
Integrating Conformational Dynamics and Perturbation-Based Network Modeling for Mutational Profiling of Binding and Allostery in the SARS-CoV-2 Spike Variant Complexes with Antibodies: Balancing Local and Global Determinants of Mutational Escape Mechanisms



Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Dynamics Simulations
2.2. Distance Fluctuations Stability and Communication Analysis
2.3. Mutational Scanning and Sensitivity Analysis
2.4. Network Analysis and Perturbation-Based Mutational Profiling of Allosteric Propensities
3. Results and Discussion
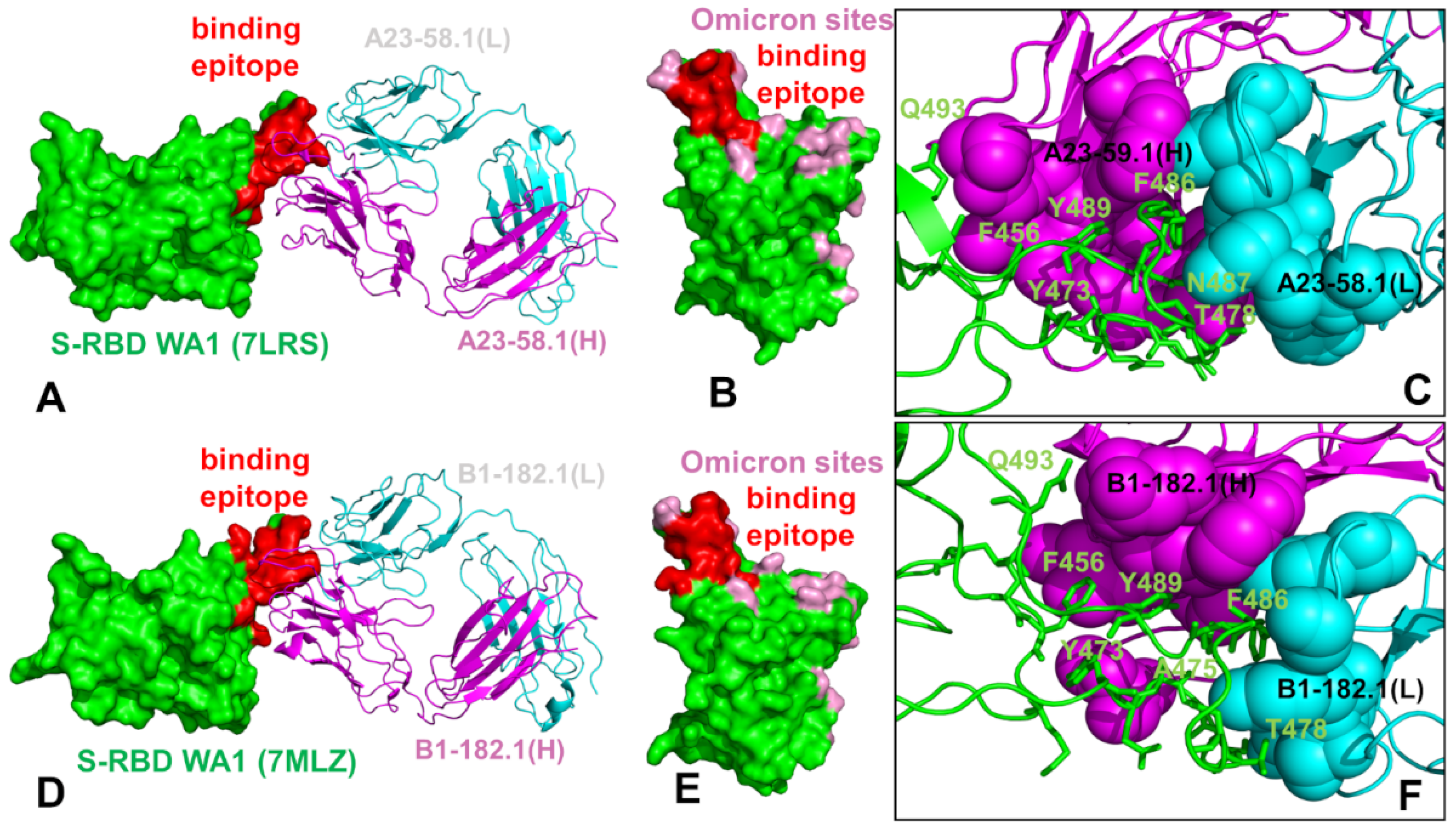
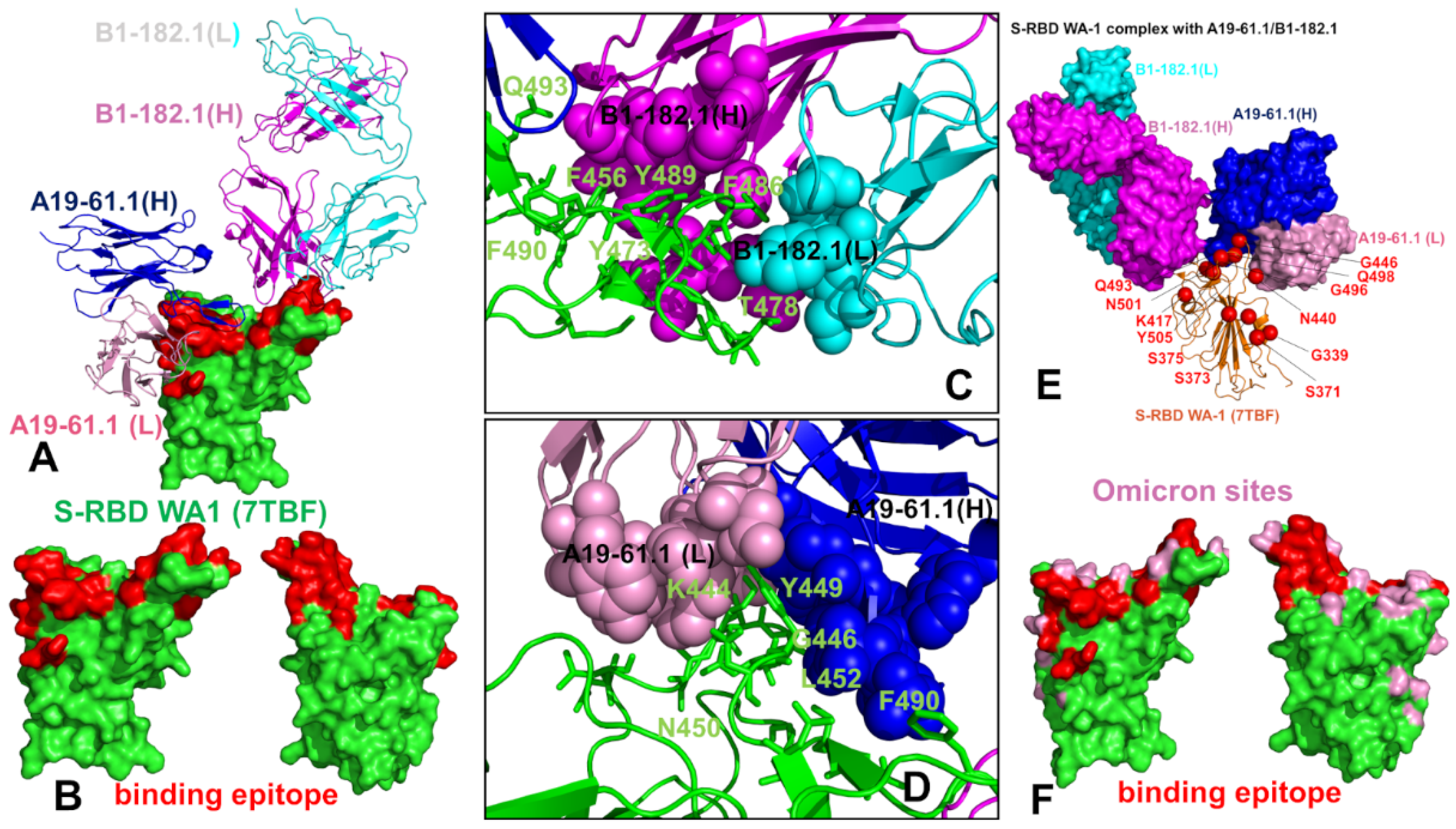
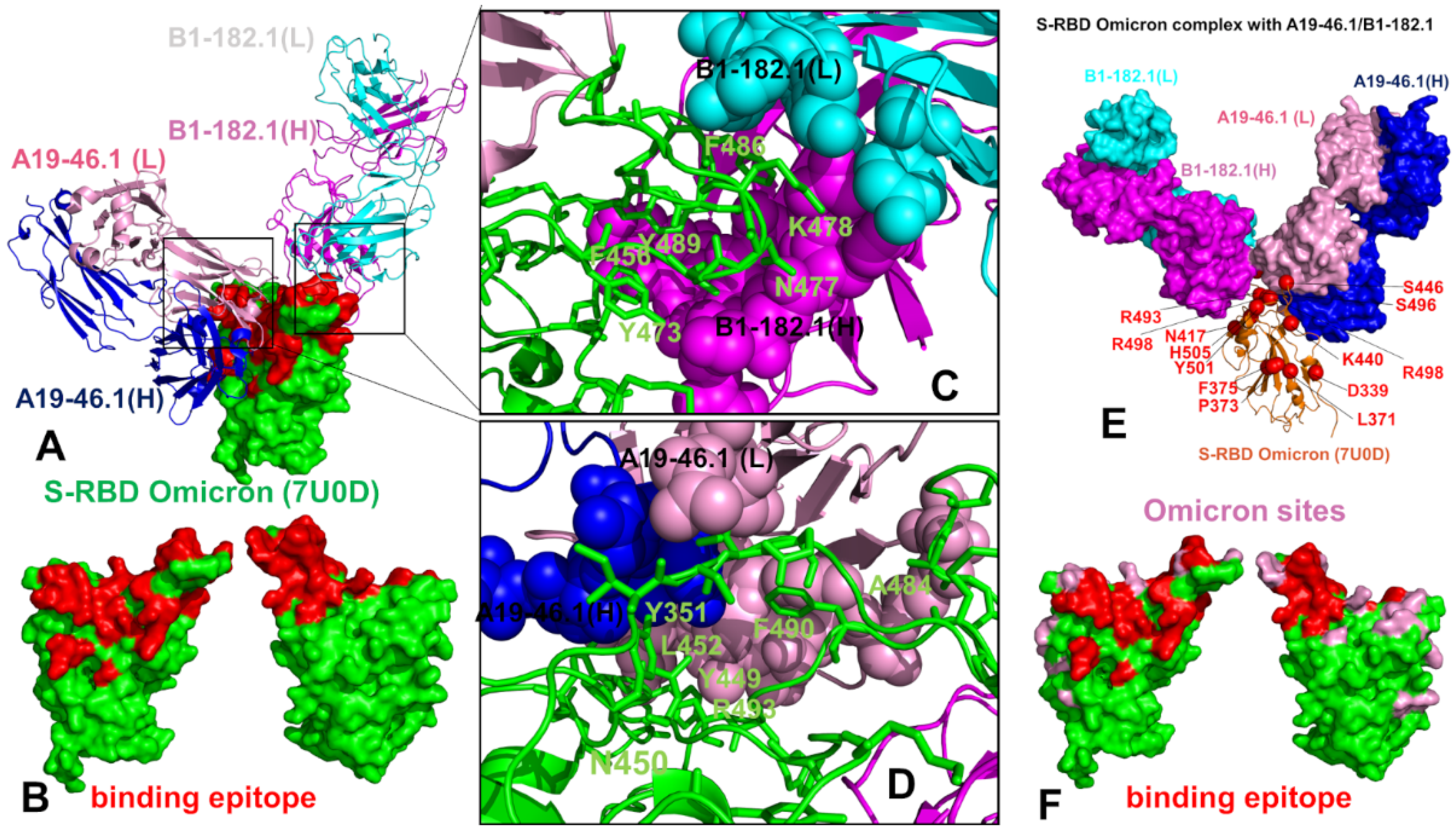
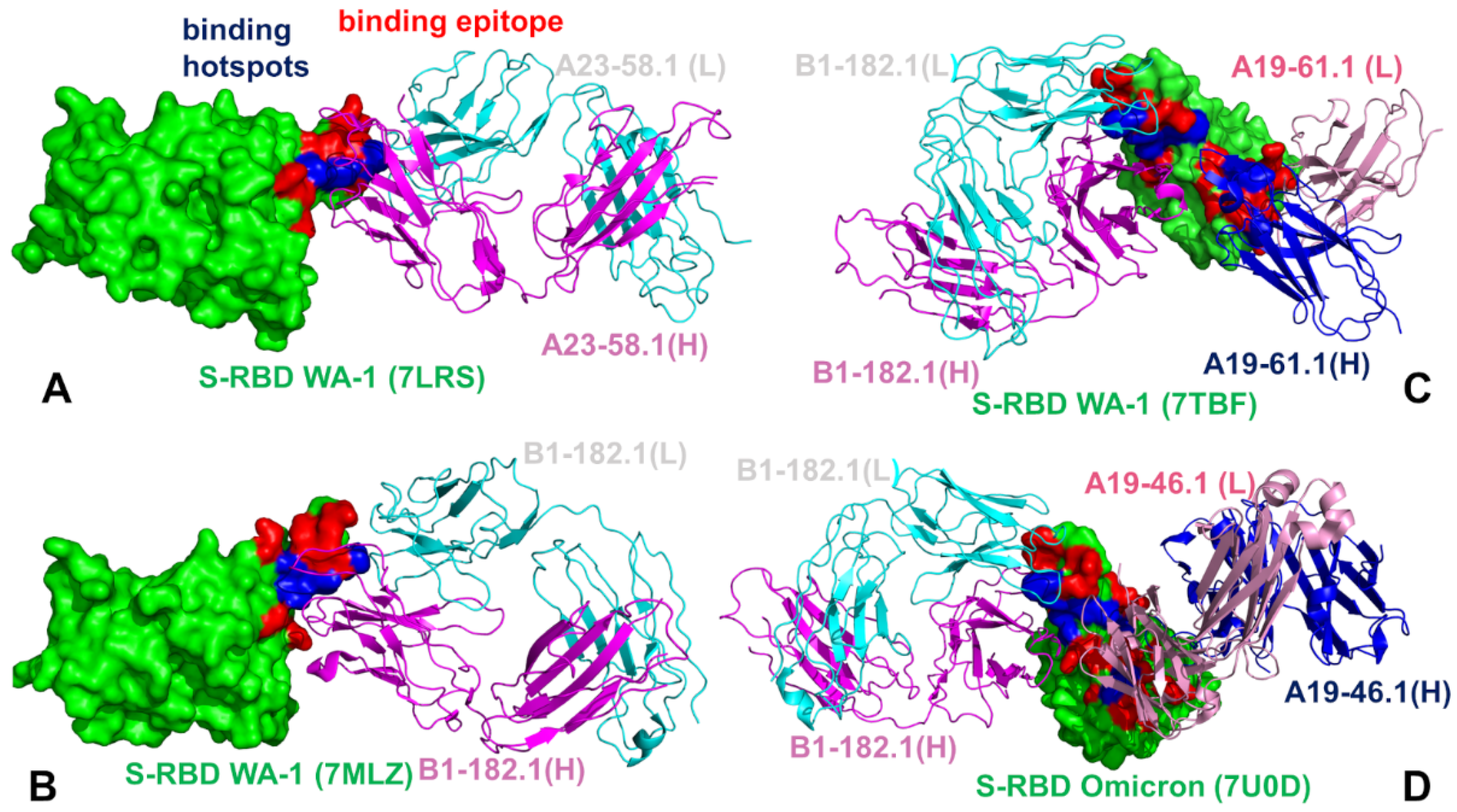
3.1. Structural Analysis of the S-RBD Complexes with Antibodies
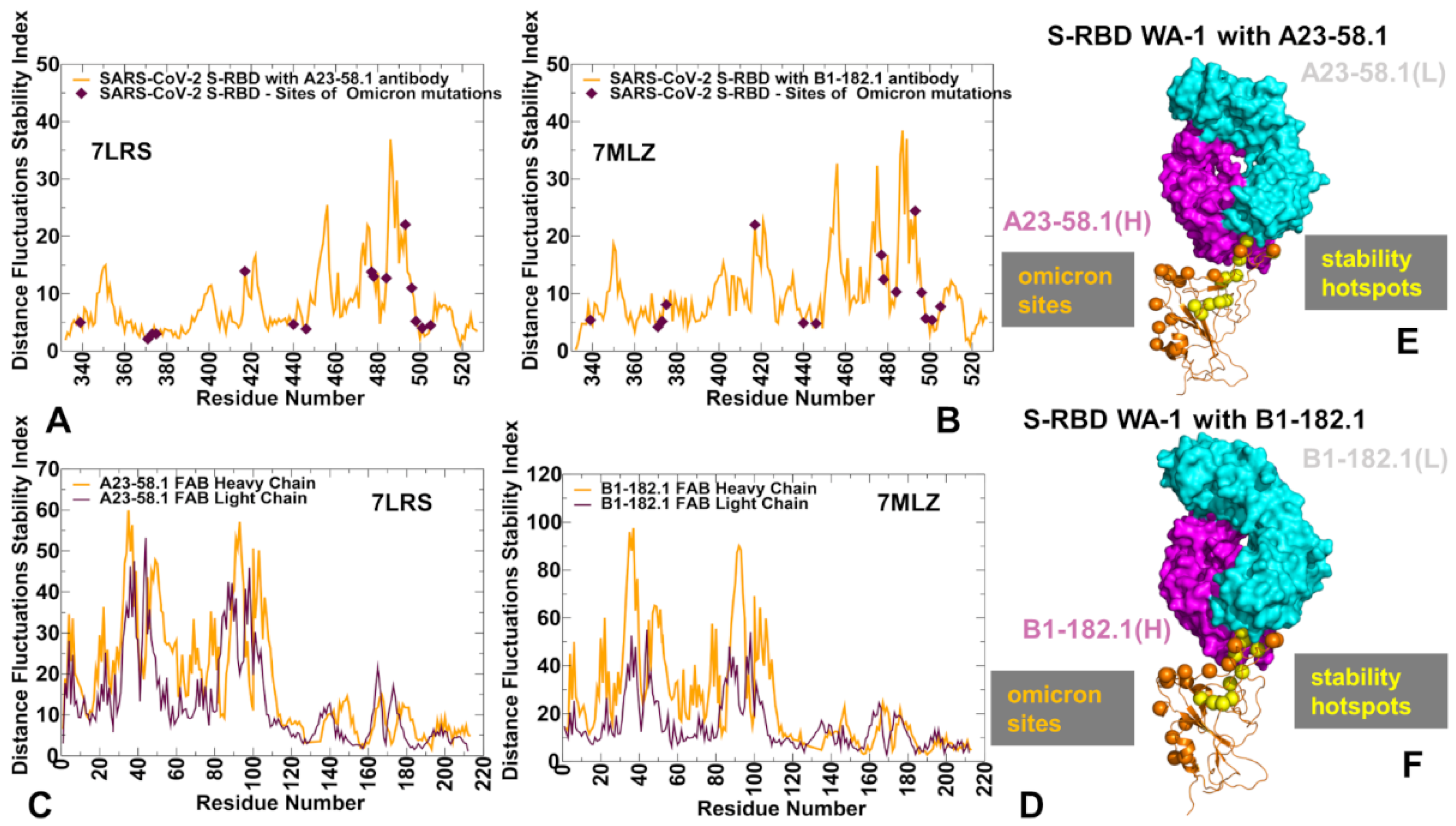
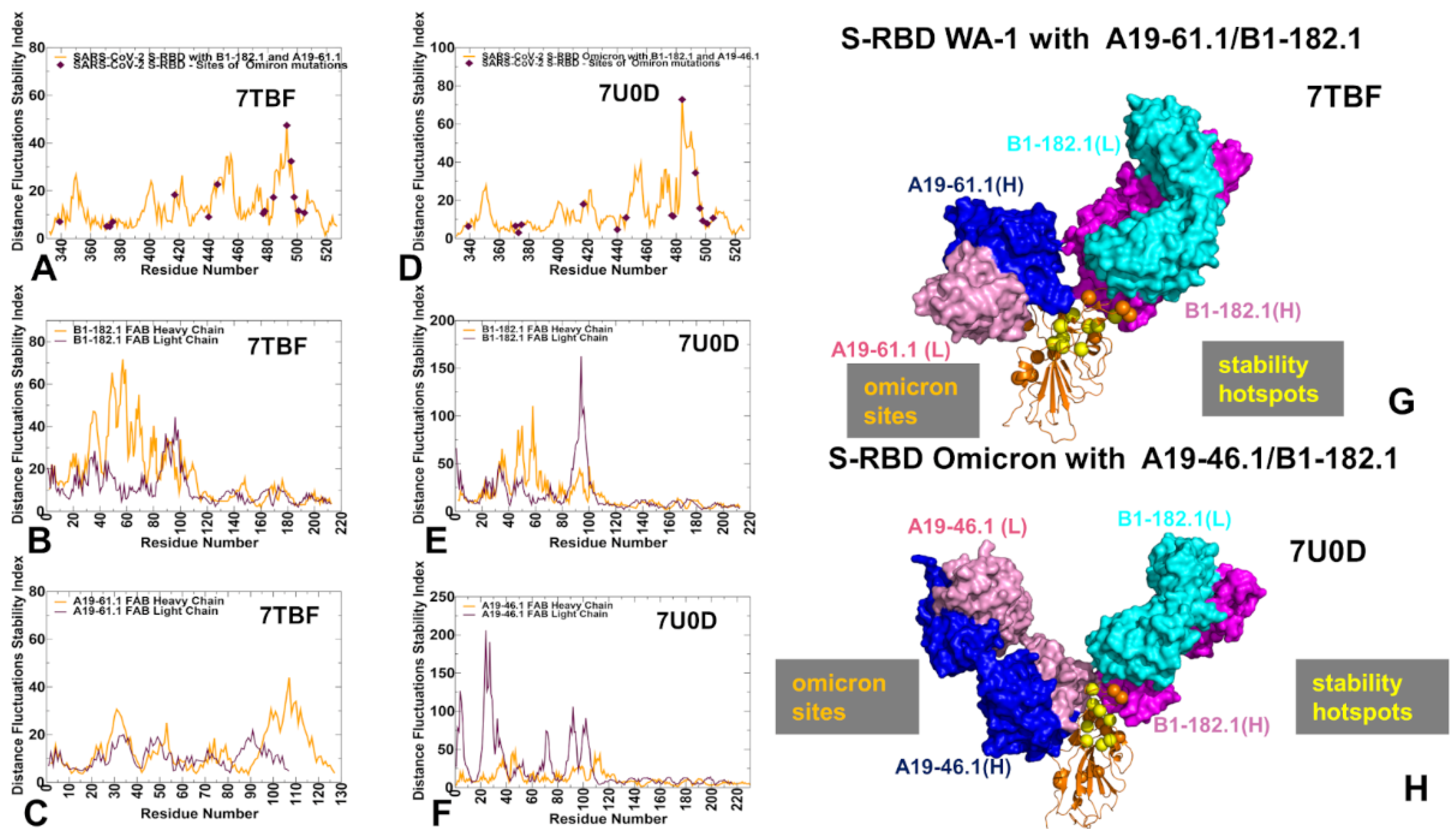
3.2. MD Simulations and Distance Fluctuation Analysis of Conformational Ensembles of the S-RBD Complexes with Antibodies Reveal Specific Dynamic Signatures and Stability Centers
3.3. Ensemble-Based Mutational Scanning and Energetic Cartography Identifies Binding Affinity Hotspots in the SARS-CoV-2 RBD Complexes with Ultrapotent Antibodies
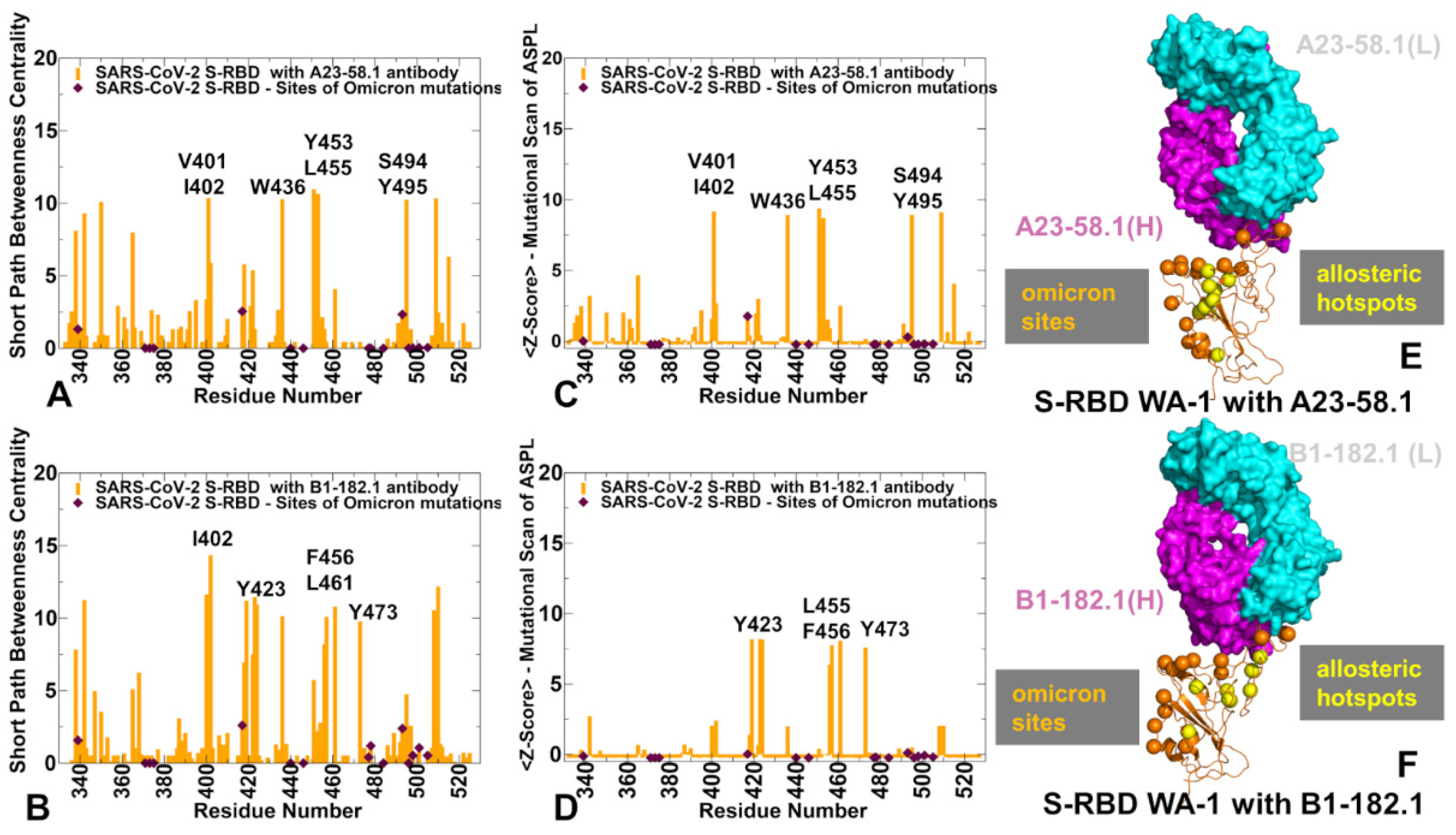
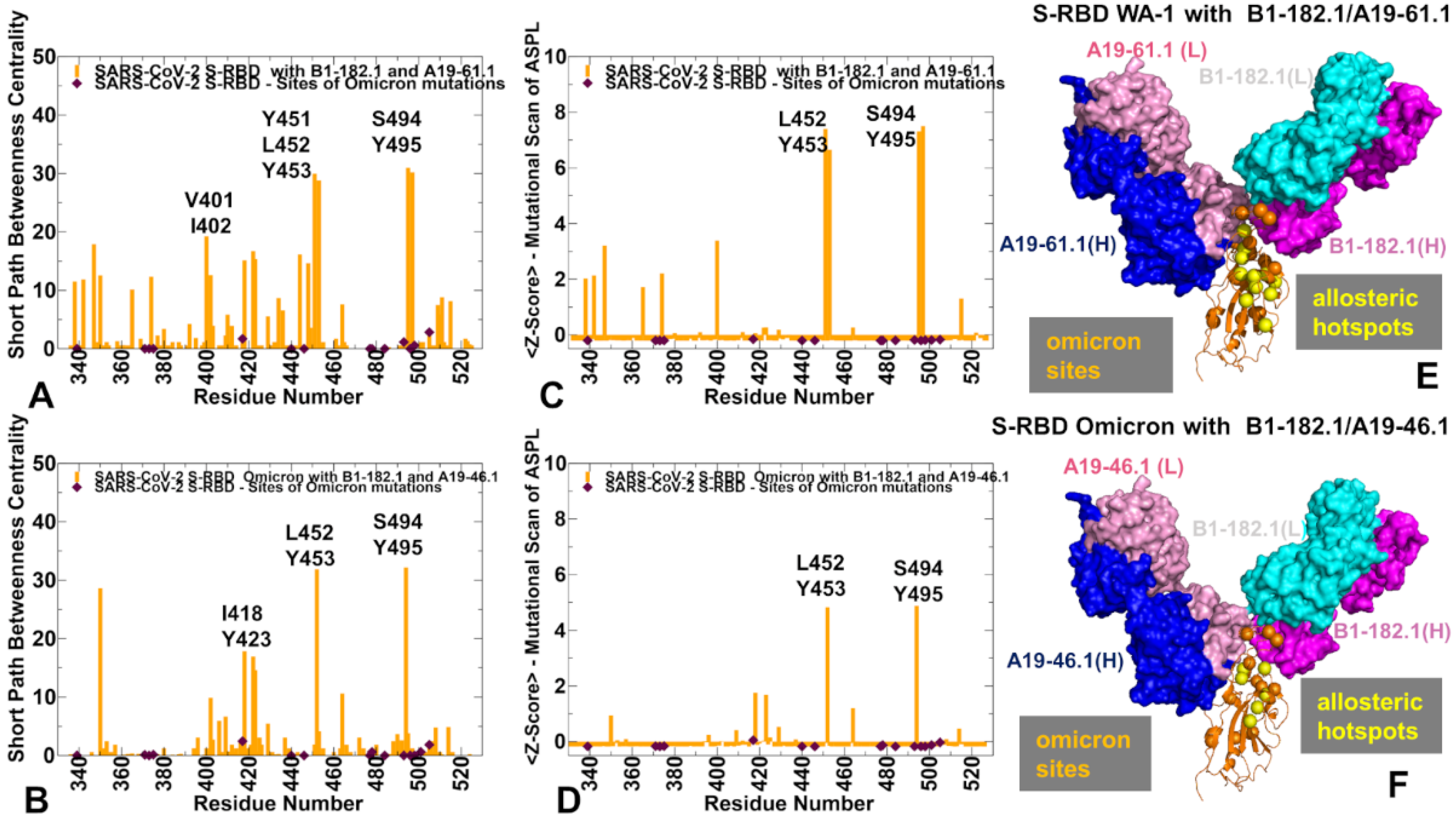
3.4. Allosteric Mutational Profiling of the Interaction Networks in the S-RBD Complexes Discern Sites and Mechanisms of Mutational Escape
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501–1505. [Google Scholar] [CrossRef]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.M.C.; Kopp, M.; Li, D.; Parks, R.; Hsu, A.L.; et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. [Google Scholar] [CrossRef]
- McCallum, M.; Walls, A.C.; Bowen, J.E.; Corti, D.; Veesler, D. Structure-guided covalent stabilization of coronavirus spike glycoprotein trimers in the closed conformation. Nat. Struct. Mol. Biol. 2020, 27, 942–949. [Google Scholar] [CrossRef]
- Xiong, X.; Qu, K.; Ciazynska, K.A.; Hosmillo, M.; Carter, A.P.; Ebrahimi, S.; Ke, Z.; Scheres, S.H.W.; Bergamaschi, L.; Grice, G.L.; et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020, 27, 934–941. [Google Scholar] [CrossRef]
- Costello, S.M.; Shoemaker, S.R.; Hobbs, H.T.; Nguyen, A.W.; Hsieh, C.L.; Maynard, J.A.; McLellan, J.S.; Pak, J.E.; Marqusee, S. The SARS-CoV-2 spike reversibly samples an open-trimer conformation exposing novel epitopes. Nat. Struct. Mol. Biol. 2022, 27, 229–238. [Google Scholar] [CrossRef]
- McCormick, K.D.; Jacobs, J.L.; Mellors, J.W. The emerging plasticity of SARS-CoV-2. Science 2021, 371, 1306–1308. [Google Scholar] [CrossRef]
- Ghimire, D.; Han, Y.; Lu, M. Structural Plasticity and Immune Evasion of SARS-CoV-2 Spike Variants. Viruses 2022, 14, 1255. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Turoňová, B.; Sikora, M.; Schürmann, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; von Bülow, S.; Gecht, M.; Bagola, K.; Hörner, C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. [Google Scholar] [CrossRef]
- Lu, M.; Uchil, P.D.; Li, W.; Zheng, D.; Terry, D.S.; Gorman, J.; Shi, W.; Zhang, B.; Zhou, T.; Ding, S.; et al. Real-time conformational dynamics of SARS-CoV-2 spikes on virus particles. Cell Host Microbe 2020, 28, 880–891.e8. [Google Scholar] [CrossRef]
- Yang, Z.; Han, Y.; Ding, S.; Shi, W.; Zhou, T.; Finzi, A.; Kwong, P.D.; Mothes, W.; Lu, M. SARS-CoV-2 Variants Increase Kinetic Stability of Open Spike Conformations as an Evolutionary Strategy. mBio 2022, 13, e0322721. [Google Scholar] [CrossRef]
- Díaz-Salinas, M.A.; Li, Q.; Ejemel, M.; Yurkovetskiy, L.; Luban, J.; Shen, K.; Wang, Y.; Munro, J.B. Conformational dynamics and allosteric modulation of the SARS-CoV-2 spike. eLife 2022, 11, e75433. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking changes in SARS-CoV-2 Spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H., 3rd; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Muik, A.; Wallisch, A.K.; Sänger, B.; Swanson, K.A.; Mühl, J.; Chen, W.; Cai, H.; Maurus, D.; Sarkar, R.; Türeci, Ö.; et al. Neutralization of SARS-CoV-2 lineage B.1.1.7 pseudovirus by BNT162b2 vaccine-elicited human sera. Science 2021, 371, 1152–1153. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Lessells, R.J.; Giandhari, J.; Pillay, S.; Msomi, N.; Mlisana, K.; Bhiman, J.N.; von Gottberg, A.; Walaza, S.; et al. Sixteen novel lineages of SARS-CoV-2 in South Africa. Nat. Med. 2021, 27, 440–446. [Google Scholar] [CrossRef]
- Hogan, C.A.; Jassem, A.N.; Sbihi, H.; Joffres, Y.; Tyson, J.R.; Noftall, K.; Taylor, M.; Lee, T.; Fjell, C.; Wilmer, A.; et al. Rapid Increase in SARS-CoV-2 P.1 Lineage Leading to Codominance with B.1.1.7 Lineage, British Columbia, Canada, January-April 2021. Emerg. Infect. Dis. 2021, 27, 2802–2809. [Google Scholar] [CrossRef]
- Javanmardi, K.; Chou, C.W.; Terrace, C.I.; Annapareddy, A.; Kaoud, T.S.; Guo, Q.; Lutgens, J.; Zorkic, H.; Horton, A.P.; Gardner, E.C.; et al. Rapid characterization of spike variants via mammalian cell surface display. Mol. Cell. 2021, 81, 5099–5111.e8. [Google Scholar] [CrossRef]
- Tortorici, M.A.; Czudnochowski, N.; Starr, T.N.; Marzi, R.; Walls, A.C.; Zatta, F.; Bowen, J.E.; Jaconi, S.; Di Iulio, J.; Wang, Z.; et al. Broad sarbecovirus neutralization by a human monoclonal antibody. Nature 2021, 597, 103–108. [Google Scholar] [CrossRef]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.H.; Porrot, F.; Staropoli, I.; Lemoine, F.; et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature 2022, 602, 671–675. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Huo, J.; Zhou, D.; Zahradník, J.; Supasa, P.; Liu, C.; Duyvesteyn, H.M.E.; Ginn, H.M.; Mentzer, A.J.; Tuekprakhon, A.; et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell 2022, 185, 467–484.e415. [Google Scholar] [CrossRef]
- Cameroni, E.; Bowen, J.E.; Rosen, L.E.; Saliba, C.; Zepeda, S.K.; Culap, K.; Pinto, D.; VanBlargan, L.A.; De Marco, A.; di Iulio, J.; et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature 2022, 602, 664–670. [Google Scholar] [CrossRef]
- Barton, M.I.; MacGowan, S.A.; Kutuzov, M.A.; Dushek, O.; Barton, G.J.; van der Merwe, P.A. Effects of common mutations in the SARS-CoV-2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. eLife 2021, 10, e70658. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef]
- Liu, L.; Iketani, S.; Guo, Y.; Chan, J.F.; Wang, M.; Liu, L.; Luo, Y.; Chu, H.; Huang, Y.; Nair, M.S.; et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature 2022, 602, 676–681. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Li, X.; Peng, Q.; Su, C.; Huang, B.; et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein-ACE2 complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef]
- Hong, Q.; Han, W.; Li, J.; Xu, S.; Wang, Y.; Xu, C.; Li, Z.; Wang, Y.; Zhang, C.; Huang, Z.; et al. Molecular basis of receptor binding and antibody neutralization of Omicron. Nature 2022, 604, 546–552. [Google Scholar] [CrossRef]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef]
- Yin, W.; Xu, Y.; Xu, P.; Cao, X.; Wu, C.; Gu, C.; He, X.; Wang, X.; Huang, S.; Yuan, Q.; et al. Structures of the Omicron Spike trimer with ACE2 and an anti-Omicron antibody. Science 2022, 375, 1048–1053. [Google Scholar] [CrossRef]
- Gobeil, S.M.; Henderson, R.; Stalls, V.; Janowska, K.; Huang, X.; May, A.; Speakman, M.; Beaudoin, E.; Manne, K.; Li, D.; et al. Structural diversity of the SARS-CoV-2 Omicron spike. Mol. Cell 2022, 82, 2050–2068.e6. [Google Scholar] [CrossRef]
- Koehler, M.; Ray, A.; Moreira, R.A.; Juniku, B.; Poma, A.B.; Alsteens, D. Molecular insights into receptor binding energetics and neutralization of SARS-CoV-2 variants. Nat. Commun. 2021, 12, 6977. [Google Scholar] [CrossRef]
- Bauer, M.S.; Gruber, S.; Hausch, A.; Gomes, P.S.F.C.; Milles, L.F.; Nicolaus, T.; Schendel, L.C.; Navajas, P.L.; Procko, E.; Lietha, D.; et al. A tethered ligand assay to probe SARS-CoV-2:ACE2 interactions. Proc. Natl. Acad. Sci. USA 2022, 119, e2114397119. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, Y.; Fei, P.; Zhang, T.; Yao, D.; Gao, Y.; Liu, J.; Chen, H.; Lu, Q.; Mudianto, T.; et al. Mechanical activation of spike fosters SARS-CoV-2 viral infection. Cell Res. 2021, 31, 1047–1060. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Zhou, D.; Ginn, H.M.; Duyvesteyn, H.M.E.; Supasa, P.; Case, J.B.; Zhao, Y.; Walter, T.S.; Mentzer, A.J.; Liu, C.; et al. The antigenic anatomy of SARS-CoV-2 receptor binding domain. Cell 2021, 184, 2183–2200.e2122. [Google Scholar] [CrossRef]
- Hastie, K.M.; Li, H.; Bedinger, D.; Schendel, S.L.; Dennison, S.M.; Li, K.; Rayaprolu, V.; Yu, X.; Mann, C.; Zandonatti, M.; et al. Defining variant-resistant epitopes targeted by SARS-CoV-2 antibodies: A global consortium study. Science 2021, 374, 472–478. [Google Scholar] [CrossRef]
- Mittal, A.; Khattri, A.; Verma, V. Structural and antigenic variations in the spike protein of emerging SARS-CoV-2 variants. PLoS Pathog. 2022, 18, e1010260. [Google Scholar] [CrossRef]
- Deshpande, A.; Harris, B.D.; Martinez-Sobrido, L.; Kobie, J.J.; Walter, M.R. Epitope Classification and RBD Binding Properties of Neutralizing Antibodies Against SARS-CoV-2 Variants of Concern. Front. Immunol. 2021, 12, 691715. [Google Scholar] [CrossRef]
- Yi, C.; Sun, X.; Lin, Y.; Gu, C.; Ding, L.; Lu, X.; Yang, Z.; Zhang, Y.; Ma, L.; Gu, W.; et al. Comprehensive mapping of binding hot spots of SARS-CoV-2 RBD-specific neutralizing antibodies for tracking immune escape variants. Genome Med. 2021, 13, 164. [Google Scholar] [CrossRef]
- Pinto, D.; Park, Y.J.; Beltramello, M.; Walls, A.C.; Tortorici, M.A.; Bianchi, S.; Jaconi, S.; Culap, K.; Zatta, F.; De Marco, A.; et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 2020, 583, 290–295. [Google Scholar] [CrossRef]
- Tortorici, M.A.; Beltramello, M.; Lempp, F.A.; Pinto, D.; Dang, H.V.; Rosen, L.E.; McCallum, M.; Bowen, J.; Minola, A.; Jaconi, S.; et al. Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms. Science 2020, 370, 950–957. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, T.; Zhang, Y.; Yang, E.S.; Schramm, C.A.; Shi, W.; Pegu, A.; Oloniniyi, O.K.; Henry, A.R.; Darko, S.; et al. Ultrapotent antibodies against diverse and highly transmissible SARS-CoV-2 variants. Science 2021, 373, eabh1766. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Wang, L.; Misasi, J.; Pegu, A.; Zhang, Y.; Harris, D.R.; Olia, A.S.; Talana, C.A.; Yang, E.S.; Chen, M.; et al. Structural basis for potent antibody neutralization of SARS-CoV-2 variants including B.1.1.529. Science 2022, 376, eabn8897. [Google Scholar] [CrossRef] [PubMed]
- Windsor, I.W.; Tong, P.; Lavidor, O.; Sanjari Moghaddam, A.; McKay, L.G.A.; Gautam, A.; Chen, Y.; MacDonald, E.A.; Yoo, D.K.; Griffiths, A.; et al. Antibodies induced by ancestral SARS-CoV-2 strain that cross-neutralize variants from Alpha to Omicron BA.1. Sci. Immunol. 2022, eabo3425. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.; Wang, X.; He, X.; Zhao, X.; Zhang, Y.; Jiang, Y.; Li, M.; Cui, Y.; Chen, Y.; Qiao, R.; et al. Antibody evasion of SARS-CoV-2 Omicron BA.1, BA.1.1, BA.2, and BA.3 sub-lineages. Cell Host Microbe. 2022, S1931-3128(22)00243-8. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhan, W.; Yang, Z.; Tu, C.; Hu, G.; Zhang, X.; Song, W.; Du, S.; Zhu, Y.; Huang, K.; et al. Broad neutralization of SARS-CoV-2 variants by an inhalable bispecific single-domain antibody. Cell 2022, 185, 1389–1401.e18. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef]
- Sztain, T.; Ahn, S.H.; Bogetti, A.T.; Casalino, L.; Goldsmith, J.A.; Seitz, E.; McCool, R.S.; Kearns, F.L.; Acosta-Reyes, F.; Maji, S.; et al. A glycan gate controls opening of the SARS-CoV-2 spike protein. Nat. Chem. 2021, 13, 963–968. [Google Scholar] [CrossRef]
- Sikora, M.; von Bülow, S.; Blanc, F.E.C.; Gecht, M.; Covino, R.; Hummer, G. Computational epitope map of SARS-CoV-2 spike protein. PLoS Comput. Biol. 2021, 17, e1008790. [Google Scholar] [CrossRef]
- Zimmerman, M.I.; Porter, J.R.; Ward, M.D.; Singh, S.; Vithani, N.; Meller, A.; Mallimadugula, U.L.; Kuhn, C.E.; Borowsky, J.H.; Wiewiora, R.P.; et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021, 13, 651–659. [Google Scholar] [CrossRef]
- Ray, D.; Le, L.; Andricioaei, I. Distant residues modulate conformational opening in SARS-CoV-2 spike protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2100943118. [Google Scholar] [CrossRef] [PubMed]
- Fallon, L.; Belfon, K.A.A.; Raguette, L.; Wang, Y.; Stepanenko, D.; Cuomo, A.; Guerra, J.; Budhan, S.; Varghese, S.; Corbo, C.P.; et al. Free Energy Landscapes from SARS-CoV-2 Spike Glycoprotein Simulations Suggest that RBD Opening can be Modulated via Interactions in an Allosteric Pocket. J. Am. Chem. Soc. 2021, 143, 11349–11360. [Google Scholar] [CrossRef] [PubMed]
- Mansbach, R.A.; Chakraborty, S.; Nguyen, K.; Montefiori, D.C.; Korber, B.; Gnanakaran, S. The SARS-CoV-2 Spike variant D614G favors an open conformational state. Sci. Adv. 2021, 7, eabf3671. [Google Scholar] [CrossRef]
- Mori, T.; Jung, J.; Kobayashi, C.; Dokainish, H.M.; Re, S.; Sugita, Y. Elucidation of interactions regulating conformational stability and dynamics of SARS-CoV-2 S-protein. Biophys. J. 2021, 120, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Dokainish, H.M.; Re, S.; Mori, T.; Kobayashi, C.; Jung, J.; Sugita, Y. The inherent flexibility of receptor binding domains in SARS-CoV-2 spike protein. eLife 2022, 11, e75720. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G.M. Molecular simulations and network modeling reveal an allosteric signaling in the SARS-CoV-2 spike proteins. J. Proteome Res. 2020, 19, 4587–4608. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Di Paola, L. Dynamic Network Modeling of Allosteric Interactions and Communication Pathways in the SARS-CoV-2 Spike Trimer Mutants: Differential Modulation of Conformational Landscapes and Signal Transmission via Cascades of Regulatory Switches. J. Phys. Chem. B 2021, 125, 850–873. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Di Paola, L. Integrated Biophysical Modeling of the SARS-CoV-2 Spike Protein Binding and Allosteric Interactions with Antibodies. J. Phys. Chem. B 2021, 125, 4596–4619. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Comparative Perturbation-Based Modeling of the SARS-CoV-2 Spike Protein Binding with Host Receptor and Neutralizing Antibodies: Structurally Adaptable Allosteric Communication Hotspots Define Spike Sites Targeted by Global Circulating Mutations. Biochemistry 2021, 60, 1459–1484. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Dynamic Profiling of Binding and Allosteric Propensities of the SARS-CoV-2 Spike Protein with Different Classes of Antibodies: Mutational and Perturbation-Based Scanning Reveals the Allosteric Duality of Functionally Adaptable Hotspots. J. Chem. Theory Comput. 2021, 17, 4578–4598. [Google Scholar] [CrossRef]
- Verkhivker, G.; Agajanian, S.; Kassab, R.; Krishnan, K. Landscape-Based Protein Stability Analysis and Network Modeling of Multiple Conformational States of the SARS-CoV-2 Spike D614G Mutant: Conformational Plasticity and Frustration-Induced Allostery as Energetic Drivers of Highly Transmissible Spike Variants. J. Chem. Inf. Model. 2022, 62, 1956–1978. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Allosteric Control of Structural Mimicry and Mutational Escape in the SARS-CoV-2 Spike Protein Complexes with the ACE2 Decoys and Miniprotein Inhibitors: A Network-Based Approach for Mutational Profiling of Binding and Signaling. J. Chem. Inf. Model. 2021, 61, 5172–5191. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; He, X.; Ren, Y.; Wang, Z.; Zhou, H.; Fan, S.; Zhu, C.; Liu, D.; Shao, B.; Liu, T.Y.; et al. Structural insights into the SARS-CoV-2 Omicron RBD-ACE2 interaction. Cell Res. 2022, 32, 593–595. [Google Scholar] [CrossRef] [PubMed]
- Omotuyi, O.; Olubiyi, O.; Nash, O.; Afolabi, E.; Oyinloye, B.; Fatumo, S.; Femi-Oyewo, M.; Bogoro, S. SARS-CoV-2 Omicron spike glycoprotein receptor binding domain exhibits super-binder ability with ACE2 but not convalescent monoclonal antibody. Comput. Biol Med. 2022, 142, 105226. [Google Scholar] [CrossRef]
- Shah, M.; Woo, H.G. Omicron: A Heavily Mutated SARS-CoV-2 Variant Exhibits Stronger Binding to ACE2 and Potently Escapes Approved COVID-19 Therapeutic Antibodies. Front. Immunol. 2022, 12, 830527. [Google Scholar] [CrossRef]
- Hossen, M.L.; Baral, P.; Sharma, T.; Gerstman, B.; Chapagain, P. Significance of the RBD mutations in the SARS-CoV-2 omicron: From spike opening to antibody escape and cell attachment. Phys. Chem. Chem. Phys. 2022, 24, 9123–9129. [Google Scholar] [CrossRef]
- Barre, A.; Klonjkowski, B.; Benoist, H.; Rougé, P. How Do Point Mutations Enhancing the Basic Character of the RBDs of SARS-CoV-2 Variants Affect Their Transmissibility and Infectivity Capacities? Viruses 2022, 14, 783. [Google Scholar] [CrossRef]
- Rochman, N.D.; Faure, G.; Wolf, Y.I.; Freddolino, P.L.; Zhang, F.; Koonin, E.V. Epistasis at the SARS-CoV-2 Receptor-Binding Domain Interface and the Propitiously Boring Implications for Vaccine Escape. mBio 2022, 13, e0013522. [Google Scholar] [CrossRef]
- Verkhivker, G.; Agajanian, S.; Kassab, R.; Krishnan, K. Computer Simulations and Network-Based Profiling of Binding and Allosteric Interactions of SARS-CoV-2 Spike Variant Complexes and the Host Receptor: Dissecting the Mechanistic Effects of the Delta and Omicron Mutations. Int. J. Mol. Sci. 2022, 23, 4376. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlic, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [CrossRef]
- Hekkelman, M.L.; Te Beek, T.A.; Pettifer, S.R.; Thorne, D.; Attwood, T.K.; Vriend, G. WIWS: A protein structure bioinformatics web service collection. Nucleic Acids Res. 2010, 38, W719–W723. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Sali, A. ModLoop: Automated modeling of loops in protein structures. Bioinformatics 2003, 19, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Fuentes, N.; Zhai, J.; Fiser, A. ArchPRED: A template based loop structure prediction server. Nucleic Acids Res. 2006, 34, W173–W176. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Lee, D.; Park, H.; Coutsias, E.A.; Lee, J.; Seok, C. The FALC-Loop web server for protein loop modeling. Nucleic Acids Res. 2011, 39, W210–W214. [Google Scholar] [CrossRef] [PubMed]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 2009, 77, 778–795. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, H.S.; Sousa, S.F.; Cerqueira, N.M.F.S.A. VMD Store-A VMD Plugin to Browse, Discover, and Install VMD Extensions. J. Chem. Inf. Model. 2019, 59, 4519–4523. [Google Scholar] [CrossRef]
- Di Pierro, M.; Elber, R.; Leimkuhler, B. A Stochastic Algorithm for the Isobaric-Isothermal Ensemble with Ewald Summations for All Long Range Forces. J. Chem. Theory Comput. 2015, 11, 5624–5637. [Google Scholar] [CrossRef] [Green Version]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Davidchack, R.L.; Handel, R.; Tretyakov, M.V. Langevin thermostat for rigid body dynamics. J. Chem. Phys. 2009, 130, 234101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacquin-Mora, S.; Lavery, R. Investigating the local flexibility of functional residues in hemoproteins. Biophys. J. 2006, 90, 2706–2717. [Google Scholar] [CrossRef] [Green Version]
- Sacquin-Mora, S.; Laforet, E.; Lavery, R. Locating the active sites of enzymes using mechanical properties. Proteins 2007, 67, 350–359. [Google Scholar] [CrossRef]
- Bocahut, A.; Bernad, S.; Sebban, P.; Sacquin-Mora, S. Frontier residues lining globin internal cavities present specific mechanical properties. J. Am. Chem. Soc. 2011, 133, 8753–8761. [Google Scholar] [CrossRef] [PubMed]
- Sacquin-Mora, S. Motions and mechanics: Investigating conformational transitions in multi-domain proteins with coarse-grain simulations. Mol. Simul. 2014, 40, 229–236. [Google Scholar] [CrossRef]
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of changes in protein-protein binding affinity on mutations. Nucleic Acids Res. 2013, 41, W333–W339. [Google Scholar] [CrossRef] [Green Version]
- Dehouck, Y.; Gilis, D.; Rooman, M. A new generation of statistical potentials for proteins. Biophys. J. 2006, 90, 4010–4017. [Google Scholar] [CrossRef]
- Dehouck, Y.; Grosfils, A.; Folch, B.; Gilis, D.; Bogaerts, P.; Rooman, M. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics 2009, 25, 2537–2543. [Google Scholar] [CrossRef] [Green Version]
- Brinda, K.V.; Vishveshwara, S. A network representation of protein structures: Implications for protein stability. Biophys. J. 2005, 89, 4159–4170. [Google Scholar] [CrossRef] [Green Version]
- Vijayabaskar, M.S.; Vishveshwara, S. Interaction energy based protein structure networks. Biophys. J. 2010, 99, 3704–3715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 6620–6625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piovesan, D.; Minervini, G.; Tosatto, S.C. The RING 2.0 Web Server for High Quality Residue Interaction Networks. Nucleic Acids Res. 2016, 44, W367–W374. [Google Scholar] [CrossRef]
- Clementel, D.; Del Conte, A.; Monzon, A.M.; Camagni, G.F.; Minervini, G.; Piovesan, D.; Tosatto, S.C.E. RING 3.0: Fast generation of probabilistic residue interaction networks from structural ensembles. Nucleic Acids Res. 2022, 50, W651–W656. [Google Scholar] [CrossRef]
- Hagberg, A.A.; Schult, D.A.; Swart, P.J. Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science Conference (SciPy2008), Pasadena, CA, USA, 19–24 August 2008; Varoquaux, G., Vaught, T., Millman, J., Eds.; Scientific Research: Atlanta, GA, USA, 2011; pp. 11–15. [Google Scholar]
- del Sol, A.; Fujihashi, H.; Amoros, D.; Nussinov, R. Residues crucial for maintaining short paths in network communication mediate signaling in proteins. Mol. Syst. Biol. 2006, 2, 2006-0019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brysbaert, G.; Mauri, T.; Lensink, M.F. Comparing protein structures with RINspector automation in Cytoscape. F1000Research 2018, 7, 563. [Google Scholar] [CrossRef]
- Vangone, A.; Bonvin, A.M. Contacts-based prediction of binding affinity in protein-protein complexes. eLife 2015, 4, e07454. [Google Scholar] [CrossRef]
- Nguyen, H.; Lan, P.D.; Nissley, D.A.; O’Brien, E.P.; Li, M.S. Electrostatic Interactions Explain the Higher Binding Affinity of the CR3022 Antibody for SARS-CoV-2 than the 4A8 Antibody. J. Phys. Chem. B 2021, 125, 7368–7379. [Google Scholar] [CrossRef]
- Ridgway, H.; Chasapis, C.T.; Kelaidonis, K.; Ligielli, I.; Moore, G.J.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; Mavromoustakos, T.; Matsoukas, J.M. Understanding the Driving Forces That Trigger Mutations in SARS-CoV-2: Mutational Energetics and the Role of Arginine Blockers in COVID-19 Therapy. Viruses 2022, 14, 1029. [Google Scholar] [CrossRef]
- Gan, H.H.; Twaddle, A.; Marchand, B.; Gunsalus, K.C. Structural Modeling of the SARS-CoV-2 Spike/Human ACE2 Complex Interface can Identify High-Affinity Variants Associated with Increased Transmissibility. J. Mol Biol. 2021, 433, 167051. [Google Scholar] [CrossRef]
- Gan, H.H.; Zinno, J.; Piano, F.; Gunsalus, K.C. Omicron Spike protein has a positive electrostatic surface that promotes ACE2 recognition and antibody escape. bioRxiv 2022. [Google Scholar] [CrossRef]
- Du, W.; Hurdiss, D.L.; Drabek, D.; Mykytyn, A.Z.; Kaiser, F.K.; González-Hernández, M.; Muñoz-Santos, D.; Lamers, M.M.; van Haperen, R.; Li, W.; et al. An ACE2-blocking antibody confers broad neutralization and protection against Omicron and other SARS-CoV-2 variants of concern. Sci. Immunol. 2022, eabp9312. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Jia, Z.; Bao, L.; Wang, L.; Cao, L.; Chi, H.; Hu, Y.; Li, Q.; Zhou, Y.; Jiang, Y.; et al. Memory B cell repertoire from triple vaccinees against diverse SARS-CoV-2 variants. Nature 2022, 603, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fu, W.; Bao, L.; Jia, Z.; Zhang, Y.; Zhou, Y.; Wu, W.; Wu, J.; Zhang, Q.; Gao, Y.; et al. Selection and structural bases of potent broadly neutralizing antibodies from 3-dose vaccinees that are highly effective against diverse SARS-CoV-2 variants, including Omicron sublineages. Cell Res. 2022, 32, 691–694. [Google Scholar] [CrossRef]
- Tsai, K.C.; Lee, Y.C.; Tseng, T.S. Comprehensive Deep Mutational Scanning Reveals the Immune-Escaping Hotspots of SARS-CoV-2 Receptor-Binding Domain Targeting Neutralizing Antibodies. Front. Microbiol. 2021, 12, 698365. [Google Scholar] [CrossRef]
- Niu, L.; Wittrock, K.N.; Clabaugh, G.C.; Srivastava, V.; Cho, M.W. A Structural Landscape of Neutralizing Antibodies Against SARS-CoV-2 Receptor Binding Domain. Front. Immunol. 2021, 12, 647934. [Google Scholar] [CrossRef]
- Alenquer, M.; Ferreira, F.; Lousa, D.; Valério, M.; Medina-Lopes, M.; Bergman, M.L.; Gonçalves, J.; Demengeot, J.; Leite, R.B.; Lilue, J.; et al. Signatures in SARS-CoV-2 spike protein conferring escape to neutralizing antibodies. PLoS Pathog. 2021, 17, e1009772. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB | System | 1 Simulation | # Simulations |
|---|---|---|---|
| 7LRS | RBD/A23-58.1 | 500 ns | 10 |
| 7MLZ | RBD/B1-182.1 | 500 ns | 10 |
| 7TBF | RBD/A19-61.1/B1-182.1 | 500 ns | 10 |
| 7U0D | RBD Omicron/A19-46.1/B1-182.1 | 500 ns | 10 |
| Interfacial Contacts | RBD A23-58.1 | RBD B1-182.1 | RBD A19-61.1/B1-182.1 | RBD Omicron A19-46.1/B1-182.1 |
|---|---|---|---|---|
| Charged–charged | 3 | 4 | 7 | 8 |
| Charged–polar | 7 | 8 | 10 | 13 |
| Charged–apolar | 2 | 5 | 17 | 11 |
| Polar–polar | 3 | 4 | 6 | 3 |
| Polar–apolar | 13 | 21 | 27 | 26 |
| Apolar–apolar | 22 | 24 | 39 | 35 |
| ΔG comput. (kcal/mol) | −8.5 | −10.3 | −12.9 | −12.6 |
| Kd (nM) experiment | 7.3 | 2.55 | 2.33 (A19-61.1) | 3.58 (A19-46.1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verkhivker, G.; Agajanian, S.; Kassab, R.; Krishnan, K. Integrating Conformational Dynamics and Perturbation-Based Network Modeling for Mutational Profiling of Binding and Allostery in the SARS-CoV-2 Spike Variant Complexes with Antibodies: Balancing Local and Global Determinants of Mutational Escape Mechanisms. Biomolecules 2022, 12, 964. https://doi.org/10.3390/biom12070964
Verkhivker G, Agajanian S, Kassab R, Krishnan K. Integrating Conformational Dynamics and Perturbation-Based Network Modeling for Mutational Profiling of Binding and Allostery in the SARS-CoV-2 Spike Variant Complexes with Antibodies: Balancing Local and Global Determinants of Mutational Escape Mechanisms. Biomolecules. 2022; 12(7):964. https://doi.org/10.3390/biom12070964
Chicago/Turabian StyleVerkhivker, Gennady, Steve Agajanian, Ryan Kassab, and Keerthi Krishnan. 2022. "Integrating Conformational Dynamics and Perturbation-Based Network Modeling for Mutational Profiling of Binding and Allostery in the SARS-CoV-2 Spike Variant Complexes with Antibodies: Balancing Local and Global Determinants of Mutational Escape Mechanisms" Biomolecules 12, no. 7: 964. https://doi.org/10.3390/biom12070964
APA StyleVerkhivker, G., Agajanian, S., Kassab, R., & Krishnan, K. (2022). Integrating Conformational Dynamics and Perturbation-Based Network Modeling for Mutational Profiling of Binding and Allostery in the SARS-CoV-2 Spike Variant Complexes with Antibodies: Balancing Local and Global Determinants of Mutational Escape Mechanisms. Biomolecules, 12(7), 964. https://doi.org/10.3390/biom12070964