.; Kovalska, M.; Tatarkova, Z.; Tomascova, A.; Lehotsky, J. Metabolic Changes Induced by Cerebral Ischemia, the Effect of Ischemic Preconditioning, and Hyperhomocysteinemia. Biomolecules 2022, 12, 554. https://doi.org/10.3390/biom12040554
Baranovicova E, Hnilicova P, Kalenska D, Kaplan P, Kovalska M, Tatarkova Z, Tomascova A, Lehotsky J. Metabolic Changes Induced by Cerebral Ischemia, the Effect of Ischemic Preconditioning, and Hyperhomocysteinemia. Biomolecules. 2022; 12(4):554. https://doi.org/10.3390/biom12040554
Chicago/Turabian StyleBaranovicova, Eva, Petra Hnilicova, Dagmar Kalenska, Peter Kaplan, Maria Kovalska, Zuzana Tatarkova, Anna Tomascova, and Jan Lehotsky. 2022. "Metabolic Changes Induced by Cerebral Ischemia, the Effect of Ischemic Preconditioning, and Hyperhomocysteinemia" Biomolecules 12, no. 4: 554. https://doi.org/10.3390/biom12040554
APA StyleBaranovicova, E., Hnilicova, P., Kalenska, D., Kaplan, P., Kovalska, M., Tatarkova, Z., Tomascova, A., & Lehotsky, J. (2022). Metabolic Changes Induced by Cerebral Ischemia, the Effect of Ischemic Preconditioning, and Hyperhomocysteinemia. Biomolecules, 12(4), 554. https://doi.org/10.3390/biom12040554