
FBA-PRCC. Partial Rank Correlation Coefficient (PRCC) Global Sensitivity Analysis (GSA) in Application to Constraint-Based Models

Abstract
:1. Introduction
2. Materials and Methods
- Define parameter space: for non-boundary irreversible reactions only one parameter is created, for reversible and boundary reactions two parameters are created for each reaction and .
- Generate a set of quasi-random low-discrepancy points in the parameter space. Update parameters (reaction bounds) and find the optimal objective value for each point in the parameter space.
- Calculate Partial Rank Correlation Coefficient (PRCC) for each parameter and objective value. The statistical significance of the PRCC value is estimated as described by Marino et al. [20]. The sufficiency of the sample size for reliable PRCC estimation is controlled by the top-down coefficient of concordance (TDCC): when TDCC between PRCC vectors calculated at different sample sizes exceeds the threshold of 0.9, the sample size is considered sufficient for analysis.
3. Results
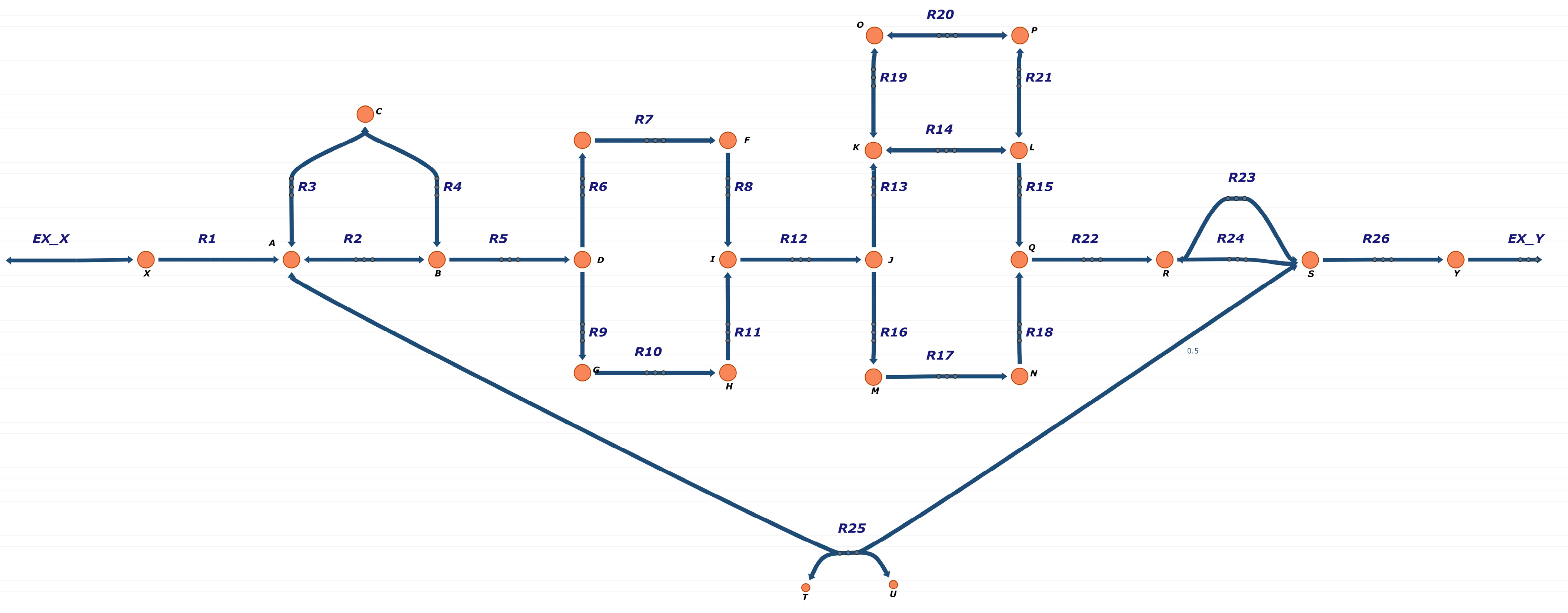
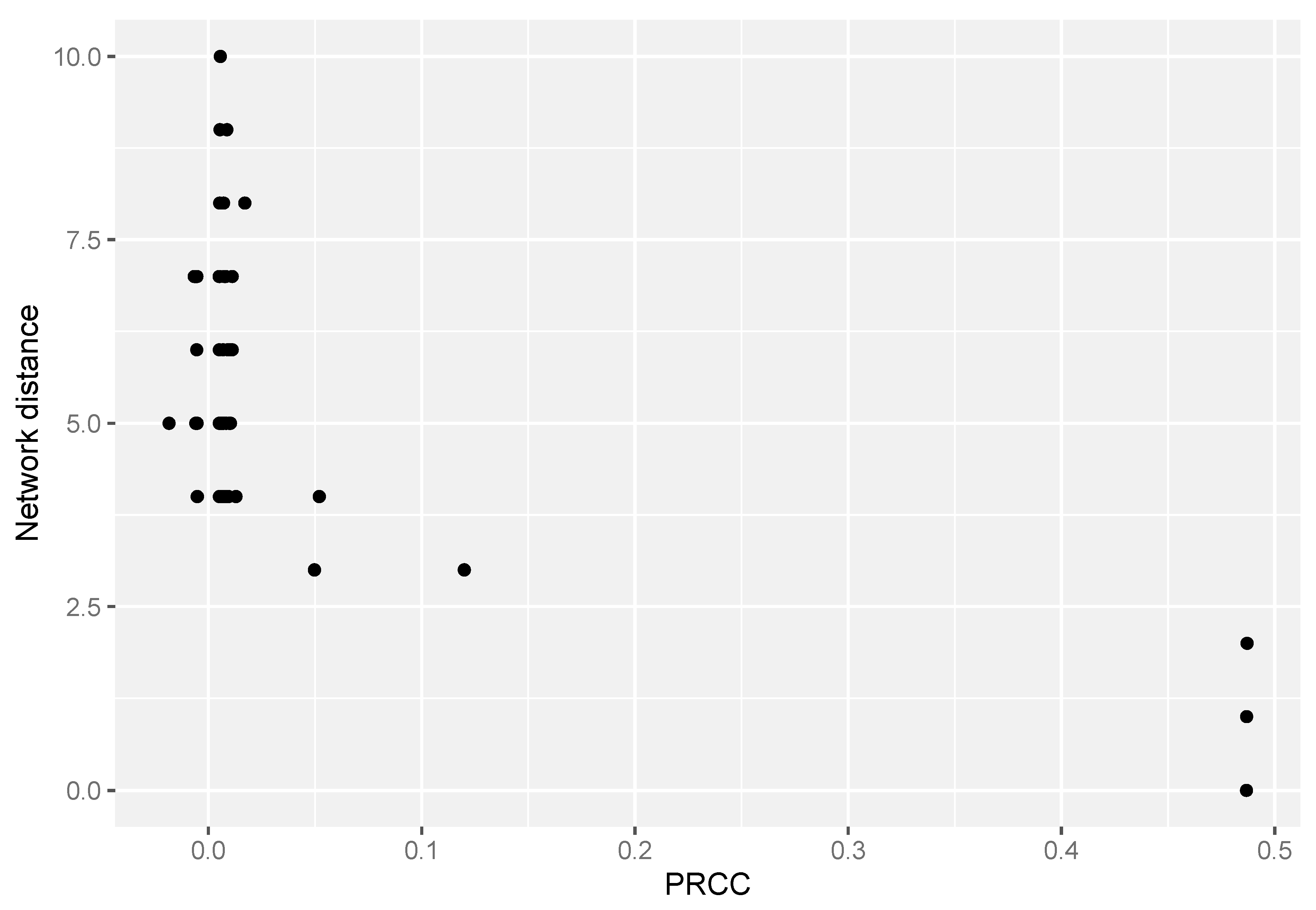
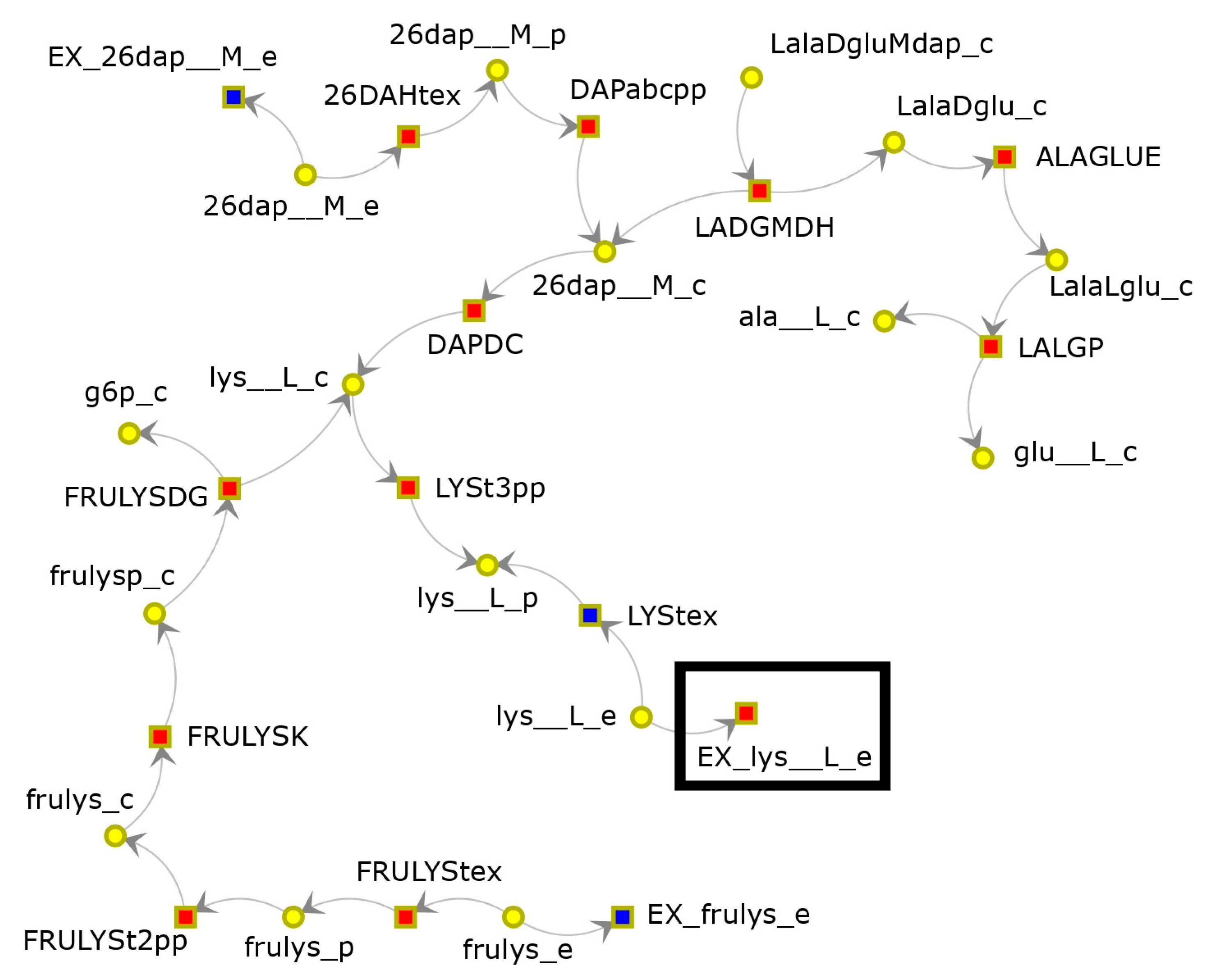
3.1. FBA-PRCC Can Identify the Backbone of the Flux-Related Network
3.2. FBA-PRCC Can Identify Controlling Steps in the Flux-Based Network
3.3. FBA-PRCC Is Computationally Efficient
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Norsigian, C.J.; Fang, X.; Seif, Y.; Monk, J.M.; Palsson, B.O. A Workflow for Generating Multi-Strain Genome-Scale Metabolic Models of Prokaryotes. Nat. Protoc. 2020, 15, 1–14. [Google Scholar] [CrossRef]
- Thiele, I.; Palsson, B.Ø. A Protocol for Generating a High-Quality Genome-Scale Metabolic Reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [Green Version]
- Burgard, A.P.; Pharkya, P.; Maranas, C.D. Optknock: A Bilevel Programming Framework for Identifying Gene Knockout Strategies for Microbial Strain Optimization. Biotechnol. Bioeng. 2003, 84, 647–657. [Google Scholar] [CrossRef]
- Cardoso, J.G.R.; Jensen, K.; Lieven, C.; Hansen, A.S.L.; Galkina, S.; Beber, M.; Özdemir, E.; Herrgård, M.J.; Redestig, H.; Sonnenschein, N. Cameo: A Python Library for Computer Aided Metabolic Engineering and Optimization of Cell Factories. ACS Synth. Biol. 2018, 7, 1163–1166. [Google Scholar] [CrossRef] [Green Version]
- McAnulty, M.J.; Yen, J.Y.; Freedman, B.G.; Senger, R.S. Genome-Scale Modeling Using Flux Ratio Constraints to Enable Metabolic Engineering of Clostridial Metabolism in Silico. BMC Syst. Biol. 2012, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-Scale Metabolic Modelling of Hepatocytes Reveals Serine Deficiency in Patients with Non-Alcoholic Fatty Liver Disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef] [Green Version]
- Thiele, I.; Swainston, N.; Fleming, R.M.T.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D.; et al. A Community-Driven Global Reconstruction of Human Metabolism. Nat. Biotechnol. 2013, 31, 419–425. [Google Scholar] [CrossRef]
- Ma, H.; Sorokin, A.; Mazein, A.; Selkov, A.; Selkov, E.; Demin, O.; Goryanin, I. The Edinburgh Human Metabolic Network Reconstruction and Its Functional Analysis. Mol. Syst. Biol. 2007, 3, 135. [Google Scholar] [CrossRef]
- Gatto, F.; Ferreira, R.; Nielsen, J. Pan-Cancer Analysis of the Metabolic Reaction Network. Metab. Eng. 2020, 57, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Özcan, E.; Çakır, T. Reconstructed Metabolic Network Models Predict Flux-Level Metabolic Reprogramming in Glioblastoma. Front. Neurosci. 2016, 10, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narad, P.; Gupta, R.; Mohanty, S.; Sharma, R.; Abbasi, N.; Sengupta, A. Systems Biology Paradigm for Exploring the Relation Between Obesity and Ovarian Cancer with a Focus on Their Genome-Scale Metabolic Models. In Emerging Technologies in Data Mining and Information Security; Dutta, P., Chakrabarti, S., Bhattacharya, A., Dutta, S., Shahnaz, C., Eds.; Lecture Notes in Networks and Systems; Springer Nature Singapore: Singapore, 2023; Volume 490, pp. 613–624. ISBN 978-981-19405-1-4. [Google Scholar]
- Norsigian, C.J.; Pusarla, N.; McConn, J.L.; Yurkovich, J.T.; Dräger, A.; Palsson, B.O.; King, Z. BiGG Models 2020: Multi-Strain Genome-Scale Models and Expansion across the Phylogenetic Tree. Nucleic Acids Res. 2019, 48, D402–D406. [Google Scholar] [CrossRef] [PubMed]
- Lieven, C.; Beber, M.E.; Olivier, B.G.; Bergmann, F.T.; Ataman, M.; Babaei, P.; Bartell, J.A.; Blank, L.M.; Chauhan, S.; Correia, K.; et al. MEMOTE for Standardized Genome-Scale Metabolic Model Testing. Nat. Biotechnol. 2020, 38, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Magnúsdóttir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P.; et al. Generation of Genome-Scale Metabolic Reconstructions for 773 Members of the Human Gut Microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiele, I.; Sahoo, S.; Heinken, A.; Hertel, J.; Heirendt, L.; Aurich, M.K.; Fleming, R.M. Personalized Whole-body Models Integrate Metabolism, Physiology, and the Gut Microbiome. Mol Syst Biol 2020, 16, e8982. [Google Scholar] [CrossRef] [PubMed]
- Machado, D.; Andrejev, S.; Tramontano, M.; Patil, K.R. Fast Automated Reconstruction of Genome-Scale Metabolic Models for Microbial Species and Communities. Nucleic Acids Res. 2018, 46, 7542–7553. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Lercher, M.J. Network Reduction Methods for Genome-Scale Metabolic Models. Cell. Mol. Life Sci. 2020, 77, 481–488. [Google Scholar] [CrossRef]
- Tefagh, M.; Boyd, S.P. Metabolic Network Reductions. bioRxiv 2018, 499251. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What Is Flux Balance Analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Marino, S.; Hogue, I.B.; Ray, C.J.; Kirschner, D.E. A Methodology for Performing Global Uncertainty and Sensitivity Analysis in Systems Biology. J. Theor. Biol. 2008, 254, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Kelk, S.M.; Olivier, B.G.; Stougie, L.; Bruggeman, F.J. Optimal Flux Spaces of Genome-Scale Stoichiometric Models Are Determined by a Few Subnetworks. Sci. Rep. 2012, 2, 580. [Google Scholar] [CrossRef] [Green Version]
- Loghmani, S.B.; Veith, N.; Sahle, S.; Bergmann, F.T.; Olivier, B.G.; Kummer, U. Inspecting the Solution Space of Genome-Scale Metabolic Models. Metabolites 2022, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Damiani, C.; Pescini, D.; Nobile, M.S. Global Sensitivity Analysis of Constraint-Based Metabolic Models. In Computational Intelligence Methods for Bioinformatics and Biostatistics; Raposo, M., Ribeiro, P., Sério, S., Staiano, A., Ciaramella, A., Eds.; Lecture Notes in Computer Science; Springer International Publishing: Cham, Switzerland, 2020; Volume 11925, pp. 179–186. ISBN 978-3-030-34584-6. [Google Scholar]
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 2013, 7, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebedeva, G.; Sorokin, A.; Faratian, D.; Mullen, P.; Goltsov, A.; Langdon, S.P.; Harrison, D.J.; Goryanin, I. Model-Based Global Sensitivity Analysis as Applied to Identification of Anti-Cancer Drug Targets and Biomarkers of Drug Resistance in the ErbB2/3 Network. Eur. J. Pharm. Sci. 2012, 46, 244–258. [Google Scholar] [CrossRef] [Green Version]
- Sorokin, A.; Sorokina, O.; Armstrong, J.D. RKappa: Statistical Sampling Suite for Kappa Models. In Hybrid Systems Biology; Maler, O., Halász, Á., Dang, T., Piazza, C., Eds.; Lecture Notes in Computer Science; Springer International Publishing: Cham, Switzerland, 2015; Volume 7699, pp. 128–142. ISBN 978-3-319-27655-7. [Google Scholar]
- Sorokin, A.; Sorokina, O.; Douglas Armstrong, J. RKappa: Software for Analyzing Rule-Based Models. In Modeling Biomolecular Site Dynamics; Methods in Molecular Biology; Hlavacek, W.S., Ed.; Springer: New York, NY, USA, 2019; Volume 1945, pp. 363–390. ISBN 978-1-4939-9100-6. [Google Scholar]
- Renardy, M.; Joslyn, L.R.; Millar, J.A.; Kirschner, D.E. To Sobol or Not to Sobol? The Effects of Sampling Schemes in Systems Biology Applications. Math. Biosci. 2021, 337, 108593. [Google Scholar] [CrossRef] [PubMed]
- King, Z.A.; Dräger, A.; Ebrahim, A.; Sonnenschein, N.; Lewis, N.E.; Palsson, B.O. Escher: A Web Application for Building, Sharing, and Embedding Data-Rich Visualizations of Biological Pathways. PLoS Comput. Biol. 2015, 11, e1004321. [Google Scholar] [CrossRef] [Green Version]
- Olivier, B.G.; Bergmann, F.T. SBML Level 3 Package: Flux Balance Constraints Version 2. J. Integr. Bioinform. 2018, 15, 20170082. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. Int. J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Iooss, B.; Veiga, S.D.; Janon, A.; Pujol, G.; Broto, B.; Boumhaout, K.; Delage, T.; Amri, R.E.; Fruth, J.; Gilquin, L.; et al. Sensitivity: Global Sensitivity Analysis of Model Outputs. 2022. Available online: https://CRAN.R-project.org/package=sensitivity (accessed on 1 March 2023).
- Weber, F.; Theers, S. ODEsensitivity: Sensitivity Analysis of Ordinary Differential Equations. 2019. Available online: https://CRAN.R-project.org/package=ODEsensitivity (accessed on 1 March 2023).
- R Core Team. R: A Language and Environment for Statistical Computing; Vienna, Austria. 2022. Available online: https://www.R-project.org/ (accessed on 1 March 2023).
- Ye, C.; Luo, Q.; Guo, L.; Gao, C.; Xu, N.; Zhang, L.; Liu, L.; Chen, X. Improving Lysine Production through Construction of an Escherichia Coli Enzyme-constrained Model. Biotechnol. Bioeng. 2020, 117, 3533–3544. [Google Scholar] [CrossRef]
- Bassalo, M.C.; Garst, A.D.; Choudhury, A.; Grau, W.C.; Oh, E.J.; Spindler, E.; Lipscomb, T.; Gill, R.T. Deep Scanning Lysine Metabolism in Escherichia coli. Mol. Syst. Biol. 2018, 14, e8371. [Google Scholar] [CrossRef]
- Orakov, A.N.; Sakenova, N.K.; Sorokin, A.; Goryanin, I.I. ASAR: Visual Analysis of Metagenomes in R. Bioinformatics 2018, 34, 1404–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Name | Value | p-Value | Name | Value | p-Value |
|---|---|---|---|---|---|
| R1u | 0.0472 | 0 | R4u | −0.00183 | 0.564 |
| R5u | 0.388 | 0 | R13u | 0.00167 | 0.597 |
| R8u | 0.0398 | 0 | R25u | −0.00162 | 0.61 |
| R12u | 0.0309 | 0 | R21u | 0.00157 | 0.62 |
| R22u | 0.0443 | 0 | R4l | 0.00114 | 0.718 |
| R26u | 0.0521 | 0 | R10u | 0.00101 | 0.749 |
| EX_Xu | 0.83 | 0 | R23l | −0.000947 | 0.765 |
| R11u | 0.0257 | 4.44 × 10−16 | R21l | 0.000698 | 0.825 |
| R9u | 0.0195 | 7.72 × 10−10 | R2l | 0.000566 | 0.858 |
| R2u | 0.00868 | 0.00607 | R14l | −0.000326 | 0.918 |
| R6u | 0.00623 | 0.049 | R19u | 0.000321 | 0.919 |
| R15u | 0.00523 | 0.0985 | EX_Xl | 0.000298 | 0.925 |
| R7u | 0.00518 | 0.102 | R14u | −0.000279 | 0.93 |
| R18u | 0.00488 | 0.123 | R3l | −0.000196 | 0.951 |
| R3u | 0.00482 | 0.127 | R20u | −0.000193 | 0.951 |
| R16u | 0.00317 | 0.316 | R20l | −0.000162 | 0.959 |
| R24u | 0.00285 | 0.368 | R26l | 5.99 × 10−6 | 0.998 |
| R17u | 0.00269 | 0.395 | R19l | 1.72 × 10−6 | 1 |
| R23u | 0.00211 | 0.505 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorokin, A.; Goryanin, I. FBA-PRCC. Partial Rank Correlation Coefficient (PRCC) Global Sensitivity Analysis (GSA) in Application to Constraint-Based Models. Biomolecules 2023, 13, 500. https://doi.org/10.3390/biom13030500
Sorokin A, Goryanin I. FBA-PRCC. Partial Rank Correlation Coefficient (PRCC) Global Sensitivity Analysis (GSA) in Application to Constraint-Based Models. Biomolecules. 2023; 13(3):500. https://doi.org/10.3390/biom13030500
Chicago/Turabian StyleSorokin, Anatoly, and Igor Goryanin. 2023. "FBA-PRCC. Partial Rank Correlation Coefficient (PRCC) Global Sensitivity Analysis (GSA) in Application to Constraint-Based Models" Biomolecules 13, no. 3: 500. https://doi.org/10.3390/biom13030500
APA StyleSorokin, A., & Goryanin, I. (2023). FBA-PRCC. Partial Rank Correlation Coefficient (PRCC) Global Sensitivity Analysis (GSA) in Application to Constraint-Based Models. Biomolecules, 13(3), 500. https://doi.org/10.3390/biom13030500