
Mitochondrial Reactive Oxygen Species, Insulin Resistance, and Nrf2-Mediated Oxidative Stress Response—Toward an Actionable Strategy for Anti-Aging



Abstract
:1. Introduction
2. Mitochondrial Dysfunction at the Common Pathway in T2D Pathogenesis
2.1. T2D Etiology
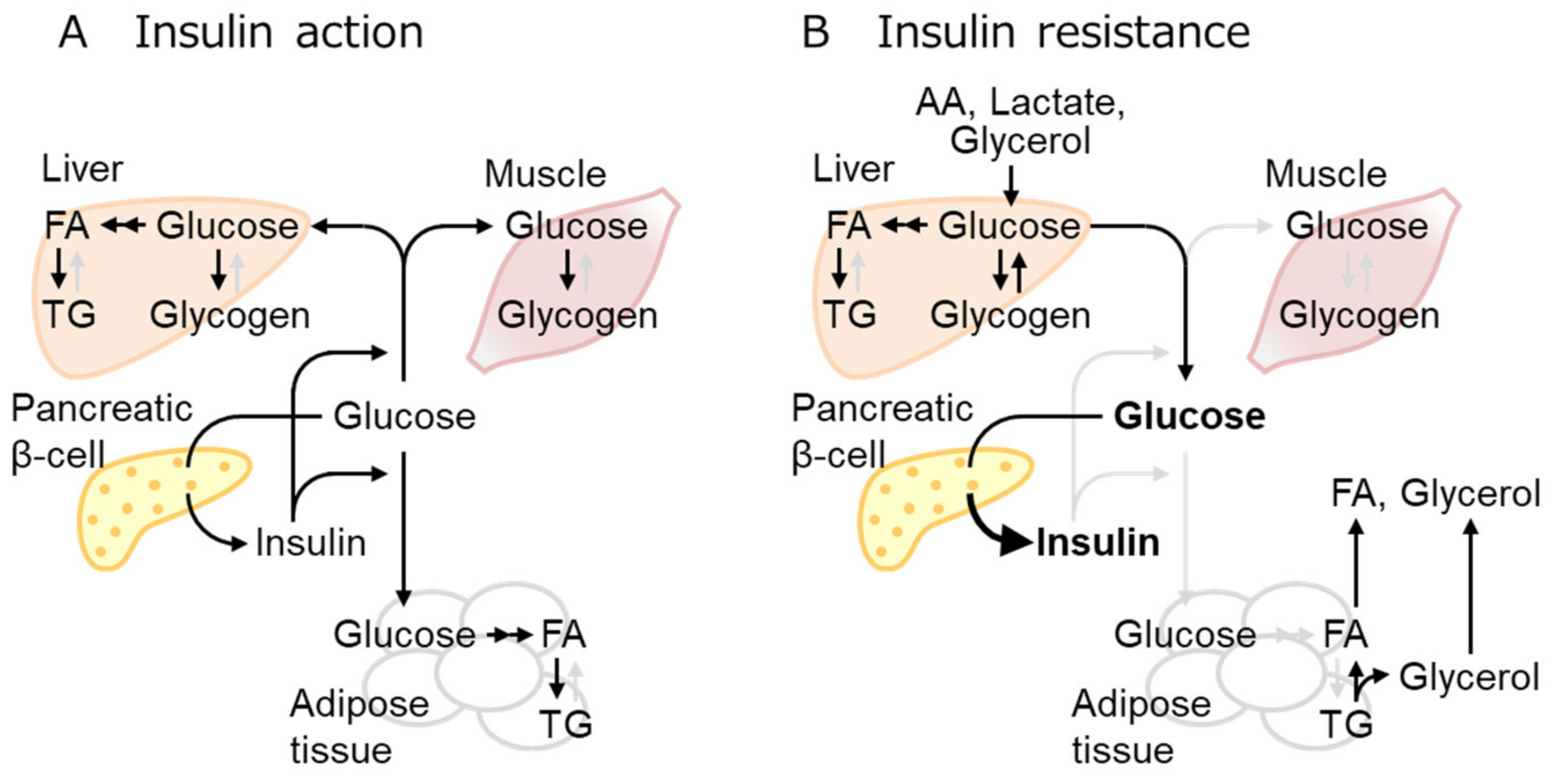
2.2. IR Mechanisms as the Early Event of Obese T2D
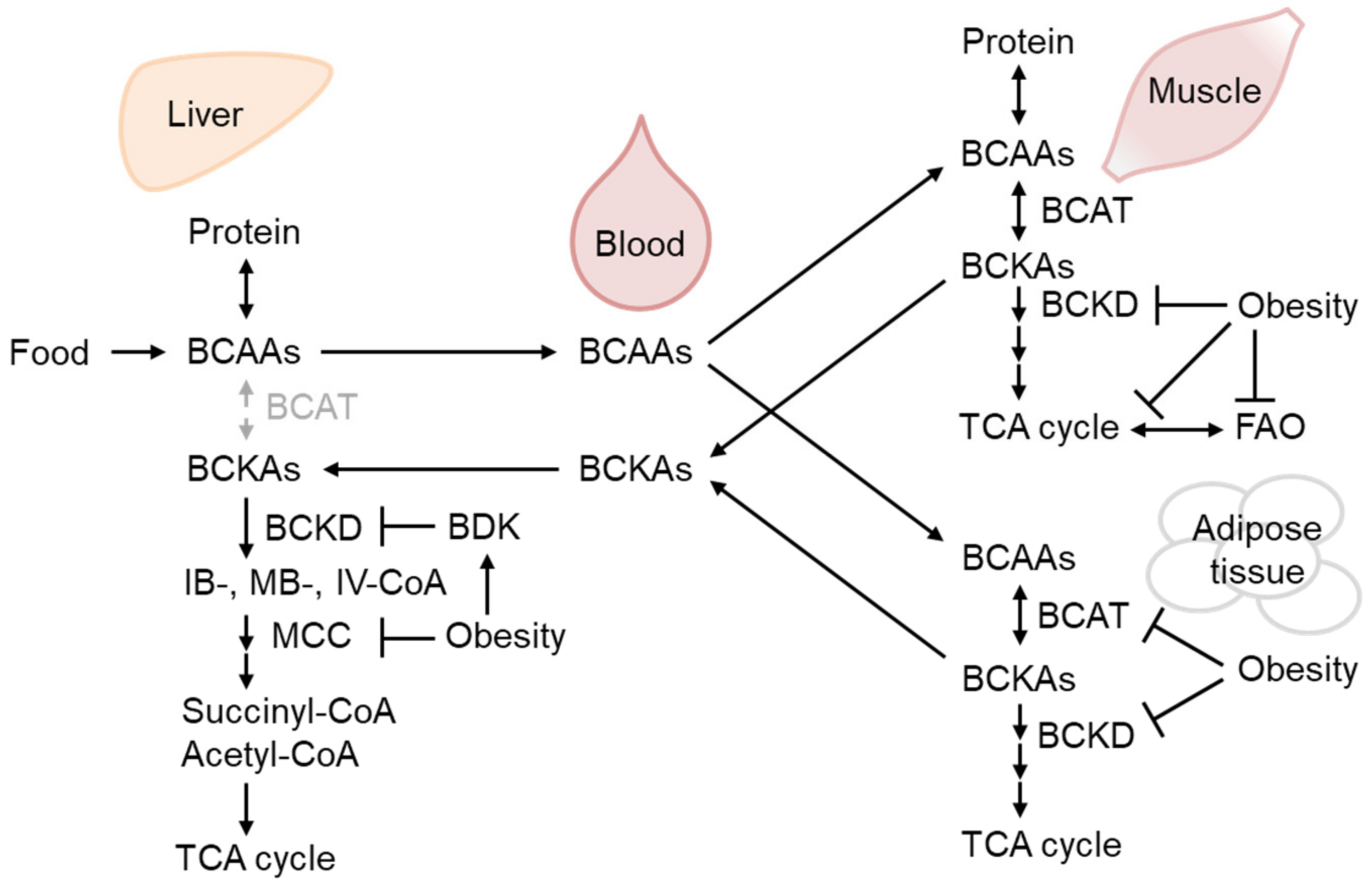
2.3. BCAA Metabolism Reduction in WAT as the Earliest Response to Obesity-Related Hyperinsulinemia and IR

3. The Role of mtROS in IR
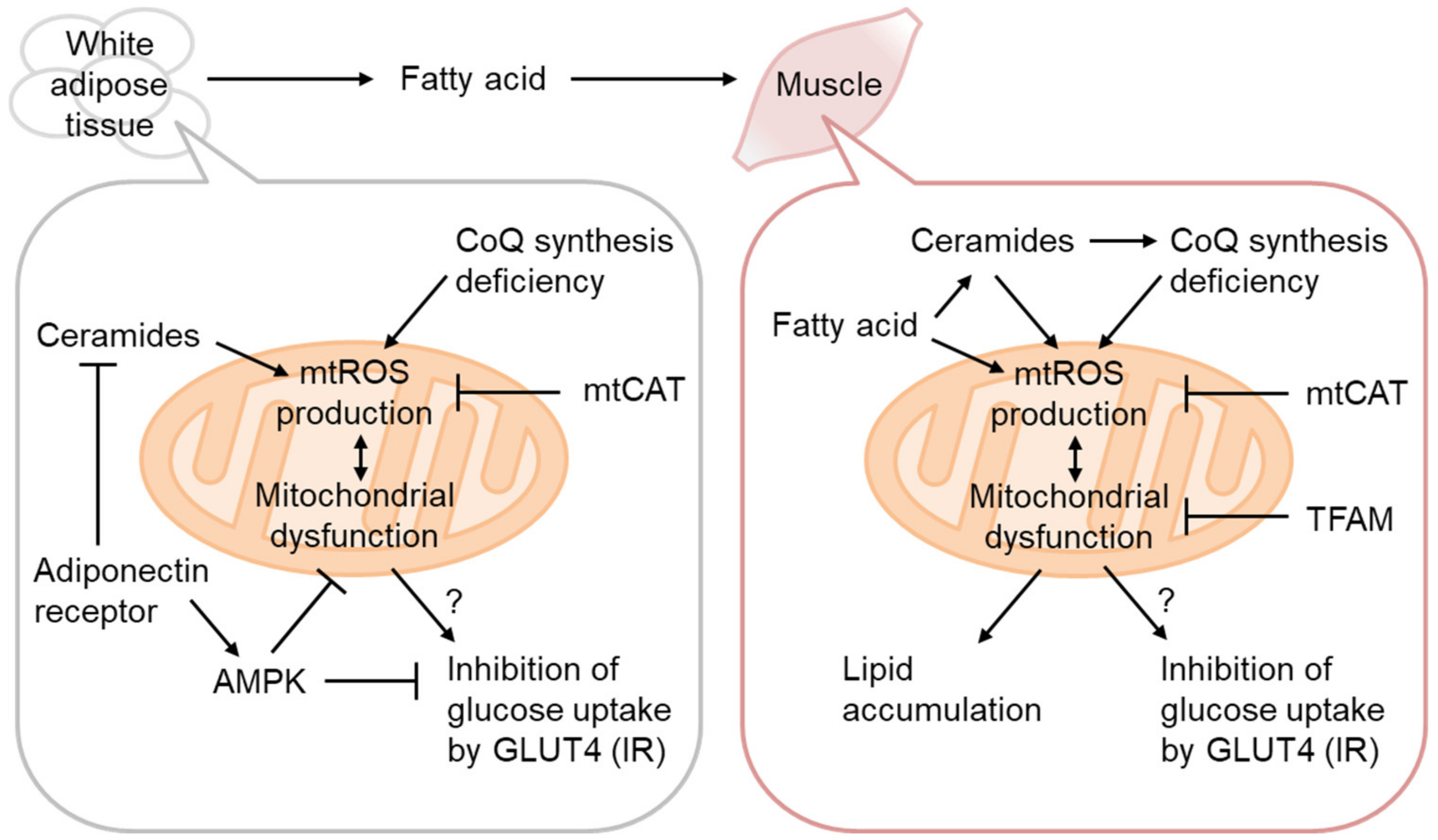
3.1. mtROS as the Cause of IR
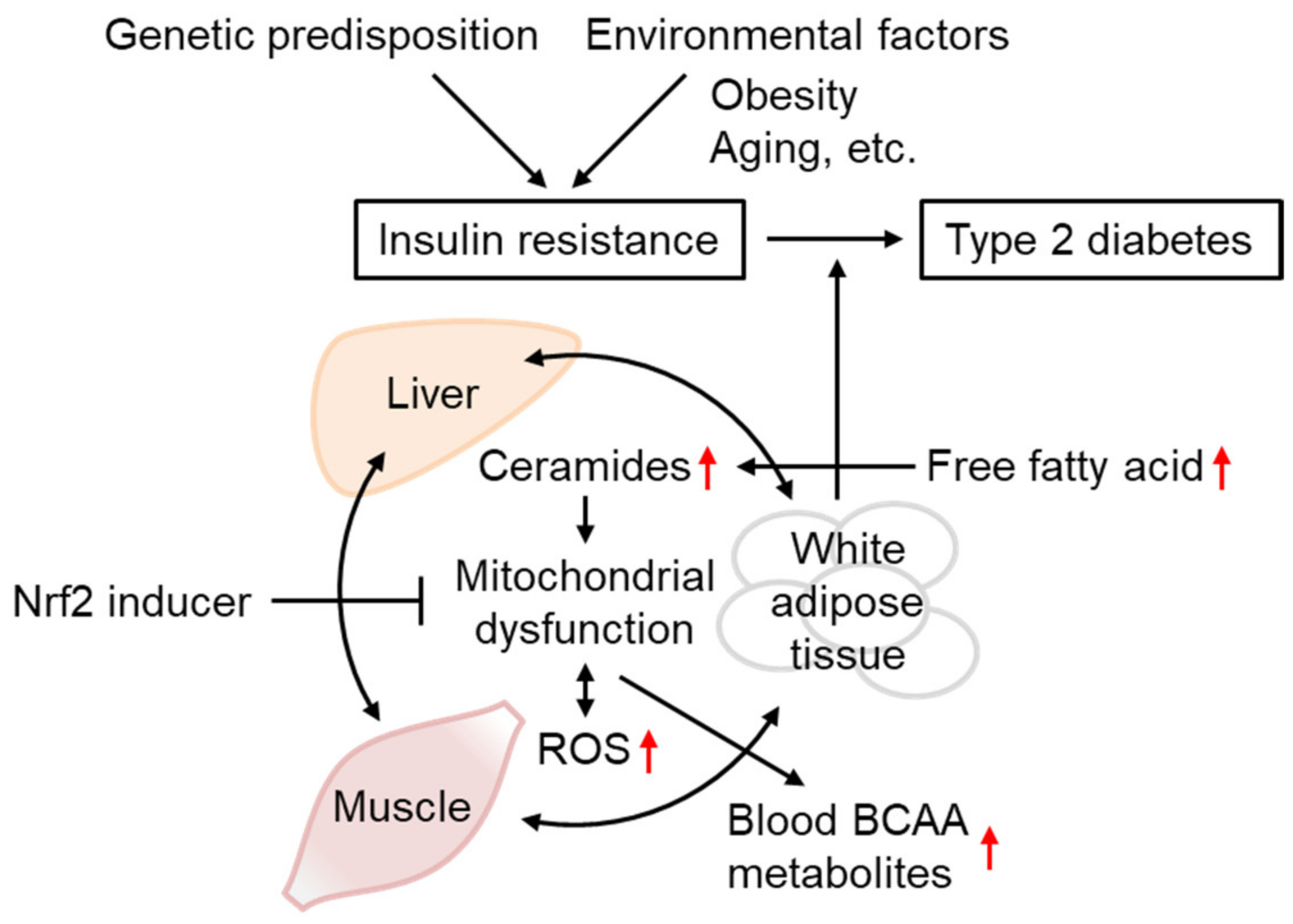
3.2. Ceramides as mtROS Sources in Insulin Resistance
4. Role of Nrf2 in Prevention of Obesity-Related IR and T2D
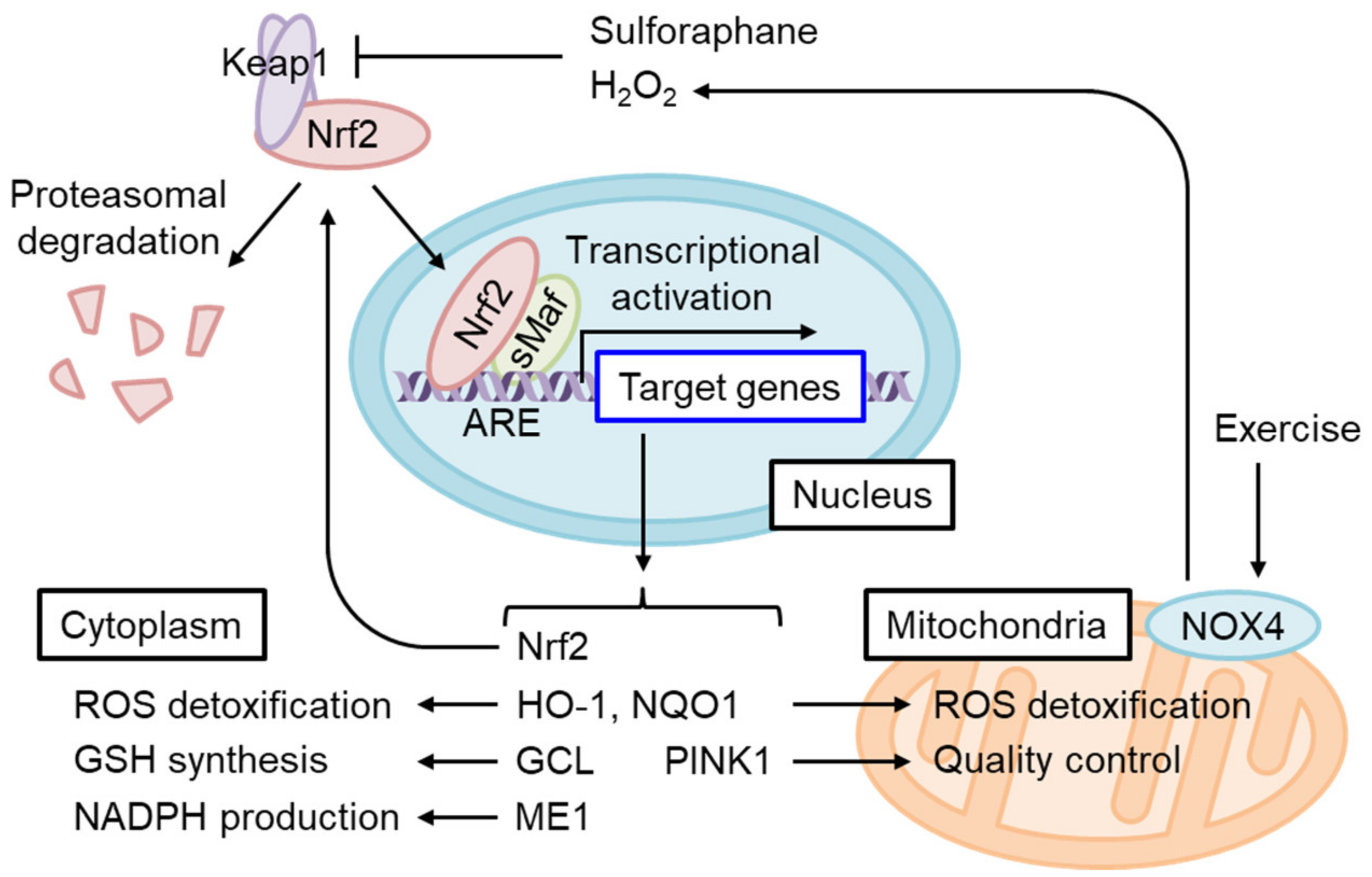
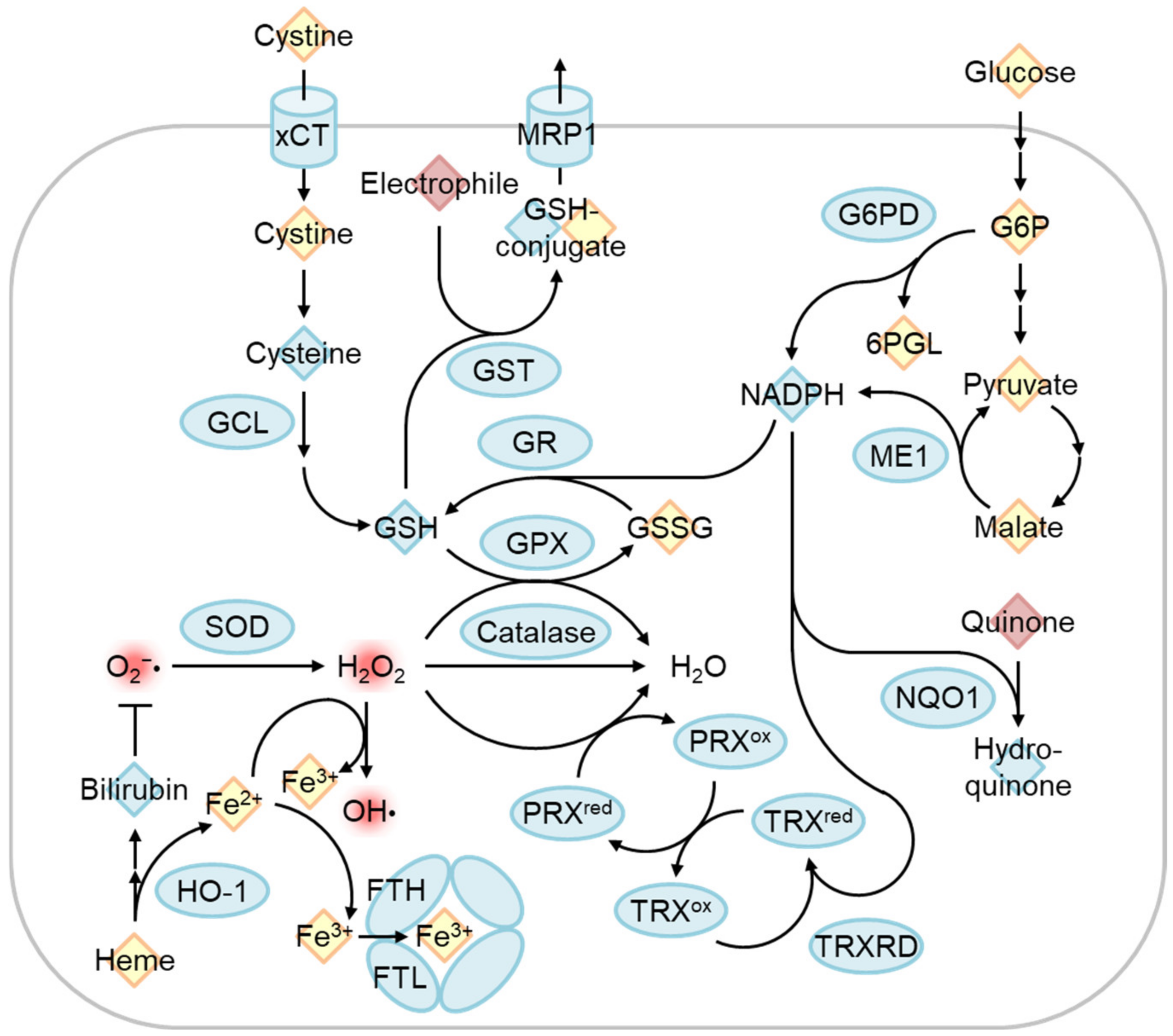
4.1. The Regulation of Oxidative Stress Response by Nrf2
4.2. The Role of Nrf2 Activation in WAT and Obesity
4.3. The Role of Nrf2 in Pancreatic β-Cells
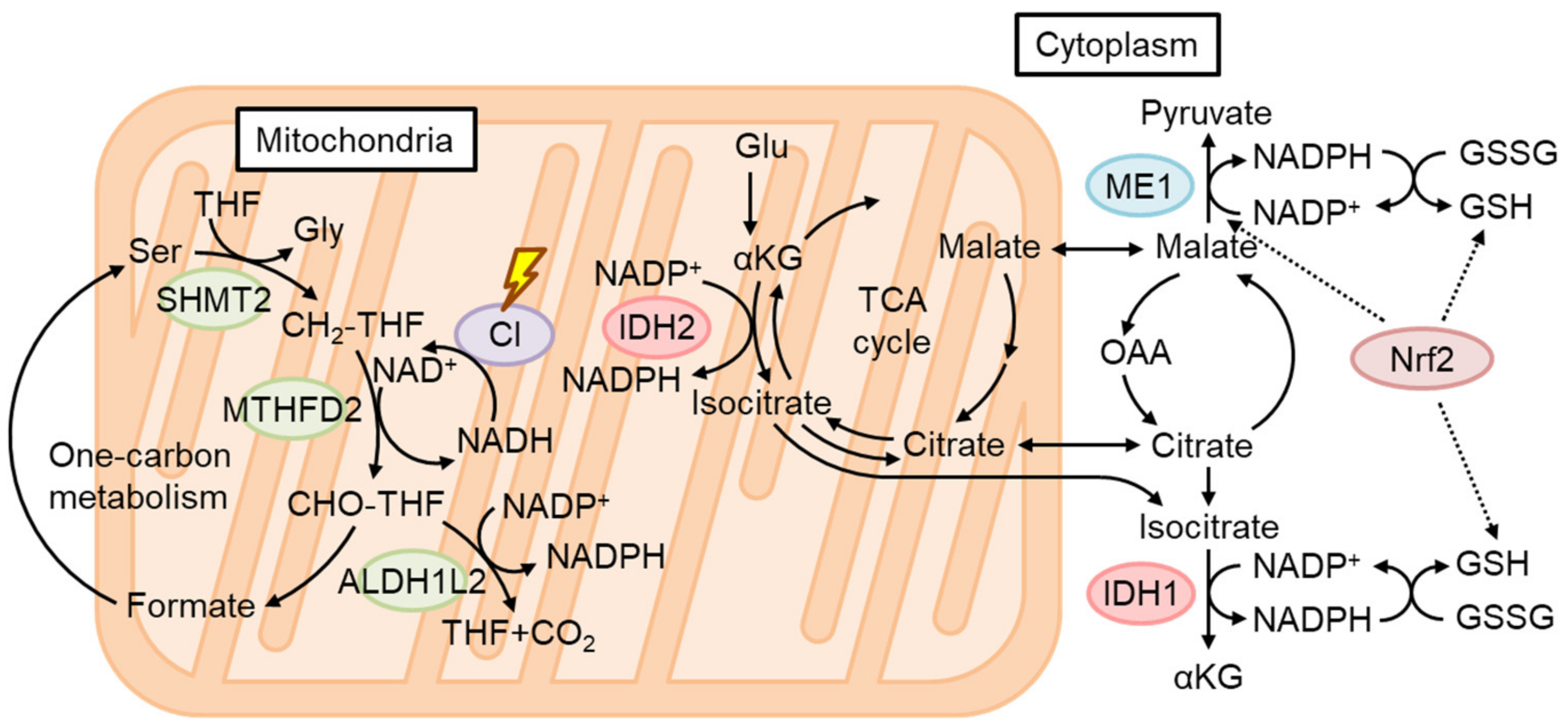
4.4. The Role of Nrf2 in Mitochondrial Function
4.5. Insulin Resistance Alleviation upon Nrf2 Activation
4.6. The Effect of Aging on T2D Etiology
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barcena, C.; Mayoral, P.; Quiros, P.M. Mitohormesis, an Antiaging Paradigm. Int. Rev. Cell Mol. Biol. 2018, 340, 35–77. [Google Scholar] [CrossRef]
- Ayer, A.; Fazakerley, D.J.; James, D.E.; Stocker, R. The role of mitochondrial reactive oxygen species in insulin resistance. Free Radic. Biol. Med. 2022, 179, 339–362. [Google Scholar] [CrossRef] [PubMed]
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Loshuertos, R.; Acin-Perez, R.; Fernandez-Silva, P.; Movilla, N.; Perez-Martos, A.; Rodriguez de Cordoba, S.; Gallardo, M.E.; Enriquez, J.A. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat. Genet. 2006, 38, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Itoh, K.; Mimura, J.; Yamamoto, M. Discovery of the negative regulator of Nrf2, Keap1: A historical overview. Antioxid. Redox. Signal. 2010, 13, 1665–1678. [Google Scholar] [CrossRef]
- Gacesa, R.; Dunlap, W.C.; Barlow, D.J.; Laskowski, R.A.; Long, P.F. Rising levels of atmospheric oxygen and evolution of Nrf2. Sci. Rep. 2016, 6, 27740. [Google Scholar] [CrossRef]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- Abdul-Muneer, P.M. Nrf2 as a Potential Therapeutic Target for Traumatic Brain Injury. J. Integr. Neurosci. 2023, 22, 81. [Google Scholar] [CrossRef]
- Tanase, D.M.; Gosav, E.M.; Anton, M.I.; Floria, M.; Seritean Isac, P.N.; Hurjui, L.L.; Tarniceriu, C.C.; Costea, C.F.; Ciocoiu, M.; Rezus, C. Oxidative Stress and NRF2/KEAP1/ARE Pathway in Diabetic Kidney Disease (DKD): New Perspectives. Biomolecules 2022, 12, 1227. [Google Scholar] [CrossRef] [PubMed]
- Tossetta, G.; Fantone, S.; Montanari, E.; Marzioni, D.; Goteri, G. Role of NRF2 in Ovarian Cancer. Antioxidants 2022, 11, 663. [Google Scholar] [CrossRef] [PubMed]
- Ghareghomi, S.; Habibi-Rezaei, M.; Arese, M.; Saso, L.; Moosavi-Movahedi, A.A. Nrf2 Modulation in Breast Cancer. Biomedicines 2022, 10, 2668. [Google Scholar] [CrossRef] [PubMed]
- Tossetta, G.; Fantone, S.; Marzioni, D.; Mazzucchelli, R. Cellular Modulators of the NRF2/KEAP1 Signaling Pathway in Prostate Cancer. Front. Biosci. 2023, 28, 143. [Google Scholar] [CrossRef]
- Hoehn, K.L.; Salmon, A.B.; Hohnen-Behrens, C.; Turner, N.; Hoy, A.J.; Maghzal, G.J.; Stocker, R.; Van Remmen, H.; Kraegen, E.W.; Cooney, G.J.; et al. Insulin resistance is a cellular antioxidant defense mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 17787–17792. [Google Scholar] [CrossRef]
- James, D.E.; Stockli, J.; Birnbaum, M.J. The aetiology and molecular landscape of insulin resistance. Nat. Rev. Mol. Cell Biol. 2021, 22, 751–771. [Google Scholar] [CrossRef]
- Gray, S.L.; Donald, C.; Jetha, A.; Covey, S.D.; Kieffer, T.J. Hyperinsulinemia precedes insulin resistance in mice lacking pancreatic beta-cell leptin signaling. Endocrinology 2010, 151, 4178–4186. [Google Scholar] [CrossRef]
- Rajan, S.; Shankar, K.; Beg, M.; Varshney, S.; Gupta, A.; Srivastava, A.; Kumar, D.; Mishra, R.K.; Hussain, Z.; Gayen, J.R.; et al. Chronic hyperinsulinemia reduces insulin sensitivity and metabolic functions of brown adipocyte. J. Endocrinol. 2016, 230, 275–290. [Google Scholar] [CrossRef]
- Perseghin, G.; Scifo, P.; De Cobelli, F.; Pagliato, E.; Battezzati, A.; Arcelloni, C.; Vanzulli, A.; Testolin, G.; Pozza, G.; Del Maschio, A.; et al. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: A 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes 1999, 48, 1600–1606. [Google Scholar] [CrossRef]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.A.; Lagou, V.; Welch, R.P.; Wheeler, E.; Montasser, M.E.; Luan, J.; Mägi, R.; Strawbridge, R.J.; Rehnberg, E.; Gustafsson, S.; et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat. Genet. 2012, 44, 991–1005. [Google Scholar] [CrossRef] [PubMed]
- Billings, L.K.; Florez, J.C. The genetics of type 2 diabetes: What have we learned from GWAS? Ann. N. Y. Acad. Sci. 2010, 1212, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Camporez, J.G.; Kursawe, R.; Titchenell, P.M.; Zhang, D.; Perry, C.J.; Jurczak, M.J.; Abudukadier, A.; Han, M.S.; Zhang, X.M.; et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 2015, 160, 745–758. [Google Scholar] [CrossRef]
- Titchenell, P.M.; Quinn, W.J.; Lu, M.; Chu, Q.; Lu, W.; Li, C.; Chen, H.; Monks, B.R.; Chen, J.; Rabinowitz, J.D.; et al. Direct Hepatocyte Insulin Signaling Is Required for Lipogenesis but Is Dispensable for the Suppression of Glucose Production. Cell Metab. 2016, 23, 1154–1166. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef]
- Biddinger, S.B.; Hernandez-Ono, A.; Rask-Madsen, C.; Haas, J.T.; Aleman, J.O.; Suzuki, R.; Scapa, E.F.; Agarwal, C.; Carey, M.C.; Stephanopoulos, G.; et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008, 7, 125–134. [Google Scholar] [CrossRef]
- Lee, Y.S.; Li, P.; Huh, J.Y.; Hwang, I.J.; Lu, M.; Kim, J.I.; Ham, M.; Talukdar, S.; Chen, A.; Lu, W.J.; et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 2011, 60, 2474–2483. [Google Scholar] [CrossRef]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef]
- Majithia, A.R.; Flannick, J.; Shahinian, P.; Guo, M.; Bray, M.A.; Fontanillas, P.; Gabriel, S.B.; Rosen, E.D.; Altshuler, D. Rare variants in PPARG with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 13127–13132. [Google Scholar] [CrossRef]
- White, P.J.; Newgard, C.B. Branched-chain amino acids in disease. Science 2019, 363, 582–583. [Google Scholar] [CrossRef] [PubMed]
- Gojda, J.; Cahova, M. Gut Microbiota as the Link between Elevated BCAA Serum Levels and Insulin Resistance. Biomolecules 2021, 11, 1414. [Google Scholar] [CrossRef] [PubMed]
- Zaganjor, E.; Yoon, H.; Spinelli, J.B.; Nunn, E.R.; Laurent, G.; Keskinidis, P.; Sivaloganathan, S.; Joshi, S.; Notarangelo, G.; Mulei, S.; et al. SIRT4 is an early regulator of branched-chain amino acid catabolism that promotes adipogenesis. Cell Rep. 2021, 36, 109345. [Google Scholar] [CrossRef] [PubMed]
- Green, C.R.; Wallace, M.; Divakaruni, A.S.; Phillips, S.A.; Murphy, A.N.; Ciaraldi, T.P.; Metallo, C.M. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol. 2016, 12, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Holecek, M. The role of skeletal muscle in the pathogenesis of altered concentrations of branched-chain amino acids (valine, leucine, and isoleucine) in liver cirrhosis, diabetes, and other diseases. Physiol. Res. 2021, 70, 293–305. [Google Scholar] [CrossRef]
- Anderson, K.A.; Huynh, F.K.; Fisher-Wellman, K.; Stuart, J.D.; Peterson, B.S.; Douros, J.D.; Wagner, G.R.; Thompson, J.W.; Madsen, A.S.; Green, M.F.; et al. SIRT4 Is a Lysine Deacylase that Controls Leucine Metabolism and Insulin Secretion. Cell Metab. 2017, 25, 838–855.e815. [Google Scholar] [CrossRef]
- Shao, J.; Liu, Y.; Zhang, X.; Shu, L.; Yu, J.; Yang, S.; Gao, C.; Wang, C.; Cao, N.; Zhou, M.; et al. BCAA catabolism drives adipogenesis via an intermediate metabolite and promotes subcutaneous adipose tissue expansion during obesity. bioRxiv 2022. [Google Scholar] [CrossRef]
- Diaz-Vegas, A.; Madsen, S.; Cooke, K.C.; Carroll, L.; Khor, J.X.Y.; Turner, N.; Lim, X.Y.; Astore, M.A.; Morris, J.; Don, A.; et al. Mitochondrial electron transport chain, ceramide and Coenzyme Q are linked in a pathway that drives insulin resistance in skeletal muscle. bioRxiv 2023. [Google Scholar] [CrossRef]
- Pietilainen, K.H.; Naukkarinen, J.; Rissanen, A.; Saharinen, J.; Ellonen, P.; Keranen, H.; Suomalainen, A.; Gotz, A.; Suortti, T.; Yki-Jarvinen, H.; et al. Global transcript profiles of fat in monozygotic twins discordant for BMI: Pathways behind acquired obesity. PLoS Med. 2008, 5, e51. [Google Scholar] [CrossRef]
- Lerin, C.; Goldfine, A.B.; Boes, T.; Liu, M.; Kasif, S.; Dreyfuss, J.M.; De Sousa-Coelho, A.L.; Daher, G.; Manoli, I.; Sysol, J.R.; et al. Defects in muscle branched-chain amino acid oxidation contribute to impaired lipid metabolism. Mol. Metab. 2016, 5, 926–936. [Google Scholar] [CrossRef]
- Zandberg, L.; van Dyk, H.C.; van der Westhuizen, F.H.; van Dijk, A.A. A 3-methylcrotonyl-CoA carboxylase deficient human skin fibroblast transcriptome reveals underlying mitochondrial dysfunction and oxidative stress. Int. J. Biochem. Cell Biol. 2016, 78, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Shao, J.; Wu, C.Y.; Shu, L.; Dong, W.; Liu, Y.; Chen, M.; Wynn, R.M.; Wang, J.; Wang, J.; et al. Targeting BCAA Catabolism to Treat Obesity-Associated Insulin Resistance. Diabetes 2019, 68, 1730–1746. [Google Scholar] [CrossRef]
- Vanweert, F.; Neinast, M.; Tapia, E.E.; van de Weijer, T.; Hoeks, J.; Schrauwen-Hinderling, V.B.; Blair, M.C.; Bornstein, M.R.; Hesselink, M.K.C.; Schrauwen, P.; et al. A randomized placebo-controlled clinical trial for pharmacological activation of BCAA catabolism in patients with type 2 diabetes. Nat. Commun. 2022, 13, 3508. [Google Scholar] [CrossRef] [PubMed]
- Menni, C.; Fauman, E.; Erte, I.; Perry, J.R.; Kastenmuller, G.; Shin, S.Y.; Petersen, A.K.; Hyde, C.; Psatha, M.; Ward, K.J.; et al. Biomarkers for type 2 diabetes and impaired fasting glucose using a nontargeted metabolomics approach. Diabetes 2013, 62, 4270–4276. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef]
- Guo, X.; Zhu, Y.; Guo, L.; Qi, Y.; Liu, X.; Wang, J.; Zhang, J.; Cui, L.; Shi, Y.; Wang, Q.; et al. BCAA insufficiency leads to premature ovarian insufficiency via ceramide-induced elevation of ROS. EMBO Mol. Med. 2023, 15, e17450. [Google Scholar] [CrossRef]
- She, P.; Van Horn, C.; Reid, T.; Hutson, S.M.; Cooney, R.N.; Lynch, C.J. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1552–E1563. [Google Scholar] [CrossRef]
- Lian, K.; Du, C.; Liu, Y.; Zhu, D.; Yan, W.; Zhang, H.; Hong, Z.; Liu, P.; Zhang, L.; Pei, H.; et al. Impaired adiponectin signaling contributes to disturbed catabolism of branched-chain amino acids in diabetic mice. Diabetes 2015, 64, 49–59. [Google Scholar] [CrossRef]
- Huynh, F.K.; Peterson, B.S.; Anderson, K.A.; Lin, Z.; Coakley, A.J.; Llaguno, F.M.S.; Nguyen, T.N.; Campbell, J.E.; Stephens, S.B.; Newgard, C.B.; et al. β-Cell-specific ablation of sirtuin 4 does not affect nutrient-stimulated insulin secretion in mice. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E805–E813. [Google Scholar] [CrossRef]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef]
- Turner, N.; Heilbronn, L.K. Is mitochondrial dysfunction a cause of insulin resistance? Trends Endocrinol. Metab. 2008, 19, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Knowles, J.W.; Xie, W.; Zhang, Z.; Chennamsetty, I.; Assimes, T.L.; Paananen, J.; Hansson, O.; Pankow, J.; Goodarzi, M.O.; Carcamo-Orive, I.; et al. Identification and validation of N-acetyltransferase 2 as an insulin sensitivity gene. J. Clin. Investig. 2015, 125, 1739–1751. [Google Scholar] [CrossRef] [PubMed]
- Camporez, J.P.; Wang, Y.; Faarkrog, K.; Chukijrungroat, N.; Petersen, K.F.; Shulman, G.I. Mechanism by which arylamine N-acetyltransferase 1 ablation causes insulin resistance in mice. Proc. Natl. Acad. Sci. USA 2017, 114, E11285–E11292. [Google Scholar] [CrossRef] [PubMed]
- Nair, K.S.; Bigelow, M.L.; Asmann, Y.W.; Chow, L.S.; Coenen-Schimke, J.M.; Klaus, K.A.; Guo, Z.K.; Sreekumar, R.; Irving, B.A. Asian Indians have enhanced skeletal muscle mitochondrial capacity to produce ATP in association with severe insulin resistance. Diabetes 2008, 57, 1166–1175. [Google Scholar] [CrossRef]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W., 3rd; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef]
- Lee, H.Y.; Lee, J.S.; Alves, T.; Ladiges, W.; Rabinovitch, P.S.; Jurczak, M.J.; Choi, C.S.; Shulman, G.I.; Samuel, V.T. Mitochondrial-Targeted Catalase Protects Against High-Fat Diet-Induced Muscle Insulin Resistance by Decreasing Intramuscular Lipid Accumulation. Diabetes 2017, 66, 2072–2081. [Google Scholar] [CrossRef]
- Koh, J.H.; Johnson, M.L.; Dasari, S.; LeBrasseur, N.K.; Vuckovic, I.; Henderson, G.C.; Cooper, S.A.; Manjunatha, S.; Ruegsegger, G.N.; Shulman, G.I.; et al. TFAM Enhances Fat Oxidation and Attenuates High-Fat Diet-Induced Insulin Resistance in Skeletal Muscle. Diabetes 2019, 68, 1552–1564. [Google Scholar] [CrossRef]
- Fazakerley, D.J.; Minard, A.Y.; Krycer, J.R.; Thomas, K.C.; Stockli, J.; Harney, D.J.; Burchfield, J.G.; Maghzal, G.J.; Caldwell, S.T.; Hartley, R.C.; et al. Mitochondrial oxidative stress causes insulin resistance without disrupting oxidative phosphorylation. J. Biol. Chem. 2018, 293, 7315–7328. [Google Scholar] [CrossRef]
- Fazakerley, D.J.; Chaudhuri, R.; Yang, P.; Maghzal, G.J.; Thomas, K.C.; Krycer, J.R.; Humphrey, S.J.; Parker, B.L.; Fisher-Wellman, K.H.; Meoli, C.C.; et al. Mitochondrial CoQ deficiency is a common driver of mitochondrial oxidants and insulin resistance. Elife 2018, 7, e32111. [Google Scholar] [CrossRef]
- Bikman, B.T.; Summers, S.A. Ceramides as modulators of cellular and whole-body metabolism. J. Clin. Investig. 2011, 121, 4222–4230. [Google Scholar] [CrossRef]
- Villa, N.Y.; Kupchak, B.R.; Garitaonandia, I.; Smith, J.L.; Alonso, E.; Alford, C.; Cowart, L.A.; Hannun, Y.A.; Lyons, T.J. Sphingolipids function as downstream effectors of a fungal PAQR. Mol. Pharmacol. 2009, 75, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Roszczyc-Owsiejczuk, K.; Zabielski, P. Sphingolipids as a Culprit of Mitochondrial Dysfunction in Insulin Resistance and Type 2 Diabetes. Front. Endocrinol. 2021, 12, 635175. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, B.; Tippetts, T.S.; Mayoral Monibas, R.; Liu, J.; Li, Y.; Wang, L.; Wilkerson, J.L.; Sweeney, C.R.; Pereira, R.F.; Sumida, D.H.; et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science 2019, 365, 386–392. [Google Scholar] [CrossRef]
- Marzec, J.M.; Christie, J.D.; Reddy, S.P.; Jedlicka, A.E.; Vuong, H.; Lanken, P.N.; Aplenc, R.; Yamamoto, T.; Yamamoto, M.; Cho, H.Y.; et al. Functional polymorphisms in the transcription factor NRF2 in humans increase the risk of acute lung injury. FASEB J. 2007, 21, 2237–2246. [Google Scholar] [CrossRef]
- Bouviere, J.; Fortunato, R.S.; Dupuy, C.; Werneck-de-Castro, J.P.; Carvalho, D.P.; Louzada, R.A. Exercise-Stimulated ROS Sensitive Signaling Pathways in Skeletal Muscle. Antioxidants 2021, 10, 537. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zuo, Z.; Li, L.; Ren, S.; Gao, T.; Fu, J.; Hou, Y.; Chen, Y.; Pi, J. Nrf2 in adipocytes. Arch. Pharm. Res. 2020, 43, 350–360. [Google Scholar] [CrossRef]
- Li, S.; Eguchi, N.; Lau, H.; Ichii, H. The Role of the Nrf2 Signaling in Obesity and Insulin Resistance. Int. J. Mol. Sci. 2020, 21, 6973. [Google Scholar] [CrossRef]
- Annie-Mathew, A.S.; Prem-Santhosh, S.; Jayasuriya, R.; Ganesh, G.; Ramkumar, K.M.; Sarada, D.V.L. The pivotal role of Nrf2 activators in adipocyte biology. Pharmacol. Res. 2021, 173, 105853. [Google Scholar] [CrossRef]
- Xu, J.; Kulkarni, S.R.; Donepudi, A.C.; More, V.R.; Slitt, A.L. Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice. Diabetes 2012, 61, 3208–3218. [Google Scholar] [CrossRef]
- Chartoumpekis, D.V.; Palliyaguru, D.L.; Wakabayashi, N.; Fazzari, M.; Khoo, N.K.H.; Schopfer, F.J.; Sipula, I.; Yagishita, Y.; Michalopoulos, G.K.; O’Doherty, R.M.; et al. Nrf2 deletion from adipocytes, but not hepatocytes, potentiates systemic metabolic dysfunction after long-term high-fat diet-induced obesity in mice. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E180–E195. [Google Scholar] [CrossRef]
- Xue, P.; Hou, Y.; Chen, Y.; Yang, B.; Fu, J.; Zheng, H.; Yarborough, K.; Woods, C.G.; Liu, D.; Yamamoto, M.; et al. Adipose deficiency of Nrf2 in ob/ob mice results in severe metabolic syndrome. Diabetes 2013, 62, 845–854. [Google Scholar] [CrossRef]
- Tanaka, Y.; Aleksunes, L.M.; Yeager, R.L.; Gyamfi, M.A.; Esterly, N.; Guo, G.L.; Klaassen, C.D. NF-E2-related factor 2 inhibits lipid accumulation and oxidative stress in mice fed a high-fat diet. J. Pharmacol. Exp. Ther. 2008, 325, 655–664. [Google Scholar] [CrossRef]
- Sahin, K.; Orhan, C.; Akdemir, F.; Tuzcu, M.; Sahin, N.; Yilmaz, I.; Juturu, V. β-Cryptoxanthin ameliorates metabolic risk factors by regulating NF-kappaB and Nrf2 pathways in insulin resistance induced by high-fat diet in rodents. Food Chem. Toxicol. 2017, 107, 270–279. [Google Scholar] [CrossRef]
- Elksnis, A.; Martinell, M.; Eriksson, O.; Espes, D. Heterogeneity of Metabolic Defects in Type 2 Diabetes and Its Relation to Reactive Oxygen Species and Alterations in Beta-Cell Mass. Front. Physiol. 2019, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Baumel-Alterzon, S.; Katz, L.S.; Brill, G.; Garcia-Ocana, A.; Scott, D.K. Nrf2: The Master and Captain of Beta Cell Fate. Trends Endocrinol. Metab. 2021, 32, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martin, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef] [PubMed]
- Baumel-Alterzon, S.; Katz, L.S.; Brill, G.; Jean-Pierre, C.; Li, Y.; Tse, I.; Biswal, S.; Garcia-Ocana, A.; Scott, D.K. Nrf2 Regulates beta-Cell Mass by Suppressing beta-Cell Death and Promoting beta-Cell Proliferation. Diabetes 2022, 71, 989–1011. [Google Scholar] [CrossRef]
- Itoh, K.; Ye, P.; Matsumiya, T.; Tanji, K.; Ozaki, T. Emerging functional cross-talk between the Keap1-Nrf2 system and mitochondria. J. Clin. Biochem. Nutr. 2015, 56, 91–97. [Google Scholar] [CrossRef]
- Pensabene, K.M.; LaMorte, J.; Allender, A.E.; Wehr, J.; Kaur, P.; Savage, M.; Eggler, A.L. Acute Oxidative Stress Can Paradoxically Suppress Human NRF2 Protein Synthesis by Inhibiting Global Protein Translation. Antioxidants 2023, 12, 1735. [Google Scholar] [CrossRef]
- Balsa, E.; Perry, E.A.; Bennett, C.F.; Jedrychowski, M.; Gygi, S.P.; Doench, J.G.; Puigserver, P. Defective NADPH production in mitochondrial disease complex I causes inflammation and cell death. Nat. Commun. 2020, 11, 2714. [Google Scholar] [CrossRef]
- Kasai, S.; Yamazaki, H.; Tanji, K.; Engler, M.J.; Matsumiya, T.; Itoh, K. Role of the ISR-ATF4 pathway and its cross talk with Nrf2 in mitochondrial quality control. J. Clin. Biochem. Nutr. 2019, 64, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Teng, W.; Li, Y.; Du, M.; Lei, X.; Xie, S.; Ren, F. Sulforaphane Prevents Hepatic Insulin Resistance by Blocking Serine Palmitoyltransferase 3-Mediated Ceramide Biosynthesis. Nutrients 2019, 11, 1185. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xie, S.; Teng, W. Sulforaphane Attenuates Nonalcoholic Fatty Liver Disease by Inhibiting Hepatic Steatosis and Apoptosis. Nutrients 2021, 14, 76. [Google Scholar] [CrossRef] [PubMed]
- Uruno, A.; Yagishita, Y.; Katsuoka, F.; Kitajima, Y.; Nunomiya, A.; Nagatomi, R.; Pi, J.; Biswal, S.S.; Yamamoto, M. Nrf2-Mediated Regulation of Skeletal Muscle Glycogen Metabolism. Mol. Cell Biol. 2016, 36, 1655–1672. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, Q.; Liu, J.; Zhang, Z.; Ma, X.; Zhang, Y.; Zhu, J.; Thring, R.W.; Wu, M.; Gao, Y.; et al. Sulforaphane alleviates high fat diet-induced insulin resistance via AMPK/Nrf2/GPx4 axis. Biomed. Pharmacother. 2022, 152, 113273. [Google Scholar] [CrossRef]
- Nagata, N.; Xu, L.; Kohno, S.; Ushida, Y.; Aoki, Y.; Umeda, R.; Fuke, N.; Zhuge, F.; Ni, Y.; Nagashimada, M.; et al. Glucoraphanin Ameliorates Obesity and Insulin Resistance through Adipose Tissue Browning and Reduction of Metabolic Endotoxemia in Mice. Diabetes 2017, 66, 1222–1236. [Google Scholar] [CrossRef]
- Xirouchaki, C.E.; Jia, Y.; McGrath, M.J.; Greatorex, S.; Tran, M.; Merry, T.L.; Hong, D.; Eramo, M.J.; Broome, S.C.; Woodhead, J.S.T.; et al. Skeletal muscle NOX4 is required for adaptive responses that prevent insulin resistance. Sci. Adv. 2021, 7, eabl4988. [Google Scholar] [CrossRef]
- Díaz-Casado, M.E.; Quiles, J.L.; Barriocanal-Casado, E.; González-García, P.; Battino, M.; López, L.C.; Varela-López, A. The Paradox of Coenzyme Q(10) in Aging. Nutrients 2019, 11, 2221. [Google Scholar] [CrossRef]
- Schmidlin, C.J.; Dodson, M.B.; Madhavan, L.; Zhang, D.D. Redox regulation by NRF2 in aging and disease. Free Radic. Biol. Med. 2019, 134, 702–707. [Google Scholar] [CrossRef]
- Rizvi, F.; Preston, C.C.; Emelyanova, L.; Yousufuddin, M.; Viqar, M.; Dakwar, O.; Ross, G.R.; Faustino, R.S.; Holmuhamedov, E.L.; Jahangir, A. Effects of Aging on Cardiac Oxidative Stress and Transcriptional Changes in Pathways of Reactive Oxygen Species Generation and Clearance. J. Am. Heart Assoc. 2021, 10, e019948. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reports | References | Section |
|---|---|---|
| Hyperinsulinemia precedes IR | [17,18] | Section 2.1 |
| Mitochondrial dysfunction and IR in lean T2D | [20,21] | Section 2.1 |
| Genetic factors associated with T2D | [22,23] | Section 2.1 |
| Selective IR in the liver | [25,26,27] | Section 2.2 |
| Impairment of BCAA metabolism by obesity | [33,35,36,37,38,39,41,42,47,48,49] | Section 2.3 |
| BCAA as a biomarker of obesity-associated IR | [44,45,46] | Section 2.3 |
| Reports | References | Section |
|---|---|---|
| Mitochondrial dysfunction in skeletal muscle is associated with IR | [21,50,51] | Section 3.1 |
| NAT2 gene variant associated with IR etiology | [52,53] | Section 3.1 |
| mtROS production is associated with IR etiology | [55,56,57,58,59] | Section 3.1 |
| Ceramide accumulation leads to mtROS production | [38,46,50,60,61,62,63] | Section 2.3 and Section 3.2 |
| Reports | References | Section |
|---|---|---|
| Background of Nrf2 and the antioxidant system | [7,64,65,66] | Section 4.1 |
| Systemic Nrf2 KO and obesity-induced IR | [66,67,68,69] | Section 4.2 |
| Adipocyte-specific Nrf2 KO worsens obesity-induced IR | [70,71] | Section 4.2 |
| Nrf2 activation improves obesity-induced IR | [72,73] | Section 4.2 |
| Role of Nrf2 in pancreatic β-cell function | [77] | Section 4.3 |
| Role of Nrf2 and its target genes in mitochondrial function | [78,81,82,83] | Section 4.4 |
| IR alleviation by the Nrf2 activator | [84,86] | Section 4.5 |
| NOX4-Nrf2 axis in muscle | [87] | Section 4.5 |
| Aging and T2D etiology | [88,89,90] | Section 4.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasai, S.; Kokubu, D.; Mizukami, H.; Itoh, K. Mitochondrial Reactive Oxygen Species, Insulin Resistance, and Nrf2-Mediated Oxidative Stress Response—Toward an Actionable Strategy for Anti-Aging. Biomolecules 2023, 13, 1544. https://doi.org/10.3390/biom13101544
Kasai S, Kokubu D, Mizukami H, Itoh K. Mitochondrial Reactive Oxygen Species, Insulin Resistance, and Nrf2-Mediated Oxidative Stress Response—Toward an Actionable Strategy for Anti-Aging. Biomolecules. 2023; 13(10):1544. https://doi.org/10.3390/biom13101544
Chicago/Turabian StyleKasai, Shuya, Daichi Kokubu, Hiroki Mizukami, and Ken Itoh. 2023. "Mitochondrial Reactive Oxygen Species, Insulin Resistance, and Nrf2-Mediated Oxidative Stress Response—Toward an Actionable Strategy for Anti-Aging" Biomolecules 13, no. 10: 1544. https://doi.org/10.3390/biom13101544
APA StyleKasai, S., Kokubu, D., Mizukami, H., & Itoh, K. (2023). Mitochondrial Reactive Oxygen Species, Insulin Resistance, and Nrf2-Mediated Oxidative Stress Response—Toward an Actionable Strategy for Anti-Aging. Biomolecules, 13(10), 1544. https://doi.org/10.3390/biom13101544