1. Introduction
The three-dimensional structure of proteins is significant in the studies of proteins since the specific shape of a protein determines its function [
1]. The protein may become denatured and not function as expected if its tertiary structure is altered due to mutations in the amino acid structure. Proteins are chains of amino acids linked by peptide bonds. However, predicting the three-dimensional structure of proteins from amino acid predictions is a challenging task [
2]. Protein secondary structure prediction is an important part of this task [
3,
4,
5,
6,
7,
8].
Protein secondary structure prediction takes primary sequences as the input, which are the amino acid sequences of proteins, to predict the secondary structure type of each amino acid. Q3 accuracy is often used to evaluate the secondary structure: helix (H), strand (E), and coil (C), where the former two are regular secondary structure states and the last one is the irregular type [
9]. Another definition of secondary structure is extends the three general states into eight fine-grained classes [
10]: 3
helix (G),
-helix (H),
-helix (I),
-stand (E),
-bridge (B),
-turn (T), high curvature loop (S), and others (L). Recently, the studies of secondary structure prediction has focused more on the prediction of 8-state secondary structure (Q8) instead of the 3-state(Q3) prediction. The reason is that a chain of 8-state secondary structure naturally contains more structural information for a variety of research and applications [
11,
12].
The methods of secondary structure prediction can be divided into template-based and template-free. Although template-based methods usually achieve better results [
13], it does not work well on proteins with very low similarity with those sequences with known structures in the PDB [
14] library. However, these proteins can be considered as newly discovered proteins, which is more like a real-world scenario. While template-free methods have used several traditional machine-learning models such as probabilistic graphical models [
15,
16], hidden Markov models [
17,
18], and Support Vector Machines (SVM) [
19,
20].
In recent years, deep learning techniques have been widely used in protein secondary structure prediction task, and achieved remarkable results compared with traditional machine learning methods. Several work explore the power of feed-forward back-propagation neural network (BPNN) with traditional machine learning models for protein secondary structure prediction [
21,
22,
23], e.g., ref. [
21] integrate BPNN with Bayesian segmentation, ref. [
22] designs an architecture of protein secondary structure prediction by combining BPNN with SVM, etc. Later, DNSS [
24] first proposes deep learning based secondary structure prediction method [
7], which utilizes a deep belief network [
25] based on restricted Boltzmann machine (RBM) [
26]. Recently, more studies seek to involve more additional features such as position-specific scoring matrix (PSSM) features to further improve prediction performance [
4].
In addition, sequence based models such as Recurrent Neural Network (RNN) encoder are used on protein sequence to predict protein secondary structures [
27] and the first application of LSTM-BRNN to secondary structure prediction can be found in [
28]; and one-dimensional Convolutional Neural Network (1d-CNN) based encoder method has also been used on such task and obtained some achievements [
29]. Moreover, some studies tackle this problem by combining the superiority of different networks, e.g., DeepACLSTM [
30] uses CNN to capture the local feature and bidirectional Long Short-term Memory Network (bLSTM) to obtain the long-distance dependency information. In such a manner, DeepACLSTM is able to obtain better amino acid sequence expression and achieve better prediction performance. Other methods that equipped with DeepCNN network [
29] or ResNet [
31] structure can also capture the long-distance dependency information from the sequence, e.g., CBRNN [
11] combines the CNN-based and RNN-based networks.
Although amino acid sequence encoders based deep learning methods have achieved great success, the relationship among the secondary structures of proteins is rarely studied. DeepCNF [
29] method employs Conditional random field (CRF) as the output layer to learn the interdependency among adjacent secondary structure labels. However, it does not take the specific characteristic of protein secondary structure into consideration, and the improvement of Q8 accuracy is limited.
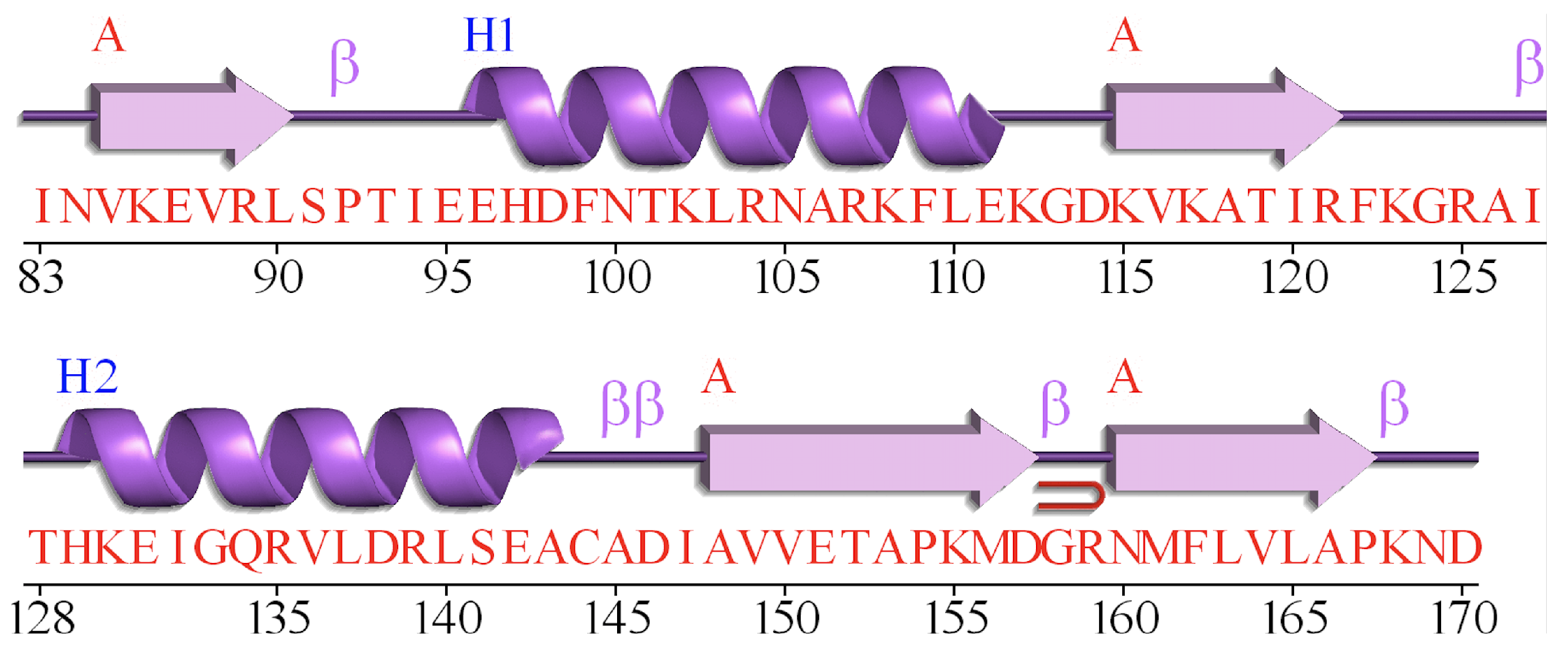
Figure 1 illustrates the secondary structure and the amino acid sequence of protein 1TIG [
14] from CB513 dataset, which is generated by PDBsum [
32]. We can observe that adjacent strings of amino acids generally contain the same secondary structure. The reason of such well-regulated feature might be caused by the characteristics of the protein secondary structure. Consequently, this problem is quite similar to the image semantic segmentation (ISS) [
33] problem. However, there exists two differences: (1) The input data of protein secondary structure task is one-dimensional sequences, while the images contains two dimensions. (2) For ISS, a pooling layer is widely used since the pooling of the adjacent pixels can effectively reduce the size of the input image [
33,
34,
35,
36]. Such implementation can reduce the network parameters while retain most of the image information. However, the amino acid information at each position is crucial for protein sequence, the pooling layer is not adoptable for the amino acid sequence.
Additionally, even various encoders have been proposed to address ISS, e.g., FastFCN [
35], GSCNN [
34], and all versions after Deeplab v2 [
33,
36], the Atrous Spatial Pyramid Pooling (ASPP) Network Structure [
33,
38] followed by the encoder still plays an important role to identify the boundaries of objects in the image.
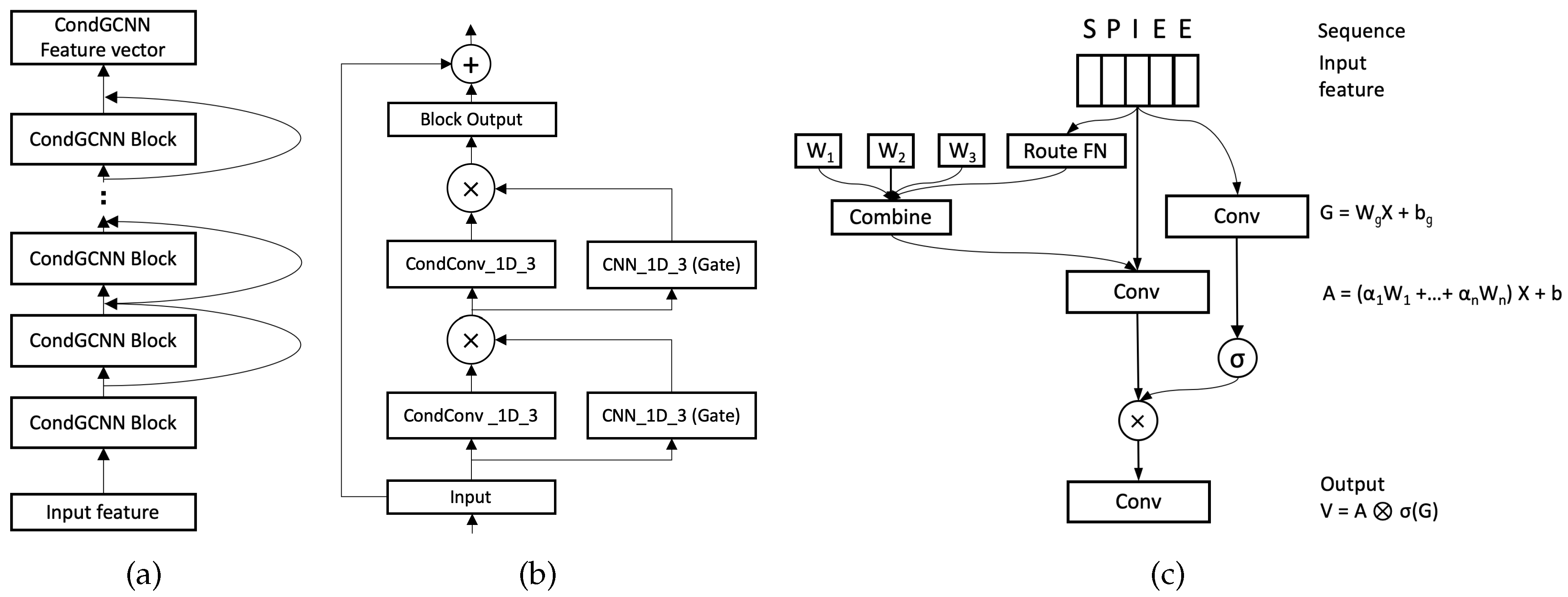
Recently, CNN-based encoding models have obtained great success on both image and language processing tasks. Gated Convolutional networks (GCNN) [
39] employs a CNN-based gating mechanism at the channel level to help the language modeling. Conditionally Parameterized Convolution (CondConv) [
40] uses extra sample-dependant modules to conditionally adjust the convolutional network, which has obtained remarkable improvement over the image processing tasks. In this paper, we present a novel protein sequence encoder, Conditionally Parameterized Gated Convolutional network (CondGCNN), which not only exploits a gating mechanism at the channel level, but also establishes a sample-dependent attention mechanism.
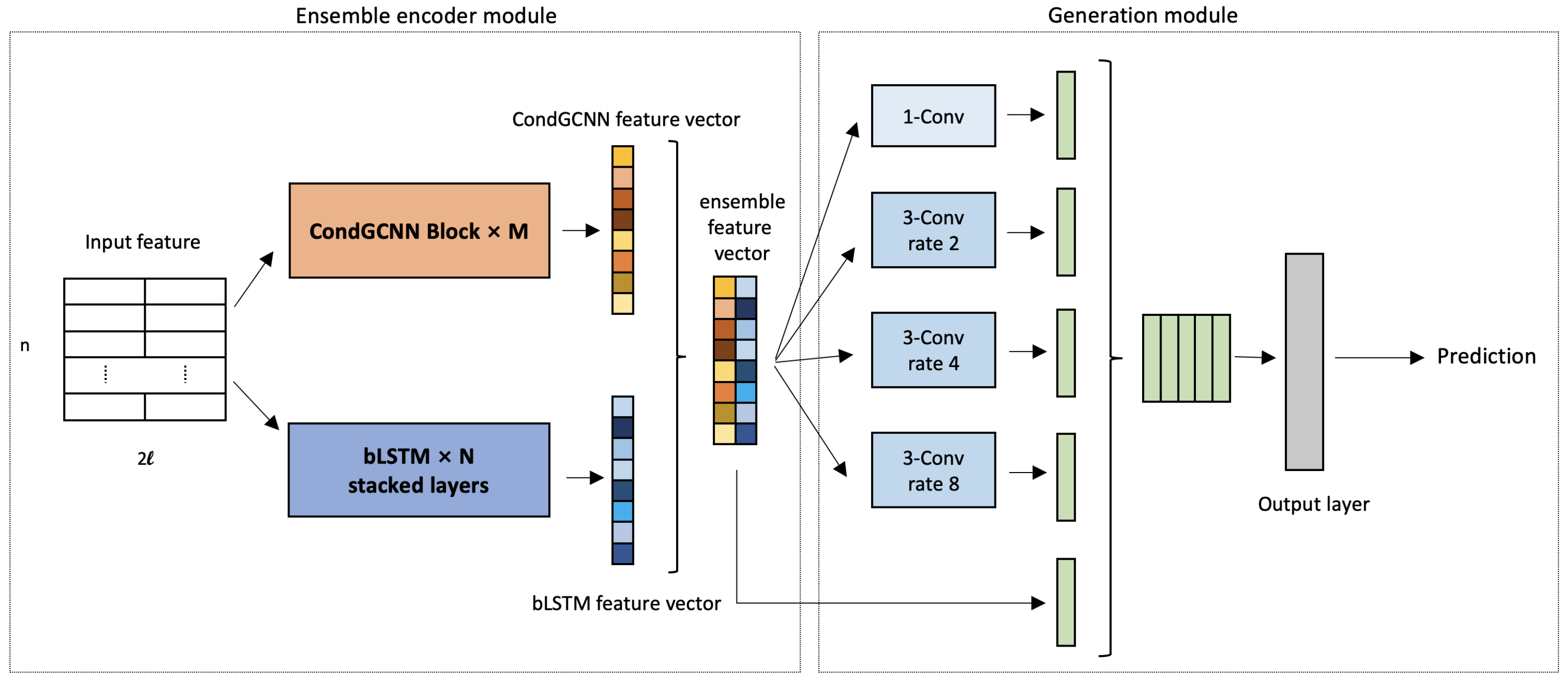
Inspired by previous work about the protein secondary structure prediction task and the ISS, we propose a protein ensemble learning method with ASP networks, which contains an ensemble amino acid sequence encoder and Atrous Spatial Pyramid Networks. Since CNN-based methods have obtained remarkable performance in language modeling and image processing tasks, and lstm-based methods are important for protein prediction [
27,
30], our amino acid sequence encoder has utilized both CondGCNN model (a new encoder we proposed) and bLSTM model. Besides, the ASP Network (optimized ASPP network for our problem) is added following the encoder.
The technical contributions of proposed method can be summarized as: (1) The work is the first to tackle protein secondary structure prediction task with image segmentation processing, which utilizes the predominance of those models applied in the segmentation area to tackle secondary structure prediction problem, e.g., employ ASPP network (optimized as ASP network in our method) to capture fine edge details in secondary structure labels. (2) We are the first to apply CondConv network on sequence processing problems, as well as embed it in the GCNN to form a novel amino acid sequence encoder. In specific, a gating mechanism is equipped at the model channel level and a sample-dependent attention is employed at the input level. (3) We construct an ensemble encoder with cnn-based and lstm-based networks, which has acquired more diverse information from amino acid sequences. (4) Through a set of extensive ablation studies, the significance of different components of the method, including architecture, features, and results, are carefully analyzed. Based on our conference version of the paper [
41], we design more experiments to verify the effect of our ASP module on the boundary residues prediction and verify the effectiveness of our framework on more datasets.
The rest of the paper is organized as follows. In
Section 2, we describe the framework and modules used in our method in detail.
Section 3 gives the experimental results to demonstrate the advantage of our method. Finally, in
Section 4, we conclude our paper and discuss its prospect.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}