
Parkin as a Molecular Bridge Linking Alzheimer’s and Parkinson’s Diseases?



Abstract
:
{kind=link}
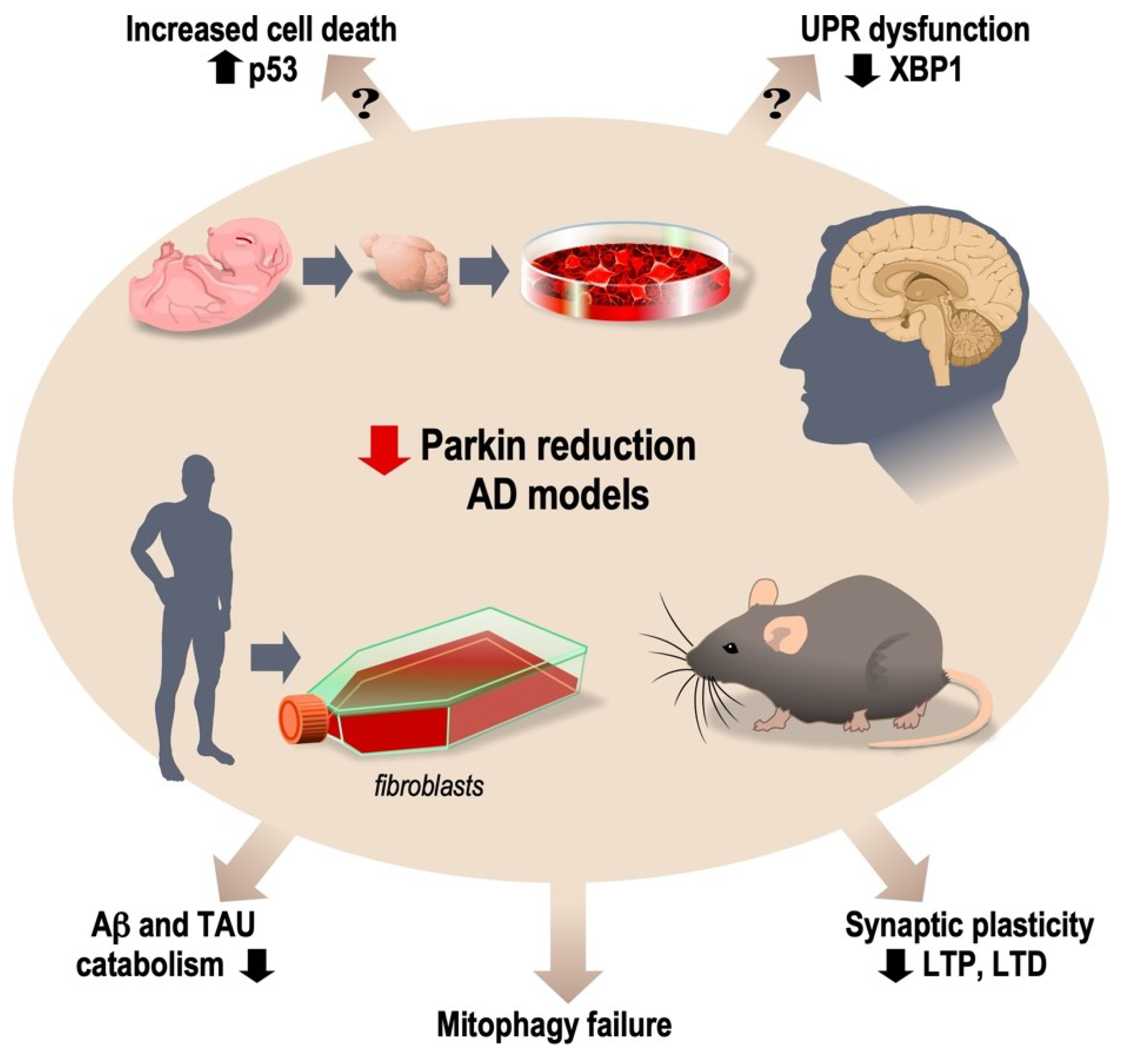{kind=link}
1. Alzheimer’s Disease in Brief
2. Parkinson’s Disease in Brief
3. Alzheimer’s and Parkinson’s Diseases: Two Distinct Pathologies with Common Dysfunctions
3.1. Proteasomal Dysfunction
3.2. Oxidative Stress Dysfunction
3.3. ER Stress and Unfolded Protein Response
3.4. Mitochondria Dysfunction
3.5. Autophagy/Mitophagy
3.6. Cell Death
4. Parkin, a Molecular Link between Alzheimer’s and Parkinson’s Diseases?
5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| ER | Endoplasmic reticulum |
| PINK1 | Phosphatase and TENsin-induced putative kinase 1 |
| XBP1s | Spliced form of X-box binding protein 1 |
| Ab peptides | Amyloid b peptides |
| PARK | Designated genes involved in familial PD |
| HTRA2 | High temperature requirement protein A2 |
| LRRK2 | Leucine rich repeat kinase 2 |
| ATP13A2 | ATPase type 13A2 |
| ERAD | ER-associated degradation |
| UPR | Unfolded protein response |
| ATF6 | Activating transcription factor 6 |
| PERK | Pancreatic ER kinase |
| GRP78 | 78 Kd Glucose-regulated protein |
| (IRE1a) | Inositol-requiring enzyme-1 alpha |
| eIF2 a | Eukaryotic initiation factor 2 alpha |
| iPSCs | Inducible pluripotent stem cells |
| PAEL-R | Parkin-associated endothelin receptor-like receptor |
| OXPHOS | Mitochondrial oxidative phosphorylation system |
| (ROS) | Reactive oxygen species |
| MIRO | Mitochondrial Rho |
| (Drp-1) | Dynamin-related-protein 1 |
| FOXO3a | Forkhead box O3 |
| PEN-2 | Presenilin enhancer 2 |
| APH-1 | Anterior pharynx-defective 1 |
References
- Selkoe, D.J. Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chetelat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, M.S.; Tewari, D.; Sharma, G.; Kabir, M.T.; Barreto, G.E.; Bin-Jumah, M.N.; Perveen, A.; Abdel-Daim, M.M.; Ashraf, G.M. Molecular Mechanisms of ER Stress and UPR in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 2902–2919. [Google Scholar] [CrossRef]
- Huang, H.C.; Tang, D.; Lu, S.Y.; Jiang, Z.F. Endoplasmic reticulum stress as a novel neuronal mediator in Alzheimer’s disease. Neurol. Res. 2015, 37, 366–374. [Google Scholar] [CrossRef]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Vaillant-Beuchot, L.; Mary, A.; Pardossi-Piquard, R.; Bourgeois, A.; Lauritzen, I.; Eysert, F.; Kinoshita, P.F.; Cazareth, J.; Badot, C.; Fragaki, K.; et al. Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer’s disease models and human brains. Acta Neuropathol. 2021, 141, 39–65. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.; Livesey, F.J. Endolysosome and autophagy dysfunction in Alzheimer disease. Autophagy 2021, 17, 3882–3883. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: Inseparable partners in a multifactorial disease. FASEB J. 2017, 31, 2729–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Weering, J.R.T.; Scheper, W. Endolysosome and Autolysosome Dysfunction in Alzheimer’s Disease: Where Intracellular and Extracellular Meet. CNS Drugs 2019, 33, 639–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauritzen, I.; Pardossi-Piquard, R.; Bourgeois, A.; Pagnotta, S.; Biferi, M.G.; Barkats, M.; Lacor, P.; Klein, W.; Bauer, C.; Checler, F. Intraneuronal aggregation of the beta-CTF fragment of APP (C99) induces Abeta-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016, 132, 257–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, J.; Savage, J.C.; Tremblay, M.E. Synaptic Loss in Alzheimer’s Disease: Mechanistic Insights Provided by Two-Photon in vivo Imaging of Transgenic Mouse Models. Front. Cell Neurosci. 2020, 14, 592607. [Google Scholar] [CrossRef]
- Colom-Cadena, M.; Spires-Jones, T.; Zetterberg, H.; Blennow, K.; Caggiano, A.; DeKosky, S.T.; Fillit, H.; Harrison, J.E.; Schneider, L.S.; Scheltens, P.; et al. The clinical promise of biomarkers of synapse damage or loss in Alzheimer’s disease. Alzheimer’s Res. Ther. 2020, 12, 21. [Google Scholar] [CrossRef]
- Lee, J.H.; Won, S.M.; Suh, J.; Son, S.J.; Moon, G.J.; Park, U.J.; Gwag, B.J. Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp. Mol. Med. 2010, 42, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Dickson, D.W. Apoptotic mechanisms in Alzheimer neurofibrillary degeneration: Cause or effect? J. Clin. Investig. 2004, 114, 23–27. [Google Scholar] [CrossRef] [Green Version]
- Chui, D.H.; Dobo, E.; Makifuchi, T.; Akiyama, H.; Kawakatsu, S.; Petit, A.; Checler, F.; Araki, W.; Takahashi, K.; Tabira, T. Apoptotic neurons in Alzheimer’s disease frequently show intracellular Abeta42 labeling. J. Alzheimer’s Dis. 2001, 3, 231–239. [Google Scholar] [CrossRef]
- Checler, F.; Alves da Costa, C. p53 in neurodegenerative diseases and brain cancers. Pharmacol. Ther. 2014, 142, 99–113. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Read, M.; Barreto, G.E.; Avila-Rodriguez, M.; Gheibi-Hayat, S.M.; Sahebkar, A. Apoptotic neurons and amyloid-beta clearance by phagocytosis in Alzheimer’s disease: Pathological mechanisms and therapeutic outlooks. Eur. J. Pharmacol. 2021, 895, 173873. [Google Scholar] [CrossRef] [PubMed]
- St George-Hyslop, P.H.; Tanzi, R.E.; Polinsky, R.J.; Haines, J.L.; Nee, L.; Watkins, P.c.; Myers, R.H.; Feldman, R.G.; Pollen, D.; Drachman, D.; et al. The genetic defect causing familial Alzheimer’s disease maps on chromosome 21. Science 1987, 235, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Tanzi, R.E. The genetics of Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2012, 107, 79–100. [Google Scholar] [CrossRef] [PubMed]
- St George-Hyslop, P.; Haines, J.; Rogaev, E.; Mortilla, M.; Vaula, G.; Pericak-Vance, M.; Foncin, J.F.; Montesi, M.; Bruni, A.; Sorbi, S.; et al. Genetic evidence for a novel fami!lial Alzheimer’s disease locus on chromosome 14. Nat. Genet. 1992, 2, 330–334. [Google Scholar] [CrossRef]
- Rogaev, E.I.; Sherrington, R.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Liang, Y.; Chi, H.; Lin, C.; Holman, K.; Tsuda, T.; et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 1995, 376, 775–778. [Google Scholar] [CrossRef]
- Levy-Lahad, E.; Wijsman, E.M.; Nemens, E.; Anderson, L.; Goddard, A.B.; Weber, J.L.; Bird, T.D.; Schellenberg, G.D. A familial Alzheimer’s disease locus on chromosome 1. Science 1995, 269, 970–973. [Google Scholar] [CrossRef]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic approach. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef]
- De Strooper, B.; Saftig, P.; Craessaerts, K.; Vanderstichele, H.; Guhde, G.; Annaert, W.; Von Figura, K.; Van Leuven, F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998, 391, 387–390. [Google Scholar] [CrossRef]
- Herreman, A.; Serneels, L.; Annaert, W.; Collen, D.; Schoonjans, L.; De Strooper, B. Total inactivation of g-secretase activity in presenilin-deficient embryonic stem cells. Nat. Cell. Biol. 2000, 2, 461–462. [Google Scholar] [CrossRef] [PubMed]
- Thinakaran, G. The role of presenilins in Alzheimers’ disease. J. Clin. Investig. 1999, 104, 1321–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassar, R.; Citron, M. Ab-generating enzymes: Recent advances in b- and g-secretases research. Neuron 2000, 27, 419–422. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Liu, K.; Qiu, Y.; Ren, Z.; Dai, R.; Deng, Y.; Qing, H. Effect of Presenilin Mutations on APP Cleavage; Insights into the Pathogenesis of FAD. Front. Aging Neurosci. 2016, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- Ancolio, K.; Marambaud, P.; Dauch, P.; Checler, F. α-secretase-derived product of b-amyloid precursor protein is decreased by presenilin 1 mutations linked to familial Alzheimer’s disease. J. Neurochem. 1997, 69, 2494–2499. [Google Scholar] [CrossRef]
- Kelleher, R.J., 3rd; Shen, J. Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 629–631. [Google Scholar] [CrossRef] [Green Version]
- Sexton, C.; Snyder, H.; Beher, D.; Boxer, A.L.; Brannelly, P.; Brion, J.P.; Buee, L.; Cacace, A.M.; Chetelat, G.; Citron, M.; et al. Current directions in tau research: Highlights from Tau 2020. Alzheimer’s Dement. 2021. [Google Scholar] [CrossRef]
- Checler, F.; Afram, E.; Pardossi-Piquard, R.; Lauritzen, I. Is gamma-secretase a beneficial inactivating enzyme of the toxic APP C-terminal fragment C99? J. Biol. Chem. 2021, 296, 100489. [Google Scholar] [CrossRef]
- Garcia-Gonzalez, L.; Pilat, D.; Baranger, K.; Rivera, S. Emerging Alternative Proteinases in APP Metabolism and Alzheimer’s Disease Pathogenesis: A Focus on MT1-MMP and MT5-MMP. Front. Aging Neurosci. 2019, 11, 244. [Google Scholar] [CrossRef]
- Baranger, K.; Marchalant, Y.; Bonnet, A.E.; Crouzin, N.; Carrete, A.; Paumier, J.M.; Py, N.A.; Bernard, A.; Bauer, C.; Charrat, E.; et al. MT5-MMP is a new pro-amyloidogenic proteinase that promotes amyloid pathology and cognitive decline in a transgenic mouse model of Alzheimer’s disease. Cell Mol. Life Sci. 2016, 73, 217–236. [Google Scholar] [CrossRef] [Green Version]
- Willem, M.; Tahirovic, S.; Busche, M.A.; Ovsepian, S.V.; Chafai, M.; Kootar, S.; Hornburg, D.; Evans, L.D.; Moore, S.; Daria, A.; et al. η-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 2015, 526, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Becker-Pauly, C.; Pietrzik, C.U. The Metalloprotease Meprin beta Is an Alternative beta-Secretase of APP. Front. Mol. Neurosci. 2016, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Valverde, A.; Dunys, J.; Lorivel, T.; Debayle, D.; Gay, A.S.; Caillava, C.; Chami, M.; Checler, F. Dipeptidyl peptidase 4 contributes to Alzheimer’s disease-like defects in a mouse model and is increased in sporadic Alzheimer’s disease brains. J. Biol. Chem. 2021, 297, 100963. [Google Scholar] [CrossRef] [PubMed]
- Valverde, A.; Dunys, J.; Lorivel, T.; Debayle, D.; Gay, A.S.; Lacas-Gervais, S.; Roques, B.P.; Chami, M.; Checler, F. Aminopeptidase A contributes to biochemical, anatomical and cognitive defects in Alzheimer’s disease (AD) mouse model and is increased at early stage in sporadic AD brain. Acta Neuropathol. 2021, 141, 823–839. [Google Scholar] [CrossRef]
- Dunys, J.; Valverde, A.; Checler, F. Are N- and C-terminally truncated Abeta species key pathological triggers in Alzheimer’s disease? J. Biol. Chem. 2018, 293, 15419–15428. [Google Scholar] [CrossRef] [Green Version]
- Wirths, O.; Zampar, S.; Weggen, S. N-Terminally Truncated Abeta Peptide Variants in Alzheimer’s Disease. In Alzheimer’s Disease; Wisniewski, T., Ed.; Exon Publications: Brisbane, Australia, 2019. [Google Scholar] [CrossRef] [Green Version]
- Bayer, T.A. N-Truncated Abeta Starting at Position Four-Biochemical Features, Preclinical Models, and Potential as Drug Target in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 710579. [Google Scholar] [CrossRef]
- Jiang, Y.; Mullaney, K.A.; Peterhoff, C.M.; Che, S.; Schmidt, S.D.; Boyer-Boiteau, A.; Ginsberg, S.D.; Cataldo, A.M.; Mathews, P.M.; Nixon, R.A. Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. USA 2010, 107, 1630–1635. [Google Scholar] [CrossRef] [Green Version]
- Bourgeois, A.; Lauritzen, I.; Lorivel, T.; Bauer, C.; Checler, F.; Pardossi-Piquard, R. Intraneuronal accumulation of C99 contributes to synaptic alterations, apathy-like behavior, and spatial learning deficits in 3xTgAD and 2xTgAD mice. Neurobiol. Aging 2018, 71, 21–31. [Google Scholar] [CrossRef]
- Kwart, D.; Gregg, A.; Scheckel, C.; Murphy, E.A.; Paquet, D.; Duffield, M.; Fak, J.; Olsen, O.; Darnell, R.B.; Tessier-Lavigne, M. A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP beta-CTFs, Not Abeta. Neuron 2019, 104, 1022. [Google Scholar] [CrossRef] [Green Version]
- Miranda, A.M.; Lasiecka, Z.M.; Xu, Y.; Neufeld, J.; Shahriar, S.; Simoes, S.; Chan, R.B.; Oliveira, T.G.; Small, S.A.; Di Paolo, G. Neuronal lysosomal dysfunction releases exosomes harboring APP C-terminal fragments and unique lipid signatures. Nat. Commun. 2018, 9, 291. [Google Scholar] [CrossRef] [Green Version]
- Lauritzen, I.; Pardossi-Piquard, R.; Bourgeois, A.; Becot, A.; Checler, F. Does Intraneuronal Accumulation of Carboxyl-terminal Fragments of the Amyloid Precursor Protein Trigger Early Neurotoxicity in Alzheimer’s Disease? Curr. Alzheimer Res. 2019, 16, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Marino, B.L.B.; de Souza, L.R.; Sousa, K.P.A.; Ferreira, J.V.; Padilha, E.C.; da Silva, C.; Taft, C.A.; Hage-Melim, L.I.S. Parkinson’s Disease: A Review from Pathophysiology to Treatment. Mini Rev. Med. Chem. 2020, 20, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Drui, G.; Carnicella, S.; Carcenac, C.; Favier, M.; Bertrand, A.; Boulet, S.; Savasta, M. Loss of dopaminergic nigrostriatal neurons accounts for the motivational and affective deficits in Parkinson’s disease. Mol. Psychiatry 2014, 19, 358–367. [Google Scholar] [CrossRef] [Green Version]
- Grosch, J.; Winkler, J.; Kohl, Z. Early Degeneration of Both Dopaminergic and Serotonergic Axons-A Common Mechanism in Parkinson’s Disease. Front. Cell Neurosci. 2016, 10, 293. [Google Scholar] [CrossRef] [Green Version]
- Masato, A.; Plotegher, N.; Boassa, D.; Bubacco, L. Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol. Neurodegener. 2019, 14, 35. [Google Scholar] [CrossRef] [Green Version]
- Surmeier, D.J. Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J. 2018, 285, 3657–3668. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Goedert, M.; Crowther, R.A.; Murrell, J.R.; Farlow, M.R.; Ghetti, B. Familial multiple system tauopathy with presenile dementia: A disease with abundant neuronal and glial tau filaments. Proc. Natl. Acad. Sci. USA 1997, 94, 4113–4118. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Pu, J. Alpha-Synuclein in Parkinson’s Disease: From Pathogenetic Dysfunction to Potential Clinical Application. Parkinson’s Dis. 2016, 2016, 1720621. [Google Scholar] [CrossRef] [Green Version]
- Lill, C.M. Genetics of Parkinson’s disease. Mol. Cell Probes 2016, 30, 386–396. [Google Scholar] [CrossRef]
- Day, J.O.; Mullin, S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021, 12, 1006. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Higgins, L.S.; Golbe, L.; Johnson, W.G.; Ide, S.E.; Di Iorio, G.; Sanges, G.; Stenroos, E.S.; Pho, L.T.; Shaffer, A.A.; et al. A gene for Parkinson’s disease maps to 4q21-q23. Science 1996, 274, 1197–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athanassiadou, A.; Voutsinas, G.; Psiouri, L.; Leroy, E.; Polymeropoulos, M.H.; Ilias, A.; Maniatis, G.M.; Papapetropoulos, T. Genetic analysis of families with Parkinson disease that carry the Ala53Thr mutation in the gene encoding alpha-synuclein. Am. J. Hum. Genet. 1999, 65, 555–558. [Google Scholar] [CrossRef] [Green Version]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Rriess, O. Ala30Pro mutation in the gene encoding a-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of a-syunuclein, causes Parkinson and Lewy bodies dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Nuytemans, K.; Theuns, J.; Cruts, M.; Van Broeckhoven, C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: A mutation update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef] [Green Version]
- Strauss, K.M.; Martins, L.M.; Plun-Favreau, H.; Marx, F.P.; Kautzmann, S.; Berg, D.; Gasser, T.; Wszolek, Z.; Müller, T.; Borneman, A.; et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum. Mol. Gen. 2005, 14, 2099–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilarino-Guell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lücking, C.B.; Dürr, A.; Bonifati, V.; Vaughan, J.; De Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denèfle, P.; Wood, N.W.; et al. Association between early-onset Parkinson’s disease and mutations in the Parkin gene. New Eng. J. Med. 2000, 342, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Lohmann-Hedrich, K. Impact of recent genetic findings in Parkinson’s disease. Curr. Opin. Neurol. 2007, 20, 453–464. [Google Scholar] [CrossRef]
- Corti, O.; Lesage, S.; Brice, A. What Genetics Tells us About the Causes and Mechanisms of Parkinson’s Disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef]
- Rogaeva, E.; Johnson, J.; Lang, A.E.; Gulick, C.; Gwinn-Hardy, K.; Kawarai, T.; Sato, C.; Morgan, A.; Werner, J.; Nussbaum, R.; et al. Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch. Neurol. 2004, 61, 1898–1904. [Google Scholar] [CrossRef]
- Bonifati, V.; Rohe, C.F.; Breedveld, G.J.; Fabrizio, E.; De Mari, M.; Tassorelli, C.; Tavella, A.; Marconi, R.; Nicholl, D.J.; Chien, H.F.; et al. Early-onset parkinsonism associated with PINK1 mutations: Frequency, genotypes, and phenotypes. Neurology 2005, 65, 87–95. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Bonifati, V.; Rizzu, P.; Squitieri, F.; Krieger, E.; Vanacore, N.; Swieten, J.C.v.; Brice, A.; Duijn, C.M.v.; Oostra, B.A.; Meco, G.; et al. DJ-1 (PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol. Sci. 2003, 24, 159–160. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, A.; Heimbach, A.; Grundemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef]
- Omura, T.; Kaneko, M.; Okuma, Y.; Matsubara, K.; Nomura, Y. Endoplasmic reticulum stress and Parkinson’s disease: The role of HRD1 in averting apoptosis in neurodegenerative disease. Oxid. Med. Cell. Longev. 2013, 2013, 239854. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.A.D.; Manaa, W.E.; Duplan, E.; Checler, F. The Endoplasmic Reticulum Stress/Unfolded Protein Response and Their Contributions to Parkinson’s Disease Physiopathology. Cells 2020, 9, 2495. [Google Scholar] [CrossRef] [PubMed]
- Dernie, F. Mitophagy in Parkinson’s disease: From pathogenesis to treatment target. Neurochem. Int. 2020, 138, 104756. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch-Day, M.A.; Mao, K.; Wang, K.; Zhao, M.; Klionsky, D.J. The role of autophagy in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef]
- Cerri, S.; Blandini, F. Role of Autophagy in Parkinson’s Disease. Curr. Med. Chem. 2019, 26, 3702–3718. [Google Scholar] [CrossRef]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [Green Version]
- Dehay, B.; Martinez-Vicente, M.; Caldwell, G.A.; Caldwell, K.A.; Yue, Z.; Cookson, M.R.; Klein, C.; Vila, M.; Bezard, E. Lysosomal impairment in Parkinson’s disease. Mov. Disord. 2013, 28, 725–732. [Google Scholar] [CrossRef]
- Nguyen, M.; Wong, Y.C.; Ysselstein, D.; Severino, A.; Krainc, D. Synaptic, Mitochondrial, and Lysosomal Dysfunction in Parkinson’s Disease. Trends Neurosci. 2019, 42, 140–149. [Google Scholar] [CrossRef]
- Klein, A.D.; Mazzulli, J.R. Is Parkinson’s disease a lysosomal disorder? Brain 2018, 141, 2255–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves Da Costa, C.; Paitel, E.; Vincent, B.; Checler, F. Alpha-synuclein lowers p53-dependent apoptotic response of neuronal cells. Abolishment by 6-hydroxydopamine and implication for Parkinson’s disease. J. Biol. Chem. 2002, 277, 50980–50984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venderova, K.; Park, D.S. Programmed cell death in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009365. [Google Scholar] [CrossRef]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef]
- Checler, F.; Alves da Costa, C.; Ancolio, K.; Lopez-Perez, E.; Marambaud, P. Role of the Proteasome in Alzheimer’s disease. Biochim. Biophys. Acta 2000, 1502, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Bonet-Costa, V.; Pomatto, L.C.; Davies, K.J. The Proteasome and Oxidative Stress in Alzheimer’s Disease. Antioxid. Redox Signal. 2016, 25, 886–901. [Google Scholar] [CrossRef] [Green Version]
- Perry, G.; Friedman, R.; Shaw, G.; Chau, V. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer’s disease brains. Proc. Natl. Acad. Sci. USA 1987, 84, 3033–3036. [Google Scholar] [CrossRef] [Green Version]
- Almeida, C.G.; Takahashi, R.H.; Gouras, G.K. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J. Neurosci. 2006, 26, 4277–4288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregori, L.; Hainfeld, J.F.; Simon, M.N.; Goldgaber, D. Binding of amyloid b protein to the 20S proteasome. J. Biol. Chem. 1997, 272, 58–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez Salon, M.; Pasquini, L.; Moreno, B.; Pasquini, J.M.; Soto, E. Relationship between b-amyloid degradation and the 26S proteasome in neural cells. Exp. Neurol. 2003, 180, 131–143. [Google Scholar] [CrossRef]
- McNaught, K.S.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 2003, 179, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Bi, M.; Du, X.; Jiao, Q.; Chen, X.; Jiang, H. Expanding the role of proteasome homeostasis in Parkinson’s disease: Beyond protein breakdown. Cell Death Dis. 2021, 12, 154. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.A.; Leurgans, S. Is there a link between cancer and Alzheimer disease? Neurology 2010, 74, 100–101. [Google Scholar] [CrossRef] [PubMed]
- Alves da Costa, C.; Dunys, J.; Brau, F.; Wilk, S.; Cappai, R.; Checler, F. 6-Hydroxydopamine but not 1-methyl-4-phenylpyridinium abolishes alpha-synuclein anti-apoptotic phenotype by inhibiting its proteasomal degradation and by promoting its aggregation. J. Biol. Chem. 2006, 281, 9824–9831. [Google Scholar] [CrossRef] [Green Version]
- Shimura, H.; Hattori, N.; Kubo, S.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Kishi, C.; Ishihara, N.; Mizushima, N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 2011, 286, 19630–19640. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Li, S.; Wu, H.; Gao, R.; Rao, G.; Wang, D.; Chen, Z.; Ma, B.; Wang, H.; Sui, N.; et al. Parkin promotes proteasomal degradation of p62: Implication of selective vulnerability of neuronal cells in the pathogenesis of Parkinson’s disease. Protein Cell 2016, 7, 114–129. [Google Scholar] [CrossRef] [Green Version]
- Betteridge, D.J. What is oxidative stress? Metab. Clin. Exp. 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Markesbery, W.R. The role of oxidative stress in Alzheimer disease. Arch. Neurol. 1999, 56, 1449–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer disease. Cell Adhes. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksenov, M.Y.; Tucker, H.M.; Nair, P.; Aksenova, M.V.; Butterfield, D.A.; Estus, S.; Markesbery, W.R. The expression of key oxidative stress-handling genes in different brain regions in Alzheimer’s disease. J. Mol. Neurosci. 1998, 11, 151–164. [Google Scholar] [CrossRef]
- Youssef, P.; Chami, B.; Lim, J.; Middleton, T.; Sutherland, G.T.; Witting, P.K. Evidence supporting oxidative stress in a moderately affected area of the brain in Alzheimer’s disease. Sci. Rep. 2018, 8, 11553. [Google Scholar] [CrossRef] [Green Version]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinson’s Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Li, X.; Li, X.; Liu, Q.; Cheng, Y. Oxidative Stress in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Mol. Neurosci. 2018, 11, 236. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Davis, R.J. Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol. 2016, 8, a006072. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Rapoport, T.A. Mechanistic insights into ER-associated protein degradation. Curr. Opin. Cell Biol. 2018, 53, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frakes, A.E.; Dillin, A. The UPR(ER): Sensor and Coordinator of Organismal Homeostasis. Mol. Cell 2017, 66, 761–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Kurtishi, A.; Rosen, B.; Patil, K.S.; Alves, G.W.; Moller, S.G. Cellular Proteostasis in Neurodegeneration. Mol. Neurobiol. 2019, 56, 3676–3689. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef]
- Hoozemans, J.J.; Veerhuis, R.; Van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef]
- Barbero-Camps, E.; Fernandez, A.; Baulies, A.; Martinez, L.; Fernandez-Checa, J.C.; Colell, A. Endoplasmic reticulum stress mediates amyloid beta neurotoxicity via mitochondrial cholesterol trafficking. Am. J. Pathol. 2014, 184, 2066–2081. [Google Scholar] [CrossRef]
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of eIF2alpha kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 2013, 16, 1299–1305. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, S.; Saido, T.C. Critical review: Involvement of endoplasmic reticulum stress in the aetiology of Alzheimer’s disease. Open Biol. 2018, 8, 180024. [Google Scholar] [CrossRef] [PubMed]
- Goswami, P.; Afjal, M.A.; Akhter, J.; Mangla, A.; Khan, J.; Parvez, S.; Raisuddin, S. Involvement of endoplasmic reticulum stress in amyloid beta (1-42)-induced Alzheimer’s like neuropathological process in rat brain. Brain Res. Bull. 2020, 165, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Nishitsuji, K.; Tomiyama, T.; Ishibashi, K.; Ito, K.; Teraoka, R.; Lambert, M.P.; Klein, W.L.; Mori, H. The E693Delta mutation in amyloid precursor protein increases intracellular accumulation of amyloid beta oligomers and causes endoplasmic reticulum stress-induced apoptosis in cultured cells. Am. J. Pathol. 2009, 174, 957–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberdi, E.; Wyssenbach, A.; Alberdi, M.; Sanchez-Gomez, M.V.; Cavaliere, F.; Rodriguez, J.J.; Verkhratsky, A.; Matute, C. Ca(2+) -dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid beta-treated astrocytes and in a model of Alzheimer’s disease. Aging Cell 2013, 12, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Castillo-Carranza, D.L.; Zhang, Y.; Guerrero-Munoz, M.J.; Kayed, R.; Rincon-Limas, D.E.; Fernandez-Funez, P. Differential activation of the ER stress factor XBP1 by oligomeric assemblies. Neurochem. Res. 2012, 37, 1707–1717. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Abisambra, J.F.; Jinwal, U.K.; Blair, L.J.; O’Leary, J.C., 3rd; Li, Q.; Brady, S.; Wang, L.; Guidi, C.E.; Zhang, B.; Nordhues, B.A.; et al. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 2013, 33, 9498–9507. [Google Scholar] [CrossRef] [Green Version]
- Mou, Z.; Yuan, Y.H.; Zhang, Z.; Song, L.K.; Chen, N.H. Endoplasmic reticulum stress, an important factor in the development of Parkinson’s disease. Toxicol Lett 2020, 324, 20–29. [Google Scholar] [CrossRef]
- Lindholm, D.; Wootz, H.; Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006, 13, 385–392. [Google Scholar] [CrossRef]
- Kovaleva, V.; Saarma, M. Endoplasmic Reticulum Stress Regulators: New Drug Targets for Parkinson’s Disease. J. Parkinson’s Dis. 2021, 11, S219–S228. [Google Scholar] [CrossRef]
- Bellucci, A.; Navarria, L.; Zaltieri, M.; Falarti, E.; Bodei, S.; Sigala, S.; Battistin, L.; Spillantini, M.; Missale, C.; Spano, P. Induction of the unfolded protein response by alpha-synuclein in experimental models of Parkinson’s disease. J. Neurochem. 2011, 116, 588–605. [Google Scholar] [CrossRef] [PubMed]
- Sugeno, N.; Takeda, A.; Hasegawa, T.; Kobayashi, M.; Kikuchi, A.; Mori, F.; Wakabayashi, K.; Itoyama, Y. Serine 129 Phosphorylation of {alpha}-Synuclein Induces Unfolded Protein Response-mediated Cell Death. J. Biol. Chem. 2008, 283, 23179–23188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbatyuk, M.S.; Shabashvili, A.; Chen, W.; Meyers, C.; Sullivan, L.F.; Salganik, M.; Lin, J.H.; Lewin, A.S.; Muzyczka, N.; Gorbatyuk, O.S. Glucose regulated protein 78 diminishes alpha-synuclein neurotoxicity in a rat model of Parkinson disease. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Bouman, L.; Schlierf, A.; Lutz, A.K.; Shan, J.; Deinlein, A.; Kast, J.; Galehdar, Z.; Palmisano, V.; Patenge, N.; Berg, D.; et al. Parkin is transcriptionally regulated by ATF4: Evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 2011, 18, 769–782. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Soda, M.; Takahashi, R. Parkin suppresses unfolded protein stress-induced cell deth through its E3 ubiquitin-protein ligase activity. J. Biol. Chem. 2002, 275, 35661–35664. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Soda, M.; Inoue, H.; Hattori, N.; Mizuno, Y.; Takahashi, R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of parkin. Cell 2001, 105, 891–902. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- El Manaa, W.; Duplan, E.; Goiran, T.; Lauritzen, I.; Vaillant Beuchot, L.; Lacas-Gervais, S.; Morais, V.A.; You, H.; Qi, L.; Salazar, M.; et al. Transcription- and phosphorylation-dependent control of a functional interplay between XBP1s and PINK1 governs mitophagy and potentially impacts Parkinson disease pathophysiology. Autophagy 2021, 17, 4363–4385. [Google Scholar] [CrossRef]
- Yuan, Y.; Cao, P.; Smith, M.A.; Kramp, K.; Huang, Y.; Hisamoto, N.; Matsumoto, K.; Hatzoglou, M.; Jin, H.; Feng, Z. Dysregulated LRRK2 signaling in response to endoplasmic reticulum stress leads to dopaminergic neuron degeneration in C. elegans. PLoS ONE 2011, 6, e22354. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Weidling, I.W.; Swerdlow, R.H. Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp. Neurol. 2020, 330, 113321. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodriguez, M.; Bacskai, B.J. Mitochondria and Calcium in Alzheimer’s Disease: From Cell Signaling to Neuronal Cell Death. Trends Neurosci. 2021, 44, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Guo, X.Q.; Chu, J.F.; Zhang, X.; Yan, Z.R.; Li, Y.Z. Potential hippocampal genes and pathways involved in Alzheimer’s disease: A bioinformatic analysis. Genet. Mol. Res. 2015, 14, 7218–7232. [Google Scholar] [CrossRef]
- Mastroeni, D.; Khdour, O.M.; Delvaux, E.; Nolz, J.; Olsen, G.; Berchtold, N.; Cotman, C.; Hecht, S.M.; Coleman, P.D. Nuclear but not mitochondrial-encoded oxidative phosphorylation genes are altered in aging, mild cognitive impairment, and Alzheimer’s disease. Alzheimer’s Dement. 2017, 13, 510–519. [Google Scholar] [CrossRef] [Green Version]
- Area-Gomez, E.; Schon, E.A. Mitochondria-associated ER membranes and Alzheimer disease. Curr. Opin. Genet. Dev. 2016, 38, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Eysert, F.; Kinoshita, P.F.; Mary, A.; Vaillant-Beuchot, L.; Checler, F.; Chami, M. Molecular Dysfunctions of Mitochondria-Associated Membranes (MAMs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9521. [Google Scholar] [CrossRef]
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease-Cause or Consequence? Biology 2019, 8, 38. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Devel. Ther. 2017, 11, 797–810. [Google Scholar] [CrossRef] [Green Version]
- Seet, R.C.; Lee, C.Y.; Lim, E.C.; Tan, J.J.; Quek, A.M.; Chong, W.L.; Looi, W.F.; Huang, S.H.; Wang, H.; Chan, Y.H.; et al. Oxidative damage in Parkinson disease: Measurement using accurate biomarkers. Free Radic. Biol. Med. 2010, 48, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Yadava, N.; Nicholls, D.G. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J. Neurosci. 2007, 27, 7310–7317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasuhn, J.; Davis, R.L.; Kumar, K.R. Targeting Mitochondrial Impairment in Parkinson’s Disease: Challenges and Opportunities. Front. Cell Dev. Biol. 2020, 8, 615461. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Petrie, T.G.; Liu, Y.; Liu, J.; Fujioka, H.; Zhu, X. Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J. Neurochem. 2012, 121, 830–839. [Google Scholar] [CrossRef]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schule, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [Green Version]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [Green Version]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef] [Green Version]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradeepkiran, J.A.; Reddy, P.H. Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101191. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [Green Version]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, G.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Ab and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.E.; Kwon, D.; Shin, N.; Kong, D.; Kim, N.G.; Kim, H.Y.; Kim, M.J.; Choi, S.W.; Kang, K.S. Accumulation of APP-CTF induces mitophagy dysfunction in the iNSCs model of Alzheimer’s disease. Cell Death Discov. 2022, 8, 1. [Google Scholar] [CrossRef]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gegg, M.E.; Cooper, J.M.; Chau, K.Y.; Rojo, M.; Schapira, A.H.; Taanman, J.W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef] [PubMed]
- Glauser, L.; Sonnay, S.; Stafa, K.; Moore, D.J. Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J. Neurochem. 2011, 118, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Tan, J.; Wang, R.; Wan, H.; He, Y.; Yan, X.; Guo, J.; Gao, Q.; Li, J.; Shang, S.; et al. PINK1 phosphorylates Drp1(S616) to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 2020, 21, e48686. [Google Scholar] [CrossRef]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagda, R.K.; Cherra, S.J., 3rd; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandebring, A.; Dehvari, N.; Perez-Manso, M.; Thomas, K.J.; Karpilovski, E.; Cookson, M.R.; Cowburn, R.F.; Cedazo-Minguez, A. Parkin deficiency disrupts calcium homeostasis by modulating phospholipase C signalling. FEBS J. 2009, 276, 5041–5052. [Google Scholar] [CrossRef] [Green Version]
- Goiran, T.; Duplan, E.; Chami, M.; Bourgeois, A.; El Manaa, W.; Rouland, L.; Dunys, J.; Lauritzen, I.; You, H.; Stambolic, V.; et al. beta-Amyloid Precursor Protein Intracellular Domain Controls Mitochondrial Function by Modulating Phosphatase and Tensin Homolog-Induced Kinase 1 Transcription in Cells and in Alzheimer Mice Models. Biol. Psychiatry 2018, 83, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef]
- Erekat, N.S. Apoptosis and its Role in Parkinson’s Disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Exon Publications: Brisbane, Australia, 2018. [Google Scholar] [CrossRef] [Green Version]
- Szybinska, A.; Lesniak, W. P53 Dysfunction in Neurodegenerative Diseases-The Cause or Effect of Pathological Changes? Aging Dis. 2017, 8, 506–518. [Google Scholar] [CrossRef] [Green Version]
- Alves da Costa, C.; Checler, F. Apoptosis in Parkinson’s disease: Is p53 the missing link between genetic and sporadic Parkinsonism? Cell Signal 2011, 23, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.D. Activation of p53 signaling initiates apoptotic death in a cellular model of Parkinson’s disease. Apoptosis 2006, 11, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Kondo, T.; Mizuno, Y.; Nagatsu, T. p53 protein, interferon-gamma, and NF-kappaB levels are elevated in the parkinsonian brain. Neurosci. Lett. 2007, 414, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Komarov, P.G.; Komarova, E.A.; Kondratov, R.V.; Christov-Tselkov, K.; Coon, J.S.; Chernov, M.V.; Gudkov, A.V. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999, 285, 1733–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.C.; Ryu, E.J.; Park, C.H.; Malagelada, C.; Greene, L.A. Puma and p53 play required roles in death-evoked in a cellular model of Parkinson disease. Neurochem. Res. 2005, 30, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Alves da Costa, C.; Duplan, E.; Checler, F. α-synuclein and p53 functional interplay in physiopathological contexts. Oncotarget 2017, 8, 9001–9002. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Davis, B.; Chiang, Y.H.; Filichia, E.; Barnett, A.; Greig, N.H.; Hoffer, B.; Luo, Y. Dopaminergic neuron-specific deletion of p53 gene is neuroprotective in an experimental Parkinson’s disease model. J. Neurochem. 2016, 138, 746–757. [Google Scholar] [CrossRef] [Green Version]
- Alves da Costa, C.; Ancolio, K.; Checler, F. Wild-type but not Parkinson’s disease-related Ala53Thr-a-synuclein protect neuronal cells from apoptotic stimuli. J. Biol. Chem 2000, 275, 24065–24069. [Google Scholar] [CrossRef] [Green Version]
- da Costa, C.A.; Masliah, E.; Checler, F. Beta-synuclein displays an antiapoptotic p53-dependent phenotype and protects neurons from 6-hydroxydopamine-induced caspase 3 activation: Cross-talk with alpha-synuclein and implication for Parkinson’s disease. J. Biol. Chem. 2003, 278, 37330–37335. [Google Scholar] [CrossRef] [Green Version]
- Duplan, E.; Giordano, C.; Checler, F.; Alves da Costa, C. Direct alpha-synuclein promoter transactivation by the tumor suppressor p53. Mol. Neurodegener. 2016, 11, 13. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Lin, C.H.; Teng, S.C. Stress-induced p53 drives BAG5 cochaperone expression to control alpha-synuclein aggregation in Parkinson’s disease. Aging 2020, 12, 20702–20727. [Google Scholar] [CrossRef] [PubMed]
- da Costa, C.A.; Sunyach, C.; Giaime, E.; West, A.; Corti, O.; Brice, A.; Safe, S.; Abou-Sleiman, P.M.; Wood, N.W.; Takahashi, H.; et al. Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson’s disease. Nat. Cell Biol. 2009, 11, 1370–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunico, C.R.; Nakamura, T.; Rockenstein, E.; Mante, M.; Adame, A.; Chan, S.F.; Newmeyer, T.F.; Masliah, E.; Nakanishi, N.; Lipton, S.A. S-Nitrosylation of parkin as a novel regulator of p53-mediated neuronal cell death in sporadic Parkinson’s disease. Mol. Neurodegener. 2013, 8, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, D.; Gu, Z.; Nakamura, T.; Shi, Z.-Q.; Ma, Y.; Gaston, B.; Palmer, L.A.; Rockenstein, E.M.; Zhang, Z.; Masliah, E.; et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10810–10814. [Google Scholar] [CrossRef] [Green Version]
- Viotti, J.; Duplan, E.; Caillava, C.; Condat, J.; Goiran, T.; Giordano, C.; Marie, Y.; Idbaih, A.; Delattre, J.Y.; Honnorat, J.; et al. Glioma tumor grade correlates with parkin depletion in mutant p53-linked tumors and results from loss of function of p53 transcriptional activity. Oncogene 2014, 33, 1764–1775. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef] [Green Version]
- Ottolini, D.; Cali, T.; Negro, A.; Brini, M. The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum. Mol. Genet. 2013, 22, 2152–2168. [Google Scholar] [CrossRef]
- Duplan, E.; Giaime, E.; Viotti, J.; Sevalle, J.; Corti, O.; Brice, A.; Ariga, H.; Qi, L.; Checler, F.; da Costa, C.A. ER-stress-associated functional link between Parkin and DJ-1 via a transcriptional cascade involving the tumor suppressor p53 and the spliced X-box binding protein XBP-1. J. Cell Sci. 2013, 126, 2124–2133. [Google Scholar] [CrossRef] [Green Version]
- Goiran, T.; Duplan, E.; Rouland, L.; El Manaa, W.; Lauritzen, I.; Dunys, J.; You, H.; Checler, F.; Alves da Costa, C. Nuclear p53-mediated repression of autophagy involves PINK1 transcriptional down-regulation. Cell Death Differ. 2018, 25, 873–884. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.J. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell 2017, 68, 281–292.e5. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Raina, A.K.; Perry, G.; Smith, M.A. Apoptosis in Alzheimer disease: A mathematical improbability. Curr. Alzheimer Res. 2006, 3, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Obulesu, M.; Lakshmi, M.J. Apoptosis in Alzheimer’s disease: An understanding of the physiology, pathology and therapeutic avenues. Neurochem. Res. 2014, 39, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Abate, G.; Frisoni, G.B.; Bourdon, J.C.; Piccirella, S.; Memo, M.; Uberti, D. The pleiotropic role of p53 in functional/dysfunctional neurons: Focus on pathogenesis and diagnosis of Alzheimer’s disease. Alzheimer’s Res. Ther. 2020, 12, 160. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Shimohama, S.; Kamoshima, W.; Matsuoka, Y.; Nomura, Y.; Taniguchi, T. Changes of p53 in the brains patients with Alzheimer disease. Biochem. Biophys. Res. Comm. 1997, 232, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Lanni, C.; Racchi, M.; Mazzini, G.; Ranzenigo, A.; Polotti, R.; Sinforiani, E.; Olivari, L.; Barcikowska, M.; Styczynska, M.; Kuznicki, J.; et al. Conformationally altered p53: A novel Alzheimer’s disease marker? Mol. Psychiatry 2008, 13, 641–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Liu, S.; Tao, Y. Regulating tumor suppressor genes: Post-translational modifications. Signal Transduct. Target. Ther. 2020, 5, 90. [Google Scholar] [CrossRef] [PubMed]
- Ohyagi, Y.; Asahara, H.; Chui, D.H.; Tsuruta, Y.; Sakae, N.; Miyoshi, K.; Yamada, T.; Kikuchi, H.; Taniwaki, T.; Murai, H.; et al. Intracellular Abeta42 activates p53 promoter: A pathway to neurodegeneration in Alzheimer’s disease. FASEB J. 2005, 19, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Checler, F.; Dunys, J.; Pardossi-Piquard, R.; Alves da Costa, C. p53 Is Regulated by and Regulates Members of the gamma-Secretase Complex. Neurodegener. Dis. 2010, 7, 50–55. [Google Scholar] [CrossRef]
- Alves da Costa, C.; Sunyach, C.; Pardossi-Piquard, R.; Sevalle, J.; Vincent, B.; Boyer, N.; Kawarai, T.; Girardot, N.; St George-Hyslop, P.; Checler, F. Presenilin-dependent gamma-secretase-mediated control of p53-associated cell death in Alzheimer’s disease. J. Neurosci. 2006, 26, 6377–6385. [Google Scholar] [CrossRef] [Green Version]
- Alves da Costa, C.; Mattson, M.P.; Ancolio, K.; Checler, F. The C-terminal fragment of presenilin 2 triggers p53-mediated staurosporine-induced apoptosis, a function independent of the presenilinase-derived N-terminal counterpart. J. Biol. Chem. 2003, 278, 12064–12069. [Google Scholar] [CrossRef] [Green Version]
- Alves da Costa, C.; Paitel, E.; Mattson, M.P.; Amson, R.; Telerman, A.; Ancolio, K.; Checler, F. Wild-type and mutated presenilins 2 trigger p53-dependent apoptosis and down-regulate presenilin 1 expression in HEK293 human cells and in murine neurons. Proc. Natl. Acad. Sci. USA 2002, 99, 4043–4048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takasugi, N.; Tomita, T.; Hayashi, I.; Tsuruoka, M.; Niimura, M.; Takahashi, Y.; Thinakaran, G.; Iwatsubo, T. The role of presenilin cofactors in the g-secretase complex. Nature 2003, 422, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Edbauer, D.; Winkler, E.; Regula, J.T.; Pesold, B.; Steiner, H.; Haass, C. Reconstitution of g-secretase activity. Nat. Cell Biol. 2003, 5, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Dunys, J.; Kawarai, T.; Sevalle, J.; Dolcini, V.; St George-Hyslop, P.; Alves da Costa, C.; Checler, F. p53-dependent Aph-1 and Pen-2 anti-apoptotic phenotype requires the integrity of the gamma-secretase complex but is independent of its activity. J. Biol. Chem. 2007, 282, 10516–10525. [Google Scholar] [CrossRef] [Green Version]
- Pardossi-Piquard, R.; Dunys, J.; Giaime, E.; Guillot-Sestier, M.V.; St George-Hyslop, P.; Checler, F.; Alves da Costa, C. p53-dependent control of cell death by nicastrin: Lack of requirement for presenilin-dependent gamma-secretase complex. J. Neurochem. 2009, 109, 225–237. [Google Scholar] [CrossRef]
- Cuesta, A.; Zambrano, A.; Royo, M.; Pascual, A. The tumor suppressor p53 regulates the expression of the beta-amyloid precursor protein. Biochem. J. 2008, 418, 643–650. [Google Scholar] [CrossRef] [Green Version]
- Pardossi-Piquard, R.; Checler, F. The physiology of the beta-amyloid precursor protein intracellular domain AICD. J. Neurochem. 2012, 120 (Suppl. 1), 109–124. [Google Scholar] [CrossRef]
- Lu, D.C.; Rabizadeh, S.; Chandra, S.; Shayya, R.F.; Ellerby, L.M.; Ye, X.; Salvesen, G.S.; Koo, E.H.; Bredesen, D.E. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat. Med. 2000, 6, 397–404. [Google Scholar] [CrossRef]
- Ozaki, T.; Li, Y.; Kikuchi, H.; Tomita, T.; Iwatsubo, T.; Nakagawara, A. The intracellular domain of the amyloid precursor protein (AICD) enhances the p53-mediated apoptosis. Biochem. Biophys. Res. Commun. 2006, 351, 57–63. [Google Scholar] [CrossRef]
- Li, M.; Pehar, M.; Liu, Y.; Bhattacharyya, A.; Zhang, S.C.; O’Riordan, K.J.; Burger, C.; D’Adamio, L.; Puglielli, L. The amyloid precursor protein (APP) intracellular domain regulates translation of p44, a short isoform of p53, through an IRES-dependent mechanism. Neurobiol. Aging 2015, 36, 2725–2736. [Google Scholar] [CrossRef] [Green Version]
- Dumanchin-Njock, C.; Alves da Costa, C.A.; Mercken, L.; Pradier, L.; Checler, F. The caspase-derived C-terminal fragment of betaAPP induces caspase-independent toxicity and triggers selective increase of Abeta42 in mammalian cells. J. Neurochem. 2001, 78, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Shaked, G.M.; Bredesen, D.E.; Koo, E.H. Mechanism of cytotoxicity mediated by the C31 fragment of the Amyloid Precursor Protein. Biochem. Biophys. Res. Commun. 2009, 388, 450–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandelwal, P.J.; Moussa, C.E. The Relationship between Parkin and Protein Aggregation in Neurodegenerative Diseases. Front. Psychiatry 2010, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.W.; Hang, L.; Yao, T.P.; Lim, K.L. Parkin Regulation and Neurodegenerative Disorders. Front. Aging Neurosci. 2015, 7, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witte, M.E.; Bol, J.G.; Gerritsen, W.H.; van der Valk, P.; Drukarch, B.; van Horssen, J.; Wilhelmus, M.M. Parkinson’s disease-associated parkin colocalizes with Alzheimer’s disease and multiple sclerosis brain lesions. Neurobiol. Dis. 2009, 36, 445–452. [Google Scholar] [CrossRef]
- Kumar, D.; Kumar, P. Abeta, Tau, and alpha-Synuclein aggregation and integrated role of PARK2 in the regulation and clearance of toxic peptides. Neuropeptides 2019, 78, 101971. [Google Scholar] [CrossRef]
- Goudarzi, S.; Hosseini, A.; Abdollahi, M.; Haghi-Aminjan, H. Insights Into Parkin-Mediated Mitophagy in Alzheimer’s Disease: A Systematic Review. Front. Aging Neurosci. 2021, 13, 674071. [Google Scholar] [CrossRef]
- Ye, X.; Sun, X.; Starovoytov, V.; Cai, Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 2015, 24, 2938–2951. [Google Scholar] [CrossRef]
- Wang, H.; Fu, J.; Xu, X.; Yang, Z.; Zhang, T. Rapamycin Activates Mitophagy and Alleviates Cognitive and Synaptic Plasticity Deficits in a Mouse Model of Alzheimer’s Disease. J. Gerontol. Ser. A 2021, 76, 1707–1713. [Google Scholar] [CrossRef]
- Martin-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; Garcia-Escudero, V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhang, T.; Ge, X.; Chen, J.; Zhao, Y.; Fu, J. Parkin overexpression attenuates Abeta-induced mitochondrial dysfunction in HEK293 cells by restoring impaired mitophagy. Life Sci. 2020, 244, 117322. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, P.J.; Herman, A.M.; Hoe, H.S.; Rebeck, G.W.; Moussa, C.E. Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum. Mol. Genet. 2011, 20, 2091–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Algarzae, N.; Hebron, M.; Miessau, M.; Moussa, C.E. Parkin prevents cortical atrophy and Abeta-induced alterations of brain metabolism: (1)(3)C NMR and magnetic resonance imaging studies in AD models. Neuroscience 2012, 225, 22–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, K.M.; Moussa, C.E.; Lee, H.K.; Kumar, P.; Kitada, T.; Qin, G.; Fu, Q.; Querfurth, H.W. Parkin reverses intracellular beta-amyloid accumulation and its negative effects on proteasome function. J. Neurosci. Res. 2010, 88, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, X.; Liu, J.; Zhu, G.; Zhuang, Y.; Suo, H.; Wang, P.; Huang, D.; Xu, J.; Huang, Y.; Yu, M.; et al. Parkin overexpression ameliorates hippocampal long-term potentiation and beta-amyloid load in an Alzheimer’s disease mouse model. Hum. Mol. Genet. 2014, 23, 1056–1072. [Google Scholar] [CrossRef] [Green Version]
- Rosen, K.M.; Veereshwarayya, V.; Moussa, C.E.; Fu, Q.; Goldberg, M.S.; Schlossmacher, M.G.; Shen, J.; Querfurth, H.W. Parkin protects against mitochondrial toxins and beta-amyloid accumulation in skeletal muscle cells. J. Biol. Chem. 2006, 281, 12809–12816. [Google Scholar] [CrossRef] [Green Version]
- Kam, M.K.; Lee, D.G.; Kim, B.; Huh, J.W.; Lee, H.J.; Park, Y.H.; Lee, D.S. Amyloid-beta oligomers induce Parkin-mediated mitophagy by reducing Miro1. Biochem J 2020, 477, 4581–4597. [Google Scholar] [CrossRef]
- Solano, R.M.; Casarejos, M.J.; Gomez, A.; Perucho, J.; de Yebenes, J.G.; Mena, M.A. Parkin null cortical neuronal/glial cultures are resistant to amyloid-beta1-42 toxicity: A role for autophagy? J. Alzheimer’s Dis. 2012, 32, 57–76. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Wang, N.; Kang, J.; Fang, Y. beta-Asarone improves learning and memory in Abeta1-42-induced Alzheimer’s disease rats by regulating PINK1-Parkin-mediated mitophagy. Metab. Brain Dis. 2020, 35, 1109–1117. [Google Scholar] [CrossRef]
- Wang, N.; Wang, H.; Li, L.; Li, Y.; Zhang, R. beta-Asarone Inhibits Amyloid-beta by Promoting Autophagy in a Cell Model of Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 1529. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, H.; Huang, P.; Li, S.; Li, B.; Huo, L.; Zhong, J.; Pan, Z.; Li, Y.; Xia, X. Panax notoginseng saponins protect PC12 cells against Abeta induced injury via promoting parkin-mediated mitophagy. J. Ethnopharmacol. 2022, 285, 114859. [Google Scholar] [CrossRef] [PubMed]
- Lonskaya, I.; Hebron, M.L.; Desforges, N.M.; Schachter, J.B.; Moussa, C.E. Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J. Mol. Med. 2014, 92, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, X.C.; Wang, Z.H.; Luo, Y.; Zhang, X.; Liu, X.P.; Feng, Q.; Wang, Q.; Yue, Z.; Chen, Z.; et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Gotz, J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019, 38, e99360. [Google Scholar] [CrossRef]
- Moussa, C.E. Parkin attenuates wild-type tau modification in the presence of beta-amyloid and alpha-synuclein. J. Mol. Neurosci. 2009, 37, 25–36. [Google Scholar] [CrossRef]
- Baxter, L.L.; Moran, T.H.; Richtsmeier, J.T.; Troncoso, J.; Reeves, R.H. Discovery and genetic localization of Down syndrome cerebellar phenotypes using the Ts65Dn mouse. Hum. Mol. Genet. 2000, 9, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Henrique, A.M.; Gianetti, N.G.; Ferrari, M.F.R. Parkin is downregulated among autophagy-related proteins prior to hyperphosphorylation of Tau in TS65DN mice. Biochem. Biophys. Res. Commun. 2021, 561, 59–64. [Google Scholar] [CrossRef]
- Guerrero, R.; Navarro, P.; Gallego, E.; Avila, J.; de Yebenes, J.G.; Sanchez, M.P. Park2-null/tau transgenic mice reveal a functional relationship between parkin and tau. J. Alzheimer’s Dis. 2008, 13, 161–172. [Google Scholar] [CrossRef]
- Menendez, J.; Rodriguez-Navarro, J.A.; Solano, R.M.; Casarejos, M.J.; Rodal, I.; Guerrero, R.; Sanchez, M.P.; Avila, J.; Mena, M.A.; de Yebenes, J.G. Suppression of Parkin enhances nigrostriatal and motor neuron lesion in mice over-expressing human-mutated tau protein. Hum. Mol. Genet. 2006, 15, 2045–2058. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Navarro, J.A.; Gomez, A.; Rodal, I.; Perucho, J.; Martinez, A.; Furio, V.; Ampuero, I.; Casarejos, M.J.; Solano, R.M.; de Yebenes, J.G.; et al. Parkin deletion causes cerebral and systemic amyloidosis in human mutated tau over-expressing mice. Hum. Mol. Genet. 2008, 17, 3128–3143. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, R.; Navarro, P.; Gallego, E.; Garcia-Cabrero, A.M.; Avila, J.; Sanchez, M.P. Hyperphosphorylated tau aggregates in the cortex and hippocampus of transgenic mice with mutant human FTDP-17 Tau and lacking the PARK2 gene. Acta Neuropathol. 2009, 117, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Vargas, D.M.; De Bastiani, M.A.; Zimmer, E.R.; Klamt, F. Alzheimer’s disease master regulators analysis: Search for potential molecular targets and drug repositioning candidates. Alzheimer’s Res. Ther. 2018, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Duplan, E.; Sevalle, J.; Viotti, J.; Goiran, T.; Bauer, C.; Renbaum, P.; Levy-Lahad, E.; Gautier, C.A.; Corti, O.; Leroudier, N.; et al. Parkin differently regulates presenilin-1 and presenilin-2 functions by direct control of their promoter transcription. J. Mol. Cell Biol. 2013, 5, 132–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauritzen, I.; Pardossi-Piquard, R.; Bauer, C.; Brigham, E.; Abraham, J.D.; Ranaldi, S.; Fraser, P.; St-George-Hyslop, P.; Le Thuc, O.; Espin, V.; et al. The beta-secretase-derived C-terminal fragment of betaAPP, C99, but not Abeta, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J. Neurosci. 2012, 32, 16243–16255. [Google Scholar] [CrossRef]
- Pulina, M.V.; Hopkins, M.; Haroutunian, V.; Greengard, P.; Bustos, V. C99 selectively accumulates in vulnerable neurons in Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 273–282. [Google Scholar] [CrossRef]
- Lauritzen, I.; Becot, A.; Bourgeois, A.; Pardossi-Piquard, R.; Biferi, M.G.; Barkats, M.; Checler, F. Targeting gamma-secretase triggers the selective enrichment of oligomeric APP-CTFs in brain extracellular vesicles from Alzheimer cell and mouse models. Transl. Neurodegener. 2019, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Rouland, L.; Duplan, E.; Ramos Dos Santos, L.; Bernardin, A.; Katula, K.S.; Manfioletti, G.; Idbaih, A.; Checler, F.; Alves da Costa, C. Therapeutic potential of parkin as a tumor suppressor via transcriptional control of cyclins in glioblastoma cell and animal models. Theranostics 2021, 11, 10047–10063. [Google Scholar] [CrossRef]
- Pardossi-Piquard, R.; Petit, A.; Kawarai, T.; Sunyach, C.; Alves da Costa, C.; Vincent, B.; Ring, S.; D’Adamio, L.; Shen, J.; Muller, U.; et al. Presenilin-dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of betaAPP and APLP. Neuron 2005, 46, 541–554. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, K.; Vogt, D.L.; Liang, M.; Shen, Y.; Lamb, B.T.; Pimplikar, S.W. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc. Natl. Acad. Sci. USA 2009, 106, 18367–18372. [Google Scholar] [CrossRef] [Green Version]
- Cisse, M.; Duplan, E.; Checler, F. The transcription factor XBP1 in memory and cognition: Implications in Alzheimer disease. Mol. Med. 2017, 22, 905–917. [Google Scholar] [CrossRef]
- Cisse, M.; Duplan, E.; Lorivel, T.; Dunys, J.; Bauer, C.; Meckler, X.; Gerakis, Y.; Lauritzen, I.; Checler, F. The transcription factor XBP1s restores hippocampal synaptic plasticity and memory by control of the Kalirin-7 pathway in Alzheimer model. Mol. Psychiatry 2017, 22, 1562–1575. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Checler, F.; Alves da Costa, C. Parkin as a Molecular Bridge Linking Alzheimer’s and Parkinson’s Diseases? Biomolecules 2022, 12, 559. https://doi.org/10.3390/biom12040559
Checler F, Alves da Costa C. Parkin as a Molecular Bridge Linking Alzheimer’s and Parkinson’s Diseases? Biomolecules. 2022; 12(4):559. https://doi.org/10.3390/biom12040559
Chicago/Turabian StyleChecler, Frédéric, and Cristine Alves da Costa. 2022. "Parkin as a Molecular Bridge Linking Alzheimer’s and Parkinson’s Diseases?" Biomolecules 12, no. 4: 559. https://doi.org/10.3390/biom12040559
APA StyleChecler, F., & Alves da Costa, C. (2022). Parkin as a Molecular Bridge Linking Alzheimer’s and Parkinson’s Diseases? Biomolecules, 12(4), 559. https://doi.org/10.3390/biom12040559