
Deciphering Cardiac Biology and Disease by Single-Cell Transcriptomic Profiling


Abstract
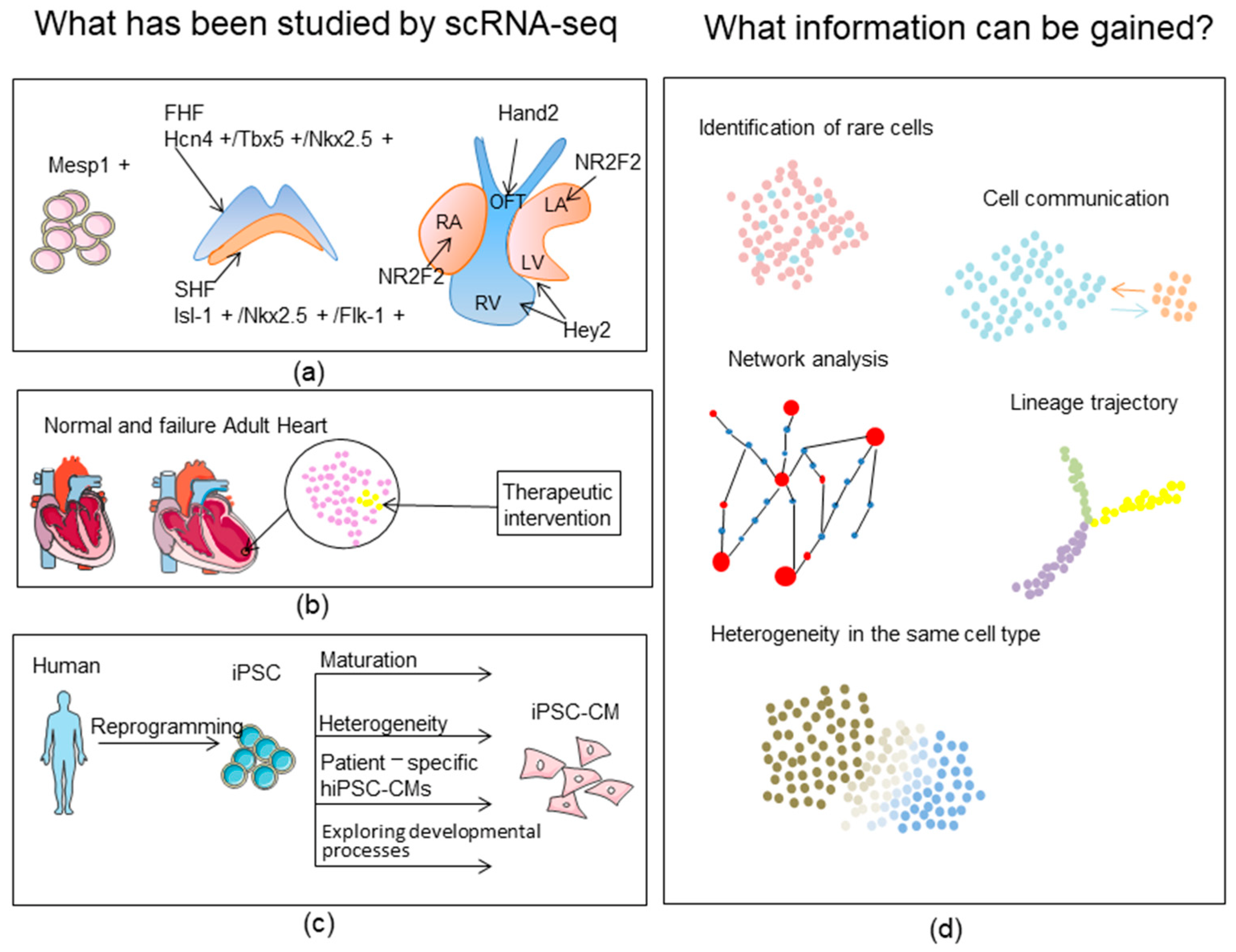
:1. Introduction
2. Single-Cell Transcriptomic Characterization of Embryonic Heart Development
2.1. ScRNA-seq Analysis of Cardiac Progenitor Differentiation
2.1.1. Mesp1
2.1.2. Isl1
2.1.3. Nkx2.5
2.1.4. Hand2
2.2. Cellular Heterogeneity in Cardiac Development
3. Cell Heterogeneity and Cell Crosstalk in Adult Heart and Cardiovascular Diseases
3.1. Cardiomyocytes
3.2. Fibroblast
3.3. Endothelial Cells
3.4. Macrophages
3.5. Smooth Muscle Cells (SMCs) and Pericytes (PCs)
3.6. Small Cell Populations
4. Modeling Human Cardiac Development and Disease with Pluripotent Stem-Cell Derived CMs
4.1. Maturation
4.2. Heterogeneity in Differentiation
4.3. Patient-Specific hiPSC-CMs
4.4. Exploring Developmental Processes
4.5. Improve Direct Reprogramming Efficiency
5. Perspectives and Significance
Author Contributions
Funding
Conflicts of Interest
References
- Kikuchi, K.; Holdway, J.E.; Werdich, A.; Anderson, R.A.; Fang, Y.; Egnaczyk, G.F.; Evans, T.; Macrae, C.A.; Stainier, D.Y.R.; Poss, K.D. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature 2010, 464, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cheng, J.; Joyner, R.W.; Wagner, M.B.; Hill, J.A. Remodeling of early-phase repolarization: A mechanism of abnormal impulse conduction in heart failure. Circulation 2006, 113, 1849–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattila, S.; Ingel, N.N., Jr.; Daughters, G.T., 2nd; Adler, S.C.; Wexler, L.; Dong, E., Jr. The effects of atrial pacing on the synergy and hemodynamics of the orthotopically transplanted canine heart. Circulation 1973, 48, 386–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emanuel, K.; Mackiewicz, U.; Lewartowski, B. On the source of Ca(2+) activating the tonic component of contraction of myocytes of guinea pig heart. Cardiovasc. Res. 2001, 52, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Doll, S.; Dreßen, M.; Geyer, P.E.; Itzhak, D.N.; Braun, C.; Doppler, S.A.; Meier, F.; Deutsch, M.-A.; Lahm, H.; Lange, R.; et al. Region and cell-type resolved quantitative proteomic map of the human heart. Nat. Commun. 2017, 8, 1469. [Google Scholar] [CrossRef]
- Li, J.; Salvador, A.M.; Li, G.; Valkov, N.; Ziegler, O.; Yeri, A.; Xiao, C.Y.; Meechoovet, B.; Alsop, E.; Rodosthenous, R.S.; et al. Mir-30d Regulates Cardiac Remodeling by Intracellular and Paracrine Signaling. Circ. Res. 2021, 128, e1–e23. [Google Scholar]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Fürth, D.; Qian, X.; Wärdell, E.; Custodio, J.; Reimegård, J.; Salmén, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660.e19. [Google Scholar] [CrossRef]
- Wang, L.; Yu, P.; Zhou, B.; Song, J.; Li, Z.; Zhang, M.; Guo, G.; Wang, Y.; Chen, X.; Han, L.; et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat. Cell Biol. 2020, 22, 108–119. [Google Scholar] [CrossRef]
- Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Wirka, R.C.; Pjanic, M.; Quertermous, T. Advances in Transcriptomics: Investigating Cardiovascular Disease at Unprecedented Resolution. Circ. Res. 2018, 122, 1200–1220. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Xu, A.; Sim, S.; Priest, J.R.; Tian, X.; Khan, T.; Quertermous, T.; Zhou, B.; Tsao, P.S.; Quake, S.R.; et al. Transcriptomic Profiling Maps Anatomically Patterned Subpopulations among Single Embryonic Cardiac Cells. Dev. Cell 2016, 39, 491–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, N.R.; Chaffin, M.; Fleming, S.J.; Hall, A.W.; Parsons, V.A.; Bedi, K.; Akkad, A.-D.; Herndon, C.N.; Arduini, A.; Papangeli, I.; et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation 2020, 142, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zheng, Y.; Liu, X.; Yan, L.; Fan, X.; Yong, J.; Hu, Y.; Dong, J.; Li, Q.; Wu, X.; et al. Single-Cell Transcriptome Analysis Maps the Developmental Track of the Human Heart. Cell Rep. 2019, 26, 1934–1950.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Z.; Yu, P.; Li, D.; Li, Z.; Liao, Y.; Wang, Y.; Bingying, Z.; Wang, L. Single-Cell Reconstruction of Progression Trajectory Reveals Intervention Principles in Pathological Cardiac Hypertrophy. Circulation 2020, 141, 1704–1719. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Wang, X.; Guo, G.; Wang, L.; Chen, S.; Yin, P.; Chen, K.; Chen, L.; Zhang, Z.; Chen, X.; et al. Resolving the intertwining of inflammation and fibrosis in human heart failure at single-cell level. Basic Res. Cardiol. 2021, 116, 55. [Google Scholar] [CrossRef]
- De Soysa, T.Y.; Ranade, S.S.; Okawa, S.; Ravichandran, S.; Huang, Y.; Salunga, H.T.; Schricker, A.; del Sol, A.; Gifford, C.A.; Srivastava, D. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature 2019, 572, 120–124. [Google Scholar] [CrossRef]
- Friedman, C.E.; Nguyen, Q.; Lukowski, S.W.; Helfer, A.H.S.; Chiu, J.; Miklas, S.; Levy, S.; Suo, J.-D.J.; Han, P.; Osteil, G.; et al. Single-Cell Transcriptomic Analysis of Cardiac Differentiation from Human PSCs Reveals HOPX-Dependent Cardiomyocyte Maturation. Cell Stem Cell 2018, 23, 586–598.e8. [Google Scholar] [CrossRef] [Green Version]
- Paik, D.T.; Cho, S.; Tian, L.; Chang, H.Y.; Wu, J.C. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat. Rev. Cardiol. 2020, 17, 457–473. [Google Scholar] [CrossRef]
- Lescroart, F.; Wang, X.; Lin, X.; Swedlund, B.; Gargouri, S.; Sànchez-Dànes, A.; Moignard, V.; Dubois, C.; Paulissen, C.; Kinston, S.; et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 2018, 359, 1177–1181. [Google Scholar] [CrossRef] [Green Version]
- Tyser, R.C.V.; Ibarra-Soria, X.; Mcdole, K.; Arcot Jayaram, S.; Godwin, J.; van den Brand Teun, A.H.; Miranda Antonio, M.A.; Scialdone, A.; Keller Philipp, J.; Marioni John, C.; et al. Characterization of a common progenitor pool of the epicardium and myocardium. Science 2021, 371, eabb2986. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Preussner, J.; Chen, X.; Yekelchyk, M.; Kuenne, C.; Looso, M.; Zhou, Y.; Teichmann, S.; Braun., T. Single cell RNA-seq and ATAC-seq analysis of cardiac progenitor cell transition states and lineage settlement. Nat. Commun. 2018, 9, 4877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, H.; Luo, Y.; Yue, Y.; Zhang, J.; Ai, S.; Li, X.; Wang, X.; Zhang, Y.-L.; Wei, Y.; Li, H.-H.; et al. Single-Cell Transcriptomics Reveals Chemotaxis-Mediated Intraorgan Crosstalk During Cardiogenesis. Circ. Res. 2019, 125, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Goodyer, W.R.; Beyersdorf, B.M.; Paik, D.T.; Tian, L.; Li, G.; Buikema, J.W.; Chirikian, O.; Choi, S.; Venkatraman, S.; Adams, E.L.; et al. Transcriptomic Profiling of the Developing Cardiac Conduction System at Single-Cell Resolution. Circ. Res. 2019, 125, 379–397. [Google Scholar] [CrossRef]
- Delaughter, D.M.; Bick, A.G.; Wakimoto, H.; McKean, D.; Gorham, J.M.; Kathiriya, I.S.; Hinson, J.T.; Homsy, J.; Gray, J.; Pu, W.; et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell. 2016, 39, 480–490. [Google Scholar] [CrossRef] [Green Version]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef]
- Yekelchyk, M.; Guenther, S.; Preussner, J.; Braun, T. Mono- and multi-nucleated ventricular cardiomyocytes constitute a transcriptionally homogenous cell population. Basic Res. Cardiol. 2019, 114, 36. [Google Scholar] [CrossRef] [Green Version]
- Nomura, S.; Satoh, M.; Fujita, T.; Higo, T.; Sumida, T.; Ko, T.; Yamaguchi, T.; Tobita, T.; Naito, A.T.; Ito, M.; et al. Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat. Commun. 2018, 9, 4435. [Google Scholar] [CrossRef]
- Gladka, M.M.; Molenaar, B.; de Ruiter, H.; van der Elst, S.; Tsui, H.; Versteeg, D.; Lacraz, G.P.A.; Huibers, M.M.H.; van Oudenaarden, A.; van Rooij, E. Single-Cell Sequencing of the Healthy and Diseased Heart Reveals Cytoskeleton-Associated Protein 4 as a New Modulator of Fibroblasts Activation. Circulation 2018, 138, 166–180. [Google Scholar] [CrossRef]
- Skelly, D.A.; Squiers, G.T.; Mclellan, M.A.; Bolisetty, M.T.; Robson, P.; Rosenthal, N.A.; Pinto, A.R. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018, 22, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Liu, W.; Hua, X.; Chen, X.; Chang, Y.; Hu, Y.; Xu, Z.; Song, J. Single-Cell Transcriptomic Atlas of Different Human Cardiac Arteries Identifies Cell Types Associated with Vascular Physiology. Arter. Thromb. Vasc. 2021, 41, 1408–1427. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, Y.; Ma, H.; Xie, Y.; Xu, J.; Near, D.; Wang, H.; Garbutt, T.; Li, Y.; Liu, J.; et al. Single cell dual-omics reveals the transcriptomic and epigenomic diversity of cardiac non-myocytes. Cardiovasc. Res. 2021, cvab134. [Google Scholar] [CrossRef] [PubMed]
- See, K.; Tan, W.L.; Lim, E.H.; Tiang, Z.; Lee, L.T.; Li, P.Y.Q.; Luu, T.D.A.; Ackers-Johnson, M.; Foo, R.S. Single cardiomyocyte nuclear transcriptomes reveal a lincRNA-regulated de-differentiation and cell cycle stress-response in vivo. Nat. Commun. 2017, 8, 225. [Google Scholar] [CrossRef] [Green Version]
- Ni, X.; Xu, K.; Zhao, Y.; Li, J.; Wang, L.; Yu, F.; Li, G. Single-cell analysis reveals the purification and maturation effects of glucose starvation in hiPSC-CMs. Biochem. Biophys. Res. Commun. 2021, 534, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Giacomelli, E.; Meraviglia, V.; Campostrini, G.; Cochrane, A.; Cao, X.; van Helden, R.W.J.; Garcia, A.K.; Mircea, M.; Kostidis, S.; Davis, R.P.; et al. Human-iPSC-Derived Cardiac Stromal Cells Enhance Maturation in 3D Cardiac Microtissues and Reveal Non-cardiomyocyte Contributions to Heart Disease. Cell Stem Cell 2020, 26, 862–879.e11. [Google Scholar] [CrossRef] [PubMed]
- Helle, E.; Ampuja, M.; Dainis, A.; Antola, L.; Temmes, E.; Tolvanen, E.; Mervaala, E.; Kivelä, R. HiPS-Endothelial Cells Acquire Cardiac Endothelial Phenotype in Co-culture with hiPS-Cardiomyocytes. Front. Cell Dev. Biol. 2021, 9, 715093. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, Z.; Welch, J.D.; Gao, X.; Wang, L.; Garbutt, T.; Keepers, B.; Ma, H.; Prins, J.F.; Shen, W.; et al. Single-Cell Transcriptomic Analyses of Cell Fate Transitions during Human Cardiac Reprogramming. Cell Stem Cell 2019, 25, 149–164.e9. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, L.; Welch, J.D.; Ma, H.; Zhou, Y.; Vaseghi, H.R.; Yu, S.; Wall, J.B.; Alimohamadi, S.; Zheng, M.; et al. Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte. Nature 2017, 551, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, Y.; Qian, Y.; Liu, J.; Qian, L. Delineating chromatin accessibility re-patterning at single cell level during early stage of direct cardiac reprogramming. J. Mol. Cell. Cardiol. 2021, 162, 62–71. [Google Scholar] [CrossRef]
- Churko, J.M.; Garg, P.; Treutlein, B.; Venkatasubramanian, M.; Wu, H.; Lee, J.; Wessells, Q.N.; Chen, S.-Y.; Chen, W.-Y.; Chetal, K.; et al. Defining human cardiac transcription factor hierarchies using integrated single-cell heterogeneity analysis. Nat. Commun. 2018, 9, 4906. [Google Scholar] [CrossRef]
- Schmid, C.; Wohnhaas, C.T.; Hildebrandt, T.; Baum, P.; Rast, G. Characterization of iCell cardiomyocytes using single-cell RNA-sequencing methods. J. Pharmacol. Toxicol. Methods 2020, 106, 106915. [Google Scholar] [CrossRef] [PubMed]
- Selewa, A.; Dohn, R.; Eckart, H.; Lozano, S.; Xie, B.; Gauchat, E.; Elorbany, R.; Rhodes, K.; Burnett, J.; Gilad, Y.; et al. Systematic Comparison of High-throughput Single-Cell and Single-Nucleus Transcriptomes during Cardiomyocyte Differentiation. Sci. Rep. 2020, 10, 1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biendarra-Tiegs, S.M.; Li, X.; Ye, D.; Brandt, E.B.; Ackerman, M.J.; Nelson, T.J. Single-Cell RNA-Sequencing and Optical Electrophysiology of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Reveal Discordance Between Cardiac Subtype-Associated Gene Expression Patterns and Electrophysiological Phenotypes. Stem Cells Dev. 2019, 28, 659–673. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liu, S.; Gao, H.; Han, L.; Chu, X.; Sheng, Y.; Shou, W.; Wang, Y.; Liu, Y.; Wan, J.; et al. Genome-wide studies reveal the essential and opposite roles of ARID1A in controlling human cardiogenesis and neurogenesis from pluripotent stem cells. Genome Biol. 2020, 21, 169. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Liao, Y.; Ren, Z.; Mao, L.; Yao, F.; Yu, P.; Ye, Y.; Zhang, Z.; Li, S.; Xu, H.; et al. Single-cell reconstruction of differentiation trajectory reveals a critical role of ETS1 in human cardiac lineage commitment. BMC Biol. 2019, 17, 89. [Google Scholar] [CrossRef] [PubMed]
- Krane, M.; Dreßen, M.; Santamaria, G.; My, I.; Schneider, C.M.; Dorn, T.; Laue, S.; Mastantuono, E.; Berutti, R.; Rawat, H.; et al. Sequential Defects in Cardiac Lineage Commitment and Maturation Cause Hypoplastic Left Heart Syndrome. Circulation 2021, 144, 1409–1428. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.-Y.; Keung, W.; Chan, C.-H.; Geng, L.; Wong, N.; Brenière-Letuffe, D.; Li, R.A.; Cheung, Y.-F. Single-Cell Transcriptomics of Engineered Cardiac Tissues from Patient-Specific Induced Pluripotent Stem Cell-Derived Cardiomyocytes Reveals Abnormal Developmental Trajectory and Intrinsic Contractile Defects in Hypoplastic Right Heart Syndrome. J. Am. Heart Assoc. 2020, 9, e016528. [Google Scholar] [CrossRef] [PubMed]
- Paige, S.L.; Galdos, F.X.; Lee, S.; Chin, E.T.; Ranjbarvaziri, S.; Feyen, D.A.M.; Darsha, A.K.; Xu, S.; Ryan, J.A.; Beck, A.L.; et al. Patient-Specific Induced Pluripotent Stem Cells Implicate Intrinsic Impaired Contractility in Hypoplastic Left Heart Syndrome. Circulation 2020, 142, 1605–1608. [Google Scholar] [CrossRef]
- Mehrabi, M.; Morris, T.A.; Cang, Z.; Nguyen, C.H.H.; Sha, Y.; Asad, M.N.; Khachikyan, N.; Greene, T.L.; Becker, D.M.; Nie, Q.; et al. A Study of Gene Expression, Structure, and Contractility of iPSC-Derived Cardiac Myocytes from a Family with Heart Disease due to LMNA Mutation. Ann. Biomed. Eng. 2021, 49, 3524–3539. [Google Scholar] [CrossRef]
- Paik, D.T.; Tian, L.; Lee, J.; Sayed, N.; Chen, I.Y.; Rhee, S.; Rhee, J.-W.; Kim, Y.; Wirka, R.C.; Buikema, J.W.; et al. Large-Scale Single-Cell RNA-Seq Reveals Molecular Signatures of Heterogeneous Populations of Human Induced Pluripotent Stem Cell-Derived Endothelial Cells. Circ. Res. 2018, 123, 443–450. [Google Scholar] [CrossRef]
- McCracken, I.R.; Taylor, R.S.; Kok, F.O.; de la Cuesta, F.; Dobie, R.; Henderson, B.E.P.; Mountford, J.C.; Caudrillier, A.; Henderson, N.C.; Ponting, C.P.; et al. Transcriptional dynamics of pluripotent stem cell-derived endothelial cell differentiation revealed by single-cell RNA sequencing. Eur. Heart J. 2020, 41, 1024–1036. [Google Scholar] [PubMed]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; He, A. Single-Cell Transcriptomic Analysis of Cardiac Progenitor Differentiation. Curr. Cardiol. Rep. 2020, 22, 38. [Google Scholar] [CrossRef] [PubMed]
- Domian, I.J.; Chiravuri, M.; van der Meer, P.; Feinberg, A.W.; Shi, X.; Shao, Y.; Wu, S.M.; Parker, K.K.; Chien, K.R. Generation of functional ventricular heart muscle from mouse ventricular progenitor cells. Science 2009, 326, 426–429. [Google Scholar] [CrossRef] [Green Version]
- Mosimann, C.; Panáková, D.; Werdich, A.A.; Musso, G.; Burger, A.; Lawson, K.L.; Carr, L.A.; Nevis, K.R.; Sabeh, M.K.; Zhou, Y.; et al. Chamber identity programs drive early functional partitioning of the heart. Nat Commun. 2015, 6, 8146. [Google Scholar] [CrossRef] [Green Version]
- Bondue, A.; Lapouge, G.; Paulissen, C.; Semeraro, C.; Iacovino, M.; Kyba, M.; Blanpain, C. Mesp1 Acts as a Master Regulator of Multipotent Cardiovascular Progenitor Specification. Cell Stem Cell 2008, 3, 69–84. [Google Scholar] [CrossRef]
- Srivastava, D.; Cserjesi, P.; Olson, E.N. A Subclass of bHLH Proteins Required for Cardiac Morphogenesis. Science 1995, 270, 1995–1999. [Google Scholar] [CrossRef]
- Tsuchihashi, T.; Maeda, J.; Shin, C.H.; Ivey, K.N.; Black, B.L.; Olson, E.N.; Yamagishi, H.; Srivastava, D. Hand2 function in second heart field progenitors is essential for cardiogenesis. Dev. Biol. 2011, 351, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.S.-K.; Chan, H.H.W.; Kyba, M. Heterogeneity of Mesp1+ mesoderm revealed by single-cell RNA-seq. Biochem. Biophys. Res. Commun. 2016, 474, 469–475. [Google Scholar] [CrossRef] [Green Version]
- Hu, P.; Liu, J.; Zhao, J.; Wilkins, B.J.; Lupino, K.; Wu, H.; Pei, L. Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev. 2018, 32, 1344–1357. [Google Scholar] [CrossRef]
- Wang, Y.; Yao, F.; Wang, L.; Li, Z.; Ren, Z.; Li, D.; Zhang, M.; Han, L.; Wang, S.-Q.; Zhou, B.; et al. Single-cell analysis of murine fibroblasts identifies neonatal to adult switching that regulates cardiomyocyte maturation. Nat. Commun. 2020, 11, 2585. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, S.; Miyagawa-Tomita, S.; Inoue, T.; Kanno, J.; Saga, Y. Mesp1-nonexpressing cells contribute to the ventricular cardiac conduction system. Dev. Dyn. 2006, 235, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Saga, Y.; Kitajima, S.; Miyagawa-Tomita, S. Mesp1 Expression Is the Earliest Sign of Cardiovascular Development. Trends Cardiovasc. Med. 2000, 10, 345–352. [Google Scholar] [CrossRef]
- Bondue, A.; Blanpain, C. Mesp1: A key regulator of cardiovascular lineage commitment. Circ. Res. 2010, 107, 1414–1427. [Google Scholar] [CrossRef] [Green Version]
- Chiapparo, G.; Lin, X.; Lescroart, F.; Chabab, S.; Paulissen, C.; Pitisci, L.; Bondue, A.; Blanpain, C. Mesp1 controls the speed, polarity, and directionality of cardiovascular progenitor migration. J. Cell Biol. 2016, 213, 463–477. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, L.; Diaz, A.D.; Benham, A.; Xu, X.; Wijaya, C.S.; Fa’ak, F.; Luo, W.; Soibam, B.; Azares, A.; et al. Mesp1 Marked Cardiac Progenitor Cells Repair Infarcted Mouse Hearts. Sci. Rep. 2016, 6, 31457. [Google Scholar] [CrossRef] [Green Version]
- Islas, J.F.; Liu, Y.; Weng, K.C.; Robertson, M.J.; Zhang, S.; Prejusa, A.; Harger, J.; Tikhomirova, D.; Chopra, M.; Iyer, D.; et al. Transcription factors ETS2 and MESP1 transdifferentiate human dermal fibroblasts into cardiac progenitors. Proc. Natl. Acad. Sci. USA 2012, 109, 13016–13021. [Google Scholar] [CrossRef] [Green Version]
- Wada, R.; Muraoka, N.; Inagawa, K.; Yamakawa, H.; Miyamoto, K.; Sadahiro, T.; Umei, T.; Kaneda, R.; Suzuki, T.; Kamiya, K.; et al. Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc. Natl. Acad. Sci. USA 2013, 110, 12667–12672. [Google Scholar] [CrossRef] [Green Version]
- Bu, L.; Jiang, X.; Martin-Puig, S.; Caron, L.; Zhu, S.; Shao, Y.; Roberts, D.J.; Huang, P.L.; Domian, I.J.; Chien, K.R. Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature 2009, 460, 113–117. [Google Scholar] [CrossRef]
- Bulatovic, I.; Månsson-Broberg, A.; Sylvén, C.; Grinnemo, K.H. Human fetal cardiac progenitors: The role of stem cells and progenitors in the fetal and adult heart. Best Pract. Res. Clin. Obstet. Gynaecol. 2016, 31, 58–68. [Google Scholar] [CrossRef]
- Moretti, A.; Caron, L.; Nakano, A.; Lam, J.T.; Bernshausen, A.; Chen, Y.; Qyang, Y.; Bu, L.; Sasaki, M.; Martin-Puig, S.; et al. Multipotent Embryonic Isl1+ Progenitor Cells Lead to Cardiac, Smooth Muscle, and Endothelial Cell Diversification. Cell 2006, 127, 1151–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Boni, A.; Urbanek, K.; Nascimbene, A.; Hosoda, T.; Zheng, H.; Delucchi, F.; Amano, K.; Gonzalez, A.; Vitale, S.; Ojaimi, C.; et al. Notch1 regulates the fate of cardiac progenitor cells. Proc. Natl. Acad. Sci. USA 2008, 105, 15529–15534. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.M.; Fujiwara, Y.; Cibulsky, S.M.; Clapham, D.E.; Lien, C.L.; Schultheiss, T.M.; Orkin, S.H. Developmental origin of a bipotential myocardial and smooth muscle cell precursor in the mammalian heart. Cell 2006, 127, 1137–1150. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.D.; Stone, N.R.; Liu, L.; Spencer, C.I.; Qian, L.; Hayashi, Y.; Delgado-Olguin, P.; Ding, S.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of human fibroblasts toward a cardiomyocyte-like state. Stem Cell Rep. 2013, 1, 235–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruneau, B.G.; Bao, Z.Z.; Tanaka, M.; Schott, J.J.; Izumo, S.; Cepko, C.L.; Seidman, J.G.; Seidman, C.E. Cardiac expression of the ventricle-specific homeobox gene Irx4 is modulated by Nkx2-5 and dHand. Dev. Biol. 2000, 217, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taubenschmid, J.; Weitzer, G. Mechanisms of cardiogenesis in cardiovascular progenitor cells. Int. Rev. Cell Mol. Biol. 2012, 293, 195–267. [Google Scholar]
- Gittenberger-de Groot, A.C.; Lie-Venema, H.; van den Akker, N.M.; Winter, E.M.; Poelmann, R.E. Coronary vascular development. Wien. Klin. Wochenschr. 2007, 119, 4–5. [Google Scholar]
- Ma, Q.; Zhou, B.; Pu, W.T. Reassessment of Isl1 and Nkx2-5 cardiac fate maps using a Gata4-based reporter of Cre activity. Dev. Biol. 2008, 323, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, E.M.; Ma, Q.; Juraszek, A.L.; Moses, K.; Schwartz, R.J.; Izumo, S.; Pu, W.T. Morphogenesis of the right ventricle requires myocardial expression of Gata4. J. Clin. Investig. 2005, 115, 1522–1531. [Google Scholar] [CrossRef] [Green Version]
- Farbehi, N.; Patrick, R.; Dorison, A.; Xaymardan, M.; Janbandhu, V.; Wystub-Lis, K.; Ho, J.W.; Nordon, R.E.; Harvey, R.P. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. eLife 2019, 8, e43882. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, F.; Isherwood, J.; Bawa, T.; Patel, D.; Gurdziel, K.; Lanfear, D.E.; Ruden, D.M.; Levy, P.D. Single-Cell RNA Sequencing of the Cardiovascular System: New Looks for Old Diseases. Front. Cardiovasc. Med. 2019, 6, 173. [Google Scholar] [CrossRef] [PubMed]
- King, K.R.; Aguirre, A.D.; Ye, Y.-X.; Sun, Y.; Roh, J.D.; Ng, R.P.; Kohler, R.H.; Arlauckas, S.P.; Iwamoto, Y.; Savol, A.; et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 2017, 23, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Schafer, S.; Viswanathan, S.; Widjaja, A.A.; Lim, W.-W.; Moreno-Moral, A.; DeLaughter, D.M.; Ng, B.; Patone, G.; Chow, K.; Khin, E.; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017, 552, 110–115. [Google Scholar] [CrossRef]
- Satoh, M.; Nomura, S.; Harada, M.; Yamaguchi, T.; Ko, T.; Sumida, T.; Toko, H.; Naito, A.T.; Takeda, N.; Tobita, T.; et al. High-throughput single-molecule RNA imaging analysis reveals heterogeneous responses of cardiomyocytes to hemodynamic overload. J. Mol. Cell. Cardiol. 2019, 128, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Gago-Lopez, N.; Li, N.; Zhang, Z.; Alver, N.; Liu, Y.; Martinson, A.M.; Mehri, A.; MacLellan, W.R. Single-cell imaging and transcriptomic analyses of endogenous cardiomyocyte dedifferentiation and cycling. Cell Discov. 2019, 5, 30. [Google Scholar] [CrossRef]
- Wolfien, M.; Galow, A.M.; Müller, P.; Bartsch, M.; Brunner, R.M.; Goldammer, T.; Wolkenhauer, O.; Hoeflich, A.; David, R. Single-Nucleus Sequencing of an Entire Mammalian Heart: Cell Type Composition and Velocity. Cells 2020, 9, 318. [Google Scholar] [CrossRef] [Green Version]
- Linscheid, N.; Logantha, S.; Poulsen, P.C.; Zhang, S.; Schrölkamp, M.; Egerod, K.L.; Thompson, J.J.; Kitmitto, A.; Galli, G.; Humphries, M.J.; et al. Quantitative proteomics and single-nucleus transcriptomics of the sinus node elucidates the foundation of cardiac pacemaking. Nat. Commun. 2019, 10, 2889. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Frangogiannis, N.G. Fibroblasts in post-infarction inflammation and cardiac repair. Biochim. Biophys. Acta 2013, 1833, 945–953. [Google Scholar] [CrossRef] [Green Version]
- Stanley, E.R.; Berg, K.L.; Einstein, D.B.; Lee, P.S.; Pixley, F.J.; Wang, Y.; Yeung, Y.G. Biology and action of colony—Stimulating factor-1. Mol. Reprod. Dev. 1997, 46, 4–10. [Google Scholar] [CrossRef]
- Hassankhani, A.; Steinhelper, M.E.; Soonpaa, M.H.; Katz, E.B.; Taylor, D.A.; Andrade-Rozental, A.; Factor, S.M.; Steinberg, J.J.; Field, L.J.; Federoff, H.J. Overexpression of NGF within the heart of transgenic mice causes hyperinnervation, cardiac enlargement, and hyperplasia of ectopic cells. Dev. Biol. 1995, 169, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Resendiz, S.; Muñoz-Vega, M.; Contreras, W.E.; Crespo-Avilan, G.E.; Rodriguez-Montesinos, J.; Arias-Carrión, O.; Pérez-Méndez, O.; Boisvert, W.A.; Preissner, K.T.; Cabrera-Fuentes, H.A. Responses of Endothelial Cells Towards Ischemic Conditioning Following Acute Myocardial Infarction. Cond. Med. 2018, 1, 247–258. [Google Scholar] [PubMed]
- Thiriot, A.; Perdomo, C.; Cheng, G.; Novitzky-Basso, I.; McArdle, S.; Kishimoto, J.K.; Barreiro, O.; Mazo, I.; Triboulet, R.; Ley, K.; et al. Differential DARC/ACKR1 expression distinguishes venular from non-venular endothelial cells in murine tissues. BMC Biol. 2017, 15, 45. [Google Scholar] [CrossRef] [PubMed]
- Lafuse, W.P.; Wozniak, D.J.; Rajaram, M.V.S. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells 2020, 10, 51. [Google Scholar] [CrossRef]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Seno, A.; Takeda, Y.; Matsui, M.; Okuda, A.; Nakano, T.; Nakada, Y.; Kumazawa, T.; Nakagawa, H.; Nishida, T.; Onoue, K.; et al. Suppressed Production of Soluble Fms-Like Tyrosine Kinase-1 Contributes to Myocardial Remodeling and Heart Failure. Hypertension 2016, 68, 678–687. [Google Scholar] [CrossRef]
- Lee, L.L.; Chintalgattu, V. Pericytes in the Heart. Adv. Exp. Med. Biol. 2019, 1122, 187–210. [Google Scholar]
- Kaplan, A.; Abidi, E.; El-Yazbi, A.; Eid, A.; Booz, G.W.; Zouein, F.A. Direct cardiovascular impact of SGLT2 inhibitors: Mechanisms and effects. Heart Fail. Rev. 2018, 23, 419–437. [Google Scholar] [CrossRef]
- Bondjers, C.; He, L.; Takemoto, M.; Norlin, J.; Asker, N.; Hellström, M.; Lindahl, P.; Betsholtz, C. Microarray analysis of blood microvessels from PDGF-B and PDGF-Rbeta mutant mice identifies novel markers for brain pericytes. FASEB J. 2006, 20, 1703–1705. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, H.; Liu, Y.; Adams, S.; Eilken, H.; Stehling, M.; Corada, M.; Dejana, E.; Zhou, B.; Adams, R.H. Endothelial cells are progenitors of cardiac pericytes and vascular smooth muscle cells. Nat. Commun. 2016, 7, 12422. [Google Scholar] [CrossRef]
- Sahara, M.; Santoro, F.; Sohlmer, J.; Zhou, C.; Witman, N.; Leung, C.Y.; Mononen, M.; Bylund, K.; Gruber, P.; Chien, K.R. Population and Single-Cell Analysis of Human Cardiogenesis Reveals Unique LGR5 Ventricular Progenitors in Embryonic Outflow Tract. Dev. Cell 2019, 48, 475–490.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Lee, J.; Lee, D.Y.; Kim, J.Y.; Lim, H.J.; Lim, S.; Cho, Y.S. Schwann Cell Precursors from Human Pluripotent Stem Cells as a Potential Therapeutic Target for Myelin Repair. Stem Cell Rep. 2017, 8, 1714–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Farid, M.; Lin, B.L.; Miyamoto, M.; Kwon, C. Transcriptomic entropy benchmarks stem cell-derived cardiomyocyte maturation against endogenous tissue at single cell level. PLoS Comput. Biol. 2021, 17, e1009305. [Google Scholar] [CrossRef] [PubMed]
- Zwi-Dantsis, L.; Winter, C.W.; Kauscher, U.; Ferrini, A.; Wang, B.; Whittaker, T.E.; Hood, S.R.; Terracciano, C.M.; Stevens, M.M. Highly purified extracellular vesicles from human cardiomyocytes demonstrate preferential uptake by human endothelial cells. Nanoscale 2020, 12, 19844–19854. [Google Scholar] [CrossRef]
- Nakano, H.; Minami, I.; Braas, D.; Pappoe, H.; Wu, X.; Sagadevan, A.; Vergnes, L.; Fu, K.; Morselli, M.; Dunham, C.; et al. Glucose inhibits cardiac muscle maturation through nucleotide biosynthesis. eLife 2017, 6, e29330. [Google Scholar] [CrossRef]
- Andrysiak, K.; Stępniewski, J.; Dulak, J. Human-induced pluripotent stem cell-derived cardiomyocytes, 3D cardiac structures, and heart-on-a-chip as tools for drug research. Pflügers Arch. 2021, 473, 1061–1085. [Google Scholar] [CrossRef]
- Ye, L.; Yu, Y.; Zhao, Z.A.; Zhao, D.; Ni, X.; Wang, Y.; Fang, X.; Yu, M.; Wang, Y.; Tang, J.M.; et al. Patient-specific iPSC-derived cardiomyocytes reveal abnormal regulation of FGF16 in a familial atrial septal defect. Cardiovasc. Res. 2022, 118, 859–871. [Google Scholar] [CrossRef]
- Mckeithan, W.L.; Feyen, D.A.M.; Bruyneel, A.A.N.; Okolotowicz, K.J.; Ryan, D.A.; Sampson, K.J.; Potet, F.; Savchenko, A.; Gómez-Galeno, J.; Vu, M.; et al. Reengineering an Antiarrhythmic Drug Using Patient hiPSC Cardiomyocytes to Improve Therapeutic Potential and Reduce Toxicity. Cell Stem Cell 2020, 27, 813–821.e6. [Google Scholar] [CrossRef]
- Lee, J.; Termglinchan, V.; Diecke, S.; Itzhaki, I.; Lam, C.K.; Garg, P.; Lau, E.; Greenhaw, M.; Seeger, T.; Wu, H.; et al. Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 2019, 572, 335–340. [Google Scholar] [CrossRef]
- Iwanski, J.; Kazmouz, S.G.; Li, S.; Stansfield, B.; Salem, T.T.; Perez-Miller, S.; Kazui, T.; Jena, L.; Uhrlaub, J.L.; Lick, S.; et al. Antihypertensive drug treatment and susceptibility to SARS-CoV-2 infection in human PSC-derived cardiomyocytes and primary endothelial cells. Stem Cell Rep. 2021, 16, 2459–2472. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Fabyanic, E.; Kwon, D.Y.; Tang, S.; Zhou, Z.; Wu, H. Dissecting Cell-Type Composition and Activity-Dependent Transcriptional State in Mammalian Brains by Massively Parallel Single-Nucleus RNA-Seq. Mol. Cell. 2017, 68, 1006–1015.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Reference | Species | Developmental Stage | Technique (Number of Cells/Nuclei) | Target Tissues/Cells |
|---|---|---|---|---|
| Chan et al. [59] | Mouse | Mouse ESC-derived embryoid bodies day 4 | scRNA-seq (42 cells) | Dissociated cells from embryoid bodies |
| Lescroart et al. [20] | Mouse | Embryonic day 6.25 and 7.5 | scRNA-seq (513 cells) | Mesp1+ or Mesp1 KO CPCs |
| Tyser et al. [21] | Mouse | Embryonic day 7.75–8.25 | scRNA-seq (3105 cells) Multiplexed RNA imaging | Manual microdissection to isolate the anterior cardiac region |
| Jia et al. [22] | Mouse | Embryonic day 7.5, 8.5, and 9.5 | scRNA-seq and scATAC-seq (>1258 cells) | Isl1+ or Nkx2.5+ CPCs |
| Li et al. [12] | Mouse | Embryonic day 8.5, 9.5, and 10.5 | scRNA-seq (2233 cells) | Microdissected embryonic heart tissues of each chamber |
| Xiong et al. [23] | Mouse | Embryonic day 7.75, 8.25, and 9.75 | scRNA-seq (1231 cells), CHIP-seq | Isl1+ or Nkx2.5+ CPCs |
| de Soysa et al. [17] | Mouse | Embryonic day 7.75, 8.25, and 9.25 | scRNA-seq (36,777 cells) | CPCs from control and Hand2-null embryos |
| DeLaughter et al. [25] | Mouse | Embryonic day 9.5, 11.5, 14.5, and 18.5; postnatal day 0, 3, and 21 | scRNA-seq (1133 cells) | Microdissected embryonic heart tissues of each chamber |
| Cui et al. [14] | Human | 5–25 weeks gestation | scRNA-seq (3842 cells) | Anatomically informed cardiac cells from human embryos |
| Goodyer et al. [24] | Mouse | Embryonic day 16.5 | scRNA-seq (22,462 cells) | Cells from three zones of microdissected hearts: sinoatrial node region, atrioventricular node/His region, and bundle branch/Purkinje fiber region |
| Asp et al. [7] | Human | 4.5–5, 6.5, and 9 weeks gestation | scRNA-seq (3717 cells), spatial barcoding, and in situ sequencing | Human embryonic and fetal cardiac cells |
| Hu et al. [60] | Mouse | postnatal immature heart | snRNA-seq (15,082 cells) | isolated nuclei from postnatal hearts (P6, P10) |
| Wang et al. [61] | Mouse | Postnatal day 1, 4, 7, 14 and 56 | scRNA-seq (2137 cells) | CMs and non-CMs from left ventricles |
| Reference | Species | Sample Category | Technique (Number of Cells/Nuclei) | Target Tissues/Cells |
|---|---|---|---|---|
| Dick et al. [26] | Mouse | adult heart | scRNA-seq (8283 cells) | Macrophages and dendritic cells from adult heart, cardiac mononuclear cells from adult heart (non-operated or D11 post-MI) |
| Tucker et al. [13] | Human | adult heart | snRNA-seq (287,269 cells) | Tissue samples taken from the lateral aspect of the four cardiac chambers from potential transplant donors |
| Litvinukova et al. [9] | Human | adult heart | scRNA-seq (123,893 cells), snRNA-seq (363,213 nuclei), and multiplexed RNA imaging | Full-thickness myocardial biopsies from the left and right atria, left and right ventricles, and interventricular septum and apex from deceased transplant organ donors |
| Wang et al. [8] | Human | adult heart | scRNA-seq (21,422 cells) | CMs and non-CMs from biopsy samples of LA and LVs of normal, failed, and recovered adult human hearts |
| Yekelchyk et al. [27] | Mouse | adult heart | scRNA-seq (>586 cells) | CMs from both healthy and hypertrophic ventricles |
| Nomura et al. [28] | Mouse and human | adult heart | scRNA-seq (396 cells) | CMs isolated from LVs of mice after sham surgery or 3 days and 1, 2, 4, and 8 weeks after TAC/DCM patients or normal control |
| Gladka et al. [29] | Mouse | adult heart | scRNA-seq (426 cells) | Cells from the infarct and border zone region from infarcted heart at day 3 post-MI or sham |
| Ren et al. [15] | Mouse and human | adult heart | scRNA-seq (11,492 cells) | CMs and non-CMs isolated from LVs of mice after sham or 2, 5, 8, and 11 weeks after TAC/end-stage DCM, HCM patients, and control |
| Skelly et al. [30] | Mouse | adult heart | scRNA-seq (10,519 cells) | Non-CMs from the heart |
| Rao et al. [16] | Human | adult heart | scRNA-seq (200,615 cells) | Non-CMs from left and right ventricle of DCM hearts and infarcted and non-infarcted area of ICM hearts |
| Hu et al. [31] | Human | adult heart | scRNA-seq (>100,000 cells) | Cells from human aorta, pulmonary artery, and coronary arteries collected from patients undergoing heart transplantation |
| King et al. [83] | Mouse | adult heart | scRNA-seq (4215 cells) | Leukocytes isolated from wild-type and Irf3-null heart at day 4 post-MI or sham |
| Wang et al. [32] | Mouse | adult heart | scRNA-seq (12,779 cells), scATAC-seq (9524 nuclei) | Heart non-myocytes |
| See et al. [33] | Mouse and human | adult heart | snRNA-seq (359 nuclei) | Nuclei of CMs isolated from LVs of mice 8 weeks after TAC or sham surgery/end-stage DCM patients or control |
| Reference | Species | Sample Category | Technique (Number of Cells/Nuclei) | Target Tissues/Cells |
|---|---|---|---|---|
| Friedman et al. [18] | Human | hESC and hiPSC and derivative | scRNA-seq (43,168 cells) | hESC- and hiPSC-derived cells (D0, D2, D5, D15, and D30) |
| Ni et al. [34] | Human | hiPSC and derivative | scRNA-seq (13,827 cells) | hiPSC-derived cells (D20) |
| Giacomelli et al. [35] | Human | hESC and hiPSC and derivative | scRNA-seq (16,307 cells) | 3D and 2D cultured hiPSC-derived cells |
| Helle et al. [36] | Human | hiPSC and derivative | scRNA-seq (4000 cells) | co-cultured hiPS-CMs and hiPS-Ecs (48 h) |
| Zhou et al. [37] | Human | hiCM | scRNA-sEq (652 cells) | cardiac fibroblast reprogramming into CMs (D0, D3, D5, D7, and D9) |
| Liu et al. [38] | Mouse | iCM | scRNA-seq (454 cells) | cardiac fibroblast reprogramming into CMs (D3) |
| Wang et al. [39] | Mouse | iCM | scATAC-seq (19,397 nuclei) | cardiac fibroblast reprogramming into CMs (D3) |
| Churko et al. [40] | Human | hiPSC and derivative | scRNA-seq (10,419 cells) | hiPSC-derived cells(D1, D5, D14, D30, and D45) |
| Schmid et al. [41] | Human | iCell | scRNA-seq (1421 cells) | Commercially available iCell cardiomyocyte (Fuijifilm Cellular Dynamics) |
| Selewa et al. [42] | Human | hiPSC and derivative | scRNA-seq (25,475 cells), snRNA-seq (22,025 nuclei) | hiPSC-derived cells or isolated nulcei (D0, D1, D3, D7, and D15), nuclei from adult heart tissue (68Y) |
| Biendarra-Tiegs et al. [43] | Human | hiPSC and derivative | scRNA-seq (85 cells) | hiPSC-CMs (D12 and D40) |
| Liu et al. [44] | hESC | scRNA-seq (20,455 cells), ATAC-seq and ChIP-seq | WT and ARID1A−/− hESCs | |
| Ruan et al. [45] | Human | hESC and derivative | scRNA-seq (6879 cells) | hESC- derived cells (D0, D2, D5, D9, D14, and D60) |
| Krane et al. [46] | Human | iPSC-CMs | scRNA-seq (10,870 cells) | iPSC-CMs from patients with HLHS and control (D14) |
| Lam et al. [47] | Human | iPSC-CMs | scRNA-seq (25,059 cells) | Time-matched D30 hiPSC-CMs, D10 hCAS, and D10 hCTS from two healthy subject and two PAIVS hiPSC lines |
| Paige et al. [48] | Human | iPSC-CMs | scRNA-seq (9899 cells) | iPSC-CMs from one control and one HLHS patient (D30) |
| Mehrabi et al. [49] | Human | iPSC-CMs | scRNA-seq (25,619 cells) | iPSC-CMs from 2 control and 2 LMNA patients with a (c.357-2A > G) |
| Paik et al. [50] | Human | iPSC-ECs and derivative | scRNA-seq (5673 cells) | iPSC-ECs differentiation (D8 and D12) |
| McCracken et al. [51] | Human | hESC-ECs and derivative | scRNA-seq (105,727 cells) | hESC-ECs differentiation (D8) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Hu, S.; Zhou, B. Deciphering Cardiac Biology and Disease by Single-Cell Transcriptomic Profiling. Biomolecules 2022, 12, 566. https://doi.org/10.3390/biom12040566
Wang L, Hu S, Zhou B. Deciphering Cardiac Biology and Disease by Single-Cell Transcriptomic Profiling. Biomolecules. 2022; 12(4):566. https://doi.org/10.3390/biom12040566
Chicago/Turabian StyleWang, Le, Shengshou Hu, and Bingying Zhou. 2022. "Deciphering Cardiac Biology and Disease by Single-Cell Transcriptomic Profiling" Biomolecules 12, no. 4: 566. https://doi.org/10.3390/biom12040566
APA StyleWang, L., Hu, S., & Zhou, B. (2022). Deciphering Cardiac Biology and Disease by Single-Cell Transcriptomic Profiling. Biomolecules, 12(4), 566. https://doi.org/10.3390/biom12040566