

RetroComposer: Composing Templates for Template-Based Retrosynthesis Prediction

Abstract
:1. Introduction
2. Related Work
3. Preliminary Knowledge
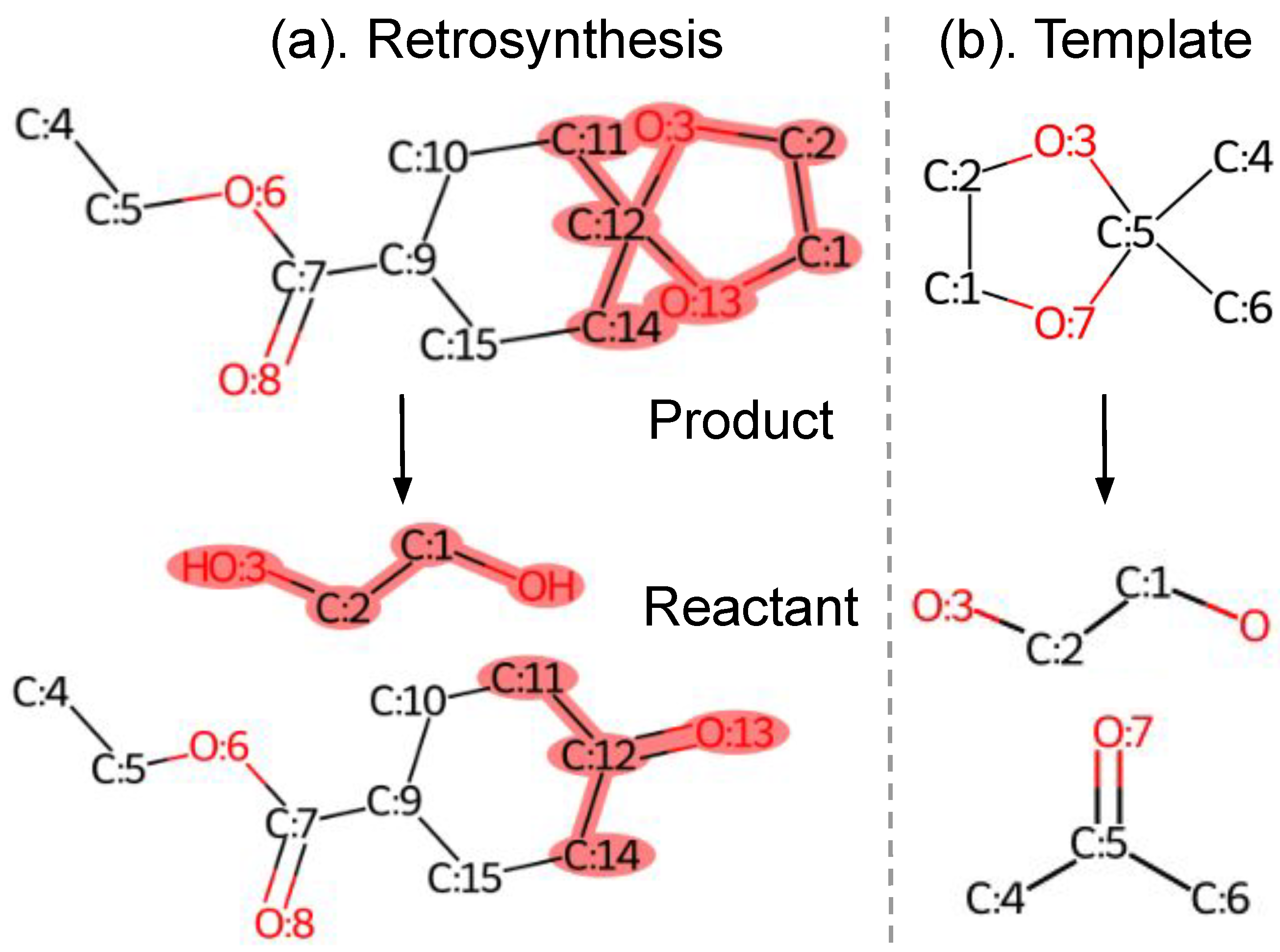
3.1. Retrosynthesis and Template
3.2. Molecule Graph Representation
3.3. Graph Attention Networks
3.4. Graph-Level Embedding
4. Methods
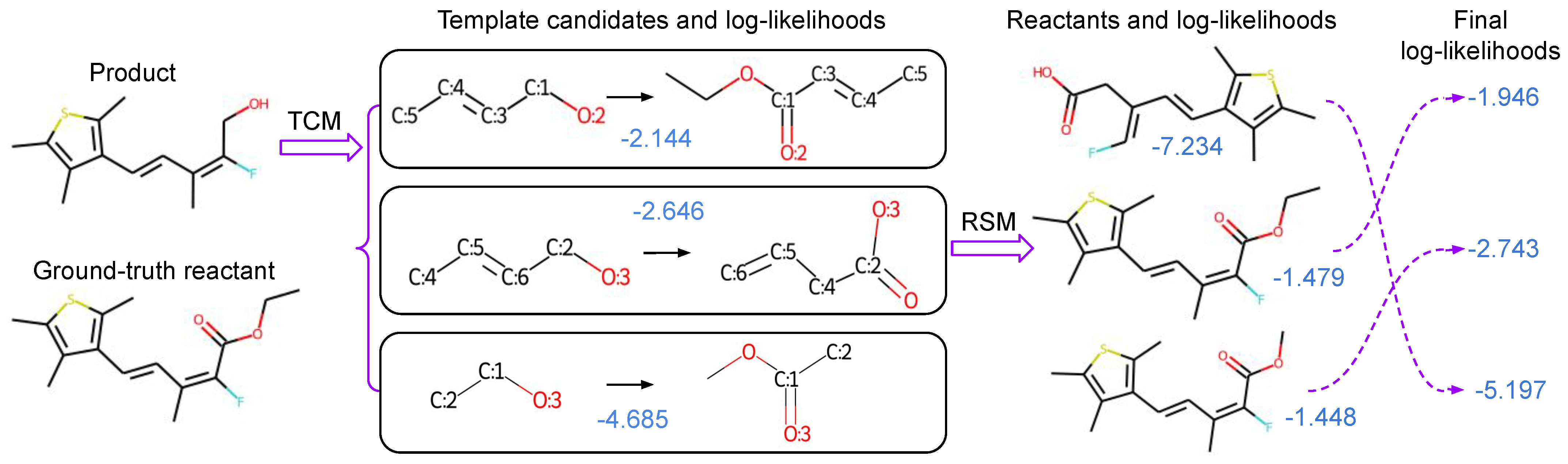
4.1. Compose Retrosynthesis Templates
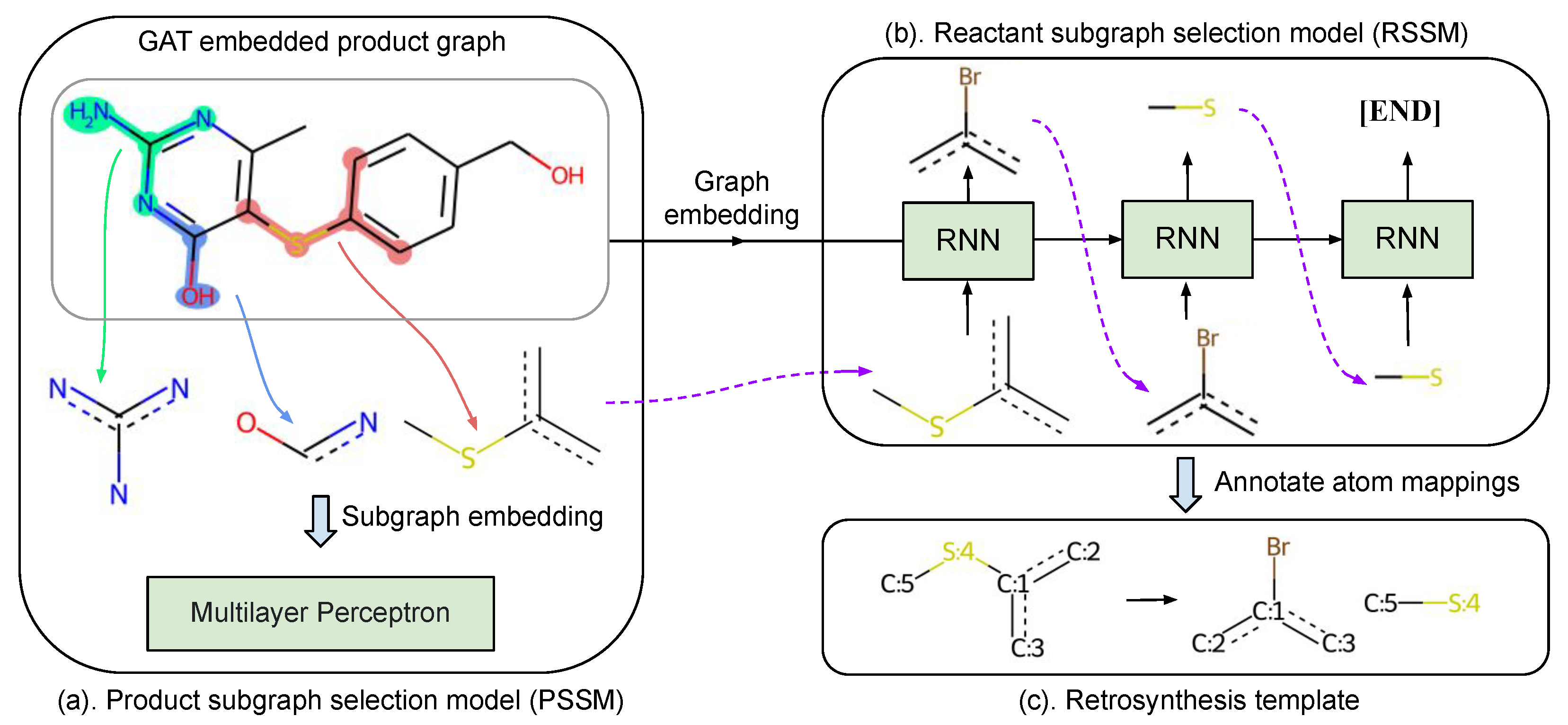
4.1.1. Subgraph Selection
4.1.2. Product Subgraph Selection
4.1.3. Reactant Subgraph Selection
4.1.4. Annotate Atom Mappings
4.2. Score Predicted Reactants
5. Experiment and Results
5.1. Dataset and Preprocessing
5.2. Evaluation
5.3. Implementation
5.4. Main Results
5.4.1. Retrosynthesis Prediction Performance
5.4.2. Ablation Study of PSSM Loss
5.4.3. Ablation Study of Hyper-Parameter λ
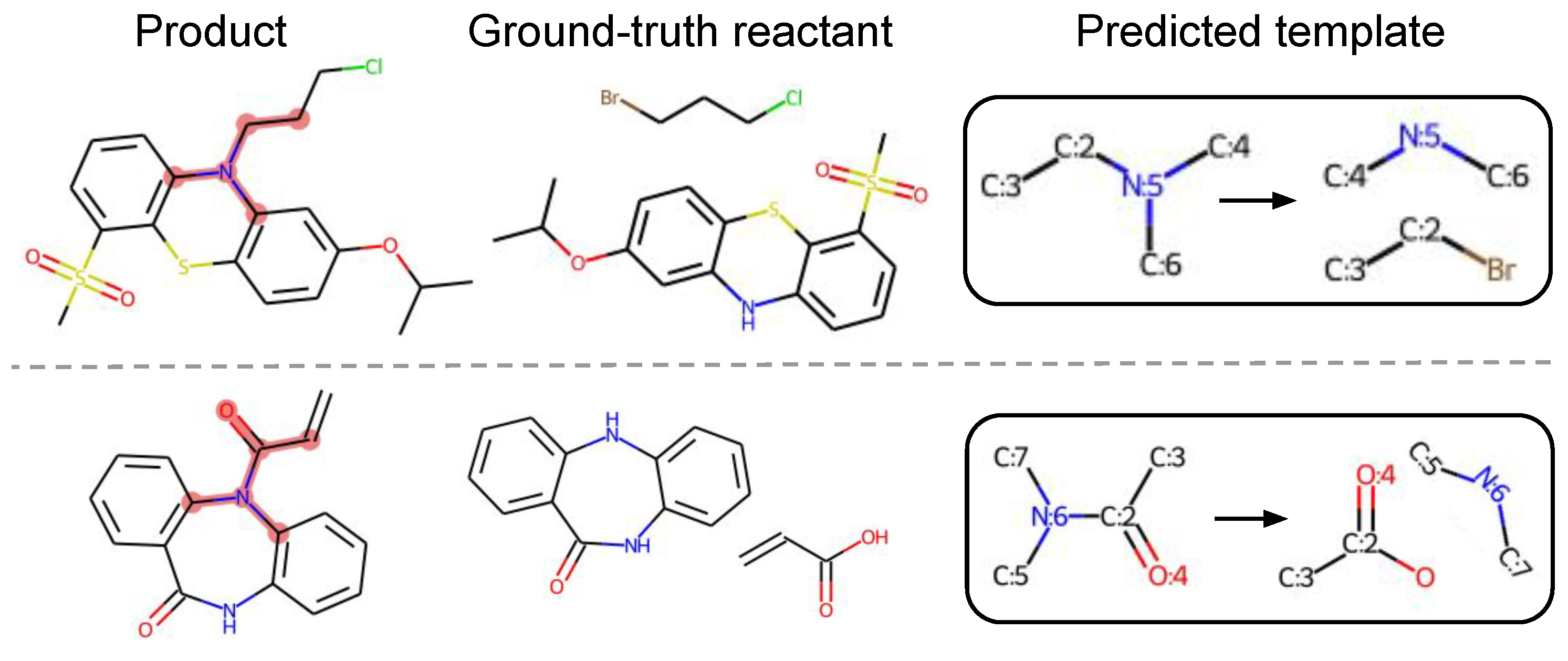
5.4.4. Novel Templates
6. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. USPTO-50K Dataset Information

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Reaction Type Name | Number of Reactions |
|---|---|---|
| 1 | Heteroatom alkylation and arylation | 15,204 |
| 2 | Acylation and related processes | 11,972 |
| 3 | C-C bond formation | 5667 |
| 4 | Heterocycle formation | 909 |
| 5 | Protections | 672 |
| 6 | Deprotections | 8405 |
| 7 | Reductions | 4642 |
| 8 | Oxidations | 822 |
| 9 | Functional group interconversion | 1858 |
| 10 | Functional group addition (FGA) | 231 |
| # total templates | 10,386 |
| # unique product subgraphs | 7766 |
| # unique reactant subgraphs | 4391 |
| Test reactions coverage by training templates | 94.08% |
| Average # contained product subgraphs per mol | 35.19 |
| Average # applicable product subgraphs per mol | 2.02 |
| Average # templates per reaction | 2.23 |
| Average # reactants per reaction | 1.71 |
Appendix A.2. Atom and Bond Features
| Feature | Description | Size |
|---|---|---|
| Bond type | Single, double, triple, or aromatic. | 4 |
| Conjugation | Whether the bond is conjugated. | 1 |
| In ring | Whether the bond is part of a ring. | 1 |
| Stereo | None, any, E/Z or cis/trans. | 6 |
| Feature | Description | Size |
|---|---|---|
| Atom type | Type of atom (ex. C, N, O), by atomic number. | 17 |
| # Bonds | Number of bonds the atom is involved in. | 6 |
| Formal charge | Integer electronic charge assigned to atom. | 5 |
| Chirality | Unspecified, tetrahedral CW/CCW, or other. | 4 |
| # Hs | Number of bonded Hydrogen atom. | 5 |
| Hybridization | sp, sp2, sp3, sp3d, or sp3d2. | 5 |
| Aromaticity | Whether this atom is part of an aromatic system. | 1 |
| Atomic mass | Mass of the atom, divided by 100. | 1 |
| Reaction type | The specified reaction type if it exists. | 10 |
References
- Corey, E.J.; Wipke, W.T. Computer-assisted design of complex organic syntheses. Science 1969, 166, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J. The logic of chemical synthesis: Multistep synthesis of complex carbogenic molecules (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1991, 30, 455–465. [Google Scholar] [CrossRef]
- Gothard, C.M.; Soh, S.; Gothard, N.A.; Kowalczyk, B.; Wei, Y.; Baytekin, B.; Grzybowski, B.A. Rewiring chemistry: Algorithmic discovery and experimental validation of one-pot reactions in the network of organic chemistry. Angew. Chem. Int. Ed. 2012, 51, 7922–7927. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Ding, Q.; Zhao, P.; Zheng, S.; Yang, J.; Yu, Y.; Huang, J. RetroXpert: Decompose Retrosynthesis Prediction Like A Chemist. Adv. Neural Inf. Process. Syst. 2020, 33, 11248–11258. [Google Scholar]
- Liu, B.; Ramsundar, B.; Kawthekar, P.; Shi, J.; Gomes, J.; Luu Nguyen, Q.; Ho, S.; Sloane, J.; Wender, P.; Pande, V. Retrosynthetic reaction prediction using neural sequence-to-sequence models. ACS Cent. Sci. 2017, 3, 1103–1113. [Google Scholar] [CrossRef]
- Zheng, S.; Rao, J.; Zhang, Z.; Xu, J.; Yang, Y. Predicting Retrosynthetic Reactions using Self-Corrected Transformer Neural Networks. J. Chem. Inf. Model. 2020, 60, 47–55. [Google Scholar] [CrossRef]
- Shi, C.; Xu, M.; Guo, H.; Zhang, M.; Tang, J. A Graph to Graphs Framework for Retrosynthesis Prediction. arXiv 2020, arXiv:2003.12725. [Google Scholar]
- Sacha, M.; Błaż, M.; Byrski, P.; Włodarczyk-Pruszyński, P.; Jastrzebski, S. Molecule Edit Graph Attention Network: Modeling Chemical Reactions as Sequences of Graph Edits. J. Chem. Inf. Model. 2021, 61, 3273–3284. [Google Scholar] [CrossRef]
- Sun, R.; Dai, H.; Li, L.; Kearnes, S.; Dai, B. Towards understanding retrosynthesis by energy-based models. Adv. Neural Inf. Process. Syst. 2021, 34, 10186–10194. [Google Scholar]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Tetko, I.V.; Karpov, P.; Van Deursen, R.; Godin, G. State-of-the-art augmented NLP transformer models for direct and single-step retrosynthesis. Nat. Commun. 2020, 11, 5575. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; Qiu, J.; Chen, G.; Liu, H.; Liao, B.; Hsieh, C.; Yao, X. RetroPrime: A Diverse, plausible and Transformer-based method for Single-Step retrosynthesis predictions. Chem. Eng. J. 2021, 420, 129845. [Google Scholar] [CrossRef]
- Somnath, V.R.; Bunne, C.; Coley, C.; Krause, A.; Barzilay, R. Learning graph models for retrosynthesis prediction. Adv. Neural Inf. Process. Syst. 2021, 34, 9405–9415. [Google Scholar]
- Szymkuć, S.; Gajewska, E.P.; Klucznik, T.; Molga, K.; Dittwald, P.; Startek, M.; Bajczyk, M.; Grzybowski, B.A. Computer-Assisted Synthetic Planning: The End of the Beginning. Angew. Chem. Int. Ed. 2016, 55, 5904–5937. [Google Scholar] [CrossRef]
- Coley, C.W.; Rogers, L.; Green, W.H.; Jensen, K.F. Computer-assisted retrosynthesis based on molecular similarity. ACS Cent. Sci. 2017, 3, 1237–1245. [Google Scholar] [CrossRef]
- Segler, M.H.; Waller, M.P. Neural-symbolic machine learning for retrosynthesis and reaction prediction. Chem.-Eur. J. 2017, 23, 5966–5971. [Google Scholar] [CrossRef]
- Segler, M.H.; Preuss, M.; Waller, M.P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. [Google Scholar] [CrossRef]
- Dai, H.; Li, C.; Coley, C.; Dai, B.; Song, L. Retrosynthesis Prediction with Conditional Graph Logic Network. In Proceedings of the Advances in Neural Information Processing Systems, Vancouver, BC, Canada, 8–14 December 2019; pp. 8870–8880. [Google Scholar]
- Segler, M.H.; Waller, M.P. Modelling chemical reasoning to predict and invent reactions. Chem.-Eur. J. 2017, 23, 6118–6128. [Google Scholar] [CrossRef]
- Baylon, J.L.; Cilfone, N.A.; Gulcher, J.R.; Chittenden, T.W. Enhancing retrosynthetic reaction prediction with deep learning using multiscale reaction classification. J. Chem. Inf. Model. 2019, 59, 673–688. [Google Scholar] [CrossRef]
- Tu, Z.; Coley, C.W. Permutation invariant graph-to-sequence model for template-free retrosynthesis and reaction prediction. arXiv 2021, arXiv:2110.09681. [Google Scholar] [CrossRef]
- Irwin, R.; Dimitriadis, S.; He, J.; Bjerrum, E.J. Chemformer: A Pre-Trained Transformer for Computational Chemistry. Mach. Learn. Sci. Technol. 2021, 3, 015022. [Google Scholar] [CrossRef]
- Mao, K.; Xiao, X.; Xu, T.; Rong, Y.; Huang, J.; Zhao, P. Molecular graph enhanced transformer for retrosynthesis prediction. Neurocomputing 2021, 457, 193–202. [Google Scholar] [CrossRef]
- Gilmer, J.; Schoenholz, S.S.; Riley, P.F.; Vinyals, O.; Dahl, G.E. Neural message passing for quantum chemistry. In Proceedings of the 34th International Conference on Machine Learning-Volume 70, Sydney, Australia, 6–11 August 2017; pp. 1263–1272. [Google Scholar]
- Veličković, P.; Cucurull, G.; Casanova, A.; Romero, A.; Liò, P.; Bengio, Y. Graph Attention Networks. In Proceedings of the 6th International Conference on Learning Representations, Vancouver, BC, Canada, 30 April–3 May 2018. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. In Proceedings of the Advances in Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; pp. 5998–6008. [Google Scholar]
- Xu, K.; Li, C.; Tian, Y.; Sonobe, T.; Kawarabayashi, K.i.; Jegelka, S. Representation learning on graphs with jumping knowledge networks. In Proceedings of the International Conference on Machine Learning, Stockholm, Sweden, 10–15 July 2018; pp. 5453–5462. [Google Scholar]
- Li, Y.; Tarlow, D.; Brockschmidt, M.; Zemel, R. Gated graph sequence neural networks. arXiv 2015, arXiv:1511.05493. [Google Scholar]
- Tillmann, C.; Ney, H. Word reordering and a dynamic programming beam search algorithm for statistical machine translation. Comput. Linguist. 2003, 29, 97–133. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics. 2021. Available online: https://github.com/rdkit/rdkit/tree/Release_2021_03_1 (accessed on 14 September 2022).
- Coley, C.W.; Green, W.H.; Jensen, K.F. RDChiral: An RDKit wrapper for handling stereochemistry in retrosynthetic template extraction and application. J. Chem. Inf. Model. 2019, 59, 2529–2537. [Google Scholar] [CrossRef]
- Jin, W.; Coley, C.; Barzilay, R.; Jaakkola, T. Predicting organic reaction outcomes with weisfeiler-lehman network. In Proceedings of the Advances in Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- Schneider, N.; Stiefl, N.; Landrum, G.A. What’s what: The (nearly) definitive guide to reaction role assignment. J. Chem. Inf. Model. 2016, 56, 2336–2346. [Google Scholar] [CrossRef]
- Lowe, D.M. Extraction of Chemical Structures and Reactions from the Literature. Ph.D. Thesis, University of Cambridge, Cambridge, UK, 2012. [Google Scholar]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. Pytorch: An imperative style, high-performance deep learning library. Adv. Neural Inf. Process. Syst. 2019, 32, 8026–8037. [Google Scholar]
- Fey, M.; Lenssen, J.E. Fast graph representation learning with PyTorch Geometric. arXiv 2019, arXiv:1903.02428. [Google Scholar]
- Hu, W.; Liu, B.; Gomes, J.; Zitnik, M.; Liang, P.; Pande, V.; Leskovec, J. Strategies for pre-training graph neural networks. arXiv 2019, arXiv:1905.12265. [Google Scholar]
- Cho, K.; Van Merriënboer, B.; Bahdanau, D.; Bengio, Y. On the properties of neural machine translation: Encoder-decoder approaches. arXiv 2014, arXiv:1409.1259. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Delving deep into rectifiers: Surpassing human-level performance on imagenet classification. In Proceedings of the IEEE International Conference on Computer Vision, Santiago, Chile, 7–13 December 2015; pp. 1026–1034. [Google Scholar]
- Ioffe, S.; Szegedy, C. Batch normalization: Accelerating deep network training by reducing internal covariate shift. In Proceedings of the International Conference on Machine Learning, Lille, France, 7–9 July 2015; pp. 448–456. [Google Scholar]
- Ba, J.L.; Kiros, J.R.; Hinton, G.E. Layer normalization. arXiv 2016, arXiv:1607.06450. [Google Scholar]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]




| Methods | Without Reaction Types | With Reaction Types | ||||||
|---|---|---|---|---|---|---|---|---|
| Top-1 | Top-3 | Top-5 | Top-10 | Top-1 | Top-3 | Top-5 | Top-10 | |
| Template-free methods | ||||||||
| SCROP [6] | 43.7 | 60.0 | 65.2 | 68.7 | 59.0 | 74.8 | 78.1 | 81.1 |
| G2Gs [7] | 48.9 | 67.6 | 72.5 | 75.5 | 61.0 | 81.3 | 86.0 | 88.7 |
| MEGAN [8] | 48.1 | 70.7 | 78.4 | 86.1 | 60.7 | 82.0 | 87.5 | 91.6 |
| RetroXpert* [4] | 50.4 | 61.1 | 62.3 | 63.4 | 62.1 | 75.8 | 78.5 | 80.9 |
| RetroPrime [12] | 51.4 | 70.8 | 74.0 | 76.1 | 64.8 | 81.6 | 85.0 | 86.9 |
| AT [11] | 53.5 | - | 81.0 | 85.7 | - | - | - | - |
| GraphRetro [13] | 53.7 | 68.3 | 72.2 | 75.5 | 63.9 | 81.5 | 85.2 | 88.1 |
| Dual model [9] | 53.6 | 70.7 | 74.6 | 77.0 | 65.7 | 81.9 | 84.7 | 85.9 |
| Template-based methods | ||||||||
| RetroSim [15] | 37.3 | 54.7 | 63.3 | 74.1 | 52.9 | 73.8 | 81.2 | 88.1 |
| NeuralSym [16] | 44.4 | 65.3 | 72.4 | 78.9 | 55.3 | 76.0 | 81.4 | 85.1 |
| GLN [18] | 52.5 | 69.0 | 75.6 | 83.7 | 64.2 | 79.1 | 85.2 | 90.0 |
| Ours | 54.5 | 77.2 | 83.2 | 87.7 | 65.9 | 85.8 | 89.5 | 91.5 |
| TCM only | 49.6 | 71.7 | 80.8 | 86.4 | 60.9 | 82.3 | 87.5 | 90.9 |
| RSM only | 51.8 | 75.7 | 82.4 | 87.3 | 64.3 | 84.8 | 88.9 | 91.4 |
| Types | Methods | Top-1 | Top-3 | Top-5 | Top-10 | |
|---|---|---|---|---|---|---|
| Without | Equation (6) | Ours | 54.5 | 77.2 | 83.2 | 87.7 |
| TCM only | 49.6 | 71.7 | 80.8 | 86.4 | ||
| RSM only | 51.8 | 75.7 | 82.4 | 87.3 | ||
| BCE | Ours | 53.1 | 77.1 | 83.8 | 89.2 | |
| TCM only | 46.5 | 69.9 | 78.5 | 86.9 | ||
| RSM only | 51.2 | 75.7 | 82.9 | 88.6 | ||
| With | Equation (6) | Ours | 65.9 | 85.8 | 89.5 | 91.5 |
| TCM only | 60.9 | 82.3 | 87.5 | 90.9 | ||
| RSM only | 64.3 | 84.8 | 88.9 | 91.4 | ||
| BCE | Ours | 65.3 | 85.9 | 90.3 | 92.6 | |
| TCM only | 58.5 | 81.8 | 87.6 | 91.5 | ||
| RSM only | 64.2 | 85.4 | 89.6 | 92.4 |
| 0 | 0.1 | 0.2 | 0.3 | 0.4 | 0.5 | 0.6 | 0.7 | 0.8 | 0.9 | 1.0 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Without types | 51.8 | 53.3 | 53.9 | 54.5 | 54.5 | 54.4 | 54.1 | 53.6 | 53.0 | 52.3 | 49.6 |
| With types | 64.3 | 65.2 | 65.6 | 65.7 | 65.9 | 65.9 | 65.6 | 65.1 | 64.7 | 64.4 | 60.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, C.; Zhao, P.; Lu, C.; Yu, Y.; Huang, J. RetroComposer: Composing Templates for Template-Based Retrosynthesis Prediction. Biomolecules 2022, 12, 1325. https://doi.org/10.3390/biom12091325
Yan C, Zhao P, Lu C, Yu Y, Huang J. RetroComposer: Composing Templates for Template-Based Retrosynthesis Prediction. Biomolecules. 2022; 12(9):1325. https://doi.org/10.3390/biom12091325
Chicago/Turabian StyleYan, Chaochao, Peilin Zhao, Chan Lu, Yang Yu, and Junzhou Huang. 2022. "RetroComposer: Composing Templates for Template-Based Retrosynthesis Prediction" Biomolecules 12, no. 9: 1325. https://doi.org/10.3390/biom12091325
APA StyleYan, C., Zhao, P., Lu, C., Yu, Y., & Huang, J. (2022). RetroComposer: Composing Templates for Template-Based Retrosynthesis Prediction. Biomolecules, 12(9), 1325. https://doi.org/10.3390/biom12091325