


The Role of Disordered Regions in Orchestrating the Properties of Multidomain Proteins: The SARS-CoV-2 Nucleocapsid Protein and Its Interaction with Enoxaparin

 ,
,  and
and 
Abstract
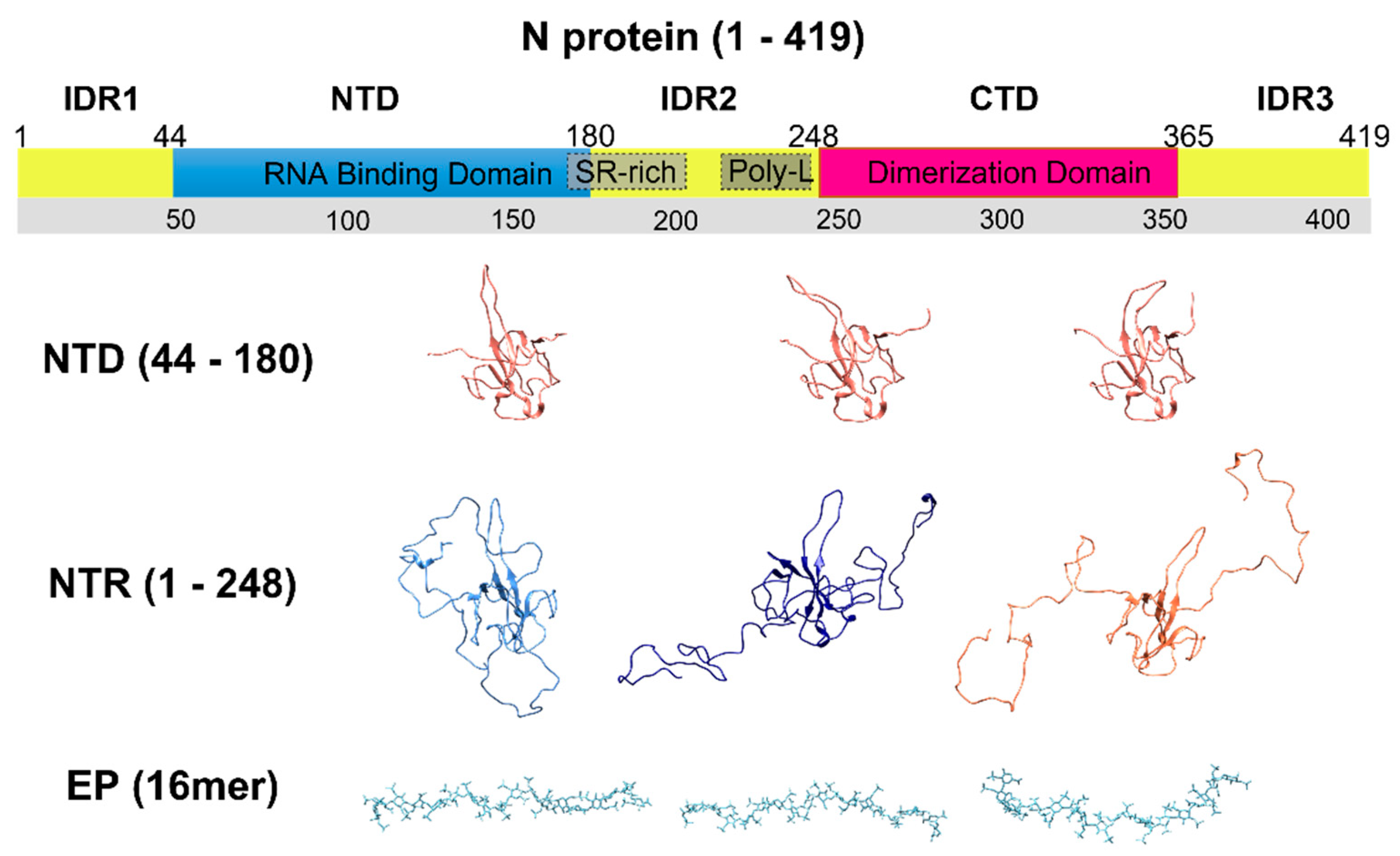
:1. Introduction
2. Materials and Methods
2.1. Protein Sample Preparation
2.2. NMR Experiments
- -
- a Bruker AVANCE III spectrometer operating at 950.20 MHz 1H, 238.93 MHz 13C, and 96.28 MHz 15N frequencies, equipped with a cryogenically cooled probe head optimized for 1H-direct detection (TCI). Namely, 950.
- -
- a Bruker AVANCE NEO spectrometer operating at 700.06 MHz 1H, 176.03 MHz 13C, and 70.94 MHz 15N frequencies equipped with a cryogenically cooled probe head optimized for 13C-direct detection (TXO). Namely, 700C.
- -
- a Bruker Avance NEO spectrometer operating at 700.13 MHz 1H, 176.05 MHz 13C, and 70.94 MHz 15N equipped with a cryogenically cooled triple resonance probe head optimized for 1H-direct detection (TXI). Namely, 700H.
- -
- a Bruker AVANCE III-HD spectrometer operating at 600.13 MHz 1H, 120.90 MHz 13C, and 60.81 MHz 15N frequencies equipped with a probe head optimized for 1H-direct detection (TXI). Namely, 600.
2.3. Kd Estimation
2.4. Protein-Ligand Docking
3. Results
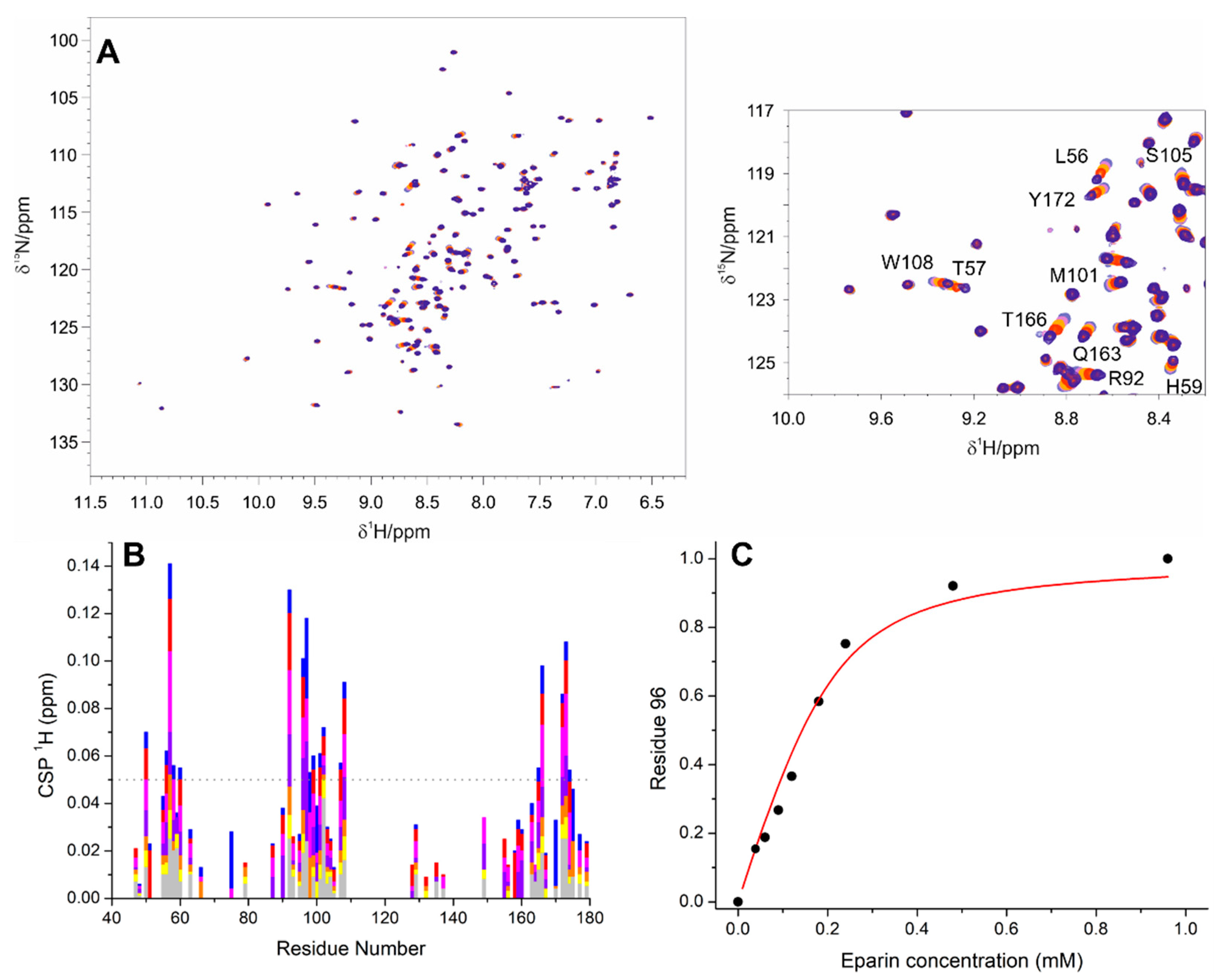
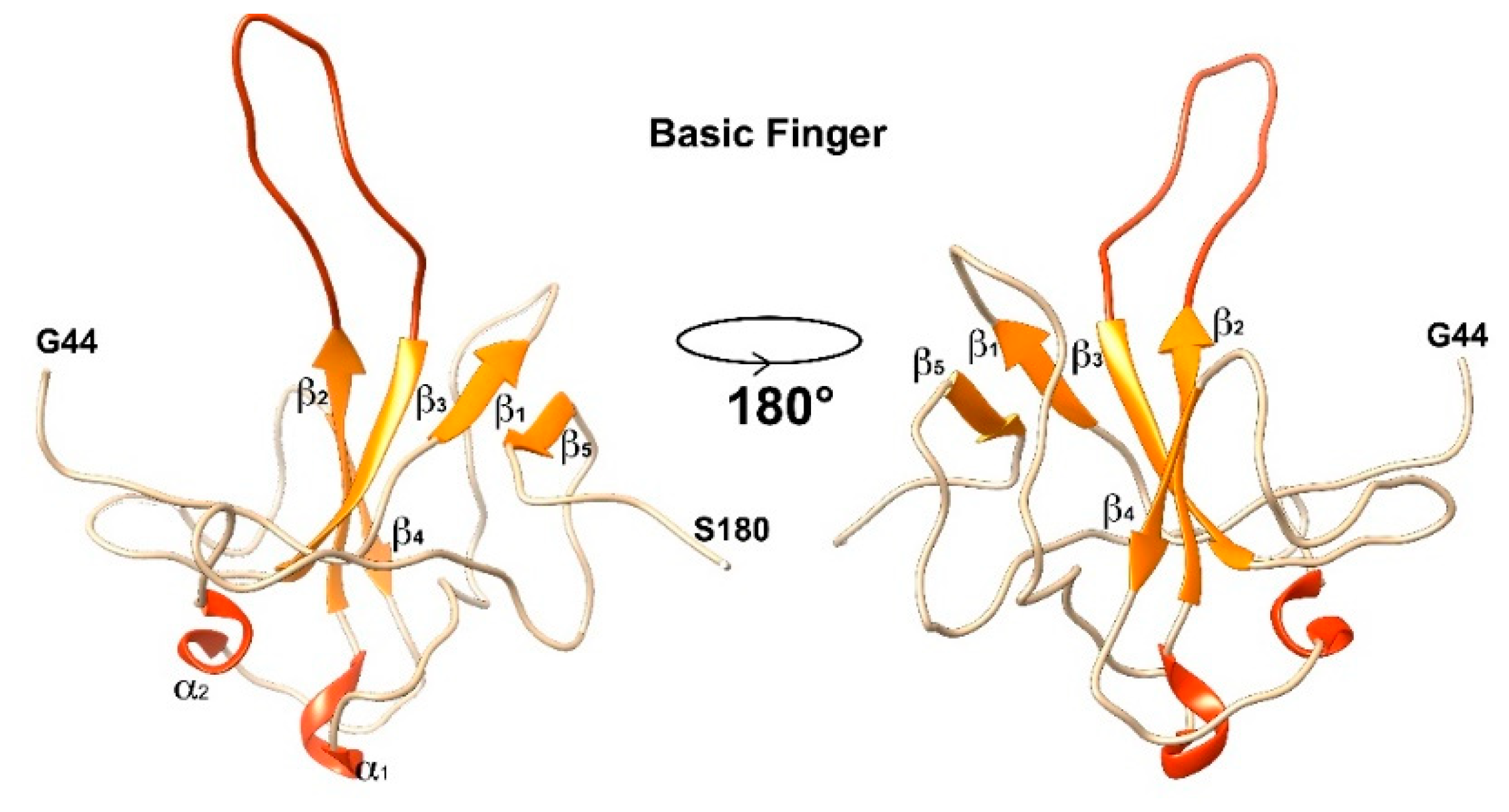
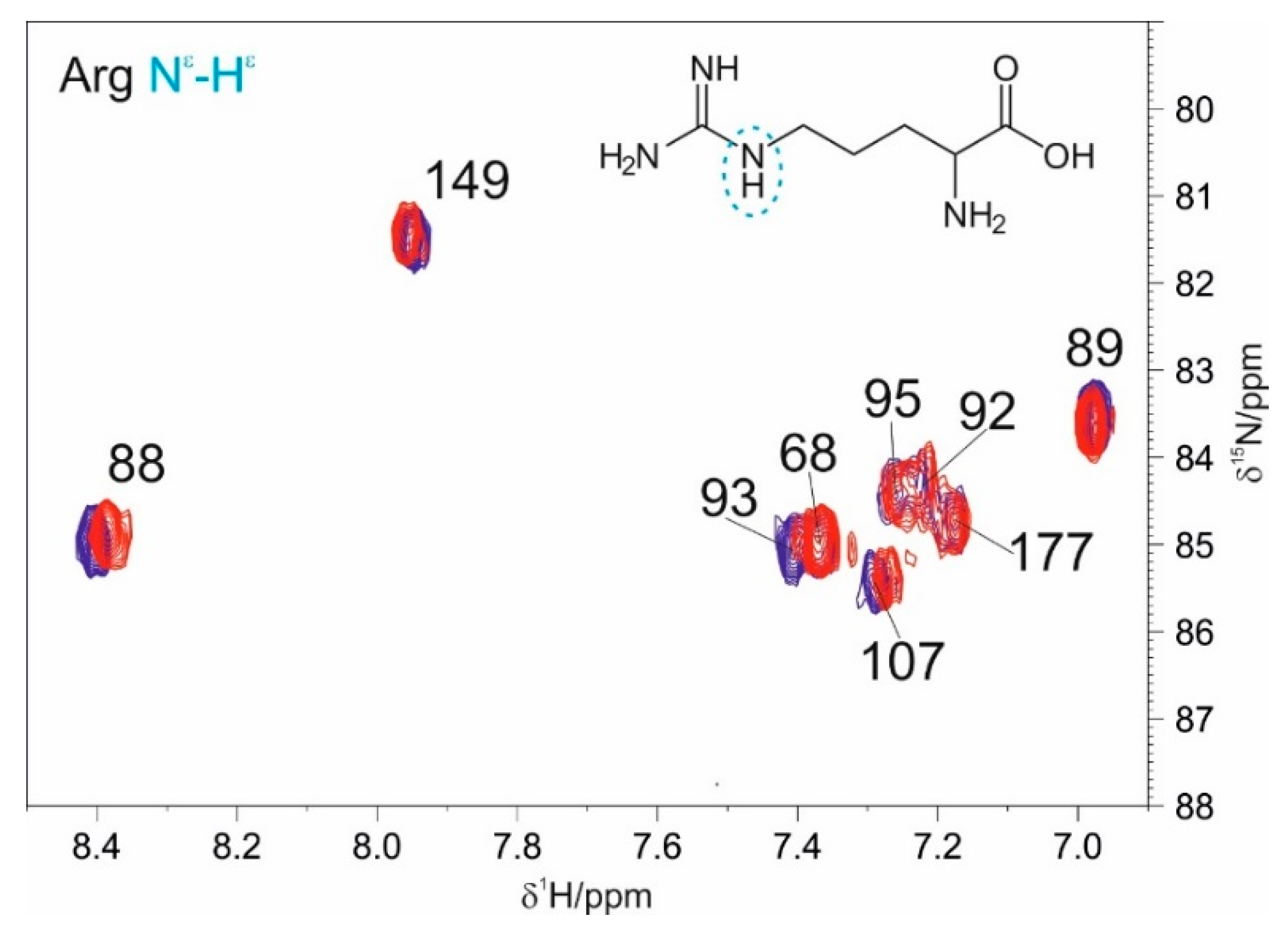
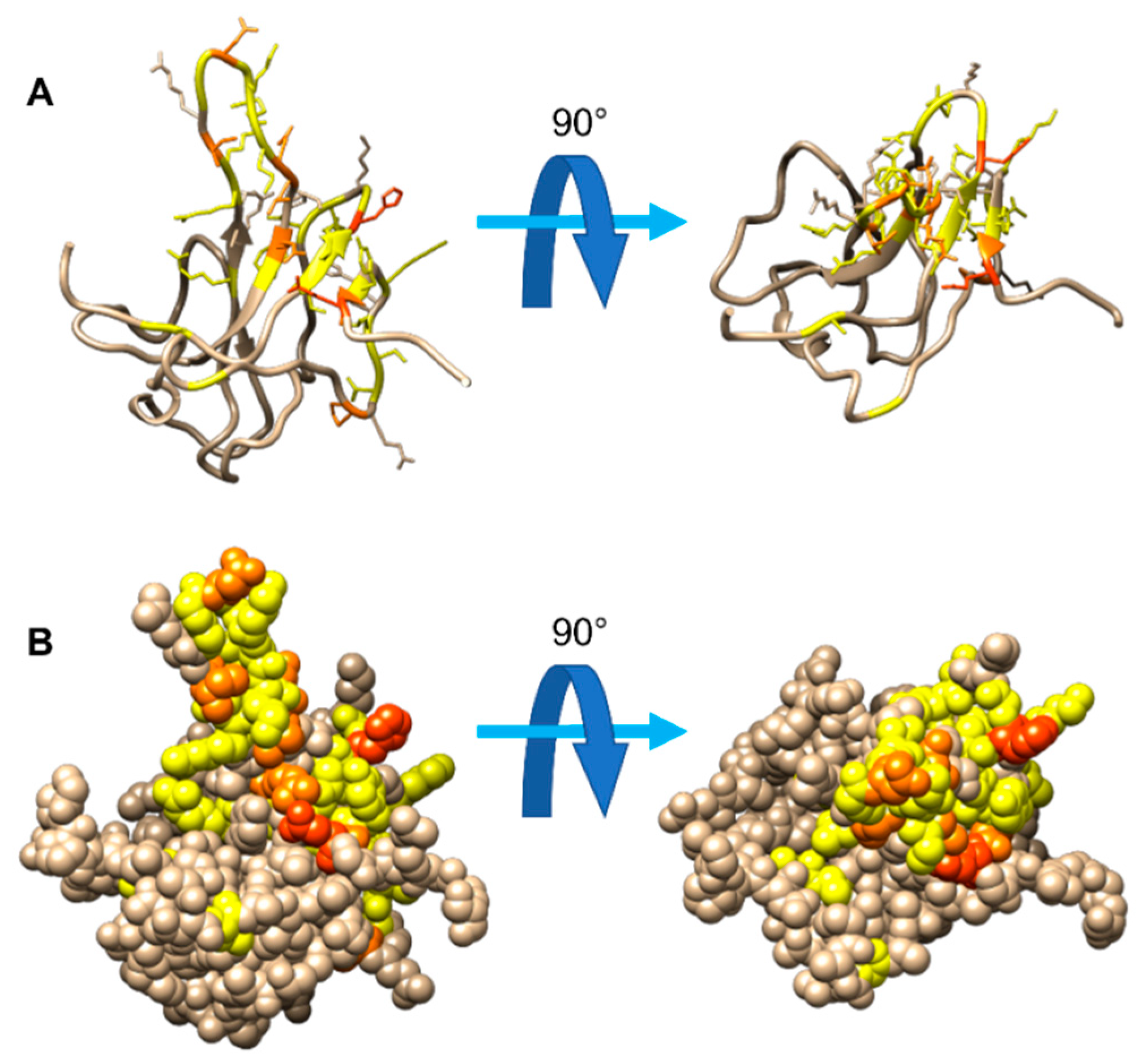
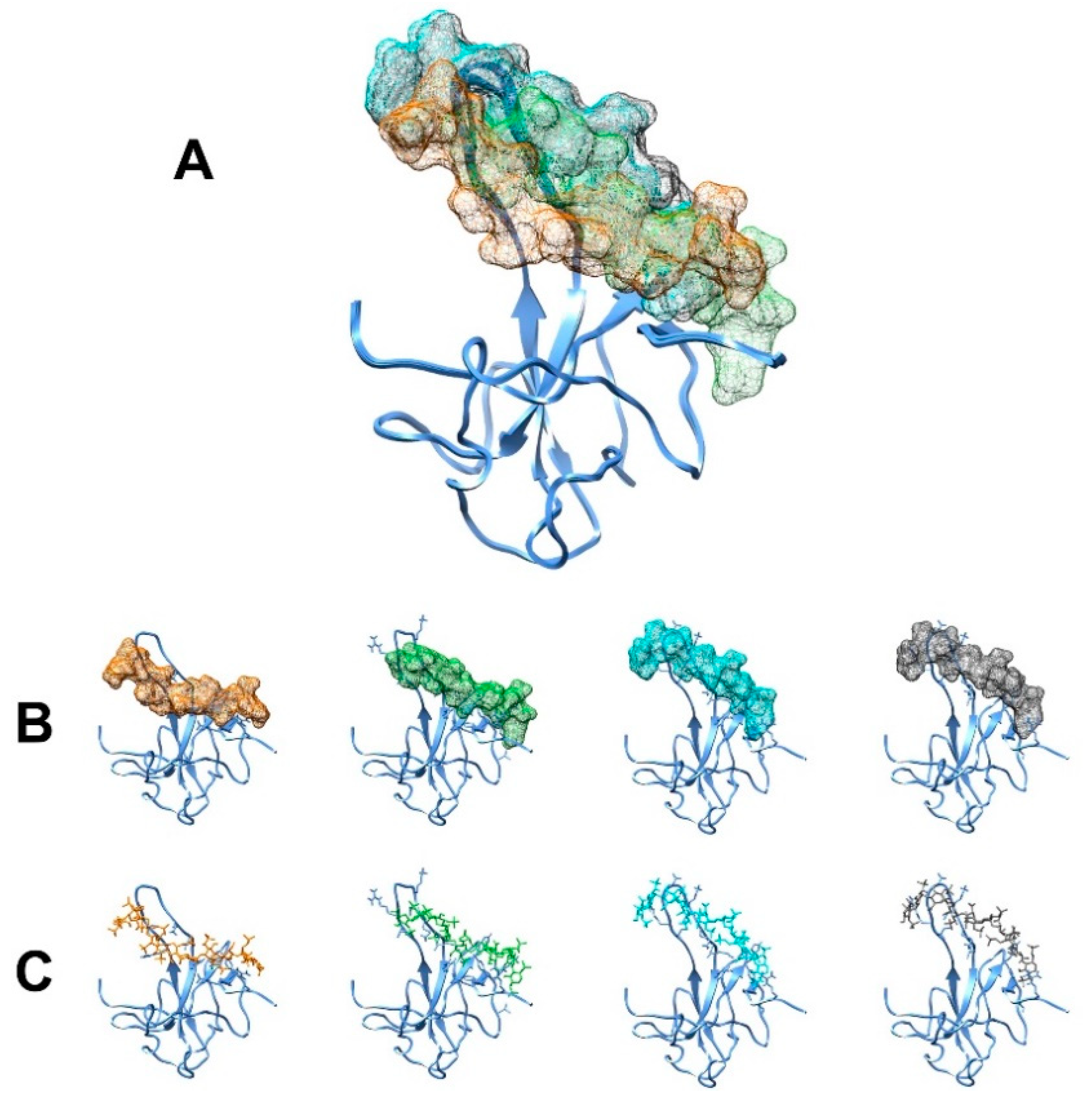
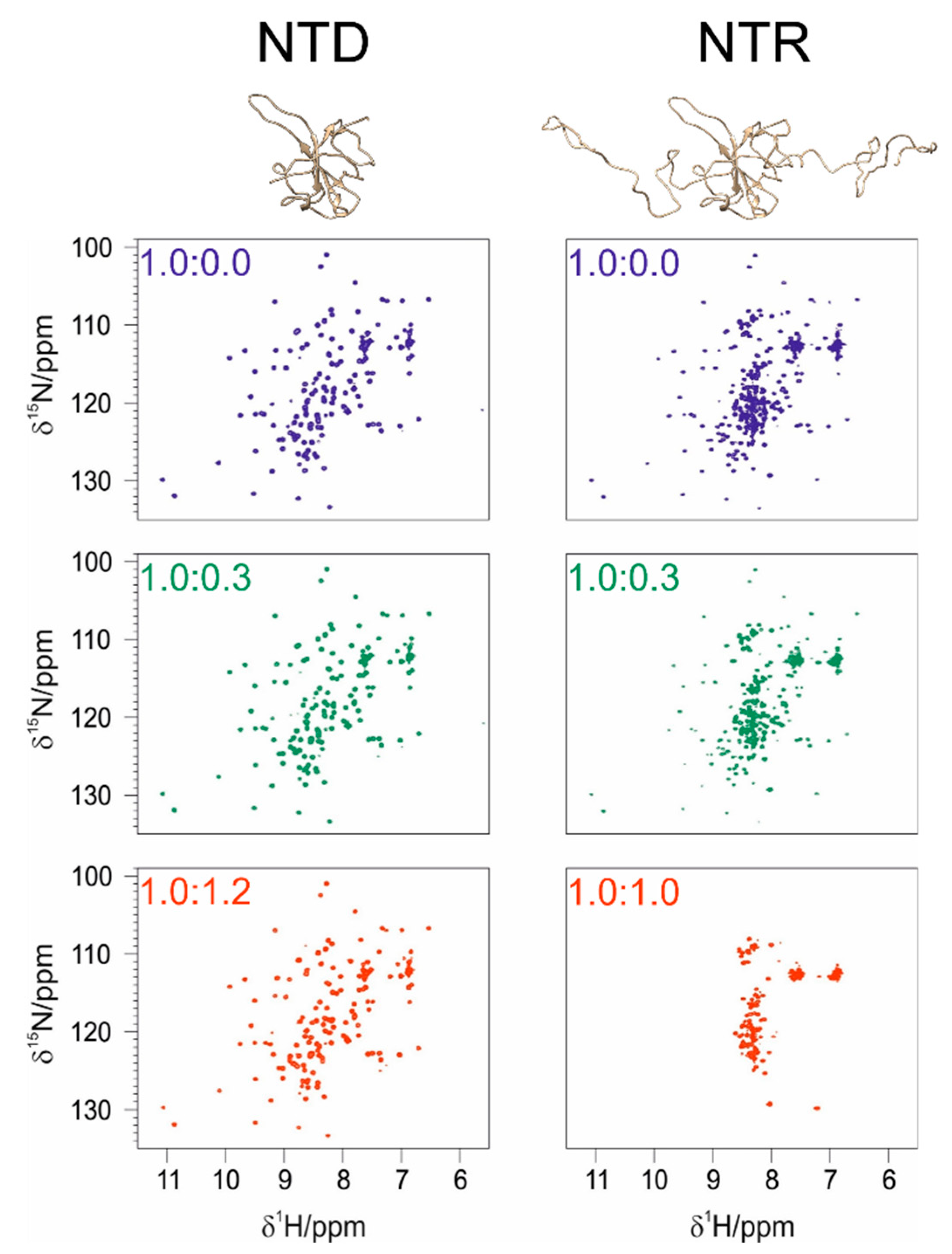
3.1. The Interaction of EP with NTD
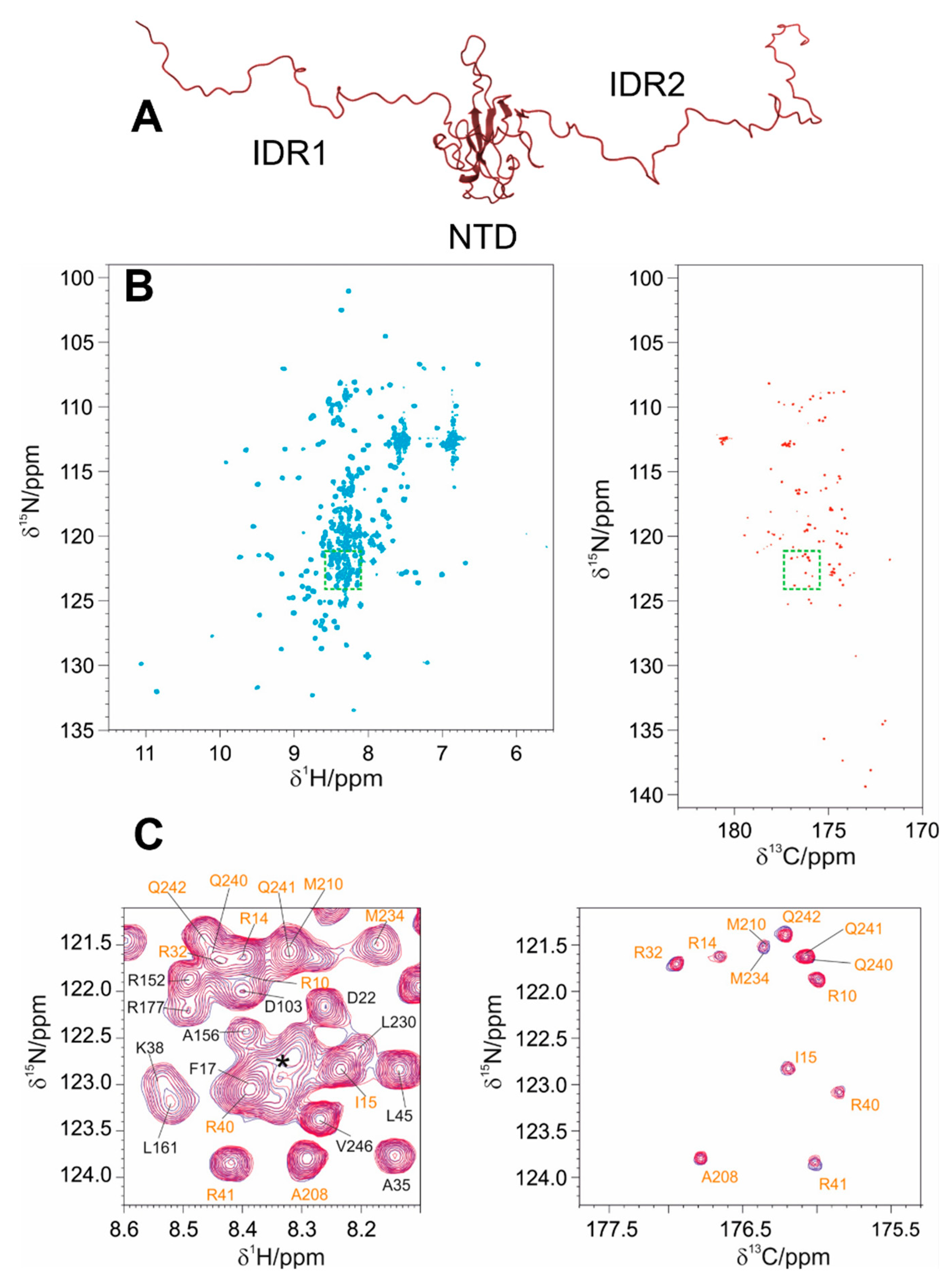
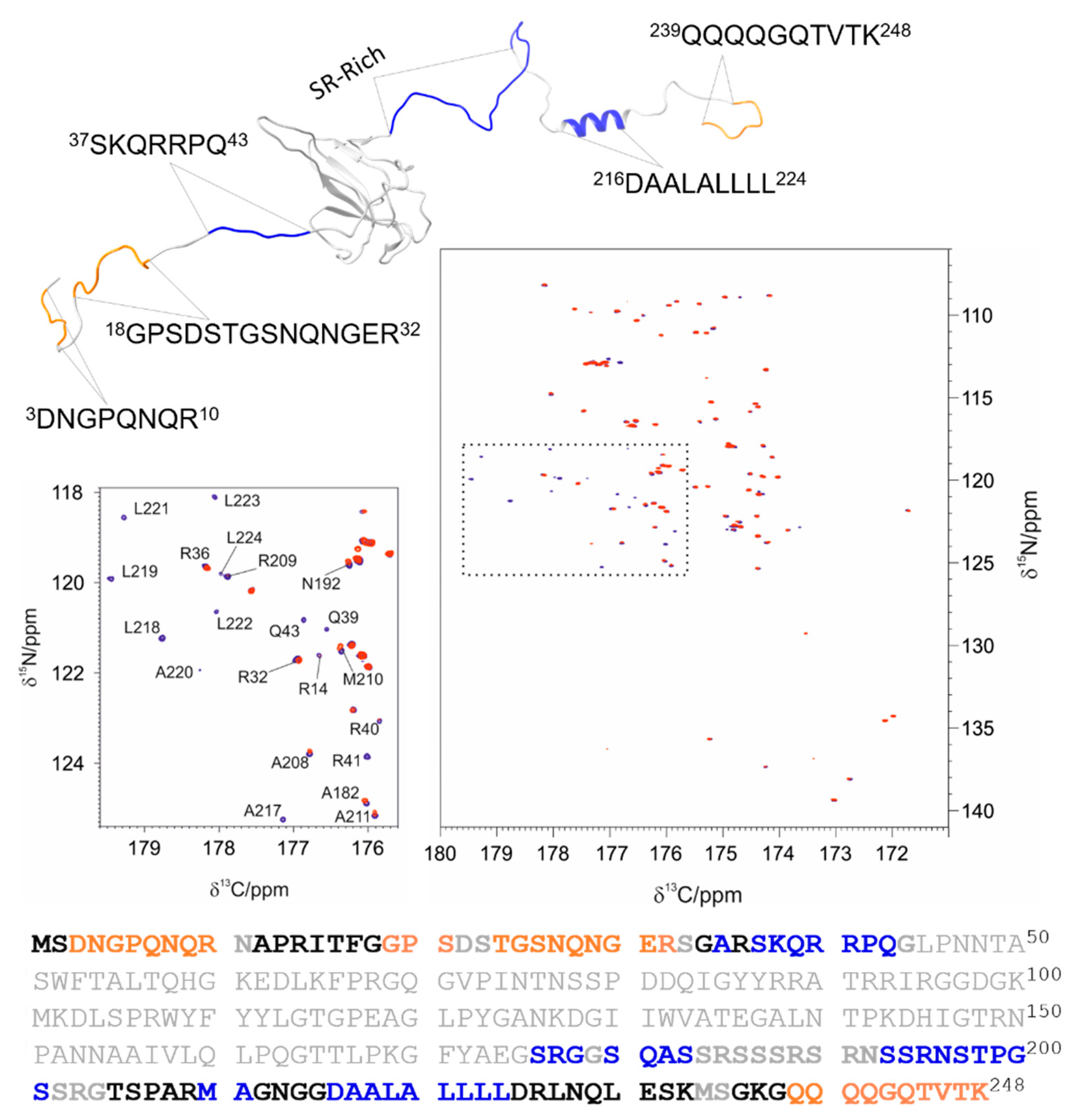
3.2. The Interaction of EP with NTR: The Role of the Intrinsically Disordered Regions
4. Discussion
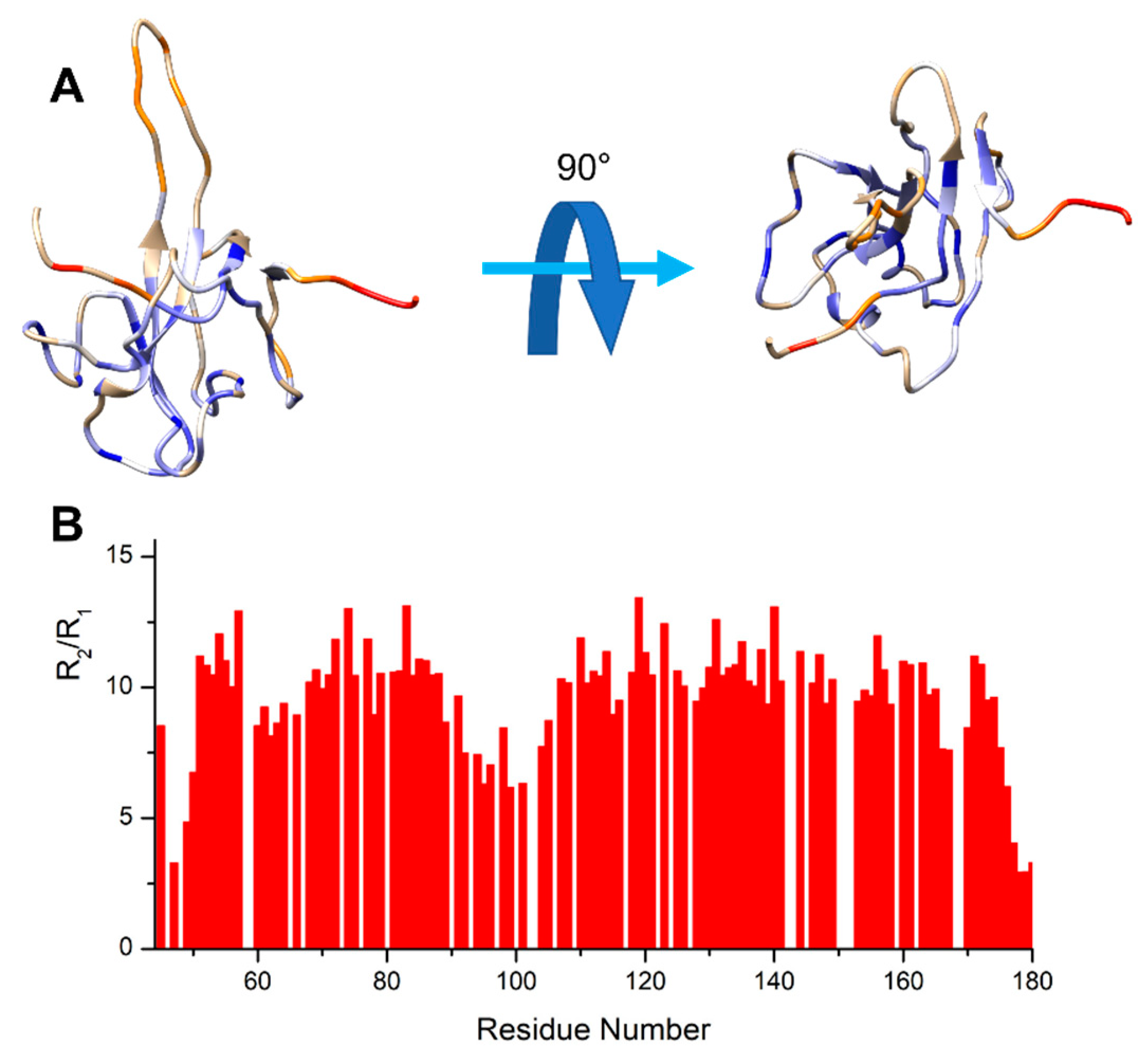
4.1. The Dynamical Binding Modes of NTD
4.2. The Role of IDRs in Orchestrating NTR-EP Interaction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tarighi, P.; Eftekhari, S.; Chizari, M.; Sabernavaei, M.; Jafari, D.; Mirzabeigi, P. A Review of Potential Suggested Drugs for Coronavirus Disease (COVID-19) Treatment. Eur. J. Pharmacol. 2021, 895, 173890. [Google Scholar] [CrossRef]
- Tabll, A.A.; Shahein, Y.E.; Omran, M.M.; Elnakib, M.M.; Ragheb, A.A.; Amer, K.E. A Review of Monoclonal Antibodies in COVID-19: Role in Immunotherapy, Vaccine Development and Viral Detection. Hum. Antibodies 2021, 29, 179–191. [Google Scholar] [CrossRef]
- Zheng, C.; Shao, W.; Chen, X.; Zhang, B.; Wang, G.; Zhang, W. Real-World Effectiveness of COVID-19 Vaccines: A Literature Review and Meta-Analysis. Int. J. Infect. Dis. 2022, 114, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Faraji, S.N.; Raee, M.J.; Hashemi, S.M.A.; Daryabor, G.; Tabrizi, R.; Dashti, F.S.; Behboudi, E.; Heidarnejad, K.; Nowrouzi-Sohrabi, P.; Hatam, G. Human Interaction Targets of SARS-CoV-2 Spike Protein: A Systematic Review. Eur. J. Inflamm. 2022, 20, 1721727X2210953. [Google Scholar] [CrossRef]
- Molina-Mora, J.A. Insights into the Mutation T1117I in the Spike and the Lineage B.1.1.389 of SARS-CoV-2 Circulating in Costa Rica. Gene Rep. 2022, 27, 101554. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 2022, 19, 409–424. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Thorne, L.G.; Bouhaddou, M.; Reuschl, A.-K.; Zuliani-Alvarez, L.; Polacco, B.; Pelin, A.; Batra, J.; Whelan, M.V.X.; Hosmillo, M.; Fossati, A.; et al. Evolution of Enhanced Innate Immune Evasion by SARS-CoV-2. Nature 2022, 602, 487–495. [Google Scholar] [CrossRef]
- Syed, A.M.; Taha, T.Y.; Tabata, T.; Chen, I.P.; Ciling, A.; Khalid, M.M.; Sreekumar, B.; Chen, P.-Y.; Hayashi, J.M.; Soczek, K.M.; et al. Rapid Assessment of SARS-CoV-2–Evolved Variants Using Virus-like Particles. Science 2021, 374, 1626–1632. [Google Scholar] [CrossRef]
- Quaglia, F.; Salladini, E.; Carraro, M.; Minervini, G.; Tosatto, S.C.E.; Le Mercier, P. SARS-CoV-2 Variants Preferentially Emerge at Intrinsically Disordered Protein Sites Helping Immune Evasion. FEBS J. 2022, 289, 4240–4250. [Google Scholar] [CrossRef]
- Chang, C.K.; Hou, M.H.; Chang, C.F.; Hsiao, C.D.; Huang, T.H. The SARS Coronavirus Nucleocapsid Protein-Forms and Functions. Antivir. Res. 2014, 103, 39–50. [Google Scholar] [CrossRef]
- Xue, B.; Blocquel, D.; Habchi, J.; Uversky, A.V.; Kurgan, L.; Uversky, V.N.; Longhi, S. Structural Disorder in Viral Proteins. Chem. Rev. 2014, 114, 6880–6911. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.K.-M.; Dunker, A.K.; Foster, J.A.; Uversky, V.N. Rigidity of the Outer Shell Predicted by a Protein Intrinsic Disorder Model Sheds Light on the COVID-19 (Wuhan-2019-NCoV) Infectivity. Biomolecules 2020, 10, 331. [Google Scholar] [CrossRef]
- Giri, R.; Bhardwaj, T.; Shegane, M.; Gehi, B.R.; Kumar, P.; Gadhave, K.; Oldfield, C.J.; Uversky, V.N. Understanding COVID-19 via Comparative Analysis of Dark Proteomes of SARS-CoV-2, Human SARS and Bat SARS-like Coronaviruses. Cell. Mol. Life Sci. 2021, 78, 1655–1688. [Google Scholar] [CrossRef] [PubMed]
- Schiavina, M.; Pontoriero, L.; Uversky, V.N.; Felli, I.C.; Pierattelli, R. The Highly Flexible Disordered Regions of the SARS-CoV-2 Nucleocapsid N Protein within the 1–248 Residue Construct: Sequence-Specific Resonance Assignments through NMR. Biomol. NMR Assign. 2021, 15, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-K.; Hsu, Y.-L.; Chang, Y.-H.; Chao, F.-A.; Wu, M.-C.; Huang, Y.-S.; Hu, C.-K.; Huang, T.-H. Multiple Nucleic Acid Binding Sites and Intrinsic Disorder of Severe Acute Respiratory Syndrome Coronavirus Nucleocapsid Protein: Implications for Ribonucleocapsid Protein Packaging. J. Virol. 2009, 83, 2255–2264. [Google Scholar] [CrossRef]
- Forsythe, H.M.; Rodriguez Galvan, J.; Yu, Z.; Pinckney, S.; Reardon, P.; Cooley, R.B.; Zhu, P.; Rolland, A.D.; Prell, J.S.; Barbar, E. Multivalent Binding of the Partially Disordered SARS-CoV-2 Nucleocapsid Phosphoprotein Dimer to RNA. Biophys. J. 2021, 120, 2890–2901. [Google Scholar] [CrossRef]
- Bessa, L.M.; Guseva, S.; Camacho-Zarco, A.R.; Salvi, N.; Maurin, D.; Perez, L.M.; Botova, M.; Malki, A.; Nanao, M.; Jensen, M.R.; et al. The Intrinsically Disordered SARS-CoV-2 Nucleoprotein in Dynamic Complex with Its Viral Partner Nsp3a. Sci. Adv. 2022, 8, eabm4034. [Google Scholar] [CrossRef]
- Pontoriero, L.; Schiavina, M.; Korn, S.M.; Schlundt, A.; Pierattelli, R.; Felli, I.C. NMR Reveals Specific Tracts within the Intrinsically Disordered Regions of the SARS-CoV-2 Nucleocapsid Protein Involved in RNA Encountering. Biomolecules 2022, 12, 929. [Google Scholar] [CrossRef]
- Peng, Y.; Du, N.; Lei, Y.; Dorje, S.; Qi, J.; Luo, T.; Gao, G.F.; Song, H. Structures of the SARS-CoV-2 Nucleocapsid and Their Perspectives for Drug Design. EMBO J. 2020, 39, e105938. [Google Scholar] [CrossRef]
- Guseva, S.; Perez, L.M.; Camacho-Zarco, A.; Bessa, L.M.; Salvi, N.; Malki, A.; Maurin, D.; Blackledge, M. 1H, 13C and 15N Backbone Chemical Shift Assignments of the n-Terminal and Central Intrinsically Disordered Domains of SARS-CoV-2 Nucleoprotein. Biomol. NMR Assign. 2021, 15, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Dinesh, D.C.; Chalupska, D.; Silhan, J.; Koutna, E.; Nencka, R.; Veverka, V.; Boura, E. Structural Basis of RNA Recognition by the SARS-CoV-2 Nucleocapsid Phosphoprotein. PLoS Pathog. 2020, 16, e1009100. [Google Scholar] [CrossRef] [PubMed]
- Savastano, A.; Ibáñez de Opakua, A.; Rankovic, M.; Zweckstetter, M. Nucleocapsid Protein of SARS-CoV-2 Phase Separates into RNA-Rich Polymerase-Containing Condensates. Nat. Commun. 2020, 11, 6041. [Google Scholar] [CrossRef] [PubMed]
- Redzic, J.S.; Lee, E.; Born, A.; Issaian, A.; Henen, M.A.; Nichols, P.J.; Blue, A.; Hansen, K.C.; D’Alessandro, A.; Vögeli, B.; et al. The Inherent Dynamics and Interaction Sites of the SARS-CoV-2 Nucleocapsid N-Terminal Region. J. Mol. Biol. 2021, 433, 167108. [Google Scholar] [CrossRef]
- Caruso, I.P.; dos Santos Almeida, V.; do Amaral, M.J.; de Andrade, G.C.; de Araújo, G.R.; de Araújo, T.S.; de Azevedo, J.M.; Barbosa, G.M.; Bartkevihi, L.; Bezerra, P.R.; et al. Insights into the Specificity for the Interaction of the Promiscuous SARS-CoV-2 Nucleocapsid Protein N-Terminal Domain with Deoxyribonucleic Acids. Int. J. Biol. Macromol. 2022, 203, 466–480. [Google Scholar] [CrossRef]
- Shi, C.; Tingting, W.; Li, J.-P.; Sullivan, M.A.; Wang, C.; Wang, H.; Deng, B.; Zhang, Y. Comprehensive Landscape of Heparin Therapy for COVID-19. Carbohydr. Polym. 2021, 254, 117232. [Google Scholar] [CrossRef]
- Shan, D.; Johnson, J.M.; Fernandes, S.C.; Suib, H.; Hwang, S.; Wuelfing, D.; Mendes, M.; Holdridge, M.; Burke, E.M.; Beauregard, K.; et al. N-Protein Presents Early in Blood, Dried Blood and Saliva during Asymptomatic and Symptomatic SARS-CoV-2 Infection. Nat. Commun. 2021, 12, 1931. [Google Scholar] [CrossRef]
- Kielstein, J.T.; Borchina, D.-N.; Fühner, T.; Hwang, S.; Mattoon, D.; Ball, A.J. Hemofiltration with the Seraph® 100 Microbind® Affinity Filter Decreases SARS-CoV-2 Nucleocapsid Protein in Critically Ill COVID-19 Patients. Crit. Care 2021, 25, 190. [Google Scholar] [CrossRef]
- López-Muñoz, A.D.; Kosik, I.; Holly, J.; Yewdell, J.W. Cell Surface SARS-CoV-2 Nucleocapsid Protein Modulates Innate and Adaptive Immunity. Sci. Adv. 2022, 8, eabp9770. [Google Scholar] [CrossRef]
- González-Motos, V.; Kropp, K.A.; Viejo-Borbolla, A. Chemokine Binding Proteins: An Immunomodulatory Strategy Going Viral. Cytokine Growth Factor Rev. 2016, 30, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Hernaez, B.; Alcamí, A. Virus-Encoded Cytokine and Chemokine Decoy Receptors. Curr. Opin. Immunol. 2020, 66, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Bernadó, P.; Mylonas, E.; Petoukhov, M.V.; Blackledge, M.; Svergun, D.I. Structural Characterization of Flexible Proteins Using Small-Angle X-ray Scattering. J. Am. Chem. Soc. 2007, 129, 5656–5664. [Google Scholar] [CrossRef] [PubMed]
- Tria, G.; Mertens, H.D.T.; Kachala, M.; Svergun, D.I. Advanced Ensemble Modelling of Flexible Macromolecules Using X-ray Solution Scattering. IUCrJ 2015, 2, 207–217. [Google Scholar] [CrossRef]
- Khan, S.; Gor, J.; Mulloy, B.; Perkins, S.J. Semi-Rigid Solution Structures of Heparin by Constrained X-ray Scattering Modelling: New Insight into Heparin–Protein Complexes. J. Mol. Biol. 2010, 395, 504–521. [Google Scholar] [CrossRef]
- Altincekic, N.; Korn, S.M.; Qureshi, N.S.; Dujardin, M.; Ninot-Pedrosa, M.; Abele, R.; Abi Saad, M.J.; Alfano, C.; Almeida, F.C.L.; Alshamleh, I.; et al. Large-Scale Recombinant Production of the SARS-CoV-2 Proteome for High-Throughput and Structural Biology Applications. Front. Mol. Biosci. 2021, 8, 653148. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Marley, J.; Lu, M.; Bracken, C. A Method for Efficient Isotopic Labeling of Recombinant Proteins. J. Biomol. NMR 2001, 20, 71–75. [Google Scholar] [CrossRef]
- Palmer, A.G.; Cavanagh, J.; Wright, P.E.; Rance, M. Sensitivity Improvement in Proton-Detected Two-Dimensional Heteronuclear Correlation NMR Spectroscopy. J. Magn. Reson. 1991, 93, 151–170. [Google Scholar] [CrossRef]
- Schleucher, J.; Schwendinger, M.; Sattler, M.; Schmidt, P.; Schedletzky, O.; Glaser, S.J.; Sorensen, O.W.; Griesinger, C. A General Enhancement Scheme in Heteronuclear Multidimensional NMR Employing Pulsed Field Gradients. J. Biomol. NMR 1994, 4, 301–306. [Google Scholar] [CrossRef]
- Pontoriero, L.; Schiavina, M.; Murrali, M.G.; Pierattelli, R.; Felli, I.C. Monitoring the Interaction of A-Synuclein with Calcium Ions through Exclusively Heteronuclear Nuclear Magnetic Resonance Experiments. Angew. Chem. Int. Ed. 2020, 59, 18537–18545. [Google Scholar] [CrossRef]
- Bermel, W.; Bertini, I.; Csizmok, V.; Felli, I.C.; Pierattelli, R.; Tompa, P. H-Start for Exclusively Heteronuclear NMR Spectroscopy: The Case of Intrinsically Disordered Proteins. J. Magn. Reson. 2009, 198, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Schiavina, M.; Murrali, M.G.; Pontoriero, L.; Sainati, V.; Kümmerle, R.; Bermel, W.; Pierattelli, R.; Felli, I.C. Taking Simultaneous Snapshots of Intrinsically Disordered Proteins in Action. Biophys. J. 2019, 117, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Geen, H.; Freeman, R. Band-Selective Radiofrequency Pulses. J. Magn. Reson. 1991, 93, 93–141. [Google Scholar] [CrossRef]
- Emsley, L.; Bodenhausen, G. Optimization of Shaped Selective Pulses for NMR Using a Quaternion Description of Their Overall Propagators. J. Magn. Reson. 1992, 97, 135–148. [Google Scholar] [CrossRef]
- Felli, I.C.; Pierattelli, R. Spin-State-Selective Methods in Solution- and Solid-State Biomolecular 13C NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2015, 84, 1–13. [Google Scholar] [CrossRef]
- Piotto, M.; Saudek, V.; Sklenar, V. Gradient-Tailored Excitation for Single-Quantum NMR Spectroscopy of Aqueous Solutions. J. Biomol. NMR 1992, 2, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Farrow, N.A.; Muhandiram, R.; Singer, A.U.; Pascal, S.M.; Kay, C.M.; Gish, G.; Shoelson, S.E.; Pawson, T.; Forman-Kay, J.D.; Kay, L.E. Backbone Dynamics of a Free and a Phosphopeptide-Complexed Src Homology 2 Domain Studied by 15N NMR Relaxation. Biochemistry 1994, 33, 5984–6003. [Google Scholar] [CrossRef]
- Johnson, C.S. Diffusion Ordered Nuclear Magnetic Resonance Spectroscopy: Principles and Applications. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 34, 203–256. [Google Scholar] [CrossRef]
- Keller, R. The Computer Aided Resonance Assignment Tutorial; Cantina Verlag: Goldau, Switzerland, 2004; pp. 1–81. [Google Scholar]
- Bartels, C.; Xia, T.H.; Billeter, M.; Güntert, P.; Wüthrich, K. The Program XEASY for Computer-Supported NMR Spectral Analysis of Biological Macromolecules. J. Biomol. NMR 1995, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.L.; Bax, A.; Arata, Y.; Hilbers, C.W.; Kaptein, R.; Sykes, B.D.; Wright, P.E.; Wuethrich, K. Recommendations for the Presentation of NMR Structures of Proteins and Nucleic Acids. Pure Appl. Chem. 1998, 70, 117–142. [Google Scholar] [CrossRef]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A Protein−Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Honorato, R.V.; Koukos, P.I.; Jiménez-García, B.; Tsaregorodtsev, A.; Verlato, M.; Giachetti, A.; Rosato, A.; Bonvin, A.M.J.J. Structural Biology in the Clouds: The WeNMR-EOSC Ecosystem. Front. Mol. Biosci. 2021, 8, 729513. [Google Scholar] [CrossRef]
- Felli, I.C.; Pierattelli, R. 13 C Direct Detected NMR for Challenging Systems. Chem. Rev. 2022, 122, 9468–9496. [Google Scholar] [CrossRef] [PubMed]
- Bertini, I.; Felli, I.C.; Gonnelli, L.; Pierattelli, R.; Spyranti, Z.; Spyroulias, G.A. Mapping Protein–Protein Interaction by 13C′-Detected Heteronuclear NMR Spectroscopy. J. Biomol. NMR 2006, 36, 111–122. [Google Scholar] [CrossRef]
- Alik, A.; Bouguechtouli, C.; Julien, M.; Bermel, W.; Ghouil, R.; Zinn-Justin, S.; Theillet, F. Sensitivity-Enhanced 13 C-NMR Spectroscopy for Monitoring Multisite Phosphorylation at Physiological Temperature and PH. Angew. Chem. Int. Ed. 2020, 59, 10411–10415. [Google Scholar] [CrossRef]
- Clarkson, M.W.; Lei, M.; Eisenmesser, E.Z.; Labeikovsky, W.; Redfield, A.; Kern, D. Mesodynamics in the SARS Nucleocapsid Measured by NMR Field Cycling. J. Biomol. NMR 2009, 45, 217–225. [Google Scholar] [CrossRef]
- Korn, S.M.; Dhamotharan, K.; Schlundt, A. The Preference Signature of the SARS-CoV-2 Nucleocapsid NTD for Its 5′-Genomic RNA Elements. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Perdikari, T.M.; Murthy, A.C.; Ryan, V.H.; Watters, S.; Naik, M.T.; Fawzi, N.L. SARS-CoV-2 Nucleocapsid Protein Phase-separates with RNA and with Human HnRNPs. EMBO J. 2020, 39, e106478. [Google Scholar] [CrossRef]
- Carlson, C.R.; Asfaha, J.B.; Ghent, C.M.; Howard, C.J.; Hartooni, N.; Safari, M.; Frankel, A.D.; Morgan, D.O. Phosphoregulation of Phase Separation by the SARS-CoV-2 N Protein Suggests a Biophysical Basis for Its Dual Functions. Mol. Cell 2020, 80, 1092–1103.e4. [Google Scholar] [CrossRef]
- Cubuk, J.; Alston, J.J.; Incicco, J.J.; Singh, S.; Stuchell-Brereton, M.D.; Ward, M.D.; Zimmerman, M.I.; Vithani, N.; Griffith, D.; Wagoner, J.A.; et al. The SARS-CoV-2 Nucleocapsid Protein Is Dynamic, Disordered, and Phase Separates with RNA. Nat. Commun. 2021, 12, 1936. [Google Scholar] [CrossRef]
- Lu, S.; Ye, Q.; Singh, D.; Cao, Y.; Diedrich, J.K.; Yates, J.R.; Villa, E.; Cleveland, D.W.; Corbett, K.D. The SARS-CoV-2 Nucleocapsid Phosphoprotein Forms Mutually Exclusive Condensates with RNA and the Membrane-Associated M Protein. Nat. Commun. 2021, 12, 502. [Google Scholar] [CrossRef]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A Meta-Predictor of Intrinsically Disordered Amino Acids. Biochim. Biophys. Acta-Proteins Proteom. 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence Complexity of Disordered Protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins in Human Diseases: Introducing the D 2 Concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Erdös, G.; Dosztányi, Z. IUPred2A: Context-Dependent Prediction of Protein Disorder as a Function of Redox State and Protein Binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef]
- Dosztányi, Z.; Csizmók, V.; Tompa, P.; Simon, I. The Pairwise Energy Content Estimated from Amino Acid Composition Discriminates between Folded and Intrinsically Unstructured Proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef]
- Kurzbach, D.; Platzer, G.; Schwarz, T.C.; Henen, M.A.; Konrat, R.; Hinderberger, D. Cooperative Unfolding of Compact Conformations of the Intrinsically Disordered Protein Osteopontin. Biochemistry 2013, 52, 5167–5175. [Google Scholar] [CrossRef]
- Hileman, R.E.; Fromm, J.R.; Weiler, J.M.; Linhardt, R.J. Glycosaminoglycan-Protein Interactions: Definition of Consensus Sites in Glycosaminoglycan Binding Proteins. BioEssays 1998, 20, 156–167. [Google Scholar] [CrossRef]
- Capila, I.; Linhardt, R.J. Heparin-Protein Interactions. Angew. Chem. Int. Ed. 2002, 41, 390–412. [Google Scholar] [CrossRef]
- Deka, P.; Rajan, P.K.; Perez-Canadillas, J.M.; Varani, G. Protein and RNA Dynamics Play Key Roles in Determining the Specific Recognition of GU-Rich Polyadenylation Regulatory Elements by Human Cstf-64 Protein. J. Mol. Biol. 2005, 347, 719–733. [Google Scholar] [CrossRef]
- Huang, Q.; Yu, L.; Petros, A.M.; Gunasekera, A.; Liu, Z.; Xu, N.; Hajduk, P.; Mack, J.; Fesik, S.W.; Olejniczak, E.T. Structure of the N-Terminal RNA-Binding Domain of the SARS CoV Nucleocapsid Protein. Biochemistry 2004, 43, 6059–6063. [Google Scholar] [CrossRef] [PubMed]
- Caruso, Í.P.; Sanches, K.; Da Poian, A.T.; Pinheiro, A.S.; Almeida, F.C.L. Dynamics of the SARS-CoV-2 Nucleoprotein N-Terminal Domain Triggers RNA Duplex Destabilization. Biophys. J. 2021, 120, 2814–2827. [Google Scholar] [CrossRef] [PubMed]
- Mukrasch, M.D.; Biernat, J.; von Bergen, M.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Sites of Tau Important for Aggregation Populate β-Structure and Bind to Microtubules and Polyanions. J. Biol. Chem. 2005, 280, 24978–24986. [Google Scholar] [CrossRef] [PubMed]
- Sottini, A.; Borgia, A.; Borgia, M.B.; Bugge, K.; Nettels, D.; Chowdhury, A.; Heidarsson, P.O.; Zosel, F.; Best, R.B.; Kragelund, B.B.; et al. Polyelectrolyte Interactions Enable Rapid Association and Dissociation in High-Affinity Disordered Protein Complexes. Nat. Commun. 2020, 11, 5736. [Google Scholar] [CrossRef]
- Teilum, K.; Olsen, J.G.; Kragelund, B.B. On the Specificity of Protein–Protein Interactions in the Context of Disorder. Biochem. J. 2021, 478, 2035–2050. [Google Scholar] [CrossRef]
- Arbesú, M.; Pons, M. Integrating Disorder in Globular Multidomain Proteins: Fuzzy Sensors and the Role of SH3 Domains. Arch. Biochem. Biophys. 2019, 677, 108161. [Google Scholar] [CrossRef]
- Arbesú, M.; Iruela, G.; Fuentes, H.; Teixeira, J.M.C.; Pons, M. Intramolecular Fuzzy Interactions Involving Intrinsically Disordered Domains. Front. Mol. Biosci. 2018, 5, 39. [Google Scholar] [CrossRef]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why Are “natively Unfolded” Proteins Unstructured under Physiologic Conditions? Proteins Struct. Funct. Genet. 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Dunker, A.K.; Babu, M.M.; Barbar, E.; Blackledge, M.; Bondos, S.E.; Dosztányi, Z.; Dyson, H.J.; Forman-Kay, J.; Fuxreiter, M.; Gsponer, J.; et al. What’s in a Name? Why These Proteins Are Intrinsically Disordered. Intrinsically Disord. Proteins 2013, 1, e24157. [Google Scholar] [CrossRef]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing Protein Intrinsic Disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [Green Version]
- Kurzbach, D.; Schwarz, T.C.; Platzer, G.; Höfler, S.; Hinderberger, D.; Konrat, R. Compensatory Adaptations of Structural Dynamics in an Intrinsically Disordered Protein Complex. Angew. Chem. 2014, 53, 3840–3843. [Google Scholar] [CrossRef] [PubMed]
- Sibille, N.; Sillen, A.; Leroy, A.; Wieruszeski, J.-M.; Mulloy, B.; Landrieu, I.; Lippens, G. Structural Impact of Heparin Binding to Full-Length Tau As Studied by NMR Spectroscopy. Biochemistry 2006, 45, 12560–12572. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Fuxreiter, M. Fuzzy Complexes: Polymorphism and Structural Disorder in Protein-Protein Interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef]
- Murrali, M.G.; Felli, I.C.; Pierattelli, R. Adenoviral E1A Exploits Flexibility and Disorder to Target Cellular Proteins. Biomolecules 2020, 10, 1541. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Construct | Experiment | Data Points | Spectral Width (Hz) | Number of Scans | Interscan Delay (s) | Field (1H MHz) | ||
|---|---|---|---|---|---|---|---|---|
| F1 | F2 | F1 | F2 | |||||
| NTD | 2D CACO | 128 | 1024 | 7407 (13Cα) | 5263 (13C′) | 32 | 1.6 | 700 |
| NTD | 2D (H)CBCACO | 174 | 1024 | 11,628 (13Cali) | 5263 (13C′) | 32 | 1.0 | 700 |
| NTD | 2D (HCA)CON | 128 | 1024 | 3413 (15N) | 5000 (13C′) | 96 | 1.1 | 700 |
| NTD | 2D HC | 256 | 1024 | 10,638 (13Caro) | 11,364 (1H) | 4 | 1.1 | 700 |
| NTD | 2D HN | 256 | 2048 | 4347 (15N) | 19,132 (1H) | 16 | 1.0 | 950 |
| NTD | 2D HεNε | 256 | 4096 | 11,627 (15Nε) | 19,132 (1Hε) | 8 | 1.0 | 950 |
| NTR | mr_CON//HN | 400 | 1024 | 2840 (15N) | 5263 (13C) | 16 | 1.9 | 700 |
| NTR | mr_CON//HN | 400 | 4096 | 3195 (15N) | 20,833 (1H) | 32 | 1.9 | 700 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiavina, M.; Pontoriero, L.; Tagliaferro, G.; Pierattelli, R.; Felli, I.C. The Role of Disordered Regions in Orchestrating the Properties of Multidomain Proteins: The SARS-CoV-2 Nucleocapsid Protein and Its Interaction with Enoxaparin. Biomolecules 2022, 12, 1302. https://doi.org/10.3390/biom12091302
Schiavina M, Pontoriero L, Tagliaferro G, Pierattelli R, Felli IC. The Role of Disordered Regions in Orchestrating the Properties of Multidomain Proteins: The SARS-CoV-2 Nucleocapsid Protein and Its Interaction with Enoxaparin. Biomolecules. 2022; 12(9):1302. https://doi.org/10.3390/biom12091302
Chicago/Turabian StyleSchiavina, Marco, Letizia Pontoriero, Giuseppe Tagliaferro, Roberta Pierattelli, and Isabella C. Felli. 2022. "The Role of Disordered Regions in Orchestrating the Properties of Multidomain Proteins: The SARS-CoV-2 Nucleocapsid Protein and Its Interaction with Enoxaparin" Biomolecules 12, no. 9: 1302. https://doi.org/10.3390/biom12091302
APA StyleSchiavina, M., Pontoriero, L., Tagliaferro, G., Pierattelli, R., & Felli, I. C. (2022). The Role of Disordered Regions in Orchestrating the Properties of Multidomain Proteins: The SARS-CoV-2 Nucleocapsid Protein and Its Interaction with Enoxaparin. Biomolecules, 12(9), 1302. https://doi.org/10.3390/biom12091302