
An Introduced RNA-Only Approach for Plasmid Curing via the CRISPR-Cpf1 System in Saccharomyces cerevisiae


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Strains, Media and Growth Conditions
2.2. Plasmid Construction and crRNA Design
2.3. Transformation and Plasmid-Curing in Yeast
2.4. Statistical Analysis
3. Results
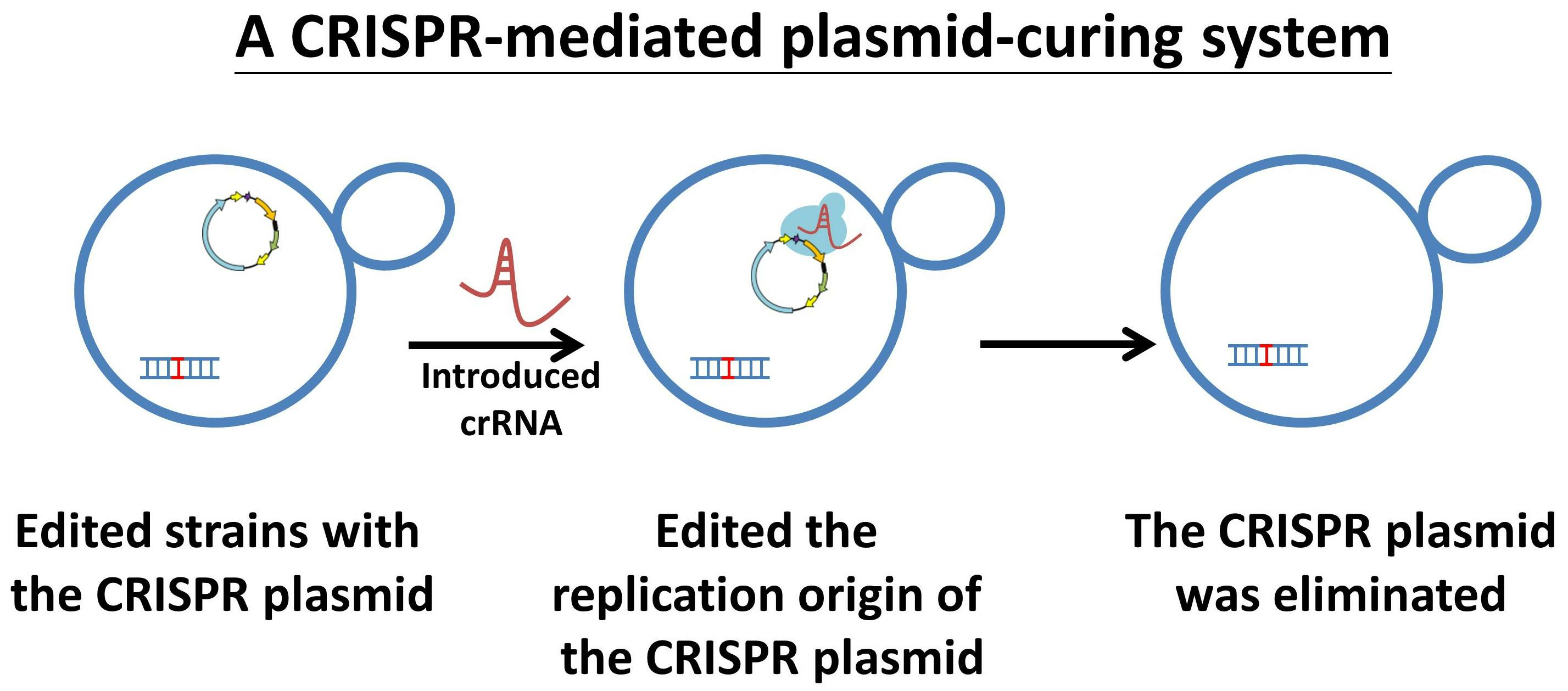
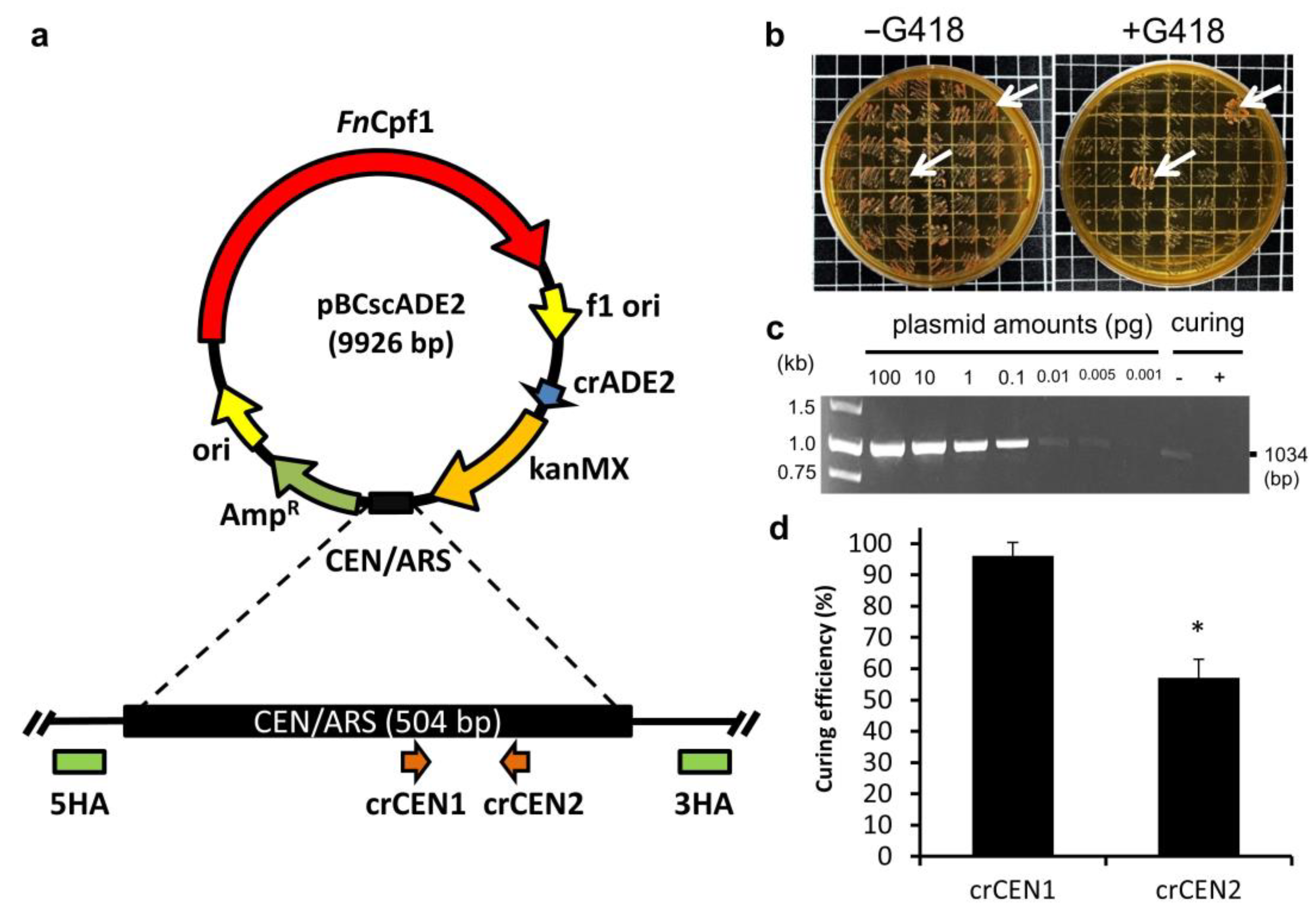
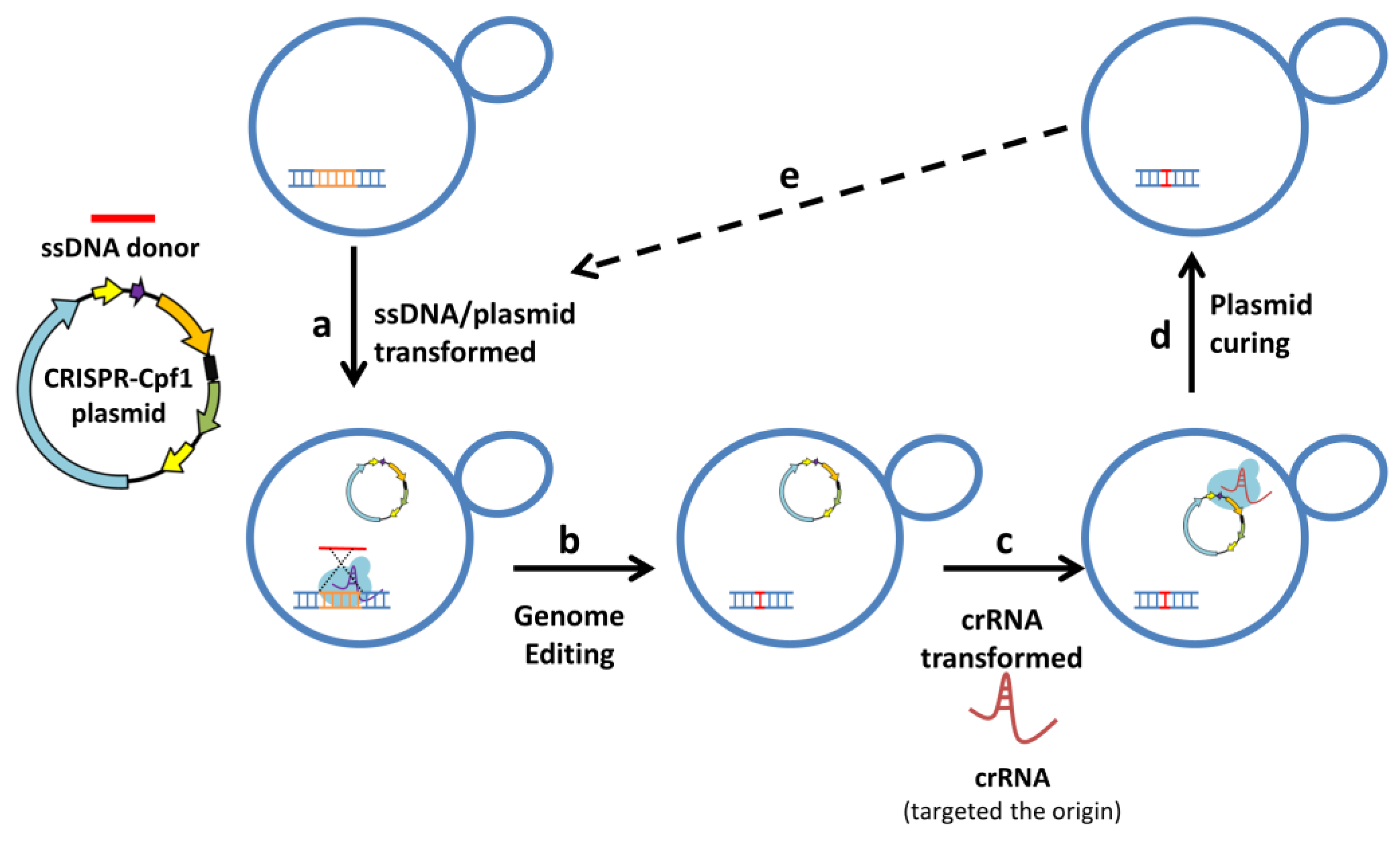
3.1. Design a Plasmid-Curing System Based on the Cleavage of the Replication Origin
3.2. CRISPR Plasmids Can Be Cured with Only a Single crRNA
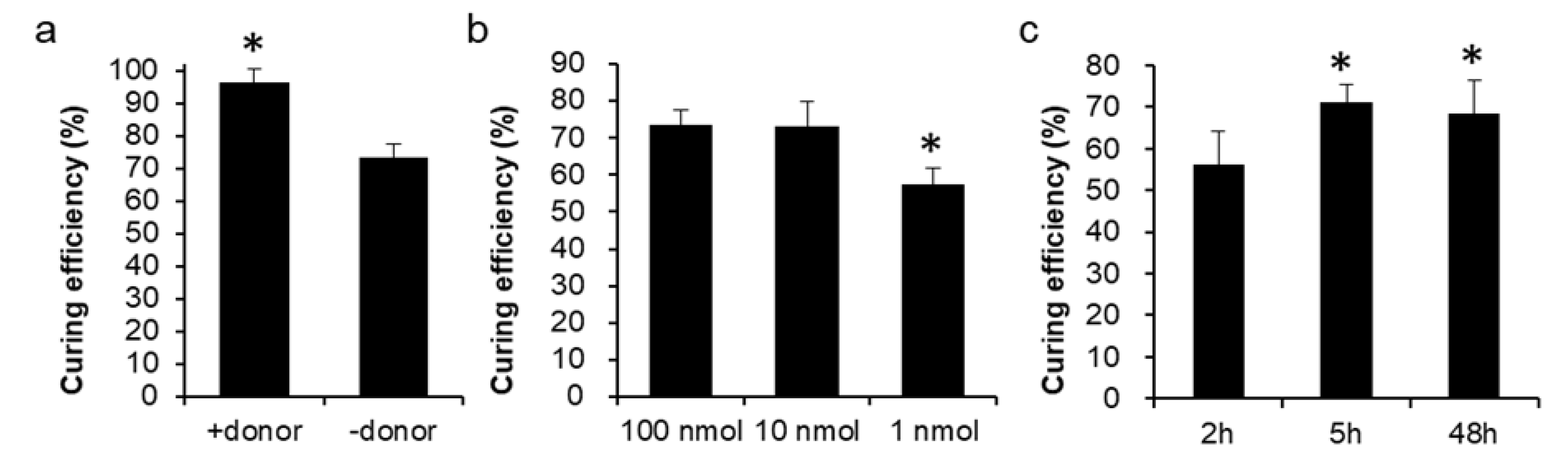
3.3. The Economic and Efficient Plasmid-Curing System
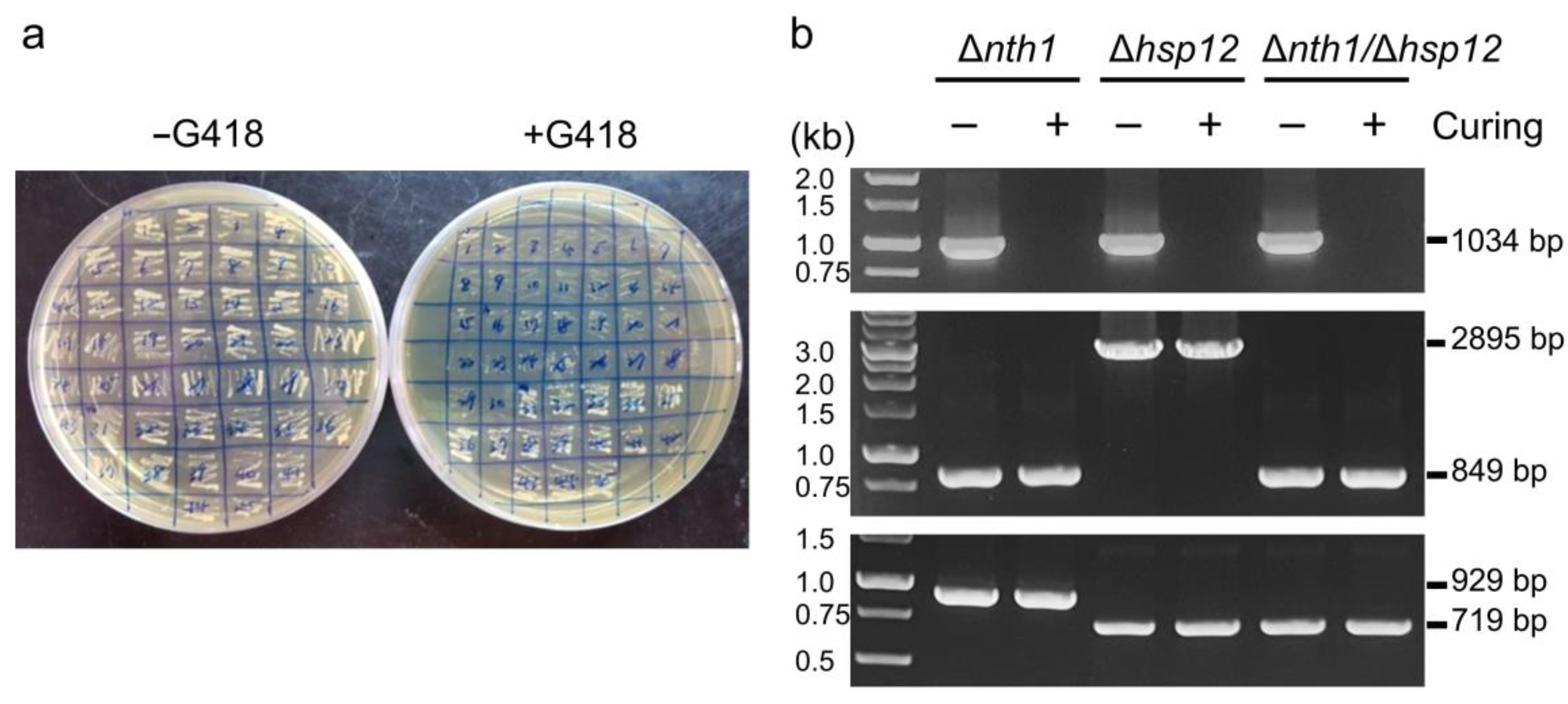
3.4. The Genome Edited and the Plasmid-Free Yeast Strains
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Margulis, L.; Chapman, M.J. Endosymbioses: Cyclical and permanent in evolution. Trends Microbiol. 1998, 6, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Guerineau, M.; Grandchamp, C.; Paoletti, C.; Slonimski, P. Characterization of a new class of circular DNA molecules in yeast. Biochem. Biophys. Res. Commun. 1971, 42, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Hülter, N.; Ilhan, J.; Wein, T.; Kadibalban, A.S.; Hammerschmidt, K.; Dagan, T. An evolutionary perspective on plasmid lifestyle modes. Curr. Opin. Microbiol. 2017, 38, 74–80. [Google Scholar] [CrossRef]
- Shintani, M.; Sanchez, Z.K.; Kimbara, K. Genomics of microbial plasmids: Classification and identification based on replication and transfer systems and host taxonomy. Front. Microbiol. 2015, 6, 242. [Google Scholar] [CrossRef]
- del Solar, G.; Giraldo, R.; Ruiz-Echevarría, M.J.; Espinosa, M.; Díaz-Orejas, R. Replication and control of circular bacterial plasmids. Microbiol. Mol. Biol. Rev. 1998, 62, 434–464. [Google Scholar] [CrossRef] [PubMed]
- Jensen, N.B.; Strucko, T.; Kildegaard, K.R.; David, F.; Maury, J.; Mortensen, U.H.; Forster, J.; Nielsen, J.; Borodina, I. EasyClone: Method for iterative chromosomal integration of multiple genes in Saccharomyces cerevisiae. FEMS Yeast Res. 2014, 14, 238–248. [Google Scholar] [CrossRef]
- Hohnholz, R.; Pohlmann, K.J.; Achstetter, T. A set of isomeric episomal plasmids for systematic examination of mitotic stability in Saccharomyces cerevisiae. Yeast 2017, 34, 267–275. [Google Scholar] [CrossRef]
- Chlebowicz-Śledziewska, E.; Śledziewski, A.Z. Construction of multicopy yeast plasmids with regulated centromere function. Gene 1985, 39, 25–31. [Google Scholar] [CrossRef]
- Shmakova, A.A.; Shmakova, O.P.; Karpukhina, A.A.; Vassetzky, Y.S. CRISPR/Cas: History and perspectives. Russ. J. Dev. Biol. 2022, 53, 272–282. [Google Scholar] [CrossRef]
- Huang, Z.; Liu, G. Current advancement in the application of prime editing. Front. Bioeng. Biotechnol. 2023, 11, 1039315. [Google Scholar] [CrossRef]
- Świat, M.A.; Dashko, S.; den Ridder, M.; Wijsman, M.; van der Oost, J.; Daran, J.-M.; Daran-Lapujade, P. FnCpf1: A novel and efficient genome editing tool for Saccharomyces cerevisiae. Nucleic Acids Res. 2017, 45, 12585–12598. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Song, M.; Lee, J.; Menon, A.V.; Jung, S.; Kang, Y.-M.; Choi, J.W.; Woo, E.; Koh, H.C.; Nam, J.-W.; et al. In vivo high-throughput profiling of CRISPR–Cpf1 activity. Nat. Methods 2017, 14, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef]
- Shmakov, S.; Smargon, A.; Scott, D.; Cox, D.; Pyzocha, N.; Yan, W.; Abudayyeh, O.O.; Gootenberg, J.S.; Makarova, K.S.; Wolf, Y.I.; et al. Diversity and evolution of class 2 CRISPR–Cas systems. Nat. Rev. Microbiol. 2017, 15, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Stella, S.; Alcón, P.; Montoya, G. Structure of the Cpf1 endonuclease R-loop complex after target DNA cleavage. Nature 2017, 546, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Fonfara, I.; Richter, H.; Bratovič, M.; LeRhun, A.; Charpentier, E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 2016, 532, 517–521. [Google Scholar] [CrossRef]
- Lauritsen, I.; Porse, A.; Sommer, M.O.A.; Nørholm, M.H.H. A versatile one-step CRISPR-Cas9 based approach to plasmid-curing. Microb. Cell Fact. 2017, 16, 135. [Google Scholar] [CrossRef]
- Spengler, G.; Molnar, A.; Schelz, Z.; Amaral, L.; Sharples, D.; Molnar, J. The mechanism of plasmid curing in bacteria. Curr. Drug Targets 2006, 7, 823–841. [Google Scholar] [CrossRef]
- Sengupta, M.; Austin, S. Prevalence and significance of plasmid maintenance functions in the virulence plasmids of pathogenic bacteria. Infect. Immun. 2011, 79, 2502. [Google Scholar] [CrossRef]
- Reyrat, J.M.; Pelicic, V.; Gicquel, B.; Rappuoli, R. Counterselectable markers: Untapped tools for bacterial genetics and pathogenesis. Infect. Immun. 1998, 66, 4011–4017. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto-Gotoh, T.; Franklin, F.C.H.; Nordheim, A.; Timmis, K.N. Specific-purpose plasmid cloning vectors I. Low copy number, temperature-sensitive, mobilization-defective pSC101-derived containment vectors. Gene 1981, 16, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Buckner, M.M.C.; Ciusa, M.L.; Piddock, L.J.V. Strategies to combat antimicrobial resistance: Anti-plasmid and plasmid curing. FEMS Microbiol. Rev. 2018, 42, 781–804. [Google Scholar] [CrossRef]
- Li, Y.; Lin, Z.; Huang, C.; Zhang, Y.; Wang, Z.; Tang, Y.; Chen, T.; Zhao, X. Metabolic engineering of Escherichia coli using CRISPR–Cas9 meditated genome editing. Metab. Eng. 2015, 31, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, B.; Duan, C.; Sun, B.; Yang, J.; Yang, S. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 2015, 81, 2506–2514. [Google Scholar] [CrossRef]
- Chen, B.C.; Chu, W.S.; Lin, H.Y. Enhancement of CRISPR-Cpf 1 genome editing efficiency using ssDNA homologous recombination in Saccharomyces cerevisiae. Taiwan Agric. Chem. Food Sci. 2021, 59, 4–10. [Google Scholar] [CrossRef]
- Chen, B.C.; Lin, H.Y. Deletion of NTH1 and HSP12 increases the freeze–thaw resistance of baker’s yeast in bread dough. Microb. Cell Fact. 2022, 21, 149. [Google Scholar] [CrossRef]
- Thompson, J.R.; Register, E.; Curotto, J.; Kurtz, M.; Kelly, R. An improved protocol for the preparation of yeast cells for transformation by electroporation. Yeast 1998, 14, 565–571. [Google Scholar] [CrossRef]
- Li, Z.H.; Meng, H.; Ma, B.; Tao, X.; Liu, M.; Wang, F.Q.; Wei, D.Z. Immediate, multiplexed and sequential genome engineering facilitated by CRISPR/Cas9 in Saccharomyces cerevisiae. J. Ind. Microbiol. Biotechnol. 2020, 47, 83–96. [Google Scholar] [CrossRef]
- Lim, H.; Choi, S.K. Programmed gRNA removal system for CRISPR-Cas9-mediated multi-round genome editing in Bacillus subtilis. Front. Microbiol. 2019, 10, 1140. [Google Scholar] [CrossRef]
- Zou, Y.; Qiu, L.; Xie, A.; Han, W.; Zhang, S.; Li, J.; Zhao, S.; Li, Y.; Liang, Y.; Hu, Y. Development and application of a rapid all-in-one plasmid CRISPR-Cas9 system for iterative genome editing in Bacillus subtilis. Microb. Cell Fact. 2022, 21, 173. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Xiang, H.; Mu, D.; Wang, D.; Wang, T. Exploiting a conjugative CRISPR/Cas9 system to eliminate plasmid harbouring the mcr-1 gene from Escherichia coli. Int. J. Antimicrob. Agents 2019, 53, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; He, Y.; Zhang, H.; Liao, X.P.; Liu, Y.H.; Sun, J.; Du, H.; Kreiswirth, B.N.; Chen, L. CRISPR-Cas9-mediated carbapenemase gene and plasmid curing in carbapenem-resistant Enterobacteriaceae. Antimicrob. Agents Chemother. 2020, 64, e00843-20. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.J.; Seo, S.O.; Kim, S.A.; Li, L.; Kim, T.J.; Kim, S.C.; Jin, Y.S.; Han, N.S. Elimination of the cryptic plasmid in Leuconostoc citreum by CRISPR/Cas9 system. J. Biotechnol. 2017, 251, 151–155. [Google Scholar] [CrossRef]
- Cao, Q.H.; Shao, H.H.; Qiu, H.; Li, T.; Zhang, Y.Z.; Tan, X.M. Using the CRISPR/Cas9 system to eliminate native plasmids of Zymomonas mobilis ZM4. Biosci. Biotechnol. Biochem. 2017, 81, 453–459. [Google Scholar] [CrossRef]
- Wang, X.; Lyu, Y.; Wang, S.; Zheng, Q.; Feng, E.; Zhu, L.; Pan, C.; Wang, S.; Wang, D.; Liu, X.; et al. Application of CRISPR/Cas9 system for plasmid elimination and bacterial killing of Bacillus cereus group strains. Front. Microbiol. 2021, 12, 536357. [Google Scholar] [CrossRef]
- Manghwar, H.; Li, B.; Ding, X.; Hussain, A.; Lindsey, K.; Zhang, X. CRISPR/Cas systems in genome editing: Methodologies and tools for sgRNA design, off-target evaluation, and strategies to mitigate off-target effects. Adv. Sci. 2020, 7, 1902312. [Google Scholar] [CrossRef]
- Batista Napotnik, T.; Polajžer, T.; Miklavčič, D. Cell death due to electroporation—A review. Bioelectrochem 2021, 141, 107871. [Google Scholar] [CrossRef]
- Moreno-Mateos, M.A.; Fernandez, J.P.; Rouet, R.; Vejnar, C.E.; Lane, M.A.; Mis, E.; Khokha, M.K.; Doudna, J.A.; Giraldez, A.J. CRISPR-Cpf1 mediates efficient homology-directed repair and temperature-controlled genome editing. Nat. Commun. 2017, 8, 2024. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| crRNA | Targeted Sequence | PAM | Primer Sequence for Colony PCR | ssDNA Template Sequence |
|---|---|---|---|---|
| crCEN1 | TTT CGT GTG TGG TCT TCT ACA CAG ACA A | TTTC | F1-F: TTT TTC GCC CTT TGA CGT TG KX-R: ATC GCG AGC CCA TTT ATA CC | CEN HA: GAG ACG AAA GGG CCT CGT GAT ACG CCT ATT TTT ATA GGT TAA TGT CAT GAT ATT TGT TTA TTT TTC TAA ATA CAT TCA AAT ATG TAT CCG CTC ATG AGA C |
| crCEN2 | TTT CCG AAG ATG TAA AAG ACT CTA GGG G | TTTC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, B.-C.; Chen, Y.-Z.; Lin, H.-Y. An Introduced RNA-Only Approach for Plasmid Curing via the CRISPR-Cpf1 System in Saccharomyces cerevisiae. Biomolecules 2023, 13, 1561. https://doi.org/10.3390/biom13101561
Chen B-C, Chen Y-Z, Lin H-Y. An Introduced RNA-Only Approach for Plasmid Curing via the CRISPR-Cpf1 System in Saccharomyces cerevisiae. Biomolecules. 2023; 13(10):1561. https://doi.org/10.3390/biom13101561
Chicago/Turabian StyleChen, Bo-Chou, Yu-Zhen Chen, and Huan-Yu Lin. 2023. "An Introduced RNA-Only Approach for Plasmid Curing via the CRISPR-Cpf1 System in Saccharomyces cerevisiae" Biomolecules 13, no. 10: 1561. https://doi.org/10.3390/biom13101561
APA StyleChen, B. -C., Chen, Y. -Z., & Lin, H. -Y. (2023). An Introduced RNA-Only Approach for Plasmid Curing via the CRISPR-Cpf1 System in Saccharomyces cerevisiae. Biomolecules, 13(10), 1561. https://doi.org/10.3390/biom13101561