

Efficacy of HDAC Inhibitors in Driving Peroxisomal β-Oxidation and Immune Responses in Human Macrophages: Implications for Neuroinflammatory Disorders
 , , ,
, , , 
 and
and 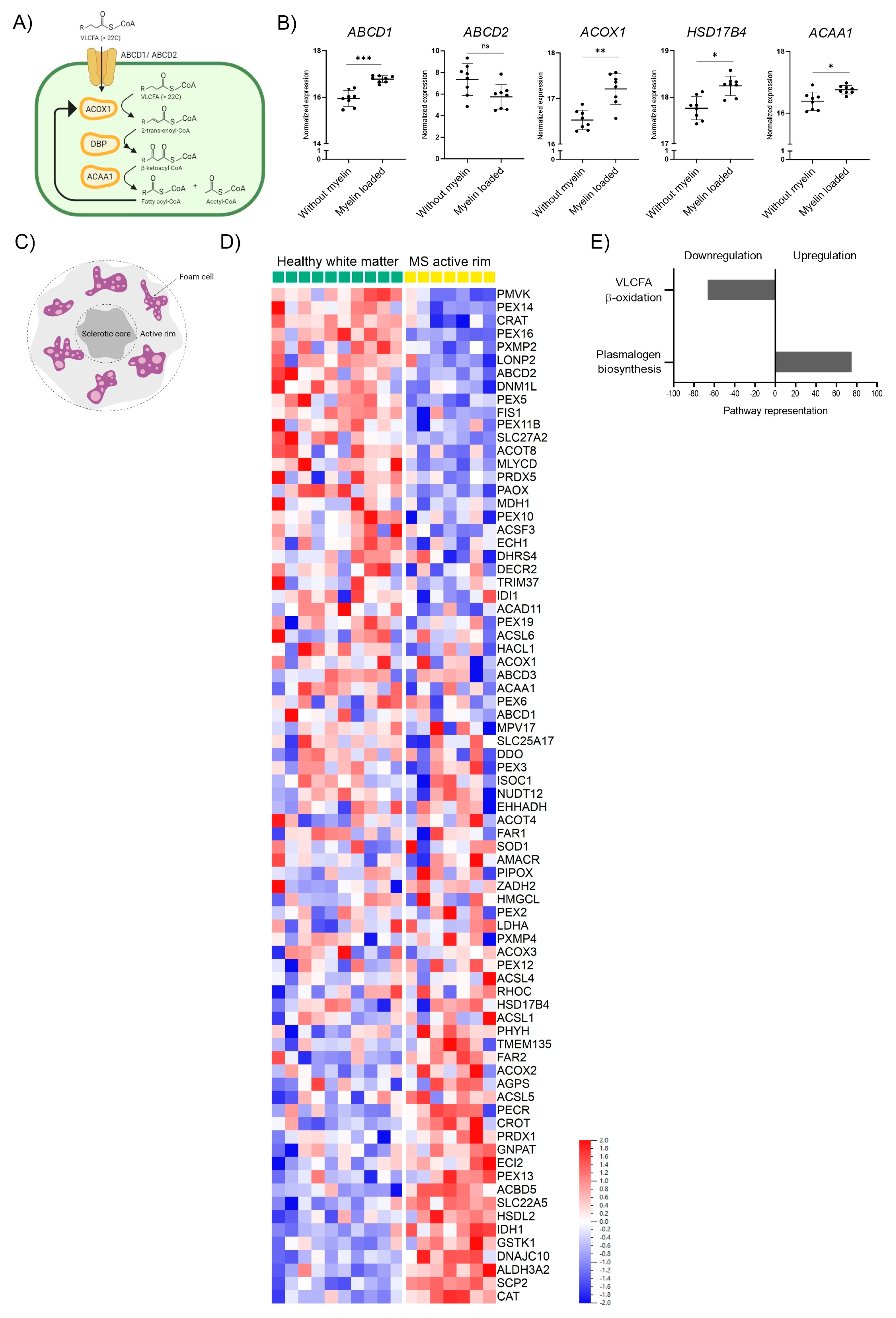{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of Human PBMCs, CD14+ Monocytes, CD3+ T Cells, and CD19+ B Cells
2.2. Differentiation and Activation of Human Macrophages
2.3. Calcein AM Viability Assay
2.4. RNA Isolation and RT-qPCR
2.5. β-Oxidation of 14C-Labelled C16:0 and C26:0
2.6. Immunoblotting
2.7. Flow Cytometry Analysis
2.8. Real-Time Myelin Phagocytosis and Migration Assay
2.9. Cytokine Measurement
2.10. Bioinformatic Analysis
2.11. Statistics
3. Results
3.1. Peroxisomal Genes Involved in VLCFA Degradation Are Induced by Myelin Phagocytosis but Downregulated in the Brain White Matter of MS Patients
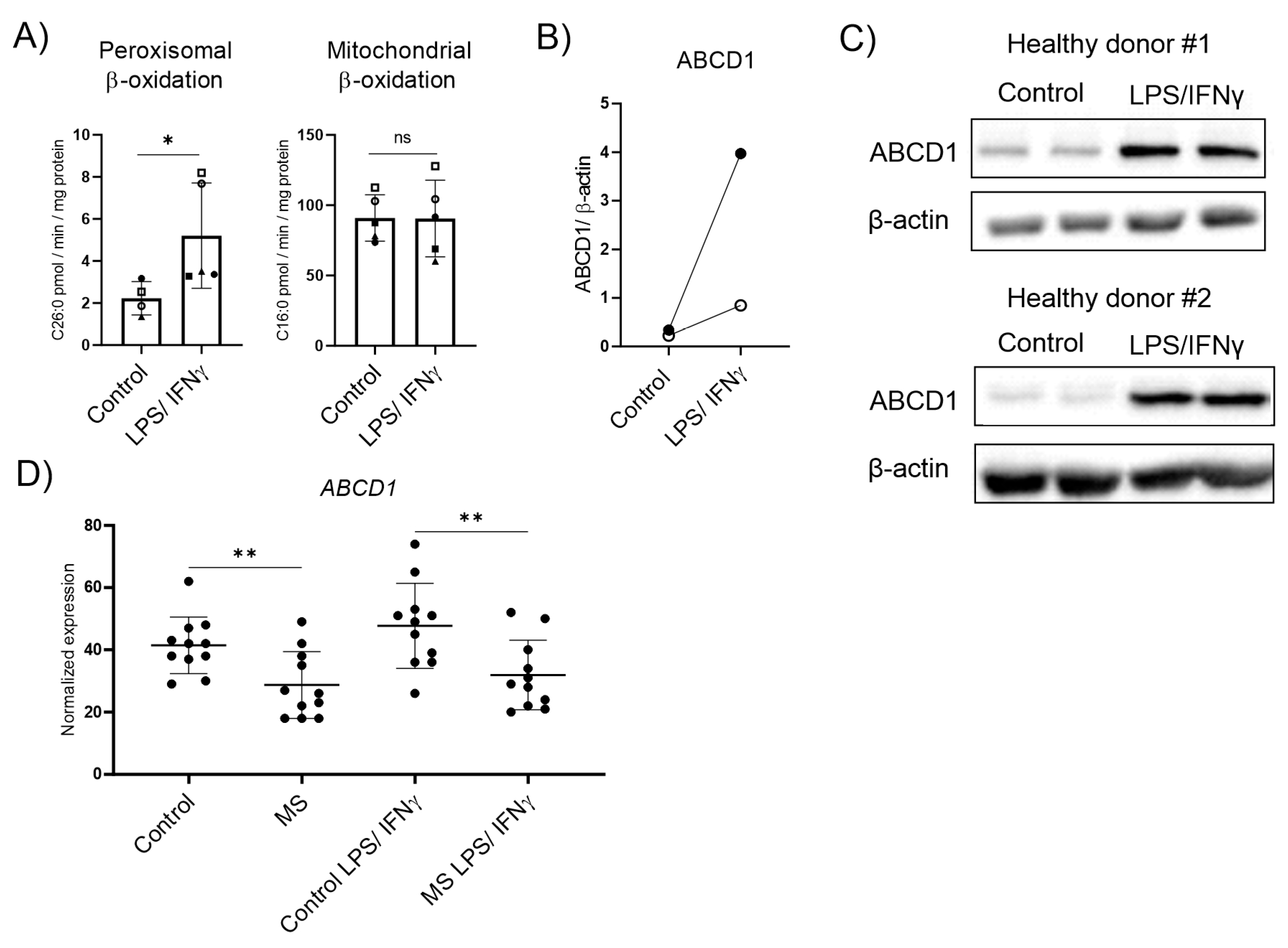
3.2. ABCD1 Encoding the Rate-Limiting Factor of VLCFA Degradation and Stimulated by Pro-Inflammatory Activation, Is Downregulated in MS Macrophages
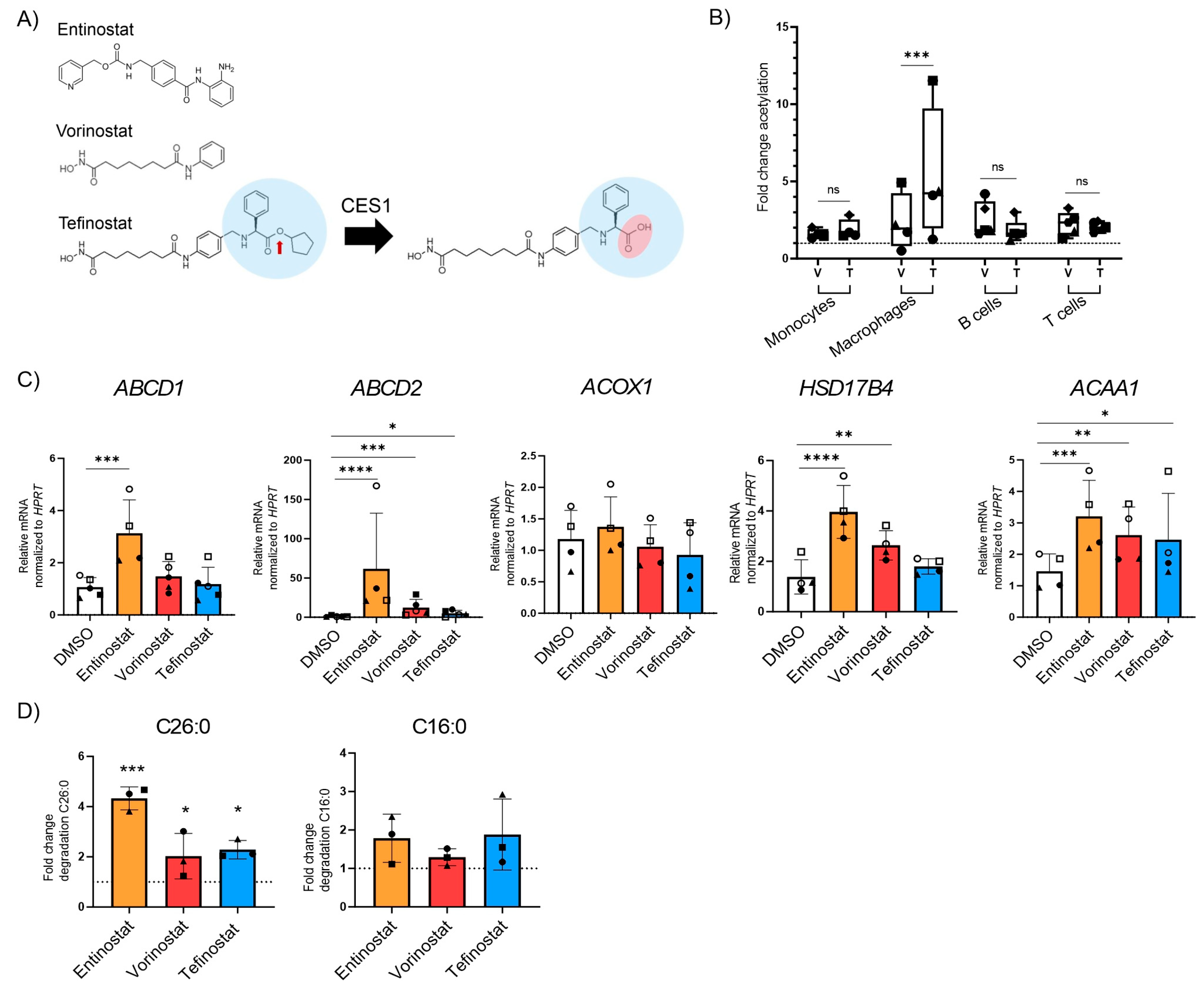
3.3. Tefinostat Modulates VLCFA Metabolism in Macrophages but It Is Less Effective than Entinostat at Inducing Peroxisomal VLCFA Transporter Expression and Degradation of VLCFAs
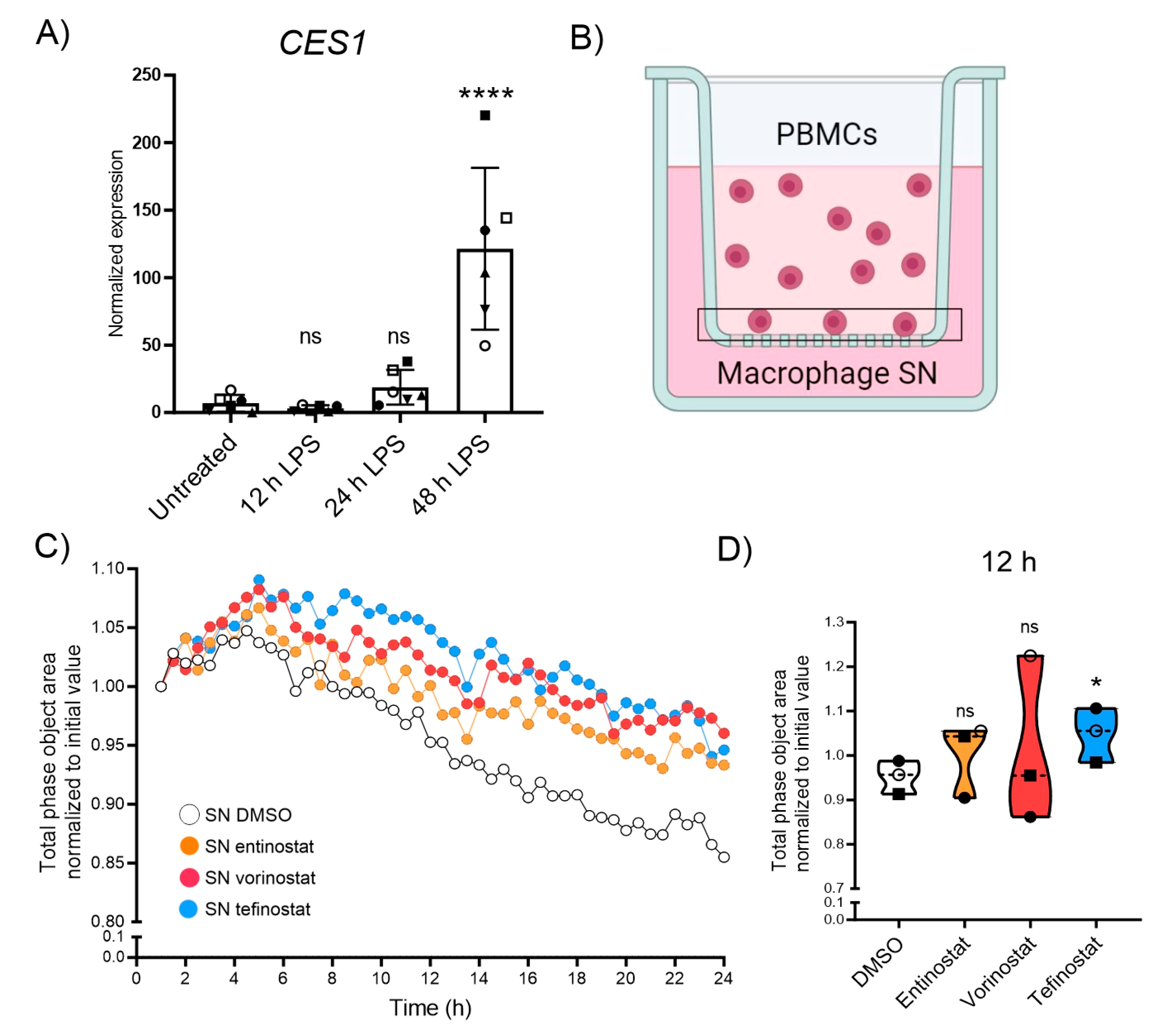
3.4. Tefinostat Interferes with the Chemotactic Recruitment of PBMCs
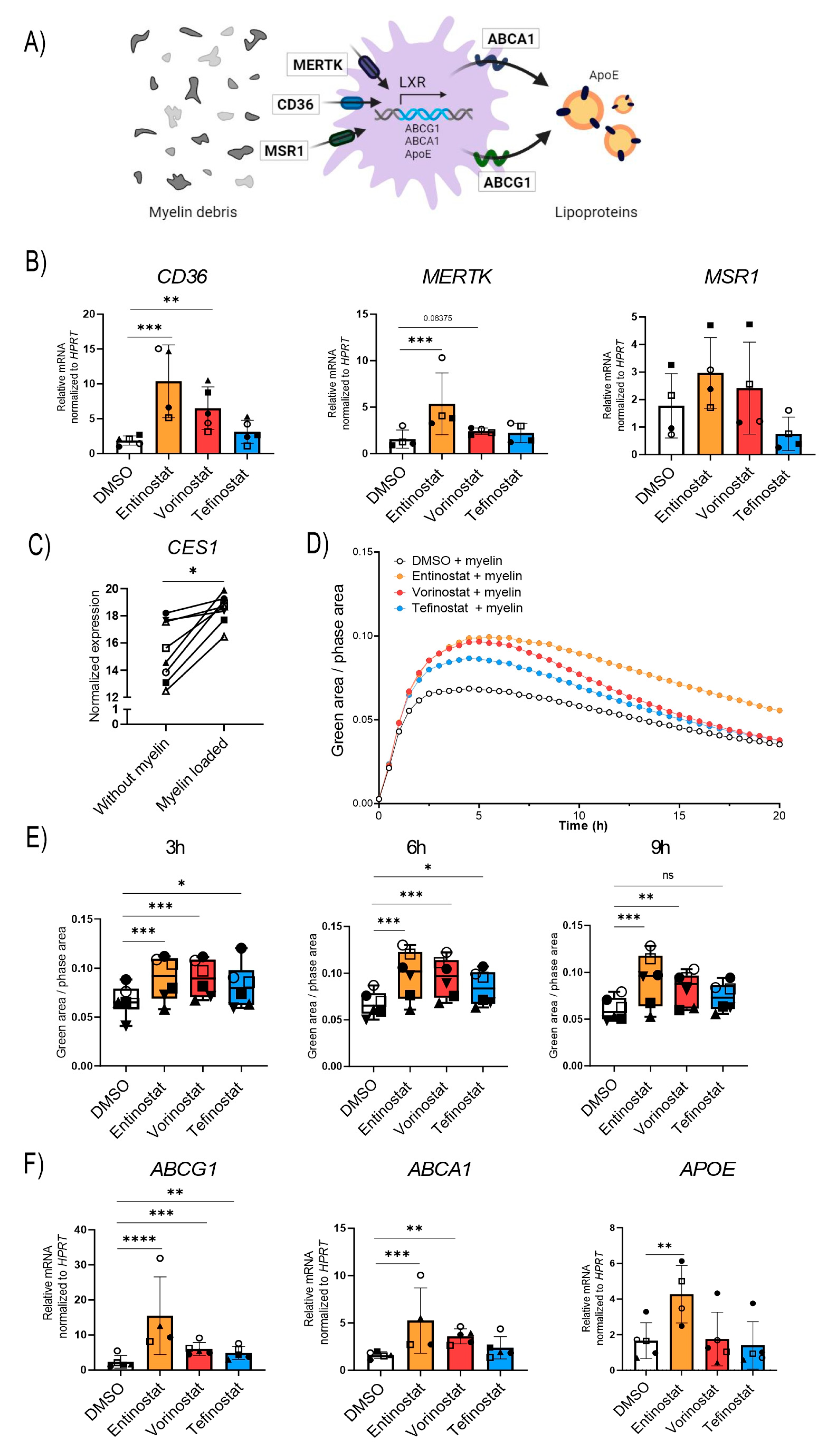
3.5. Entinostat Has the Highest Capacity to Promote Uptake of Myelin Debris and Expression of Proteins Involved in Lipid Export
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grajchen, E.; Hendriks, J.J.A.; Bogie, J.F.J. The physiology of foamy phagocytes in multiple sclerosis. Acta Neuropathol. Commun. 2018, 6, 124. [Google Scholar] [CrossRef] [PubMed]
- Weinhofer, I.; Zierfuss, B.; Hametner, S.; Wagner, M.; Popitsch, N.; Machacek, C.; Bartolini, B.; Zlabinger, G.; Ohradanova-Repic, A.; Stockinger, H.; et al. Impaired plasticity of macrophages in X-linked adrenoleukodystrophy. Brain 2018, 141, 2329–2342. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, S.A.; Spieth, L.; Saher, G. Local cholesterol metabolism orchestrates remyelination. Trends Neurosci. 2022, 45, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Sastry, P.S. Lipids of nervous tissue: Composition and metabolism. Prog. Lipid Res. 1985, 24, 69–176. [Google Scholar] [CrossRef] [PubMed]
- Buda, A.; Forss-Petter, S.; Hua, R.; Jaspers, Y.; Lassnig, M.; Waidhofer-Sollner, P.; Kemp, S.; Kim, P.; Weinhofer, I.; Berger, J. ABCD1 transporter deficiency results in altered cholesterol homeostasis. Biomolecules 2023, 13, 1333. [Google Scholar] [CrossRef]
- Baarine, M.; Andreoletti, P.; Athias, A.; Nury, T.; Zarrouk, A.; Ragot, K.; Vejux, A.; Riedinger, J.M.; Kattan, Z.; Bessede, G.; et al. Evidence of oxidative stress in very long chain fatty acid--treated oligodendrocytes and potentialization of ROS production using RNA interference-directed knockdown of ABCD1 and ACOX1 peroxisomal proteins. Neuroscience 2012, 213, 1–18. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Fitzner, D.; Bosch-Queralt, M.; Weil, M.T.; Su, M.; Sen, P.; Ruhwedel, T.; Mitkovski, M.; Trendelenburg, G.; Lutjohann, D.; et al. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 2018, 359, 684–688. [Google Scholar] [CrossRef]
- Parisi, L.R.; Li, N.; Atilla-Gokcumen, G.E. Very long chain fatty acids are functionally involved in necroptosis. Cell Chem. Biol. 2017, 24, 1445–1454.e1448. [Google Scholar] [CrossRef] [PubMed]
- Schneiter, R.; Brugger, B.; Amann, C.M.; Prestwich, G.D.; Epand, R.F.; Zellnig, G.; Wieland, F.T.; Epand, R.M. Identification and biophysical characterization of a very-long-chain-fatty-acid-substituted phosphatidylinositol in yeast subcellular membranes. Biochem. J. 2004, 381, 941–949. [Google Scholar] [CrossRef]
- Ho, J.K.; Moser, H.; Kishimoto, Y.; Hamilton, J.A. Interactions of a very long chain fatty acid with model membranes and serum albumin. Implications for the pathogenesis of adrenoleukodystrophy. J. Clin. Investig. 1995, 96, 1455–1463. [Google Scholar] [CrossRef]
- Zierfuss, B.; Buda, A.; Villoria-Gonzalez, A.; Logist, M.; Fabjan, J.; Parzer, P.; Battin, C.; Vandersteene, S.; Dijkstra, I.M.E.; Waidhofer-Sollner, P.; et al. Saturated very long-chain fatty acids regulate macrophage plasticity and invasiveness. J. Neuroinflamm. 2022, 19, 305. [Google Scholar] [CrossRef]
- Raas, Q.; Tawbeh, A.; Tahri-Joutey, M.; Gondcaille, C.; Keime, C.; Kaiser, R.; Trompier, D.; Nasser, B.; Leoni, V.; Bellanger, E.; et al. Peroxisomal defects in microglial cells induce a disease-associated microglial signature. Fronti. Mol. Neurosci. 2023, 16, 1170313. [Google Scholar] [CrossRef] [PubMed]
- Kassmann, C.M. Myelin peroxisomes-essential organelles for the maintenance of white matter in the nervous system. Biochimie 2014, 98, 111–118. [Google Scholar] [CrossRef]
- Turk, B.R.; Theda, C.; Fatemi, A.; Moser, A.B. X-linked adrenoleukodystrophy: Pathology, pathophysiology, diagnostic testing, newborn screening and therapies. Int. J. Dev. Neurosci. 2020, 80, 52–72. [Google Scholar] [CrossRef]
- Berger, J.; Forss-Petter, S.; Eichler, F.S. Pathophysiology of X-linked adrenoleukodystrophy. Biochimie 2014, 98, 135–142. [Google Scholar] [CrossRef]
- Powers, J.M.; Liu, Y.; Moser, A.B.; Moser, H.W. The inflammatory myelinopathy of adrenoleukodystrophy: Cells, effector molecules, and pathogenetic implications. J. Neuropath. Exp. Neur. 1992, 51, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Weinhofer, I.; Buda, A.; Kunze, M.; Palfi, Z.; Traunfellner, M.; Hesse, S.; Villoria-Gonzalez, A.; Hofmann, J.; Hametner, S.; Regelsberger, G.; et al. Peroxisomal very long-chain fatty acid transport is targeted by herpesviruses and the antiviral host response. Commun. Biol. 2022, 5, 944. [Google Scholar] [CrossRef] [PubMed]
- Weinhofer, I.; Rommer, P.; Zierfuss, B.; Altmann, P.; Foiani, M.; Heslegrave, A.; Zetterberg, H.; Gleiss, A.; Musolino, P.L.; Gong, Y.; et al. Neurofilament light chain as a potential biomarker for monitoring neurodegeneration in X-linked adrenoleukodystrophy. Nat. Commun. 2021, 12, 1816. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Rice, C.; Hares, K.; Redondo, J.; Kemp, K.; Williams, M.; Brown, A.; Scolding, N.; Wilkins, A. Reductions in neuronal peroxisomes in multiple sclerosis grey matter. Mult. Scler. 2014, 20, 651–659. [Google Scholar] [CrossRef]
- Roczkowsky, A.; Doan, M.A.L.; Hlavay, B.; Mamik, M.K.; Branton, W.G.; McKenzie, B.A.; Saito, L.B.; Schmitt, L.; Eitzen, G.; Di Cara, F.; et al. Peroxisome injury in multiple sclerosis: Protective effects of 4-phenylbutyrate in CNS-associated macrophages. J. Neurosci. 2022, 42, 7152–7165. [Google Scholar] [CrossRef]
- Mapunda, J.A.; Tibar, H.; Regragui, W.; Engelhardt, B. How does the immune system enter the brain? Front. Immunol. 2022, 13, 805657. [Google Scholar] [CrossRef] [PubMed]
- Reali, C.; Magliozzi, R.; Roncaroli, F.; Nicholas, R.; Howell, O.W.; Reynolds, R. B cell rich meningeal inflammation associates with increased spinal cord pathology in multiple sclerosis. Brain Pathol. 2020, 30, 779–793. [Google Scholar] [CrossRef] [PubMed]
- Melhem, E.R.; Loes, D.J.; Georgiades, C.S.; Raymond, G.V.; Moser, H.W. X-linked adrenoleukodystrophy: The role of contrast-enhanced MR imaging in predicting disease progression. AJNR Am. J. Neuroradiol. 2000, 21, 839–844. [Google Scholar] [PubMed]
- Van der Voorn, J.P.; Pouwels, P.J.; Powers, J.M.; Kamphorst, W.; Martin, J.J.; Troost, D.; Spreeuwenberg, M.D.; Barkhof, F.; van der Knaap, M.S. Correlating quantitative MR imaging with histopathology in X-linked adrenoleukodystrophy. AJNR Am. J. Neuroradiol. 2011, 32, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Munoz, U.; Sebal, C.; Escudero, E.; Esiri, M.; Tzartos, J.; Sloan, C.; Sadaba, M.C. Main role of antibodies in demyelination and axonal damage in multiple sclerosis. Cell Mol. Neurobiol. 2022, 42, 1809–1827. [Google Scholar] [CrossRef] [PubMed]
- Cartier, N.; Aubourg, P. Hematopoietic stem cell transplantation and hematopoietic stem cell gene therapy in X-linked adrenoleukodystrophy. Brain Pathol. 2010, 20, 857–862. [Google Scholar] [CrossRef]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Muraro, P.A.; Martin, R. Immunological questions on hematopoietic stem cell transplantation for multiple sclerosis. Bone Marrow Transpl. 2003, 32 (Suppl. S1), S41–S44. [Google Scholar] [CrossRef]
- Weber, F.D.; Wiesinger, C.; Forss-Petter, S.; Regelsberger, G.; Einwich, A.; Weber, W.H.; Kohler, W.; Stockinger, H.; Berger, J. X-linked adrenoleukodystrophy: Very long-chain fatty acid metabolism is severely impaired in monocytes but not in lymphocytes. Hum. Mole Genet. 2014, 23, 2542–2550. [Google Scholar] [CrossRef]
- Bergner, C.G.; van der Meer, F.; Winkler, A.; Wrzos, C.; Turkmen, M.; Valizada, E.; Fitzner, D.; Hametner, S.; Hartmann, C.; Pfeifenbring, S.; et al. Microglia damage precedes major myelin breakdown in X-linked adrenoleukodystrophy and metachromatic leukodystrophy. Glia 2019, 67, 1196–1209. [Google Scholar] [CrossRef]
- Absinta, M.; Maric, D.; Gharagozloo, M.; Garton, T.; Smith, M.D.; Jin, J.; Fitzgerald, K.C.; Song, A.; Liu, P.; Lin, J.P.; et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 2021, 597, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, S.; Fransen, N.L.; van Eden, C.G.; Ramaglia, V.; Mason, M.; Huitinga, I. Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: A retrospective autopsy cohort analysis. Acta Neuropathol. 2018, 135, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Bruck, W.; Lassmann, H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Frischer, J.M.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Bruck, W.; et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Chueh, A.C.; Tse, J.W.; Togel, L.; Mariadason, J.M. Mechanisms of histone deacetylase inhibitor-regulated gene expression in cancer cells. Antioxid. Redox Sign. 2015, 23, 66–84. [Google Scholar] [CrossRef] [PubMed]
- Suliman, B.A.; Xu, D.; Williams, B.R. HDACi: Molecular mechanisms and therapeutic implications in the innate immune system. Immunol. Cell Biol. 2012, 90, 23–32. [Google Scholar] [CrossRef]
- Dai, Y.; Wei, T.; Shen, Z.; Bei, Y.; Lin, H.; Dai, H. Classical HDACs in the regulation of neuroinflammation. Neurochem. Int. 2021, 150, 105182. [Google Scholar] [CrossRef]
- Hamminger, P.; Rica, R.; Ellmeier, W. Histone deacetylases as targets in autoimmune and autoinflammatory diseases. Adv. Immunol. 2020, 147, 1–59. [Google Scholar] [CrossRef]
- Ge, Z.; Da, Y.; Xue, Z.; Zhang, K.; Zhuang, H.; Peng, M.; Li, Y.; Li, W.; Simard, A.; Hao, J.; et al. Vorinostat, a histone deacetylase inhibitor, suppresses dendritic cell function and ameliorates experimental autoimmune encephalomyelitis. Exp. Neurol. 2013, 241, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Zierfuss, B.; Weinhofer, I.; Kuhl, J.S.; Kohler, W.; Bley, A.; Zauner, K.; Binder, J.; Martinovic, K.; Seiser, C.; Hertzberg, C.; et al. Vorinostat in the acute neuroinflammatory form of X-linked adrenoleukodystrophy. Ann. Clin. Transl. Neur. 2020, 7, 639–652. [Google Scholar] [CrossRef]
- Zierfuss, B.; Weinhofer, I.; Buda, A.; Popitsch, N.; Hess, L.; Moos, V.; Hametner, S.; Kemp, S.; Kohler, W.; Forss-Petter, S.; et al. Targeting foam cell formation in inflammatory brain diseases by the histone modifier MS-275. Ann. Clin. Transl. Neur. 2020, 7, 2161–2177. [Google Scholar] [CrossRef] [PubMed]
- Needham, L.A.; Davidson, A.H.; Bawden, L.J.; Belfield, A.; Bone, E.A.; Brotherton, D.H.; Bryant, S.; Charlton, M.H.; Clark, V.L.; Davies, S.J.; et al. Drug targeting to monocytes and macrophages using esterase-sensitive chemical motifs. J. Pharmacol. Exp. Ther. 2011, 339, 132–142. [Google Scholar] [CrossRef]
- Ossenkoppele, G.J.; Lowenberg, B.; Zachee, P.; Vey, N.; Breems, D.; Van de Loosdrecht, A.A.; Davidson, A.H.; Wells, G.; Needham, L.; Bawden, L.; et al. A phase I first-in-human study with tefinostat—A monocyte/macrophage targeted histone deacetylase inhibitor—In patients with advanced haematological malignancies. Brit. J. Haematol. 2013, 162, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Fransson, J.; Gomez-Conde, A.I.; Romero-Imbroda, J.; Fernandez, O.; Leyva, L.; de Fonseca, F.R.; Chun, J.; Louapre, C.; Van-Evercooren, A.B.; Zujovic, V.; et al. Activation of macrophages by lysophosphatidic acid through the lysophosphatidic acid receptor 1 as a novel mechanism in multiple sclerosis pathogenesis. Mol. Neurobiol. 2021, 58, 470–482. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, D.A.E.; van Scheppingen, J.; van der Poel, M.; Bossers, K.; Schuurman, K.G.; van Eden, C.G.; Hol, E.M.; Hamann, J.; Huitinga, I. Gene expression profiling of multiple sclerosis pathology identifies early patterns of demyelination surrounding chronic active lesions. Front. Immunol. 2017, 8, 1810. [Google Scholar] [CrossRef]
- Netik, A.; Forss-Petter, S.; Holzinger, A.; Molzer, B.; Unterrainer, G.; Berger, J. Adrenoleukodystrophy-related protein can compensate functionally for adrenoleukodystrophy protein deficiency (X-ALD): Implications for therapy. Hum. Mol. Genet. 1999, 8, 907–913. [Google Scholar] [CrossRef]
- Gore, L.; Rothenberg, M.L.; O’Bryant, C.L.; Schultz, M.K.; Sandler, A.B.; Coffin, D.; McCoy, C.; Schott, A.; Scholz, C.; Eckhardt, S.G. A phase I and pharmacokinetic study of the oral histone deacetylase inhibitor, MS-275, in patients with refractory solid tumors and lymphomas. Clin. Cancer Res. 2008, 14, 4517–4525. [Google Scholar] [CrossRef] [PubMed]
- Batlevi, C.L.; Kasamon, Y.; Bociek, R.G.; Lee, P.; Gore, L.; Copeland, A.; Sorensen, R.; Ordentlich, P.; Cruickshank, S.; Kunkel, L.; et al. Engage- 501: Phase II study of entinostat (SNDX-275) in relapsed and refractory hodgkin lymphoma. Haematologica 2016, 101, 968–975. [Google Scholar] [CrossRef]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G.; et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Song, H.; Yin, L.; Rizzo, M.G.; Sidhu, R.; Covey, D.F.; Ory, D.S.; Semenkovich, C.F. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature 2016, 539, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Garcia Corrales, A.V.; Verberk, S.G.S.; Haidar, M.; Grajchen, E.; Dehairs, J.; Vanherle, S.; Loix, M.; Weytjens, T.; Gervois, P.; Matsuzaka, T.; et al. Fatty acid elongation by ELOVL6 hampers remyelination by promoting inflammatory foam cell formation during demyelination. Proc. Natl. Acad. Sci. USA 2023, 120, e2301030120. [Google Scholar] [CrossRef] [PubMed]
- Zierfuss, B.; Wang, Z.; Jackson, A.N.; Moezzi, D.; Yong, V.W. Iron in multiple sclerosis—Neuropathology, immunology, and real-world considerations. Mult. Scler. Relat. Dis. 2023, 78, 104934. [Google Scholar] [CrossRef] [PubMed]
- Yong, V.W. Microglia in multiple sclerosis: Protectors turn destroyers. Neuron 2022, 110, 3534–3548. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Fahimi, H.D. Peroxisomes and oxidative stress. Biochim. Biophys. Acta 2006, 1763, 1755–1766. [Google Scholar] [CrossRef]
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020, 585, 603–608. [Google Scholar] [CrossRef]
- Knapper, S.; Dennis, M.; Drummond, M.W.; Hills, R.K.; Dillon, R.J.; Pocock, C.; Culligan, D.J.; Vyas, P.; Wiseman, D.; Zabkiewicz, J.; et al. Results of a phase 2 trial of the monocyte-targeted histone deacetylase inhibitor tefinostat (CHR-2845) in chronic myelomonocytic leukemia (CMML)—The uk monocle study. Blood 2018, 132, 1818. [Google Scholar] [CrossRef]
- Hu, E.; Dul, E.; Sung, C.M.; Chen, Z.; Kirkpatrick, R.; Zhang, G.F.; Johanson, K.; Liu, R.; Lago, A.; Hofmann, G.; et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J. Pharmacol. Exp. Ther. 2003, 307, 720–728. [Google Scholar] [CrossRef]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Qian, X.; Mills, E.; Berghs, S.C.; Carey, N.; et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–589. [Google Scholar] [CrossRef]
- Fischer, A.; Sananbenesi, F.; Mungenast, A.; Tsai, L.H. Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol. Sci. 2010, 31, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.L.; Yen, J.L.; Kuo, Y.C.; Kang, J.J.; Cheng, Y.W.; Huang, W.J.; Hsiao, G. HDAC8 inhibitor WK2-16 therapeutically targets lipopolysaccharide-induced mouse model of neuroinflammation and microglial activation. Int. J. Mol. Sci. 2019, 20, 410. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, C.; Wang, G.; Wang, H.; Liu, X.; Huang, C.; Chen, Y.; Fan, L.; Zhou, L.; Tong, A. HDAC3 inhibitor (BRD3308) modulates microglial pyroptosis and neuroinflammation through PPARgamma/NLRP3/GSDMD to improve neurological function after intraventricular hemorrhage in mice. Neuropharmacology 2023, 237, 109633. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villoria-González, A.; Zierfuss, B.; Parzer, P.; Heuböck, E.; Zujovic, V.; Waidhofer-Söllner, P.; Ponleitner, M.; Rommer, P.; Göpfert, J.; Forss-Petter, S.; et al. Efficacy of HDAC Inhibitors in Driving Peroxisomal β-Oxidation and Immune Responses in Human Macrophages: Implications for Neuroinflammatory Disorders. Biomolecules 2023, 13, 1696. https://doi.org/10.3390/biom13121696
Villoria-González A, Zierfuss B, Parzer P, Heuböck E, Zujovic V, Waidhofer-Söllner P, Ponleitner M, Rommer P, Göpfert J, Forss-Petter S, et al. Efficacy of HDAC Inhibitors in Driving Peroxisomal β-Oxidation and Immune Responses in Human Macrophages: Implications for Neuroinflammatory Disorders. Biomolecules. 2023; 13(12):1696. https://doi.org/10.3390/biom13121696
Chicago/Turabian StyleVilloria-González, Andrea, Bettina Zierfuss, Patricia Parzer, Elisabeth Heuböck, Violetta Zujovic, Petra Waidhofer-Söllner, Markus Ponleitner, Paulus Rommer, Jens Göpfert, Sonja Forss-Petter, and et al. 2023. "Efficacy of HDAC Inhibitors in Driving Peroxisomal β-Oxidation and Immune Responses in Human Macrophages: Implications for Neuroinflammatory Disorders" Biomolecules 13, no. 12: 1696. https://doi.org/10.3390/biom13121696
APA StyleVilloria-González, A., Zierfuss, B., Parzer, P., Heuböck, E., Zujovic, V., Waidhofer-Söllner, P., Ponleitner, M., Rommer, P., Göpfert, J., Forss-Petter, S., Berger, J., & Weinhofer, I. (2023). Efficacy of HDAC Inhibitors in Driving Peroxisomal β-Oxidation and Immune Responses in Human Macrophages: Implications for Neuroinflammatory Disorders. Biomolecules, 13(12), 1696. https://doi.org/10.3390/biom13121696