

The Interplay between Dysregulated Metabolism and Epigenetics in Cancer


Abstract
:1. Introduction
2. The Mitochondrion—Discovery and Structure
3. Mitochondrial Cellular Roles
3.1. Oxidative Phosphorylation and Energy Production
3.2. Iron–Sulfur (Fe-S) Cluster Formation
3.3. Calcium Homeostatic Control
3.4. Mitochondria and Cell Death
4. Apoptosis
5. Necrosis
6. Parthanatos
7. Autophagy
7.1. Cell Signaling and ROS
7.2. Lipid Metabolism
8. Mitochondria and Disease
Cellular Consequences of Mitochondrial Dysfunction
9. Mitochondrial Dysfunction and Cancer Initiation/Progression
10. Metabolic Dysfunction and Genomic Instability
11. Metabolic Dysfunction and Aberrant Growth
12. Mitochondrial Dysfunction and the Tumorigenic Phenotype
13. Mitochondrial Horizontal Transfer Experiments
14. Investigations of Altered Metabolism in Cancers
15. MRC Dysfunction and Cancer
15.1. C-I Dysfunction and Cancer
15.2. C-II Dysfunction and Cancer
15.3. C-III Dysfunction and Cancer
15.4. C-IV Dysfunction and Cancer
15.5. C-V Dysfunction and Cancer
16. TCA Cycle Dysfunction and Cancer
16.1. Isocitrate Dehydrogenase Dysfunction and Cancer
16.2. Fumarate Hydratase and Cancer
16.3. Citrate Synthase and Cancer
16.4. Aconitate Hydratase and Cancer
16.5. Malic Enzyme and Cancer
17. ROS and Cancer
18. Cellular Consequences of Metabolic Reprogramming
19. Epigenetics and Epigenome
20. DNA Packaging and Histones
21. DNA Methylation
22. Histone Methylation
23. Histone Acetylation
24. Histone Phosphorylation
25. Deposition of Epigenetic Marks Is Dependent on Mitochondrial Metabolites
25.1. ACoA and Epigenetics
25.2. NNMT and Epigenetics
25.3. NAD+ and Epigenetics
25.4. SAM and Epigenetics
25.5. FAD and Epigenetics
25.6. α-KG, 2-HG, and Epigenetics
26. Therapies Targeting Dysregulated Metabolics and Epigenetics in Cancers
27. Concluding Remarks
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Schulz, T.J.; Thierbach, R.; Voigt, A.; Drewes, G.; Mietzner, B.; Steinberg, P.; Pfeiffer, A.F.; Ristow, M. Induction of oxidative metabolism by mitochondrial frataxin inhibits cancer growth: Otto Warburg revisited. J. Biol. Chem. 2006, 281, 977–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab. 2010, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Altenberg, B.; Greulich, K.O. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 2004, 84, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Ananieva, E. Targeting amino acid metabolism in cancer growth and anti-tumor immune response. World J. Biol. Chem. 2015, 6, 281–289. [Google Scholar] [CrossRef]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Brusselmans, K.; Swinnen, J.V. The Lipogenic Switch in Cancers. In Mitochondria and Cancer, 1st ed.; Singh, K.K., Costello, L.C., Eds.; Springer: New York, NY. USA, 2009; pp. 39–60. [Google Scholar] [CrossRef] [Green Version]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [Green Version]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baysal, B.E. On the association of succinate dehydrogenase mutations with hereditary paraganglioma. Trends Endocrinol. Metab. 2003, 14, 453–459. [Google Scholar] [CrossRef]
- Bonello, S.; Zahringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Gorlach, A. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnartz, B.; Anglmayer, R.; Zanssen, S. Comprehensive scanning of somatic mitochondrial DNA alterations in acute leukemia developing from myelodysplastic syndromes. Cancer Res. 2004, 64, 1966–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, G.; Adebanjo, O.A.; Freedman, B.D.; Anandatheerthavarada, H.K.; Vijayasarathy, C.; Zaidi, M.; Kotlikoff, M.; Avadhani, N.G. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: A novel mode of inter-organelle crosstalk. EMBO J. 1999, 18, 522–533. [Google Scholar] [CrossRef] [Green Version]
- Amuthan, G.; Biswas, G.; Zhang, S.Y.; Klein-Szanto, A.; Vijayasarathy, C.; Avadhani, N.G. Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. EMBO J. 2001, 20, 1910–1920. [Google Scholar] [CrossRef] [Green Version]
- Erol, A. Retrograde regulation due to mitochondrial dysfunction may be an important mechanism for carcinogenesis. Med. Hypotheses 2005, 65, 525–529. [Google Scholar] [CrossRef]
- Miceli, M.V.; Jazwinski, S.M. Common and cell type-specific responses of human cells to mitochondrial dysfunction. Exp. Cell Res. 2005, 302, 270–280. [Google Scholar] [CrossRef]
- Tuppen, H.A.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 2010, 1797, 113–128. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.D.C.S.; Yan, G. Detection of Mitochondrial Mutations in Cancer by Next Generation Sequencing. J. Next Gener. Seq. Appl. 2014, 1, 1. [Google Scholar] [CrossRef] [Green Version]
- Reznik, E.; Wang, Q.; La, K.; Schultz, N.; Sander, C. Mitochondrial respiratory gene expression is suppressed in many cancers. Elife 2017, 6, e21592. [Google Scholar] [CrossRef]
- Ley, T.J.; Mardis, E.R.; Ding, L.; Fulton, B.; McLellan, M.D.; Chen, K.; Dooling, D.; Dunford-Shore, B.H.; McGrath, S.; Hickenbotham, M.; et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008, 456, 66–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Genetics: Mitochondrial DNA in evolution and disease. Nature 2016, 535, 498–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef]
- Dang, C.V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Grzybowska-Szatkowska, L.; Slaska, B. Mitochondrial NADH dehydrogenase polymorphisms are associated with breast cancer in Poland. J. Appl. Genet. 2014, 55, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, G.K.; Sheafor, B.A. Red Blood Cells: Centerpiece in the Evolution of the Vertebrate Circulatory System. Am. Zool. 1999, 39, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Palade, G.E. The fine structure of mitochondria. Anat. Rec. 1952, 114, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Sottocasa, G.L.; Kuylenstierna, B.; Ernster, L.; Bergstrand, A. An electron-transport system associated with the outer membrane of liver mitochondria. a biochemical and morphological study. J. Cell Biol. 1967, 32, 415–438. [Google Scholar] [CrossRef]
- Parsons, D.F.; Williams, G.R.; Chance, B. Characteristics of isolated and purified preparations of the outer and inner membranes of mitochondria. Ann. N. Y. Acad. Sci. 1966, 137, 643–666. [Google Scholar] [CrossRef]
- Schnaitman, C.; Greenawalt, J.W. Enzymatic properties of the inner and outer membranes of rat liver mitochondria. J. Cell Biol. 1968, 38, 158–175. [Google Scholar] [CrossRef]
- Schnaitman, C.; Erwin, V.G.; Greenawalt, J.W. The submitochondrial localization of monoamine oxidase. an enzymatic marker for the outer membrane of rat liver mitochondria. J. Cell Biol. 1967, 32, 719–735. [Google Scholar] [CrossRef] [Green Version]
- Perkins, G.A.; Frey, T.G. Recent structural insight into mitochondria gained by microscopy. Micron 2000, 31, 97–111. [Google Scholar] [CrossRef]
- Logan, D.C. The mitochondrial compartment. J. Exp. Bot. 2006, 57, 1225–1243. [Google Scholar] [CrossRef]
- Comte, J.; Maisterrena, B.; Gautheron, D.C. Lipid composition and protein profiles of outer and inner membranes from pig heart mitochondria. Comparison with microsomes. Biochim. Biophys. Acta 1976, 419, 271–284. [Google Scholar] [CrossRef]
- Hallermayer, G.; Neupert, W. Lipid composition of mitochondrial outer and inner membranes of Neurospora crassa. Biol. Chem. 1974, 355, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Colombini, M. The VDAC channel: Molecular basis for selectivity. Biochim. Biophys. Acta 2016, 1863, 2498–2502. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.F.; Bonner, W.D., Jr.; Verboon, J.G. Electron microscopy of isolated plant mitochondria and plastids using both the thin-section and negative-staining techniques. Can. J. Bot. 1965, 43, 647–655. [Google Scholar] [CrossRef]
- Werkheiser, W.C.; Bartley, W. The study of steady-state concentrations of internal solutes of mitochondria by rapid centrifugal transfer to a fixation medium. Biochem. J. 1957, 66, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Getz, G.S.; Bartley, W.; Lurie, D.; Notton, B.M. The phospholipids of various sheep organs, rat liver and of their subcellular fractions. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1968, 152, 325–339. [Google Scholar] [CrossRef]
- Chicco, A.J.; Sparagna, G.C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fry, M.; Green, D.E. Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J. Biol. Chem. 1981, 256, 1874–1880. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, E.; Klingenberg, M.; Heldt, H.W. Unspecific permeation and specific exchange of adenine nucleotides in liver mitochondria. Biochim. Biophys. Acta 1965, 104, 312–315. [Google Scholar] [CrossRef]
- Ernster, L.; Schatz, G. Mitochondria: A historical review. J. Cell Biol. 1981, 91, 227s–255s. [Google Scholar] [CrossRef]
- Formosa, L.E.; Ryan, M.T. Mitochondrial fusion: Reaching the end of mitofusin’s tether. J. Cell Biol. 2016, 215, 597–598. [Google Scholar] [CrossRef] [Green Version]
- Richter, V.; Singh, A.P.; Kvansakul, M.; Ryan, M.T.; Osellame, L.D. Splitting up the powerhouse: Structural insights into the mechanism of mitochondrial fission. Cell. Mol. Life Sci. 2015, 72, 3695–3707. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B. Introduction to Mitochondrial Oxidative Phosphorylation. In Advances in Experimental Medicine and Biology; Cohen, I.R., Lajtha, A., Lambris, J.D., Paoletti, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 748, pp. 1–12. [Google Scholar]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferree, A.; Shirihai, O. Mitochondrial Dynamics: The Intersection of Form and Function. In Mitochondrial Oxidative Phosphorylation; Kabenbach, B., Ed.; Springer: New York, NY, USA, 2012; pp. 13–40. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Reviews. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Opferman, J.T.; Iwasaki, H.; Ong, C.C.; Suh, H.; Mizuno, S.; Akashi, K.; Korsmeyer, S.J. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 2005, 307, 1101–1104. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.B.; Aon, M.A.; Hsu, Y.T.; Soane, L.; Teng, X.; McCaffery, J.M.; Cheng, W.C.; Qi, B.; Li, H.; Alavian, K.N.; et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J. Cell Biol. 2011, 195, 263–276. [Google Scholar] [CrossRef]
- Alavian, K.N.; Li, H.; Collis, L.; Bonanni, L.; Zeng, L.; Sacchetti, S.; Lazrove, E.; Nabili, P.; Flaherty, B.; Graham, M.; et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat. Cell Biol. 2011, 13, 1224–1233. [Google Scholar] [CrossRef]
- Perciavalle, R.M.; Stewart, D.P.; Koss, B.; Lynch, J.; Milasta, S.; Bathina, M.; Temirov, J.; Cleland, M.M.; Pelletier, S.; Schuetz, J.D.; et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 2012, 14, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Nass, M.M.K.; Nass, S. Intramitochondrial fibers with DNA characteristics. I. Fixation and Electron Staining Reactions. J. Cell Biol. 1963, 19, 593–611. [Google Scholar] [CrossRef] [Green Version]
- Schatz, G.; Haslbrunner, E.; Tuppy, H. Deoxyribonucleic acid associated with yeast mitochondria. Biochem. Biophys. Res. Commun. 1964, 15, 127–132. [Google Scholar] [CrossRef]
- Luck, D.J.L.; Reich, E. DNA in mitochondria of neurospora crassa. Proc. Natl. Acad. Sci. USA 1964, 52, 931–938. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondrial protein import: From proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 2010, 11, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Simcox, E.; Reeve, A. An Introduction to Mitochondria, Their Structure and Functions. In Mitochondrial Dysfunction in Neurodegenerative Disorders; Reeve, A., Simcox, E., Duchen, M., Turnbull, D., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 3–31. [Google Scholar] [CrossRef]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodson, J.D.; Chory, J. Coordination of gene expression between organellar and nuclear genomes. Nat. Rev. Genet. 2008, 9, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Castillo, S.; Baertling, F.; Kownatzki, D.; Wessels, H.J.; Arnold, S.; Brandt, U.; Nijtmans, L. The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab. 2016, 25, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, P.; Pohl, C.; Calles, C.; Marks, C.; Wild, S.; Krutmann, J. Cellular response to infrared radiation involves retrograde mitochondrial signaling. Free Radic. Biol. Med. 2007, 43, 128–135. [Google Scholar] [CrossRef]
- Singh, K.K.; Kulawiec, M.; Still, I.; Desouki, M.M.; Geradts, J.; Matsui, S. Inter-genomic cross talk between mitochondria and the nucleus plays an important role in tumorigenesis. Gene 2005, 354, 140–146. [Google Scholar] [CrossRef]
- Butow, R.A.; Avadhani, N.G. Mitochondrial signaling: The retrograde response. Mol. Cell 2004, 14, 1–15. [Google Scholar] [CrossRef]
- Siekevitz, P. Powerhouse of the Cell. Sci. Am. 1957, 197, 131–144. [Google Scholar] [CrossRef]
- Sands, R.H.; Beinert, H. Studies on mitochondria and submitochondrial particles by paramagnetic resonance (EPR) spectroscopy. Biochem. Biophys. Res. Commun. 1960, 3, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Hajnóczky, G.; Csordás, G.; Das, S.; Garcia-Perez, C.; Saotome, M.; Sinha Roy, S.; Yi, M. Mitochondrial calcium signalling and cell death: Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 2006, 40, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R. Apoptotic Pathways. Cell 1998, 94, 695–698. [Google Scholar] [CrossRef] [Green Version]
- Marzo, I.; Brenner, C.; Kroemer, G. The central role of the mitochondrial megachannel in apoptosis: Evidence obtained with intact cells, isolated mitochondria, and purified protein complexes. Biomed. Pharmacother. 1998, 52, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borkenhagen, L.F.; Kennedy, E.P.; Fielding, L. Enzymatic Formation and Decarboxylation of Phosphatidylserine. J. Biol. Chem. 1961, 236, PC28–PC30. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Rees, D.M.; Leslie, A.G.; Walker, J.E. The structure of the membrane extrinsic region of bovine ATP synthase. Proc. Natl. Acad. Sci. USA 2009, 106, 21597–21601. [Google Scholar] [CrossRef] [Green Version]
- Chance, B.; Williams, G.R. Respiratory enzymes in oxidative phosphorylation. III. The steady state. J. Biol. Chem. 1955, 217, 409–427. [Google Scholar] [CrossRef]
- Hackenbrock, C.R.; Chazotte, B.; Gupte, S.S. The random collision model and a critical assessment of diffusion and collision in mitochondrial electron transport. J. Bioenerg. Biomembr. 1986, 18, 331–368. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Enriquez, J.A. The function of the respiratory supercomplexes: The plasticity model. Biochim. Biophys. Acta 2014, 1837, 444–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruciat, C.M.; Brunner, S.; Baumann, F.; Neupert, W.; Stuart, R.A. The cytochrome bc1 and cytochrome c oxidase complexes associate to form a single supracomplex in yeast mitochondria. J. Biol. Chem. 2000, 275, 18093–18098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schagger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [Green Version]
- Dudkina, N.V.; Kouril, R.; Peters, K.; Braun, H.P.; Boekema, E.J. Structure and function of mitochondrial supercomplexes. Biochim. Biophys. Acta 2010, 1797, 664–670. [Google Scholar] [CrossRef] [Green Version]
- Vartak, R.; Porras, C.A.; Bai, Y. Respiratory supercomplexes: Structure, function and assembly. Protein Cell 2013, 4, 582–590. [Google Scholar] [CrossRef] [Green Version]
- Genova, M.L.; Lenaz, G. Functional role of mitochondrial respiratory supercomplexes. Biochim. Biophys. Acta 2014, 1837, 427–443. [Google Scholar] [CrossRef] [Green Version]
- Acin-Perez, R.; Fernandez-Silva, P.; Peleato, M.L.; Perez-Martos, A.; Enriquez, J.A. Respiratory active mitochondrial supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef]
- Shinzawa-Itoh, K.; Shimomura, H.; Yanagisawa, S.; Shimada, S.; Takahashi, R.; Oosaki, M.; Ogura, T.; Tsukihara, T. Purification of Active Respiratory Supercomplex from Bovine Heart Mitochondria Enables Functional Studies. J. Biol. Chem. 2016, 291, 4178–4184. [Google Scholar] [CrossRef] [Green Version]
- Letts, J.A.; Fiedorczuk, K.; Sazanov, L.A. The architecture of respiratory supercomplexes. Nature 2016, 537, 644–648. [Google Scholar] [CrossRef]
- Stuart, R.A. Supercomplex organization of the oxidative phosphorylation enzymes in yeast mitochondria. J. Bioenerg. Biomembr. 2008, 40, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Wu, M.; Guo, R.; Yan, K.; Lei, J.; Gao, N.; Yang, M. The architecture of the mammalian respirasome. Nature 2016, 537, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Schafer, E.; Seelert, H.; Reifschneider, N.H.; Krause, F.; Dencher, N.A.; Vonck, J. Architecture of active mammalian respiratory chain supercomplexes. J. Biol. Chem. 2006, 281, 15370–15375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.C.; Dean, D.R.; Smith, A.D.; Johnson, M.K. Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 2005, 74, 247–281. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.-H.; Jameson, G.N.L.; Huynh, B.H.; Rouault, T.A. Subcellular compartmentalization of human Nfu, an iron–sulfur cluster scaffold protein, and its ability to assemble a [4Fe–4S] cluster. Proc. Natl. Acad. Sci. USA 2003, 100, 9762–9767. [Google Scholar] [CrossRef] [Green Version]
- Lange, H.; Kaut, A.; Kispal, G.; Lill, R. A mitochondrial ferredoxin is essential for biogenesis of cellular iron-sulfur proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 1050–1055. [Google Scholar] [CrossRef] [Green Version]
- Lill, R.; Muhlenhoff, U. Iron-sulfur-protein biogenesis in eukaryotes. Trends Biochem. Sci. 2005, 30, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Intracellular calcium homeostasis and signaling. Met. Ions Life Sci. 2013, 12, 119–168. [Google Scholar] [CrossRef]
- Carafoli, E.; Crompton, M. The regulation of intracellular calcium by mitochondria. Ann. N. Y. Acad. Sci. 1978, 307, 269–284. [Google Scholar] [CrossRef]
- McCormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 1990, 70, 391–425. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, H.; Barrett, P.Q. Calcium messenger system: An integrated view. Physiol. Rev. 1984, 64, 938–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [Green Version]
- Keinan, N.; Pahima, H.; Ben-Hail, D.; Shoshan-Barmatz, V. The role of calcium in VDAC1 oligomerization and mitochondria-mediated apoptosis. Biochim. Biophys. Acta 2013, 1833, 1745–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A Forty-Kilodalton Protein of the Inner Membrane is the Mitochondrial Calcium Uniporter. Nature 2011, 476, 336–340. Available online: http://www.nature.com/nature/journal/v476/n7360/abs/nature10230.html#supplementary-information (accessed on 24 April 2023). [CrossRef] [PubMed] [Green Version]
- Palty, R.; Hershfinkel, M.; Sekler, I. Molecular Identity and Functional Properties of the Mitochondrial Na+/Ca2+ Exchanger. J. Biol. Chem. 2012, 287, 31650–31657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palty, R.; Sekler, I. The mitochondrial Na+/Ca2+ exchanger. Cell Calcium 2012, 52, 9–15. [Google Scholar] [CrossRef]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta (BBA)-Bioenerg. 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef] [Green Version]
- Szabadkai, G.; Duchen, M.R. Mitochondria: The hub of cellular Ca2+ signaling. Physiology 2008, 23, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Trapani, J.A.; Smyth, M.J. Killing by cytotoxic T cells and natural killer cells: Multiple granule serine proteases as initiators of DNA fragmentation. Immunol. Cell Biol. 1993, 71, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Kraut, R.P.; Aebersold, R.; Greenberg, A.H. A natural killer cell granule protein that induces DNA fragmentation and apoptosis. J. Exp. Med. 1992, 175, 553–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Estaquier, J.; Vallette, F.; Vayssiere, J.L.; Mignotte, B. The mitochondrial pathways of apoptosis. Adv. Exp. Med. Biol. 2012, 942, 157–183. [Google Scholar] [CrossRef]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]
- Westphal, D.; Dewson, G.; Czabotar, P.E.; Kluck, R.M. Molecular biology of Bax and Bak activation and action. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2011, 1813, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Hockings, C.; Anwari, K.; Kratina, T.; Fennell, S.; Lazarou, M.; Ryan, M.T.; Kluck, R.M.; Dewson, G. Assembly of the Bak apoptotic pore: A critical role for the Bak protein alpha6 helix in the multimerization of homodimers during apoptosis. J. Biol. Chem. 2013, 288, 26027–26038. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Yang, J.; Jones, D.P. Mitochondrial control of apoptosis: The role of cytochrome c. Biochim. Biophys. Acta 1998, 1366, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a Mitochondrial Protein that Promotes Cytochrome c–Dependent Caspase Activation by Eliminating IAP Inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef] [Green Version]
- van Loo, G.; Saelens, X.; van Gurp, M.; MacFarlane, M.; Martin, S.J.; Vandenabeele, P. The role of mitochondrial factors in apoptosis: A Russian roulette with more than one bullet. Cell Death Differ. 2002, 9, 1031–1042. [Google Scholar] [CrossRef]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budihardjo, I.; Oliver, H.; Lutter, M.; Luo, X.; Wang, X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999, 15, 269–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995, 14, 5579–5588. [Google Scholar] [CrossRef]
- Young, M.M.; Takahashi, Y.; Khan, O.; Park, S.; Hori, T.; Yun, J.; Sharma, A.K.; Amin, S.; Hu, C.D.; Zhang, J.; et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 2012, 287, 12455–12468. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Pradelli, L.A.; Beneteau, M.; Ricci, J.E. Mitochondrial control of caspase-dependent and -independent cell death. Cell. Mol. Life Sci. 2010, 67, 1589–1597. [Google Scholar] [CrossRef]
- Russell, J.H.; Ley, T.J. Lymphocyte-mediated cytotoxicity. Annu. Rev. Immunol. 2002, 20, 323–370. [Google Scholar] [CrossRef]
- Barry, M.; Bleackley, R.C. Cytotoxic T lymphocytes: All roads lead to death. Nat. Rev. Immunol. 2002, 2, 401–409. [Google Scholar] [CrossRef]
- Linkermann, A.; Vanden Berghe, T.; Takahashi, N.; Kunzendorf, U.; Krautwald, S.; Vandenabeele, P. The Potential Role of Necroptosis in Diseases. In Necrotic Cell Death; Shen, H., Vandenabeele, P., Eds.; Springer Science: New York, NY, USA, 2014; Volume 2, pp. 1–21. [Google Scholar]
- Kroemer, G.; Dallaporta, B.; Resche-Rigon, M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 1998, 60, 619–642. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997, 57, 1835–1840. [Google Scholar] [PubMed]
- Crompton, M.; Barksby, E.; Johnson, N.; Capano, M. Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie 2002, 84, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Vanlangenakker, N.; Vanden Berghe, T.; Krysko, D.V.; Festjens, N.; Vandenabeele, P. Molecular mechanisms and pathophysiology of necrotic cell death. Curr. Mol. Med. 2008, 8, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [Green Version]
- Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Mitochondrial and nuclear cross talk in cell death: Parthanatos. Ann. N. Y. Acad. Sci. 2008, 1147, 233–241. [Google Scholar] [CrossRef]
- Virag, L.; Robaszkiewicz, A.; Rodriguez-Vargas, J.M.; Oliver, F.J. Poly(ADP-ribose) signaling in cell death. Mol. Asp. Med. 2013, 34, 1153–1167. [Google Scholar] [CrossRef]
- De Duve, C. The lysosome. Sci. Am. 1963, 208, 64–72. [Google Scholar] [CrossRef]
- Katheder, N.S.; Khezri, R.; O’Farrell, F.; Schultz, S.W.; Jain, A.; Rahman, M.M.; Schink, K.O.; Theodossiou, T.A.; Johansen, T.; Juhasz, G.; et al. Microenvironmental autophagy promotes tumour growth. Nature 2017, 541, 417–420. [Google Scholar] [CrossRef] [Green Version]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef]
- De Duve, C. Lysosomes revisited. Eur. J. Biochem. 1983, 137, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Kondo-Okamoto, N. Mitochondria and autophagy: Critical interplay between the two homeostats. Biochim. Biophys. Acta 2012, 1820, 595–600. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; De Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxid. Med. Cell. Longev. 2016, 2016, 3907147. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Zhong, W.; Zhang, W.; Zhou, Z. Defect of mitochondrial respiratory chain is a mechanism of ROS overproduction in a rat model of alcoholic liver disease: Role of zinc deficiency. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 310, G205–G214. [Google Scholar] [CrossRef] [Green Version]
- Kumari, U.; Ya Jun, W.; Huat Bay, B.; Lyakhovich, A. Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in Fanconi anemia cells. Oncogene 2014, 33, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Sharma, L.K.; Fang, H.; Liu, J.; Vartak, R.; Deng, J.; Bai, Y. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum. Mol. Genet. 2011, 20, 4605–4616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwiecien, S.; Jasnos, K.; Magierowski, M.; Sliwowski, Z.; Pajdo, R.; Brzozowski, B.; Mach, T.; Wojcik, D.; Brzozowski, T. Lipid peroxidation, reactive oxygen species and antioxidative factors in the pathogenesis of gastric mucosal lesions and mechanism of protection against oxidative stress—Induced gastric injury. J. Physiol. Pharmacol. 2014, 65, 613–622. [Google Scholar] [PubMed]
- Mylonas, C.; Kouretas, D. Lipid peroxidation and tissue damage. In Vivo 1999, 13, 295–309. [Google Scholar] [PubMed]
- Kwon, J.; Lee, S.R.; Yang, K.S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.R.; Yang, K.S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef] [Green Version]
- Samper, E.; Nicholls, D.G.; Melov, S. Mitochondrial oxidative stress causes chromosomal instability of mouse embryonic fibroblasts. Aging Cell 2003, 2, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother. 2004, 7, 97–110. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Owusu-Ansah, E.; Banerjee, U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 2009, 461, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Waypa, G.B.; Smith, K.A.; Schumacker, P.T. O2 sensing, mitochondria and ROS signaling: The fog is lifting. Mol. Asp. Med. 2016, 47, 76–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Liu, Y.; Bae, N.; Nimer, S.D. The p53 tumor suppressor protein regulates hematopoietic stem cell fate. J. Cell. Physiol. 2011, 226, 2215–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, K.; Fitzpatrick, F.A. Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2, and 3 and antagonizes their transcriptional repressor function. J. Biol. Chem. 2010, 285, 17417–17424. [Google Scholar] [CrossRef] [Green Version]
- Le Belle, J.E.; Orozco, N.M.; Paucar, A.A.; Saxe, J.P.; Mottahedeh, J.; Pyle, A.D.; Wu, H.; Kornblum, H.I. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 2011, 8, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Oberley, L.W. Inhibition of tumor cell growth by overexpression of manganese-containing superoxide dismutase. Age 1998, 21, 95–97. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.G.; Weydert, C.J.; Zhang, Y.; Yu, L.; Liu, J.; Spitz, D.R.; Cullen, J.J.; Oberley, L.W. Superoxide Enhances the Antitumor Combination of AdMnSOD Plus BCNU in Breast Cancer. Cancers 2010, 2, 68–87. [Google Scholar] [CrossRef] [Green Version]
- Daum, G.; Lees, N.D.; Bard, M.; Dickson, R. Biochemistry, cell biology and molecular biology of lipids of Saccharomyces cerevisiae. Yeast 1998, 14, 1471–1510. [Google Scholar] [CrossRef]
- Tatsuta, T.; Scharwey, M.; Langer, T. Mitochondrial lipid trafficking. Trends Cell Biol. 2013, 24, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Harada, Y.; Nishikawa, S.; Yamano, K.; Kamiya, M.; Shiota, T.; Kuroda, T.; Kuge, O.; Sesaki, H.; Imai, K.; et al. Tam41 is a CDP-diacylglycerol synthase required for cardiolipin biosynthesis in mitochondria. Cell Metab. 2013, 17, 709–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osman, C.; Voelker, D.R.; Langer, T. Making heads or tails of phospholipids in mitochondria. J. Cell Biol. 2011, 192, 7–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connerth, M.; Tatsuta, T.; Haag, M.; Klecker, T.; Westermann, B.; Langer, T. Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science 2012, 338, 815–818. [Google Scholar] [CrossRef]
- Lee, H.C.; Wei, Y.H. Mitochondria and aging. Adv. Exp. Med. Biol. 2012, 942, 311–327. [Google Scholar] [CrossRef]
- Miquel, J.; Economos, A.C.; Fleming, J.; Johnson, J.E., Jr. Mitochondrial role in cell aging. Exp. Gerontol. 1980, 15, 575–591. [Google Scholar] [CrossRef]
- Muller-Hocker, J. Cytochrome-c-oxidase deficient cardiomyocytes in the human heart--an age-related phenomenon. A histochemical ultracytochemical study. Am. J. Pathol. 1989, 134, 1167–1173. [Google Scholar]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef]
- Pedersen, P.L. Tumor mitochondria and the bioenergetics of cancer cells. Prog. Exp. Tumor Res. 1978, 22, 190–274. [Google Scholar]
- Hunter, R.L.; Dragicevic, N.; Seifert, K.; Choi, D.Y.; Liu, M.; Kim, H.C.; Cass, W.A.; Sullivan, P.G.; Bing, G. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J. Neurochem. 2007, 100, 1375–1386. [Google Scholar] [CrossRef]
- Rous, P. Surmise and fact on the nature of cancer. Nature 1959, 183, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.E.; Kenyon, D.H. A unifying concept of carcinogenesis and its therapeutic implications. Oncology 1973, 27, 459–479. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.M.; Venkatachalam, M.A.; Roeser, N.F.; Nissim, I. Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc. Natl. Acad. Sci. USA 2000, 97, 2826–2831. [Google Scholar] [CrossRef] [Green Version]
- Shoubridge, E.A. Nuclear genetic defects of oxidative phosphorylation. Hum. Mol. Genet. 2001, 10, 2277–2284. [Google Scholar] [CrossRef] [Green Version]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef]
- Simonnet, H.; Alazard, N.; Pfeiffer, K.; Gallou, C.; Beroud, C.; Demont, J.; Bouvier, R.; Schagger, H.; Godinot, C. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis 2002, 23, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Simonnet, H.; Demont, J.; Pfeiffer, K.; Guenaneche, L.; Bouvier, R.; Brandt, U.; Schägger, H.; Godinot, C. Mitochondrial complex I is deficient in renal oncocytomas. Carcinogenesis 2003, 24, 1461–1466. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, V.; Kriete, A.; Sacan, A.; Jazwinski, S.M. Comparing the yeast retrograde response and NF-kappaB stress responses: Implications for aging. Aging Cell 2010, 9, 933–941. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Butow, R.A. Mitochondrial retrograde signaling. Annu. Rev. Genet. 2006, 40, 159–185. [Google Scholar] [CrossRef]
- Rothermel, B.A.; Shyjan, A.W.; Etheredge, J.L.; Butow, R.A. Transactivation by Rtg1p, a basic helix-loop-helix protein that functions in communication between mitochondria and the nucleus in yeast. J. Biol. Chem. 1995, 270, 29476–29482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekito, T.; Thornton, J.; Butow, R.A. Mitochondria-to-nuclear signaling is regulated by the subcellular localization of the transcription factors Rtg1p and Rtg3p. Mol. Biol. Cell 2000, 11, 2103–2115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothermel, B.A.; Thornton, J.L.; Butow, R.A. Rtg3p, a basic helix-loop-helix/leucine zipper protein that functions in mitochondrial-induced changes in gene expression, contains independent activation domains. J. Biol. Chem. 1997, 272, 19801–19807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borghouts, C.; Benguria, A.; Wawryn, J.; Jazwinski, S.M. Rtg2 protein links metabolism and genome stability in yeast longevity. Genetics 2004, 166, 765–777. [Google Scholar] [CrossRef]
- Jazwinski, S.M. The retrograde response: A conserved compensatory reaction to damage from within and from without. Prog. Mol. Biol. Transl. Sci. 2014, 127, 133–154. [Google Scholar] [CrossRef] [Green Version]
- Dilova, I.; Aronova, S.; Chen, J.C.; Powers, T. Tor signaling and nutrient-based signals converge on Mks1p phosphorylation to regulate expression of Rtg1.Rtg3p-dependent target genes. J. Biol. Chem. 2004, 279, 46527–46535. [Google Scholar] [CrossRef] [Green Version]
- Dilova, I.; Chen, C.Y.; Powers, T. Mks1 in concert with TOR signaling negatively regulates RTG target gene expression in S. cerevisiae. Curr. Biol. 2002, 12, 389–395. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Sekito, T.; Spirek, M.; Thornton, J.; Butow, R.A. Retrograde signaling is regulated by the dynamic interaction between Rtg2p and Mks1p. Mol. Cell 2003, 12, 401–411. [Google Scholar] [CrossRef]
- Giannattasio, S.; Liu, Z.; Thornton, J.; Butow, R.A. Retrograde response to mitochondrial dysfunction is separable from TOR1/2 regulation of retrograde gene expression. J. Biol. Chem. 2005, 280, 42528–42535. [Google Scholar] [CrossRef] [Green Version]
- Amuthan, G.; Biswas, G.; Ananadatheerthavarada, H.K.; Vijayasarathy, C.; Shephard, H.M.; Avadhani, N.G. Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene 2002, 21, 7839–7849. [Google Scholar] [CrossRef] [Green Version]
- Lv, X.; Li, J.; Zhang, C.; Hu, T.; Li, S.; He, S.; Yan, H.; Tan, Y.; Lei, M.; Wen, M.; et al. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 2017, 4, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.C.; Rathore, A.; Younas, H.; Gilkes, D.; Polotsky, V.Y. Hypoxia-Inducible Factors and Cancer. Curr. Sleep Med. Rep. 2017, 3, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, S.J.; Pan, J.; Lee, M.H. Roles of p53, MYC and HIF-1 in regulating glycolysis—The seventh hallmark of cancer. Cell. Mol. Life Sci. 2008, 65, 3981–3999. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.M.; Kim, J.S.; Yoo, Y.D. A novel protein, Romo1, induces ROS production in the mitochondria. Biochem. Biophys. Res. Commun. 2006, 347, 649–655. [Google Scholar] [CrossRef]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Seoane, M.; Mosquera-Miguel, A.; Gonzalez, T.; Fraga, M.; Salas, A.; Costoya, J.A. The mitochondrial genome is a “genetic sanctuary” during the oncogenic process. PLoS ONE 2011, 6, e23327. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, A.K.; Chatterjee, A.; Rasmussen, L.J.; Singh, K.K. Mitochondria-mediated nuclear mutator phenotype in Saccharomyces cerevisiae. Nucleic Acids Res. 2003, 31, 3909–3917. [Google Scholar] [CrossRef]
- Matoba, S.; Kang, J.-G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 Regulates Mitochondrial Respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Corcoran, C.A.; Huang, Y.; Sheikh, M.S. Energy Generating Pathways and the Tumor Suppressor p53. In Mitochondria and Cancer; Springer: New York, NY, USA, 2009; pp. 131–150. [Google Scholar] [CrossRef]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic Enzymes Can Modulate Cellular Life Span. Cancer Res. 2005, 65, 177–185. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.P.; Lim, T.T.; Chan, Y.L.; Song, A.C.M.; Yeo, B.H.; Vojtesek, B.; Coomber, D.; Rajagopal, G.; Lane, D. The p53 knowledgebase: An integrated information resource for p53 research. Oncogene 2006, 26, 1517–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petitjean, A.; Achatz, M.I.W.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Li, G.; Liu, G.; Lee, S.W.; Aaronson, S.A. p53 induction of heparin-binding EGF-like growth factor counteracts p53 growth suppression through activation of MAPK and PI3K/Akt signaling cascades. EMBO J. 2001, 20, 1931–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct Activation of Bax by p53 Mediates MitochondrialMembrane Permeabilization and Apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [Green Version]
- Erster, S.; Moll, U.M. Stress-induced p53 runs a transcription-independent death program. Biochem. Biophys. Res. Commun. 2005, 331, 843–850. [Google Scholar] [CrossRef]
- Leu, J.I.J.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D.L. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450. [Google Scholar] [CrossRef]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 Has a Direct Apoptogenic Role at the Mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Perfettini, J.-L.; Kroemer, R.T.; Kroemer, G. Fatal liaisons of p53 with Bax and Bak. Nat. Cell Biol. 2004, 6, 386–388. [Google Scholar] [CrossRef]
- Karuman, P.; Gozani, O.; Odze, R.D.; Zhou, X.C.; Zhu, H.; Shaw, R.; Brien, T.P.; Bozzuto, C.D.; Ooi, D.; Cantley, L.C.; et al. The Peutz-Jegher Gene Product LKB1 Is a Mediator of p53-Dependent Cell Death. Mol. Cell 2001, 7, 1307–1319. [Google Scholar] [CrossRef]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.-H.; Bardeesy, N.; DePinho, R.A.; Montminy, M.; Cantley, L.C. The Kinase LKB1 Mediates Glucose Homeostasis in Liver and Therapeutic Effects of Metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Amos, C.I.; Stephens, L.C.; Campos, I.; Deng, J.M.; Behringer, R.R.; Rashid, A.; Frazier, M.L. Mutation of Lkb1 and p53 Genes Exert a Cooperative Effect on Tumorigenesis. Cancer Res. 2005, 65, 11297–11303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, L.; Passegue, E. Surviving change: The metabolic journey of hematopoietic stem cells. Trends Cell Biol. 2014, 24, 479–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [Green Version]
- Gasche, C.; Chang, C.L.; Rhees, J.; Goel, A.; Boland, C.R. Oxidative stress increases frameshift mutations in human colorectal cancer cells. Cancer Res. 2001, 61, 7444–7448. [Google Scholar]
- Kulawiec, M.; Safina, A.F.; Desouki, M.M.; Still, I.; Matsui, S.-I.; Bakin, A.; Singh, K.K. Tumorigenic transformation of human breast epithelial cells induced by mitochondrial DNA depletion. Cancer Biol. Ther. 2008, 7, 1732–1743. [Google Scholar] [CrossRef] [Green Version]
- Di Donato, S. Disorders related to mitochondrial membranes: Pathology of the respiratory chain and neurodegeneration. J. Inherit. Metab. Dis. 2000, 23, 247–263. [Google Scholar] [CrossRef]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef] [Green Version]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Cha, M.Y.; Kim, D.K.; Mook-Jung, I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp. Mol. Med. 2015, 47, e150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hroudov, J.; Singh, N.; Fi, Z. Mitochondrial Dysfunctions in Neurodegenerative Diseases: Relevance to Alzheimer’s Disease. BioMed Res. Int. 2014, 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christodoulou, J. Genetic defects causing mitochondrial respiratory chain disorders and disease. Hum. Reprod. 2000, 15 (Suppl. S2), 28–43. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, M.D.; Hopkins, B.D.; Cantley, L.C. Dietary Fat and Sugar in Promoting Cancer Development and Progression. Annu. Rev. Cancer Biol. 2019, 3, 255–273. [Google Scholar] [CrossRef]
- Modica-Napolitano, J.S.; Singh, K. Mitochondria as targets for detection and treatment of cancer. Expert Rev. Mol. Med. 2002, 4, 1–19. [Google Scholar] [CrossRef]
- Tallini, G. Oncocytic tumours. Virchows Arch. 1998, 433, 5–12. [Google Scholar] [CrossRef]
- Hruban, Z.; Mochizuki, Y.; Morris, H.P.; Slesers, A. Ultrastructure of Morris renal tumors. J. Natl. Cancer Inst. 1973, 50, 1487–1495. [Google Scholar] [CrossRef]
- White, M.T.; Arya, D.V.; Tewari, K.K. Biochemical properties of neoplastic cell mitochondria. J. Natl. Cancer Inst. 1974, 53, 553–559. [Google Scholar] [CrossRef]
- Sriskanthadevan, S.; Jeyaraju, D.V.; Chung, T.E.; Prabha, S.; Xu, W.; Skrtic, M.; Jhas, B.; Hurren, R.; Gronda, M.; Wang, X.; et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood 2015, 125, 2120–2130. [Google Scholar] [CrossRef] [Green Version]
- Arismendi-Morillo, G. Electron microscopy morphology of the mitochondrial network in gliomas and their vascular microenvironment. Biochim. Biophys. Acta (BBA)-Bioenerg. 2011, 1807, 602–608. [Google Scholar] [CrossRef] [Green Version]
- Strunck, E.; Frank, K.; Vollmer, G. Regulation of Gene Expression in Endometrial Cancer Cells: Role of Extracellular Matrix in Mitochondrial Gene Expression. In Cell and Molecular Biology of Endometrial Carcinoma; Kuramoto, H., Nishida, M., Eds.; Springer: Tokyo, Japan, 2003; pp. 221–231. [Google Scholar] [CrossRef]
- Zhang, Q.; Liang, Z.; Gao, Y.; Teng, M.; Niu, L. Differentially expressed mitochondrial genes in breast cancer cells: Potential new targets for anti-cancer therapies. Gene 2016, 596, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sun, K.; Jiao, S.; Cai, N.; Zhao, X.; Zou, H.; Xie, Y.; Wang, Z.; Zhong, M.; Wei, L. High levels of SIRT1 expression enhance tumorigenesis and associate with a poor prognosis of colorectal carcinoma patients. Sci. Rep. 2014, 4, 7481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, M.G.; Adams, S.M.; Walker, R.A.; Brammar, W.J.; Varley, J.M. Differential expression of the mitochondrial gene cytochrome oxidase II in benign and malignant breast tissue. J. Pathol. 1992, 168, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Dakubo, G.D. Mitochondrial Genetic Alterations in Cancer I. In Mitochondrial Genetics and Cancer; Dakubo, G.D., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 135–165. [Google Scholar] [CrossRef]
- Larman, T.C.; DePalma, S.R.; Hadjipanayis, A.G.; Cancer Genome Atlas Research, N.; Protopopov, A.; Zhang, J.; Gabriel, S.B.; Chin, L.; Seidman, C.E.; Kucherlapati, R.; et al. Spectrum of somatic mitochondrial mutations in five cancers. Proc. Natl. Acad. Sci. USA 2012, 109, 14087–14091. [Google Scholar] [CrossRef] [Green Version]
- Czarnecka, A.; Bartnik, E. Mitochondrial DNA Mutations in Tumors. In Cellular Respiration and Carcinogenesis; Sarangarajan, R., Apte, S., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 119–130. [Google Scholar] [CrossRef]
- Damm, F.; Bunke, T.; Thol, F.; Markus, B.; Wagner, K.; Gohring, G.; Schlegelberger, B.; Heil, G.; Reuter, C.W.; Pullmann, K.; et al. Prognostic implications and molecular associations of NADH dehydrogenase subunit 4 (ND4) mutations in acute myeloid leukemia. Leukemia 2012, 26, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Komiyama, T.; Inomoto, C.; Kamiguchi, H.; Kajiwara, H.; Kobayashi, H.; Nakamura, N.; Terachi, T. Mutations in the Mitochondrial ND1 Gene Are Associated with Postoperative Prognosis of Localized Renal Cell Carcinoma. Int. J. Mol. Sci. 2016, 17, 2049. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Bermudez, A.; Vallejo, C.G.; Vicente-Blanco, R.J.; Gallardo, M.E.; Fernandez-Moreno, M.A.; Quintanilla, M.; Garesse, R. Enhanced tumorigenicity by mitochondrial DNA mild mutations. Oncotarget 2015, 6, 13628–13643. [Google Scholar] [CrossRef] [Green Version]
- Reznik, E.; Miller, M.L.; Senbabaoglu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA copy number variation across human cancers. Elife 2016, 5, e10769. [Google Scholar] [CrossRef]
- Lee, H.C.; Yin, P.H.; Lin, J.C.; Wu, C.C.; Chen, C.Y.; Wu, C.W.; Chi, C.W.; Tam, T.N.; Wei, Y.H. Mitochondrial genome instability and mtDNA depletion in human cancers. Ann. N. Y. Acad. Sci. 2005, 1042, 109–122. [Google Scholar] [CrossRef]
- Selvanayagam, P.; Rajaraman, S. Detection of mitochondrial genome depletion by a novel cDNA in renal cell carcinoma. Lab. Investig. 1996, 74, 592–599. [Google Scholar]
- Wu, C.-W.; Yin, P.-H.; Hung, W.-Y.; Li, A.F.-Y.; Li, S.-H.; Chi, C.-W.; Wei, Y.-H.; Lee, H.-C. Mitochondrial DNA mutations and mitochondrial DNA depletion in gastric cancer. Genes Chromosom. Cancer 2005, 44, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Koura, M.; Isaka, H.; Yoshida, M.C.; Tosu, M.; Sekiguchi, T. Suppression of tumorigenicity in interspecific reconstituted cells and cybrids. Gan 1982, 73, 574–580. [Google Scholar] [PubMed]
- Israel, B.A.; Schaeffer, W.I. Cytoplasmic suppression of malignancy. Vitr. Cell. Dev. Biol. 1987, 23, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Israel, B.A.; Schaeffer, W.I. Cytoplasmic mediation of malignancy. Vitr. Cell. Dev. Biol. 1988, 24, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Liu, Y.N.; Werbin, H. Cytoplasmic suppression of tumor progression in reconstituted cells. Somat. Cell Mol. Genet. 1988, 14, 345–350. [Google Scholar] [CrossRef]
- McKinnell, R.G.; Deggins, B.A.; Labat, D.D. Transplantation of pluripotential nuclei from triploid frog tumors. Science 1969, 165, 394–396. [Google Scholar] [CrossRef]
- Shay, J.W.; Lorkowski, G.; Clark, M.A. Suppression of tumorigenicity in cybrids. J. Supramol. Struct. Cell. Biochem. 1981, 16, 75–82. [Google Scholar] [CrossRef]
- Shay, J.W.; Werbin, H. Cytoplasmic suppression of tumorigenicity in reconstructed mouse cells. Cancer Res. 1988, 48, 830–833. [Google Scholar]
- Howell, A.N.; Sager, R. Tumorigenicity and its suppression in cybrids of mouse and Chinese hamster cell lines. Proc. Natl. Acad. Sci. USA 1978, 75, 2358–2362. [Google Scholar] [CrossRef] [Green Version]
- Hochedlinger, K.; Blelloch, R.; Brennan, C.; Yamada, Y.; Kim, M.; Chin, L.; Jaenisch, R. Reprogramming of a melanoma genome by nuclear transplantation. Genes Dev. 2004, 18, 1875–1885. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Connelly, M.C.; Wetmore, C.; Curran, T.; Morgan, J.I. Mouse embryos cloned from brain tumors. Cancer Res. 2003, 63, 2733–2736. [Google Scholar] [PubMed]
- Mintz, B.; Illmensee, K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc. Natl. Acad. Sci. USA 1975, 72, 3585–3589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonasson, J.; Harris, H. The analysis of malignancy by cell fusion. VIII. Evidence for the intervention of an extra-chromosomal element. J. Cell Sci. 1977, 24, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N. Mitochondria: The Ultimate Tumor Suppressor. In Cancer as a Metabolic Disease; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 195–205. [Google Scholar] [CrossRef]
- Bose, S.; Deininger, M.; Gora-Tybor, J.; Goldman, J.M.; Melo, J.V. The presence of typical and atypical BCR-ABL fusion genes in leukocytes of normal individuals: Biologic significance and implications for the assessment of minimal residual disease. Blood 1998, 92, 3362–3367. [Google Scholar] [CrossRef] [PubMed]
- Kaipparettu, B.A.; Ma, Y.; Park, J.H.; Lee, T.-L.; Zhang, Y.; Yotnda, P.; Creighton, C.J.; Chan, W.-Y.; Wong, L.-J.C. Crosstalk from Non-Cancerous Mitochondria Can Inhibit Tumor Properties of Metastatic Cells by Suppressing Oncogenic Pathways. PLoS ONE 2013, 8, e61747. [Google Scholar] [CrossRef] [Green Version]
- Elliott, R.L.; Jiang, X.P.; Head, J.F. Mitochondria organelle transplantation: Introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity. Breast Cancer Res. Treat. 2012, 136, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Wahlestedt, M.; Ameur, A.; Moraghebi, R.; Norddahl, G.L.; Sten, G.; Woods, N.B.; Bryder, D. Somatic cells with a heavy mitochondrial DNA mutational load render induced pluripotent stem cells with distinct differentiation defects. Stem Cells 2014, 32, 1173–1182. [Google Scholar] [CrossRef]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.S.; Baty, J.W.; Dong, L.-F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial Genome Acquisition Restores Respiratory Function and Tumorigenic Potential of Cancer Cells without Mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.V.; Dong, L.; Neuzil, J. Mitochondrial DNA in Tumor Initiation, Progression, and Metastasis: Role of Horizontal mtDNA Transfer. Cancer Res. 2015, 75, 3203–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Gerdes, H.H. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015, 22, 1181–1191. [Google Scholar] [CrossRef] [Green Version]
- Pyle, A.; Anugrha, H.; Kurzawa-Akanbi, M.; Yarnall, A.; Burn, D.; Hudson, G. Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol. Aging 2016, 38, 216.e7–216.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Tan, L.; Shen, R.; Zhang, L.; Zuo, H.; Wang, D.W. Decreased Peripheral Mitochondrial DNA Copy Number is Associated with the Risk of Heart Failure and Long-term Outcomes. Medicine 2016, 95, e3323. [Google Scholar] [CrossRef]
- Chen, S.; Li, Z.; He, Y.; Zhang, F.; Li, H.; Liao, Y.; Wei, Z.; Wan, G.; Xiang, X.; Hu, M.; et al. Elevated mitochondrial DNA copy number in peripheral blood cells is associated with childhood autism. BMC Psychiatry 2015, 15, 50. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Mukherjee, P. Targeting energy metabolism in brain cancer: Review and hypothesis. Nutr. Metab. 2005, 2, 30. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Cairns, R.; Papandreou, I.; Koong, A.; Denko, N.C. Oxygen Consumption Can Regulate the Growth of Tumors, a New Perspective on the Warburg Effect. PLoS ONE 2009, 4, e7033. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, A.; Wang, C.; Schreiber, S.L. Perturbational profiling of a cell-line model of tumorigenesis by using metabolic measurements. Proc. Natl. Acad. Sci. USA 2005, 102, 5992–5997. [Google Scholar] [CrossRef] [Green Version]
- John, A.P. Dysfunctional mitochondria, not oxygen insufficiency, cause cancer cells to produce inordinate amounts of lactic acid: The impact of this on the treatment of cancer. Med. Hypotheses 2001, 57, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Vitale, I.; Rigoni, A.; Vacchelli, E.; Michaud, M.; Zischka, H.; Castedo, M.; Kroemer, G. Mitochondrial gateways to cancer. Mol. Asp. Med. 2010, 31, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Cuezva, J.M.; Krajewska, M.; de Heredia, M.L.; Krajewski, S.; Santamaria, G.; Kim, H.; Zapata, J.M.; Marusawa, H.; Chamorro, M.; Reed, J.C. The bioenergetic signature of cancer: A marker of tumor progression. Cancer Res. 2002, 62, 6674–6681. [Google Scholar]
- Cho, Y.M.; Kim, J.H.; Kim, M.; Park, S.J.; Koh, S.H.; Ahn, H.S.; Kang, G.H.; Lee, J.-B.; Park, K.S.; Lee, H.K. Mesenchymal Stem Cells Transfer Mitochondria to the Cells with Virtually No Mitochondrial Function but Not with Pathogenic mtDNA Mutations. PLoS ONE 2012, 7, e32778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, E.; Fujisawa, S.; Morozov, A.; Barlas, A.; Romin, Y.; Dogan, Y.; Gholami, S.; Moreira, A.L.; Manova-Todorova, K.; Moore, M.A.S. Tunneling Nanotubes Provide a Unique Conduit for Intercellular Transfer of Cellular Contents in Human Malignant Pleural Mesothelioma. PLoS ONE 2012, 7, e33093. [Google Scholar] [CrossRef] [Green Version]
- Carew, J.S.; Huang, P. Mitochondrial defects in cancer. Mol. Cancer 2002, 1, 9. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.K.; Costello, L.C. (Eds.) Mitochondria and Cancer, 1st ed.; Springer: New York, NY, USA, 2009; p. 294. [Google Scholar] [CrossRef] [Green Version]
- Samuels, D.C.; Li, C.; Li, B.; Song, Z.; Torstenson, E.; Boyd Clay, H.; Rokas, A.; Thornton-Wells, T.A.; Moore, J.H.; Hughes, T.M.; et al. Recurrent tissue-specific mtDNA mutations are common in humans. PLoS Genet. 2013, 9, e1003929. [Google Scholar] [CrossRef] [Green Version]
- Yeung, K.Y.; Dickinson, A.; John, J.C.S. The Role of Mitochondrial DNA in Tumorigenesis. In Mitochondrial DNA, Mitochondria, Disease and Stem Cells; St. John, J.C., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 119–155. [Google Scholar] [CrossRef]
- Chatterjee, A.; Mambo, E.; Sidransky, D. Mitochondrial DNA mutations in human cancer. Oncogene 2006, 25, 4663–4674. [Google Scholar] [CrossRef] [Green Version]
- Cline, S.D. Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim. Biophys. Acta 2012, 1819, 979–991. [Google Scholar] [CrossRef] [Green Version]
- Gasparre, G.; Porcelli, A.M.; Bonora, E.; Pennisi, L.F.; Toller, M.; Iommarini, L.; Ghelli, A.; Moretti, M.; Betts, C.M.; Martinelli, G.N.; et al. Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc. Natl. Acad. Sci. USA 2007, 104, 9001–9006. [Google Scholar] [CrossRef] [Green Version]
- Gasparre, G.; Hervouet, E.; de Laplanche, E.; Demont, J.; Pennisi, L.F.; Colombel, M.; Mege-Lechevallier, F.; Scoazec, J.Y.; Bonora, E.; Smeets, R.; et al. Clonal expansion of mutated mitochondrial DNA is associated with tumor formation and complex I deficiency in the benign renal oncocytoma. Hum. Mol. Genet. 2008, 17, 986–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J.-I. ROS-Generating Mitochondrial DNA Mutations Can Regulate Tumor Cell Metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Zhang, J.; Gao, Y.; Ding, K.; Wang, N.; Zhou, D.; Jen, J.; Cheng, S. Relationship between mitochondrial DNA mutations and clinical characteristics in human lung cancer. Mitochondrion 2007, 7, 347–353. [Google Scholar] [CrossRef]
- Kretzschmar, C.; Roolf, C.; Timmer, K.; Sekora, A.; Knubel, G.; Escobar, H.M.; Fuellen, G.; Ibrahim, S.M.; Tiedge, M.; Baltrusch, S.; et al. Polymorphisms of the murine mitochondrial ND4, CYTB and COX3 genes impact hematopoiesis during aging. Oncotarget 2016, 7, 74460–74472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Zu, X.L.; Guppy, M. Cancer metabolism: Facts, fantasy, and fiction. Biochem. Biophys. Res. Commun. 2004, 313, 459–465. [Google Scholar] [CrossRef]
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 2013, 9, 712. [Google Scholar] [CrossRef]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Chi, T.Y.; Chen, G.G.; Ho, L.K.; Lai, P.B. Establishment of a doxycycline-regulated cell line with inducible, doubly-stable expression of the wild-type p53 gene from p53-deleted hepatocellular carcinoma cells. Cancer Cell Int. 2005, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Politi, K.; Zakowski, M.F.; Fan, P.D.; Schonfeld, E.A.; Pao, W.; Varmus, H.E. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006, 20, 1496–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dow, L.E.; Fisher, J.; O’Rourke, K.P.; Muley, A.; Kastenhuber, E.R.; Livshits, G.; Tschaharganeh, D.F.; Socci, N.D.; Lowe, S.W. Inducible in vivo genome editing with CRISPR-Cas9. Nat. Biotech. 2015, 33, 390–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahler, E.; Sullivan, W.J.; Cass, A.; Braas, D.; York, A.G.; Bensinger, S.J.; Graeber, T.G.; Christofk, H.R. Doxycycline alters metabolism and proliferation of human cell lines. PLoS ONE 2013, 8, e64561. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.Y.; Rhee, Y.H.; Yi, S.H.; Lee, S.J.; Kim, R.K.; Kim, H.; Park, C.H.; Lee, S.H. Doxycycline enhances survival and self-renewal of human pluripotent stem cells. Stem Cell Rep. 2014, 3, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim. Biophys. Acta 2009, 1787, 384–392. [Google Scholar] [CrossRef]
- Hirst, J.; King, M.S.; Pryde, K.R. The production of reactive oxygen species by complex I. Biochem. Soc. Trans. 2008, 36, 976–980. [Google Scholar] [CrossRef]
- Gonzalez-Halphen, D.; Ghelli, A.; Iommarini, L.; Carelli, V.; Esposti, M.D. Mitochondrial Complex I and Cell Death: A Semi-automatic Shotgun Model. Cell Death Dis. 2011, 2, e222. Available online: http://www.nature.com/cddis/journal/v/2/n10/suppinfo/cddis2011107s1.html (accessed on 24 April 2023). [CrossRef] [Green Version]
- Saada, A. Complex Subunits and Assembly Genes: Complex I. In Mitochondrial Disorders Caused by Nuclear Genes; Wong, L., Ed.; Springer: New York, NY, USA, 2013; pp. 186–202. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Alzahrani, A.S.; Zou, M.; Shi, Y. High frequency of somatic mitochondrial DNA mutations in human thyroid carcinomas and complex I respiratory defect in thyroid cancer cell lines. Oncogene 2005, 24, 1455–1460. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, F.A.; Mayr, J.A.; Neureiter, D.; Feichtinger, R.; Alinger, B.; Jones, N.D.; Eder, W.; Sperl, W.; Kofler, B. Lack of complex I is associated with oncocytic thyroid tumours. Br. J. Cancer 2009, 100, 1434–1437. [Google Scholar] [CrossRef]
- Mayr, J.A.; Meierhofer, D.; Zimmermann, F.; Feichtinger, R.; Kogler, C.; Ratschek, M.; Schmeller, N.; Sperl, W.; Kofler, B. Loss of complex I due to mitochondrial DNA mutations in renal oncocytoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 2270–2275. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Luo, L.; Proctor, S.J.; Middleton, P.G.; Blakely, E.L.; Taylor, R.W.; Turnbull, D.M. Somatic mitochondrial DNA mutations in adult-onset leukaemia. Leukemia 2003, 17, 2487–2491. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Chan, F.L.; Hong, B.F.; Chan, L.W.; Chan, P.S. Mitochondrial DNA mutations in chemical carcinogen-induced rat bladder and human bladder cancer. Oncol. Rep. 2004, 12, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, H.; Hattori, K.; Wada, R.; Ishikawa, K.; Fukuda, S.; Takenaga, K.; Nakada, K.; Hayashi, J. Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PLoS ONE 2011, 6, e23401. [Google Scholar] [CrossRef] [Green Version]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife 2014, 3, e02935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Qin, W.; Sauter, E.R. Large-scale mitochondrial DNA deletion mutations and nuclear genome instability in human breast cancer. Cancer Detect. Prev. 2004, 28, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Bassal, M.A.; Samaraweera, S.E.; Lim, K.; Benard, B.A.; Bailey, S.; Kaur, S.; Leo, P.; Toubia, J.; Thompson-Peach, C.; Nguyen, T.; et al. Germline mutations in mitochondrial complex I reveal genetic and targetable vulnerability in IDH1-mutant acute myeloid leukaemia. Nat. Commun. 2022, 13, 2614. [Google Scholar] [CrossRef]
- He, X.; Zhou, A.; Lu, H.; Chen, Y.; Huang, G.; Yue, X.; Zhao, P.; Wu, Y. Suppression of Mitochondrial Complex I Influences Cell Metastatic Properties. PLoS ONE 2013, 8, e61677. [Google Scholar] [CrossRef] [Green Version]
- Iommarini, L.; Kurelac, I.; Capristo, M.; Calvaruso, M.A.; Giorgio, V.; Bergamini, C.; Ghelli, A.; Nanni, P.; De Giovanni, C.; Carelli, V.; et al. Different mtDNA mutations modify tumor progression in dependence of the degree of respiratory complex I impairment. Hum. Mol. Genet. 2013, 23, 1453–1466. [Google Scholar] [CrossRef] [Green Version]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, C.; Iommarini, L.; Kurelac, I.; Calvaruso, M.A.; Capristo, M.; Lollini, P.; Nanni, P.; Bergamini, C.; Nicoletti, G.; De Giovanni, C.; et al. Respiratory complex I is essential to induce a Warburg profile in mitochondria-defective tumor cells. Cancer Metab. 2013, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Evenepoel, L.; Papathomas, T.G.; Krol, N.; Korpershoek, E.; de Krijger, R.R.; Persu, A.; Dinjens, W.N.M. Toward an improved definition of the genetic and tumor spectrum associated with SDH germ-line mutations. Genet. Med. 2014, 17, 610–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH Mutations Establish a Hypermethylator Phenotype in Paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eng, C.; Kiuru, M.; Fernandez, M.J.; Aaltonen, L.A. A role for mitochondrial enzymes in inherited neoplasia and beyond. Nat. Rev. Cancer 2003, 3, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.; Woodward, E.R.; Killick, P.; Morris, M.R.; Astuti, D.; Latif, F.; Maher, E.R. Germline SDHB Mutations and Familial Renal Cell Carcinoma. JNCI J. Natl. Cancer Inst. 2008, 100, 1260–1262. [Google Scholar] [CrossRef] [PubMed]
- Janeway, K.A.; Kim, S.Y.; Lodish, M.; Nosé, V.; Rustin, P.; Gaal, J.; Dahia, P.L.M.; Liegl, B.; Ball, E.R.; Raygada, M.; et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 314–318. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, D.H.; Jung, W.-H.; Koo, J.S. Succinate dehydrogenase expression in breast cancer. SpringerPlus 2013, 2, 299. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Thompson, C.B. Metabolic regulation of epigenetics. Cell Metab. 2012, 16, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Rose, N.R.; Ng, S.S.; Mecinovic, J.; Lienard, B.M.; Bello, S.H.; Sun, Z.; McDonough, M.A.; Oppermann, U.; Schofield, C.J. Inhibitor scaffolds for 2-oxoglutarate-dependent histone lysine demethylases. J. Med. Chem. 2008, 51, 7053–7056. [Google Scholar] [CrossRef]
- Killian, J.K.; Kim, S.Y.; Miettinen, M.; Smith, C.; Merino, M.; Tsokos, M.; Quezado, M.; Smith, W.I.; Jahromi, M.S.; Xekouki, P.; et al. Succinate Dehydrogenase Mutation Underlies Global Epigenomic Divergence in Gastrointestinal Stromal Tumor. Cancer Discov. 2013, 3, 648–657. [Google Scholar] [CrossRef] [Green Version]
- Cervera, A.M.; Bayley, J.-P.; Devilee, P.; McCreath, K.J. Inhibition of succinate dehydrogenase dysregulates histone modification in mammalian cells. Mol. Cancer 2009, 8, 89. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Qian, J.; Yao, D.M.; Qian, S.X.; Qian, W.; Lin, J.; Xiao, G.F.; Wang, C.Z.; Deng, Z.Q.; Ma, J.C.; et al. SF3B1 mutation is a rare event in Chinese patients with acute and chronic myeloid leukemia. Clin. Biochem. 2013, 46, 701–703. [Google Scholar] [CrossRef] [PubMed]
- Hung, W.-Y.; Wu, C.-W.; Yin, P.-H.; Chang, C.-J.; Li, A.F.-Y.; Chi, C.-W.; Wei, Y.-H.; Lee, H.-C. Somatic mutations in mitochondrial genome and their potential roles in the progression of human gastric cancer. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2010, 1800, 264–270. [Google Scholar] [CrossRef]
- Polyak, K.; Li, Y.; Zhu, H.; Lengauer, C.; Willson, J.K.V.; Markowitz, S.D.; Trush, M.A.; Kinzler, K.W.; Vogelstein, B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat. Genet. 1998, 20, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Liu, V.W.S.; Shi, H.H.; Cheung, A.N.Y.; Chiu, P.M.; Leung, T.W.; Nagley, P.; Wong, L.C.; Ngan, H.Y.S. High Incidence of Somatic Mitochondrial DNA Mutations in Human Ovarian Carcinomas. Cancer Res. 2001, 61, 5998–6001. [Google Scholar] [PubMed]
- Máximo, V.; Soares, P.; Lima, J.; Cameselle-Teijeiro, J.; Sobrinho-Simões, M. Mitochondrial DNA Somatic Mutations (Point Mutations and Large Deletions) and Mitochondrial DNA Variants in Human Thyroid Pathology: A Study with Emphasis on Hürthle Cell Tumors. Am. J. Pathol. 2002, 160, 1857–1865. [Google Scholar] [CrossRef]
- Owens, K.M.; Kulawiec, M.; Desouki, M.M.; Vanniarajan, A.; Singh, K.K. Impaired OXPHOS Complex III in Breast Cancer. PLoS ONE 2011, 6, e23846. [Google Scholar] [CrossRef] [Green Version]
- Fliss, M.S.; Usadel, H.; Caballero, O.L.; Wu, L.; Buta, M.R.; Eleff, S.M.; Jen, J.; Sidransky, D. Facile Detection of Mitochondrial DNA Mutations in Tumors and Bodily Fluids. Science 2000, 287, 2017. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, S.; Hoque, M.O.; Upadhyay, S.; Sidransky, D. Mitochondrial Cytochrome B Gene Mutation Promotes Tumor Growth in Bladder Cancer. Cancer Res. 2008, 68, 700–706. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, S.; Hoque, M.O.; Upadhyay, S.; Sidransky, D. Forced cytochrome B gene mutation expression induces mitochondrial proliferation and prevents apoptosis in human uroepithelial SV-HUC-1 cells. Int. J. Cancer 2009, 125, 2829–2835. [Google Scholar] [CrossRef] [Green Version]
- Pierron, D.; Wildman, D.E.; Huttemann, M.; Markondapatnaikuni, G.C.; Aras, S.; Grossman, L.I. Cytochrome c oxidase: Evolution of control via nuclear subunit addition. Biochim. Biophys. Acta 2012, 1817, 590–597. [Google Scholar] [CrossRef] [Green Version]
- Huttemann, M.; Helling, S.; Sanderson, T.H.; Sinkler, C.; Samavati, L.; Mahapatra, G.; Varughese, A.; Lu, G.; Liu, J.; Ramzan, R.; et al. Regulation of mitochondrial respiration and apoptosis through cell signaling: Cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim. Biophys. Acta 2012, 1817, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Okamura, S.; Ng, C.C.; Koyama, K.; Takei, Y.; Arakawa, H.; Monden, M.; Nakamura, Y. Identification of seven genes regulated by wild-type p53 in a colon cancer cell line carrying a well-controlled wild-type p53 expression system. Oncol. Res. 1999, 11, 281–285. [Google Scholar] [PubMed]
- Horng, Y.-C.; Leary, S.C.; Cobine, P.A.; Young, F.B.J.; George, G.N.; Shoubridge, E.A.; Winge, D.R. Human Sco1 and Sco2 Function as Copper-binding Proteins. J. Biol. Chem. 2005, 280, 34113–34122. [Google Scholar] [CrossRef] [Green Version]
- Permuth-Wey, J.; Chen, Y.A.; Tsai, Y.-Y.; Chen, Z.; Qu, X.; Lancaster, J.M.; Stockwell, H.; Dagne, G.; Iversen, E.; Risch, H.; et al. Inherited Variants in Mitochondrial Biogenesis Genes May Influence Epithelial Ovarian Cancer Risk. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1131–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.X.; Pervaiz, S. Involvement of cytochrome c oxidase subunits Va and Vb in the regulation of cancer cell metabolism by Bcl-2. Cell Death Differ. 2010, 17, 408–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telang, S.; Nelson, K.K.; Siow, D.L.; Yalcin, A.; Thornburg, J.M.; Imbert-Fernandez, Y.; Klarer, A.C.; Farghaly, H.; Clem, B.F.; Eaton, J.W.; et al. Cytochrome c oxidase is activated by the oncoprotein Ras and is required for A549 lung adenocarcinoma growth. Mol. Cancer 2012, 11, 60. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabó, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [Green Version]
- Nishi, T.; Forgac, M. The vacuolar (H+)-ATPases--nature’s most versatile proton pumps. Nat. Rev. Mol. Cell Biol. 2002, 3, 94–103. [Google Scholar] [CrossRef]
- Jones, J.B.; Song, J.J.; Hempen, P.M.; Parmigiani, G.; Hruban, R.H.; Kern, S.E. Detection of Mitochondrial DNA Mutations in Pancreatic Cancer Offers a “Mass”-ive Advantage over Detection of Nuclear DNA Mutations. Cancer Res. 2001, 61, 1299–1304. [Google Scholar]
- Shidara, Y.; Yamagata, K.; Kanamori, T.; Nakano, K.; Kwong, J.Q.; Manfredi, G.; Oda, H.; Ohta, S. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005, 65, 1655–1663. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Pansuriya, T.C.; van Eijk, R.; d’Adamo, P.; van Ruler, M.A.; Kuijjer, M.L.; Oosting, J.; Cleton-Jansen, A.M.; van Oosterwijk, J.G.; Verbeke, S.L.; Meijer, D.; et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat. Genet. 2011, 43, 1256–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef]
- Liu, X.; Kato, Y.; Kaneko, M.K.; Sugawara, M.; Ogasawara, S.; Tsujimoto, Y.; Naganuma, Y.; Yamakawa, M.; Tsuchiya, T.; Takagi, M. Isocitrate dehydrogenase 2 mutation is a frequent event in osteosarcoma detected by a multi-specific monoclonal antibody MsMab-1. Cancer Med. 2013, 2, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Goyal, L.; Yau, T.; Poon, R.T.; Ancukiewicz, M.; Deshpande, V.; Christiani, D.C.; Liebman, H.M.; Yang, H.; Kim, H.; et al. Circulating Oncometabolite 2-Hydroxyglutarate Is a Potential Surrogate Biomarker in Patients with Isocitrate Dehydrogenase-Mutant Intrahepatic Cholangiocarcinoma. Clin. Cancer Res. 2014, 20, 1884–1890. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Seo, S.I.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int. J. Cancer 2009, 125, 353–355. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Losman, J.-A.; Looper, R.E.; Koivunen, P.; Lee, S.; Schneider, R.K.; McMahon, C.; Cowley, G.S.; Root, D.E.; Ebert, B.L.; Kaelin, W.G. (R)-2-Hydroxyglutarate Is Sufficient to Promote Leukemogenesis and Its Effects Are Reversible. Science 2013, 339, 1621–1625. [Google Scholar] [CrossRef]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terunuma, A.; Putluri, N.; Mishra, P.; Mathé, E.A.; Dorsey, T.H.; Yi, M.; Wallace, T.A.; Issaq, H.J.; Zhou, M.; Killian, J.K.; et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J. Clin. Investig. 2014, 124, 398–412. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Ward, P.S.; Shay, J.E.S.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [Green Version]
- Rendina, A.R.; Pietrak, B.; Smallwood, A.; Zhao, H.; Qi, H.; Quinn, C.; Adams, N.D.; Concha, N.; Duraiswami, C.; Thrall, S.H.; et al. Mutant IDH1 enhances the production of 2-hydroxyglutarate due to its kinetic mechanism. Biochemistry 2013, 52, 4563–4577. [Google Scholar] [CrossRef]
- Ward, P.S.; Lu, C.; Cross, J.R.; Abdel-Wahab, O.; Levine, R.L.; Schwartz, G.K.; Thompson, C.B. The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J. Biol. Chem. 2013, 288, 3804–3815. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009, 324, 261–265. [Google Scholar] [CrossRef] [Green Version]
- Bleeker, F.E.; Atai, N.A.; Lamba, S.; Jonker, A.; Rijkeboer, D.; Bosch, K.S.; Tigchelaar, W.; Troost, D.; Vandertop, W.P.; Bardelli, A.; et al. The prognostic IDH1 (R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010, 119, 487–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guitart, A.V.; Panagopoulou, T.I.; Villacreces, A.; Vukovic, M.; Sepulveda, C.; Allen, L.; Carter, R.N.; van de Lagemaat, L.N.; Morgan, M.; Giles, P.; et al. Fumarate hydratase is a critical metabolic regulator of hematopoietic stem cell functions. J. Exp. Med. 2017, 214, 719–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, I.; Alam, A.; Rowan, A.; Barclay, E.; Jaeger, E.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410. [Google Scholar]
- Castro-Vega, L.J.; Buffet, A.; De Cubas, A.A.; Cascón, A.; Menara, M.; Khalifa, E.; Amar, L.; Azriel, S.; Bourdeau, I.; Chabre, O.; et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum. Mol. Genet. 2013, 23, 2440–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, A.A. Biomarker discovery: A proteomic approach for brain cancer profiling. Cancer Sci. 2007, 98, 201–213. [Google Scholar] [CrossRef]
- Sudarshan, S.; Shanmugasundaram, K.; Naylor, S.L.; Lin, S.; Livi, C.B.; O’Neill, C.F.; Parekh, D.J.; Yeh, I.T.; Sun, L.-Z.; Block, K. Reduced Expression of Fumarate Hydratase in Clear Cell Renal Cancer Mediates HIF-2α Accumulation and Promotes Migration and Invasion. PLoS ONE 2011, 6, e21037. [Google Scholar] [CrossRef] [Green Version]
- Fieuw, A.; Kumps, C.; Schramm, A.; Pattyn, F.; Menten, B.; Antonacci, F.; Sudmant, P.; Schulte, J.H.; Van Roy, N.; Vergult, S.; et al. Identification of a novel recurrent 1q42.2-1qter deletion in high risk MYCN single copy 11q deleted neuroblastomas. Int. J. Cancer 2012, 130, 2599–2606. [Google Scholar] [CrossRef] [Green Version]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.-L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; MacKenzie, E.D.; Karim, S.A.; Hedley, A.; Blyth, K.; Kalna, G.; Watson, D.G.; Szlosarek, P.; Frezza, C.; Gottlieb, E. Reversed argininosuccinate lyase activity in fumarate hydratase-deficient cancer cells. Cancer Metab. 2013, 1, 12. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-C.; Cheng, T.-L.; Tsai, W.-H.; Tsai, H.-J.; Hu, K.-H.; Chang, H.-C.; Yeh, C.-W.; Chen, Y.-C.; Liao, C.-C.; Chang, W.-T. Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci. Rep. 2012, 2, 785. [Google Scholar] [CrossRef] [Green Version]
- Schlichtholz, B.; Turyn, J.; Goyke, E.; Biernacki, M.; Jaskiewicz, K.; Sledzinski, Z.; Swierczynski, J. Enhanced citrate synthase activity in human pancreatic cancer. Pancreas 2005, 30, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Coquerel, A.; Wu, Z.; Gligorov, J.; Fuks, D.; Fournel, L.; Lincet, H.; Simula, L. Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update. Int. J. Mol. Sci. 2021, 22, 6587. [Google Scholar] [CrossRef] [PubMed]
- Ternette, N.; Yang, M.; Laroyia, M.; Kitagawa, M.; O’Flaherty, L.; Wolhulter, K.; Igarashi, K.; Saito, K.; Kato, K.; Fischer, R.; et al. Inhibition of Mitochondrial Aconitase by Succination in Fumarate Hydratase Deficiency. Cell Rep. 2013, 3, 689–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Mai, C.; Wei, Y.-L.; Zhao, J.-J.; Hu, Y.-M.; Zeng, Z.-L.; Yang, J.; Lu, W.-H.; Xu, R.-H.; Huang, P. Decreased expression of the mitochondrial metabolic enzyme aconitase (ACO2) is associated with poor prognosis in gastric cancer. Med. Oncol. 2013, 30, 552. [Google Scholar] [CrossRef]
- Hansford, R.G.; Lehninger, A.L. Active oxidative decarboxylation of malate by mitochondria isolated from L-1210 ascites tumor cells. Biochem. Biophys. Res. Commun. 1973, 51, 480–486. [Google Scholar] [CrossRef]
- Ren, J.-G.; Seth, P.; Everett, P.; Clish, C.B.; Sukhatme, V.P. Induction of Erythroid Differentiation in Human Erythroleukemia Cells by Depletion of Malic Enzyme 2. PLoS ONE 2010, 5, e12520. [Google Scholar] [CrossRef] [Green Version]
- Linnane, A.W.; Eastwood, H. Cellular redox regulation and prooxidant signaling systems: A new perspective on the free radical theory of aging. Ann. N. Y. Acad. Sci. 2006, 1067, 47–55. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Gabrilovich, D.I. Inhibition of myeloid cell differentiation in cancer: The role of reactive oxygen species. J. Leukoc. Biol. 2003, 74, 186–196. [Google Scholar] [CrossRef]
- Poli, G.; Leonarduzzi, G.; Biasi, F.; Chiarpotto, E. Oxidative stress and cell signalling. Curr. Med. Chem. 2004, 11, 1163–1182. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G. Mitochondria and reactive oxygen species. Which role in physiology and pathology? Adv. Exp. Med. Biol. 2012, 942, 93–136. [Google Scholar] [CrossRef] [PubMed]
- Battisti, V.; Maders, L.D.; Bagatini, M.D.; Santos, K.F.; Spanevello, R.M.; Maldonado, P.A.; Brule, A.O.; Araujo Mdo, C.; Schetinger, M.R.; Morsch, V.M. Measurement of oxidative stress and antioxidant status in acute lymphoblastic leukemia patients. Clin. Biochem. 2008, 41, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Fan, J.; Rassool, F.V. Genomic instability in myeloid malignancies: Increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008, 270, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hole, P.S.; Darley, R.L.; Tonks, A. Do reactive oxygen species play a role in myeloid leukemias? Blood 2011, 117, 5816–5826. [Google Scholar] [CrossRef] [Green Version]
- Clerkin, J.S.; Naughton, R.; Quiney, C.; Cotter, T.G. Mechanisms of ROS modulated cell survival during carcinogenesis. Cancer Lett. 2008, 266, 30–36. [Google Scholar] [CrossRef]
- Nieborowska-Skorska, M.; Flis, S.; Skorski, T. AKT-induced reactive oxygen species generate imatinib-resistant clones emerging from chronic myeloid leukemia progenitor cells. Leukemia 2014, 28, 2416–2418. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, M. Reactive oxygen species in tumor metastasis. Cancer Lett. 2008, 266, 53–59. [Google Scholar] [CrossRef]
- de Carvalho, D.D.; Sadok, A.; Bourgarel-Rey, V.; Gattacceca, F.; Penel, C.; Lehmann, M.; Kovacic, H. Nox1 downstream of 12-lipoxygenase controls cell proliferation but not cell spreading of colon cancer cells. Int. J. Cancer 2008, 122, 1757–1764. [Google Scholar] [CrossRef] [Green Version]
- Bolton-Gillespie, E.; Schemionek, M.; Klein, H.U.; Flis, S.; Hoser, G.; Lange, T.; Nieborowska-Skorska, M.; Maier, J.; Kerstiens, L.; Koptyra, M.; et al. Genomic instability may originate from imatinib-refractory chronic myeloid leukemia stem cells. Blood 2013, 121, 4175–4183. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anso, E.; Mullen, A.R.; Felsher, D.W.; Matés, J.M.; DeBerardinis, R.J.; Chandel, N.S. Metabolic changes in cancer cells upon suppression of MYC. Cancer Metab. 2013, 1, 7. [Google Scholar] [CrossRef] [Green Version]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Erez, A.; DeBerardinis, R.J. Metabolic dysregulation in monogenic disorders and cancer—Finding method in madness. Nat. Rev. Cancer 2015, 15, 440–448. [Google Scholar] [CrossRef]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef]
- Mishra, D.; Banerjee, D. Metabolic Interactions Between Tumor and Stromal Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2021, 1350, 101–121. [Google Scholar] [CrossRef] [PubMed]
- Scharping, N.E.; Delgoffe, G.M. Tumor Microenvironment Metabolism: A New Checkpoint for Anti-Tumor Immunity. Vaccines 2016, 4, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassim, S.; Pouyssegur, J. Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int. J. Mol. Sci. 2019, 21, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Kuang, H.; Ke, J.; Pi, M.; Yang, D.H. Metabolic Reprogramming Induces Immune Cell Dysfunction in the Tumor Microenvironment of Multiple Myeloma. Front. Oncol. 2020, 10, 591342. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.S.; Metallo, C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Kim, A. Mitochondria in Cancer Energy Metabolism: Culprits or Bystanders? Toxicol. Res. 2015, 31, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emadi, A. Exploiting AML vulnerability: Glutamine dependency. Blood 2015, 126, 1269–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [Green Version]
- Reitzer, L.J.; Wice, B.M.; Kennell, D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 1979, 254, 2669–2676. [Google Scholar] [CrossRef] [PubMed]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef]
- Jeltsch, A. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem 2002, 3, 274–293. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Lan, F.; Shi, Y. Epigenetic regulation: Methylation of histone and non-histone proteins. Sci. China C Life Sci. 2009, 52, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Gurley, L.R.; D’Anna, J.A.; Barham, S.S.; Deaven, L.L.; Tobey, R.A. Histone phosphorylation and chromatin structure during mitosis in Chinese hamster cells. Eur. J. Biochem. 1978, 84, 1–15. [Google Scholar] [CrossRef]
- Zee, B.M.; Levin, R.S.; Xu, B.; LeRoy, G.; Wingreen, N.S.; Garcia, B.A. In vivo residue-specific histone methylation dynamics. J. Biol. Chem. 2010, 285, 3341–3350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Xu, M.; Zhu, B. Epigenetic inheritance mediated by histone lysine methylation: Maintaining transcriptional states without the precise restoration of marks? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20110332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chestier, A.; Yaniv, M. Rapid turnover of acetyl groups in the four core histones of simian virus 40 minichromosomes. Proc. Natl. Acad. Sci. USA 1979, 76, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Jackson, V.; Shires, A.; Chalkley, R.; Granner, D.K. Studies on highly metabolically active acetylation and phosphorylation of histones. J. Biol. Chem. 1975, 250, 4856–4863. [Google Scholar] [CrossRef]
- Mishra, P.; Beura, S.; Ghosh, R.; Modak, R. Nutritional Epigenetics: How Metabolism Epigenetically Controls Cellular Physiology, Gene Expression and Disease. Subcell. Biochem. 2022, 100, 239–267. [Google Scholar] [CrossRef]
- Choubey, P.; Kaur, H.; Bansal, K. Modulation of DNA/RNA Methylation Signaling Mediating Metabolic Homeostasis in Cancer. Subcell. Biochem. 2022, 100, 201–237. [Google Scholar] [CrossRef]
- Mondal, P.; Tiwary, N.; Sengupta, A.; Dhang, S.; Roy, S.; Das, C. Epigenetic Reprogramming of the Glucose Metabolic Pathways by the Chromatin Effectors During Cancer. Subcell. Biochem. 2022, 100, 269–336. [Google Scholar] [CrossRef]
- Adhikari, S.; Guha, D.; Mohan, C.; Mukherjee, S.; Tyler, J.K.; Das, C. Reprogramming Carbohydrate Metabolism in Cancer and Its Role in Regulating the Tumor Microenvironment. Subcell. Biochem. 2022, 100, 3–65. [Google Scholar] [CrossRef] [PubMed]
- Back, F. The variable condition of euchromatin and heterochromatin. Int. Rev. Cytol. 1976, 45, 25–64. [Google Scholar] [CrossRef] [PubMed]
- North, J.A.; Simon, M.; Ferdinand, M.B.; Shoffner, M.A.; Picking, J.W.; Howard, C.J.; Mooney, A.M.; van Noort, J.; Poirier, M.G.; Ottesen, J.J. Histone H3 phosphorylation near the nucleosome dyad alters chromatin structure. Nucleic Acids Res. 2014, 42, 4922–4933. [Google Scholar] [CrossRef] [Green Version]
- Szerlong, H.J.; Hansen, J.C. Nucleosome distribution and linker DNA: Connecting nuclear function to dynamic chromatin structure. Biochem. Cell Biol. 2011, 89, 24–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Ray-Gallet, D.; Almouzni, G. The Histone H3 Family and Its Deposition Pathways. Adv. Exp. Med. Biol. 2021, 1283, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Arents, G.; Burlingame, R.W.; Wang, B.C.; Love, W.E.; Moudrianakis, E.N. The nucleosomal core histone octamer at 3.1 A resolution: A tripartite protein assembly and a left-handed superhelix. Proc. Natl. Acad. Sci. USA 1991, 88, 10148–10152. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, A.; Ioshikhes, I. Chapter Four—Dynamics of Modeled Oligonucleosomes and the Role of Histone Variant Proteins in Nucleosome Organization. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 90, pp. 119–149. [Google Scholar]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.J.; Clark, D.J.; Wolffe, A.P. Histone contributions to the structure of DNA in the nucleosome. Proc. Natl. Acad. Sci. USA 1991, 88, 6829–6833. [Google Scholar] [CrossRef] [Green Version]
- Tomschik, M.; Karymov, M.A.; Zlatanova, J.; Leuba, S.H. The archaeal histone-fold protein HMf organizes DNA into bona fide chromatin fibers. Structure 2001, 9, 1201–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doenecke, D.; Albig, W.; Bode, C.; Drabent, B.; Franke, K.; Gavenis, K.; Witt, O. Histones: Genetic diversity and tissue-specific gene expression. Histochem. Cell Biol. 1997, 107, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Farrelly, L.A.; Thompson, R.E.; Zhao, S.; Lepack, A.E.; Lyu, Y.; Bhanu, N.V.; Zhang, B.; Loh, Y.-H.E.; Ramakrishnan, A.; Vadodaria, K.C.; et al. Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 2019, 567, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Xhemalce, B.; Dawson, M.A.; Bannister, A.J. Histone Modifications. In Reviews in Cell Biology and Molecular Medicine; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011. [Google Scholar] [CrossRef]
- Zentner, G.E.; Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 259–266. [Google Scholar] [CrossRef]
- Ehrlich, M.; Wang, R.Y. 5-Methylcytosine in eukaryotic DNA. Science 1981, 212, 1350–1357. [Google Scholar] [CrossRef]
- Razin, A.; Riggs, A.D. DNA methylation and gene function. Science 1980, 210, 604–610. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, Y.; Wu, Q.; Li, J.; Sun, S.; Sun, S. S-adenosylmethionine: A metabolite critical to the regulation of autophagy. Cell Prolif. 2020, 53, e12891. [Google Scholar] [CrossRef]
- Jurkowska, R.Z.; Jeltsch, A. Mechanisms and Biological Roles of DNA Methyltransferases and DNA Methylation: From Past Achievements to Future Challenges. Adv. Exp. Med. Biol. 2022, 1389, 1–19. [Google Scholar] [CrossRef]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Okano, M.; Xie, S.; Li, E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998, 19, 219–220. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bestor, T.; Laudano, A.; Mattaliano, R.; Ingram, V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J. Mol. Biol. 1988, 203, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Goyal, R.; Jeltsch, A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 2004, 279, 48350–48359. [Google Scholar] [CrossRef] [Green Version]
- Dan, J.; Chen, T. Genetic Studies on Mammalian DNA Methyltransferases. Adv. Exp. Med. Biol. 2016, 945, 123–150. [Google Scholar] [CrossRef]
- Rhee, I.; Bachman, K.E.; Park, B.H.; Jair, K.W.; Yen, R.W.; Schuebel, K.E.; Cui, H.; Feinberg, A.P.; Lengauer, C.; Kinzler, K.W.; et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 2002, 416, 552–556. [Google Scholar] [CrossRef]
- Chen, T.; Ueda, Y.; Dodge, J.E.; Wang, Z.; Li, E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 2003, 23, 5594–5605. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.J.; Ahn, H.; Jeong, D.; Pak, M.; Moon, J.H.; Kim, S. Impact of mutations in DNA methylation modification genes on genome-wide methylation landscapes and downstream gene activations in pan-cancer. BMC Med. Genom. 2020, 13, 27. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Shen, L. The Molecular Basis of DNA Demethylation. In DNA and Histone Methylation as Cancer Targets; Kaneda, A., Tsukada, Y.-I., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 53–73. [Google Scholar] [CrossRef]
- Bolli, N.; Manes, N.; McKerrel, T.; Chi, J.; Park, N.; Gundem, G.; Quail, M.A.; Sathiaseelan, V.; Herman, B.; Crawley, C.; et al. Characterization of gene mutations and copy number changes in acute myeloid leukemia using a rapid target enrichment protocol. Haematologica 2014, 100, 214–222. [Google Scholar] [CrossRef]
- Heby, O. DNA methylation and polyamines in embryonic development and cancer. Int. J. Dev. Biol. 1995, 39, 737–757. [Google Scholar] [PubMed]
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Riggs, A.D. X inactivation, differentiation, and DNA methylation. Cytogenet. Cell Genet. 1975, 14, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Tazi, J.; Bird, A. Alternative chromatin structure at CpG islands. Cell 1990, 60, 909–920. [Google Scholar] [CrossRef]
- Jeltsch, A. Molecular biology. Phylogeny of methylomes. Science 2010, 328, 837–838. [Google Scholar] [CrossRef]
- Lim, W.J.; Kim, K.H.; Kim, J.Y.; Jeong, S.; Kim, N. Identification of DNA-Methylated CpG Islands Associated With Gene Silencing in the Adult Body Tissues of the Ogye Chicken Using RNA-Seq and Reduced Representation Bisulfite Sequencing. Front. Genet. 2019, 10, 346. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [Green Version]
- De Smet, C.; Lurquin, C.; Lethe, B.; Martelange, V.; Boon, T. DNA methylation is the primary silencing mechanism for a set of germ line- and tumor-specific genes with a CpG-rich promoter. Mol. Cell. Biol. 1999, 19, 7327–7335. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Nunez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 2021, 184, 2503–2519.e17. [Google Scholar] [CrossRef]
- Sakamoto, A.; Akiyama, Y.; Shimada, S.; Zhu, W.G.; Yuasa, Y.; Tanaka, S. DNA Methylation in the Exon 1 Region and Complex Regulation of Twist1 Expression in Gastric Cancer Cells. PLoS ONE 2015, 10, e0145630. [Google Scholar] [CrossRef] [PubMed]
- Hodges, E.; Molaro, A.; Dos Santos, C.O.; Thekkat, P.; Song, Q.; Uren, P.J.; Park, J.; Butler, J.; Rafii, S.; McCombie, W.R.; et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol. Cell 2011, 44, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Vlodrop, I.J.; Niessen, H.E.; Derks, S.; Baldewijns, M.M.; van Criekinge, W.; Herman, J.G.; van Engeland, M. Analysis of promoter CpG island hypermethylation in cancer: Location, location, location! Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 4225–4231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, M.; Sun, D.; Luo, M.; Huang, Y.; Challen, G.A.; Rodriguez, B.; Zhang, X.; Chavez, L.; Wang, H.; Hannah, R.; et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat. Genet. 2014, 46, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zheng, H.; Wang, Q.; Zhou, C.; Wei, L.; Liu, X.; Zhang, W.; Zhang, Y.; Du, Z.; Wang, X.; et al. Genome-wide analyses reveal a role of Polycomb in promoting hypomethylation of DNA methylation valleys. Genome Biol. 2018, 19, 18. [Google Scholar] [CrossRef] [Green Version]
- Xuan Lin, Q.X.; Sian, S.; An, O.; Thieffry, D.; Jha, S.; Benoukraf, T. MethMotif: An integrative cell specific database of transcription factor binding motifs coupled with DNA methylation profiles. Nucleic Acids Res. 2019, 47, D145–D154. [Google Scholar] [CrossRef] [Green Version]
- Wiles, E.T.; Selker, E.U. H3K27 methylation: A promiscuous repressive chromatin mark. Curr. Opin. Genet. Dev. 2017, 43, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [Green Version]
- Karmodiya, K.; Krebs, A.R.; Oulad-Abdelghani, M.; Kimura, H.; Tora, L. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genom. 2012, 13, 424. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharchenko, P.V.; Alekseyenko, A.A.; Schwartz, Y.B.; Minoda, A.; Riddle, N.C.; Ernst, J.; Sabo, P.J.; Larschan, E.; Gorchakov, A.A.; Gu, T.; et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 2011, 471, 480–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zentner, G.E.; Tesar, P.J.; Scacheri, P.C. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011, 21, 1273–1283. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.X.X.; Thieffry, D.; Jha, S.; Benoukraf, T. TFregulomeR reveals transcription factors’ context-specific features and functions. Nucleic Acids Res. 2020, 48, e10. [Google Scholar] [CrossRef]
- Hashimoto, H.; Wang, D.; Horton, J.R.; Zhang, X.; Corces, V.G.; Cheng, X. Structural Basis for the Versatile and Methylation-Dependent Binding of CTCF to DNA. Mol. Cell 2017, 66, 711–720.e713. [Google Scholar] [CrossRef] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [Green Version]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [Green Version]
- Maiti, A.; Drohat, A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011, 286, 35334–35338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, L.M.; Zhang, D.; Burroughs, A.M.; Aravind, L. Computational identification of novel biochemical systems involved in oxidation, glycosylation and other complex modifications of bases in DNA. Nucleic Acids Res. 2013, 41, 7635–7655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Xu, Y. Structure and Function of TET Enzymes. Adv. Exp. Med. Biol. 2016, 945, 275–302. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.L.; Veenstra, G.J.C.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef]
- Vinson, C.; Chatterjee, R. CG methylation. Epigenomics 2012, 4, 655–663. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000, 406, 593–599. [Google Scholar] [CrossRef]
- Herz, H.M.; Garruss, A.; Shilatifard, A. SET for life: Biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem. Sci. 2013, 38, 621–639. [Google Scholar] [CrossRef] [Green Version]
- Qian, C.; Zhou, M.M. SET domain protein lysine methyltransferases: Structure, specificity and catalysis. Cell. Mol. Life Sci. 2006, 63, 2755–2763. [Google Scholar] [CrossRef]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, A.J.; Zegerman, P.; Partridge, J.F.; Miska, E.A.; Thomas, J.O.; Allshire, R.C.; Kouzarides, T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001, 410, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Buratowski, S. Dimethylation of H3K4 by Set1 recruits the Set3 histone deacetylase complex to 5’ transcribed regions. Cell 2009, 137, 259–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Histone modifications for human epigenome analysis. J. Hum. Genet. 2013, 58, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Garcia, B.A. Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harb. Perspect. Biol. 2015, 7, a025064. [Google Scholar] [CrossRef] [Green Version]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Steger, D.J.; Lefterova, M.I.; Ying, L.; Stonestrom, A.J.; Schupp, M.; Zhuo, D.; Vakoc, A.L.; Kim, J.E.; Chen, J.; Lazar, M.A.; et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol. Cell. Biol. 2008, 28, 2825–2839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziller, M.J.; Gu, H.; Muller, F.; Donaghey, J.; Tsai, L.T.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenouil, R.; Cauchy, P.; Koch, F.; Descostes, N.; Cabeza, J.Z.; Innocenti, C.; Ferrier, P.; Spicuglia, S.; Gut, M.; Gut, I.; et al. CpG islands and GC content dictate nucleosome depletion in a transcription-independent manner at mammalian promoters. Genome Res. 2012, 22, 2399–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almamun, M.; Kholod, O.; Stuckel, A.J.; Levinson, B.T.; Johnson, N.T.; Arthur, G.L.; Davis, J.W.; Taylor, K.H. Inferring a role for methylation of intergenic DNA in the regulation of genes aberrantly expressed in precursor B-cell acute lymphoblastic leukemia. Leuk. Lymphoma 2017, 58, 2156–2164. [Google Scholar] [CrossRef]
- van Nuland, R.; Schram, A.W.; van Schaik, F.M.; Jansen, P.W.; Vermeulen, M.; Marc Timmers, H.T. Multivalent engagement of TFIID to nucleosomes. PLoS ONE 2013, 8, e73495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeulen, M.; Mulder, K.W.; Denissov, S.; Pijnappel, W.W.; van Schaik, F.M.; Varier, R.A.; Baltissen, M.P.; Stunnenberg, H.G.; Mann, M.; Timmers, H.T. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 2007, 131, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Wozniak, G.G.; Strahl, B.D. Hitting the ‘mark’: Interpreting lysine methylation in the context of active transcription. Biochim. Biophys. Acta 2014, 1839, 1353–1361. [Google Scholar] [CrossRef]
- Orford, K.; Kharchenko, P.; Lai, W.; Dao, M.C.; Worhunsky, D.J.; Ferro, A.; Janzen, V.; Park, P.J.; Scadden, D.T. Differential H3K4 methylation identifies developmentally poised hematopoietic genes. Dev. Cell 2008, 14, 798–809. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef] [Green Version]
- Zhou, V.W.; Goren, A.; Bernstein, B.E. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 2011, 12, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Kamal, M.; Lindblad-Toh, K.; Bekiranov, S.; Bailey, D.K.; Huebert, D.J.; McMahon, S.; Karlsson, E.K.; Kulbokas, E.J., 3rd; Gingeras, T.R.; et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 2005, 120, 169–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokholok, D.K.; Harbison, C.T.; Levine, S.; Cole, M.; Hannett, N.M.; Lee, T.I.; Bell, G.W.; Walker, K.; Rolfe, P.A.; Herbolsheimer, E.; et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 2005, 122, 517–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, R.; Bannister, A.J.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat. Cell Biol. 2004, 6, 73–77. [Google Scholar] [CrossRef]
- Rougeulle, C.; Chaumeil, J.; Sarma, K.; Allis, C.D.; Reinberg, D.; Avner, P.; Heard, E. Differential histone H3 Lys-9 and Lys-27 methylation profiles on the X chromosome. Mol. Cell. Biol. 2004, 24, 5475–5484. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Ninova, M.; Fejes Toth, K.; Aravin, A.A. The control of gene expression and cell identity by H3K9 trimethylation. Development 2019, 146, dev181180. [Google Scholar] [CrossRef] [Green Version]
- Peters, A.H.; Mermoud, J.E.; O’Carroll, D.; Pagani, M.; Schweizer, D.; Brockdorff, N.; Jenuwein, T. Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat. Genet. 2002, 30, 77–80. [Google Scholar] [CrossRef]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [Green Version]
- Cui, K.; Zang, C.; Roh, T.Y.; Schones, D.E.; Childs, R.W.; Peng, W.; Zhao, K. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 2009, 4, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Vakoc, C.R.; Sachdeva, M.M.; Wang, H.; Blobel, G.A. Profile of histone lysine methylation across transcribed mammalian chromatin. Mol. Cell. Biol. 2006, 26, 9185–9195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, K.J.; Scelfo, A.; Jammula, S.; Cuomo, A.; Barozzi, I.; Stutzer, A.; Fischle, W.; Bonaldi, T.; Pasini, D. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 2014, 53, 49–62. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, Y.B.; Pirrotta, V. Polycomb silencing mechanisms and the management of genomic programmes. Nat. Rev. Genet. 2007, 8, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Leeuwen, F.; Gafken, P.R.; Gottschling, D.E. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 2002, 109, 745–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guccione, E.; Bassi, C.; Casadio, F.; Martinato, F.; Cesaroni, M.; Schuchlautz, H.; Luscher, B.; Amati, B. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature 2007, 449, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Vishwanath, S.N.; Erdjument-Bromage, H.; Tempst, P.; Sif, S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell. Biol. 2004, 24, 9630–9645. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Xu, W. Histone H3R17me2a mark recruits human RNA polymerase-associated factor 1 complex to activate transcription. Proc. Natl. Acad. Sci. USA 2012, 109, 5675–5680. [Google Scholar] [CrossRef] [Green Version]
- Casadio, F.; Lu, X.; Pollock, S.B.; LeRoy, G.; Garcia, B.A.; Muir, T.W.; Roeder, R.G.; Allis, C.D. H3R42me2a is a histone modification with positive transcriptional effects. Proc. Natl. Acad. Sci. USA 2013, 110, 14894–14899. [Google Scholar] [CrossRef] [Green Version]
- Shoaib, M.; Chen, Q.; Shi, X.; Nair, N.; Prasanna, C.; Yang, R.; Walter, D.; Frederiksen, K.S.; Einarsson, H.; Svensson, J.P.; et al. Histone H4 lysine 20 mono-methylation directly facilitates chromatin openness and promotes transcription of housekeeping genes. Nat. Commun. 2021, 12, 4800. [Google Scholar] [CrossRef]
- Bassing, C.H.; Suh, H.; Ferguson, D.O.; Chua, K.F.; Manis, J.; Eckersdorff, M.; Gleason, M.; Bronson, R.; Lee, C.; Alt, F.W. Histone H2AX: A dosage-dependent suppressor of oncogenic translocations and tumors. Cell 2003, 114, 359–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, V.; Mazumder, A.; Surface, L.E.; Butty, V.L.; Fields, P.A.; Alwan, A.; Torrey, L.; Thai, K.K.; Levine, S.S.; Bathe, M.; et al. H2A.Z acidic patch couples chromatin dynamics to regulation of gene expression programs during ESC differentiation. PLoS Genet. 2013, 9, e1003725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raisner, R.M.; Hartley, P.D.; Meneghini, M.D.; Bao, M.Z.; Liu, C.L.; Schreiber, S.L.; Rando, O.J.; Madhani, H.D. Histone variant H2A.Z marks the 5’ ends of both active and inactive genes in euchromatin. Cell 2005, 123, 233–248. [Google Scholar] [CrossRef] [Green Version]
- Loppin, B.; Bonnefoy, E.; Anselme, C.; Laurencon, A.; Karr, T.L.; Couble, P. The histone H3.3 chaperone HIRA is essential for chromatin assembly in the male pronucleus. Nature 2005, 437, 1386–1390. [Google Scholar] [CrossRef]
- Erson, A.E.; Petty, E.M. MicroRNAs in development and disease. Clin. Genet. 2008, 74, 296–306. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Shukla, G.C.; Singh, J.; Barik, S. MicroRNAs: Processing, Maturation, Target Recognition and Regulatory Functions. Mol. Cell. Pharmacol. 2011, 3, 83–92. [Google Scholar]
- Fernandes, J.C.R.; Acuna, S.M.; Aoki, J.I.; Floeter-Winter, L.M.; Muxel, S.M. Long Non-Coding RNAs in the Regulation of Gene Expression: Physiology and Disease. NonCoding RNA 2019, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Cheetham, S.W.; Gruhl, F.; Mattick, J.S.; Dinger, M.E. Long noncoding RNAs and the genetics of cancer. Br. J. Cancer 2013, 108, 2419–2425. [Google Scholar] [CrossRef] [Green Version]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamane, K.; Toumazou, C.; Tsukada, Y.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 2006, 125, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, R.; Marmorstein, R. Structure and mechanism of lysine-specific demethylase enzymes. J. Biol. Chem. 2007, 282, 35425–35429. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.L.; Shi, Y.; Tainer, J.A. How substrate specificity is imposed on a histone demethylase—Lessons from KDM2A. Genes Dev. 2014, 28, 1735–1738. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Nady, N.; Qi, C.; Allali-Hassani, A.; Zhu, H.; Pan, P.; Adams-Cioaba, M.A.; Amaya, M.F.; Dong, A.; Vedadi, M.; et al. Methylation-state-specific recognition of histones by the MBT repeat protein L3MBTL2. Nucleic Acids Res. 2009, 37, 2204–2210. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, S.A.; Khorasanizadeh, S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science 2002, 295, 2080–2083. [Google Scholar] [CrossRef]
- Huang, Y.; Fang, J.; Bedford, M.T.; Zhang, Y.; Xu, R.M. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science 2006, 312, 748–751. [Google Scholar] [CrossRef]
- Musselman, C.A.; Lalonde, M.E.; Cote, J.; Kutateladze, T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227. [Google Scholar] [CrossRef] [Green Version]
- Musselman, C.A.; Avvakumov, N.; Watanabe, R.; Abraham, C.G.; Lalonde, M.E.; Hong, Z.; Allen, C.; Roy, S.; Nunez, J.K.; Nickoloff, J.; et al. Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nat. Struct. Mol. Biol. 2012, 19, 1266–1272. [Google Scholar] [CrossRef]
- Wysocka, J.; Swigut, T.; Xiao, H.; Milne, T.A.; Kwon, S.Y.; Landry, J.; Kauer, M.; Tackett, A.J.; Chait, B.T.; Badenhorst, P.; et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 2006, 442, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Hong, T.; Walter, K.L.; Ewalt, M.; Michishita, E.; Hung, T.; Carney, D.; Pena, P.; Lan, F.; Kaadige, M.R.; et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 2006, 442, 96–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.J.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer Epigenetics: From Mechanism to Therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.J. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 2004, 32, 959–976. [Google Scholar] [CrossRef] [Green Version]
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin. Epigenetics 2016, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Pogo, B.G.; Allfrey, V.G.; Mirsky, A.E. RNA synthesis and histone acetylation during the course of gene activation in lymphocytes. Proc. Natl. Acad. Sci. USA 1966, 55, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Clayton, A.L.; Hebbes, T.R.; Thorne, A.W.; Crane-Robinson, C. Histone acetylation and gene induction in human cells. FEBS Lett. 1993, 336, 23–26. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; McCaffery, J.M.; Irizarry, R.A.; Boeke, J.D. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol. Cell 2006, 23, 207–217. [Google Scholar] [CrossRef]
- Cai, L.; Sutter, B.M.; Li, B.; Tu, B.P. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol. Cell 2011, 42, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Galdieri, L.; Vancura, A. Acetyl-CoA carboxylase regulates global histone acetylation. J. Biol. Chem. 2012, 287, 23865–23876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeneke, E.; Witt, O.; Oehme, I. HDAC Family Members Intertwined in the Regulation of Autophagy: A Druggable Vulnerability in Aggressive Tumor Entities. Cells 2015, 4, 135–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaquero, A.; Sternglanz, R.; Reinberg, D. NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene 2007, 26, 5505–5520. [Google Scholar] [CrossRef] [Green Version]
- Sauve, A.A.; Youn, D.Y. Sirtuins: NAD+-dependent deacetylase mechanism and regulation. Curr. Opin. Chem. Biol. 2012, 16, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhang, S.; Lin, S.; Guo, Y.; Deng, W.; Zhang, Y.; Xue, Y. WERAM: A database of writers, erasers and readers of histone acetylation and methylation in eukaryotes. Nucleic Acids Res. 2017, 45, D264–D270. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef]
- Pandey, R.; Muller, A.; Napoli, C.A.; Selinger, D.A.; Pikaard, C.S.; Richards, E.J.; Bender, J.; Mount, D.W.; Jorgensen, R.A. Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes. Nucleic Acids Res. 2002, 30, 5036–5055. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef]
- Khan, A.; Zhang, X. dbSUPER: A database of super-enhancers in mouse and human genome. Nucleic Acids Res. 2016, 44, D164–D171. [Google Scholar] [CrossRef] [Green Version]
- Ghisletti, S.; Barozzi, I.; Mietton, F.; Polletti, S.; De Santa, F.; Venturini, E.; Gregory, L.; Lonie, L.; Chew, A.; Wei, C.L.; et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity 2010, 32, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.V.; Bassal, M.A.; Lin, Q.X.X.; Wu, C.-S.; Kwon, J.; Zhou, Q.; Tan, H.K.; Ebralidze, A.K.; Chai, L.; Benoukraf, T.; et al. Targeted intragenic demethylation initiates chromatin rewiring for gene activation. BioRxiv 2020. [Google Scholar] [CrossRef]
- Cohen, P. The structure and regulation of protein phosphatases. Annu. Rev. Biochem. 1989, 58, 453–508. [Google Scholar] [CrossRef] [PubMed]
- Beard, P. Mobility of histones on the chromosome of simian virus 40. Cell 1978, 15, 955–967. [Google Scholar] [CrossRef]
- Meersseman, G.; Pennings, S.; Bradbury, E.M. Mobile nucleosomes—A general behavior. EMBO J. 1992, 11, 2951–2959. [Google Scholar] [CrossRef]
- Flaus, A.; Owen-Hughes, T. Dynamic properties of nucleosomes during thermal and ATP-driven mobilization. Mol. Cell. Biol. 2003, 23, 7767–7779. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, H.; Somers, J.; Webster, R.; Flaus, A.; Owen-Hughes, T. Histone tails and the H3 alphaN helix regulate nucleosome mobility and stability. Mol. Cell. Biol. 2007, 27, 4037–4048. [Google Scholar] [CrossRef] [Green Version]
- Sawicka, A.; Seiser, C. Sensing core histone phosphorylation—A matter of perfect timing. Biochim. Biophys. Acta 2014, 1839, 711–718. [Google Scholar] [CrossRef] [Green Version]
- Fischle, W.; Tseng, B.S.; Dormann, H.L.; Ueberheide, B.M.; Garcia, B.A.; Shabanowitz, J.; Hunt, D.F.; Funabiki, H.; Allis, C.D. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 2005, 438, 1116–1122. [Google Scholar] [CrossRef]
- Cook, P.J.; Ju, B.G.; Telese, F.; Wang, X.; Glass, C.K.; Rosenfeld, M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009, 458, 591–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, D.; Keogh, M.C.; Ishii, H.; Peterson, C.L.; Buratowski, S.; Lieberman, J. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol. Cell 2005, 20, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Keogh, M.C.; Kim, J.A.; Downey, M.; Fillingham, J.; Chowdhury, D.; Harrison, J.C.; Onishi, M.; Datta, N.; Galicia, S.; Emili, A.; et al. A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature 2006, 439, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Moussaieff, A.; Rouleau, M.; Kitsberg, D.; Cohen, M.; Levy, G.; Barasch, D.; Nemirovski, A.; Shen-Orr, S.; Laevsky, I.; Amit, M.; et al. Glycolysis-Mediated Changes in Acetyl-CoA and Histone Acetylation Control the Early Differentiation of Embryonic Stem Cells. Cell Metab. 2015, 21, 392–402. [Google Scholar] [CrossRef] [Green Version]
- McGarry, J.D.; Foster, D.W. Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem. 1980, 49, 395–420. [Google Scholar] [CrossRef]
- Robinson, M.B.; Lee, M.L.; DaSilva, S. Glutamate Transporters and Mitochondria: Signaling, Co-compartmentalization, Functional Coupling, and Future Directions. Neurochem. Res. 2020, 45, 526–540. [Google Scholar] [CrossRef]
- Counihan, J.L.; Grossman, E.A.; Nomura, D.K. Cancer Metabolism: Current Understanding and Therapies. Chem. Rev. 2018, 118, 6893–6923. [Google Scholar] [CrossRef]
- Hynes, M.J.; Murray, S.L. ATP-citrate lyase is required for production of cytosolic acetyl coenzyme A and development in Aspergillus nidulans. Eukaryot. Cell 2010, 9, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Friis, R.M.; Wu, B.P.; Reinke, S.N.; Hockman, D.J.; Sykes, B.D.; Schultz, M.C. A glycolytic burst drives glucose induction of global histone acetylation by picNuA4 and SAGA. Nucleic Acids Res. 2009, 37, 3969–3980. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, V.; Williams, J.S.; Robinson, K.M.; Sopko, R.L.; Schultz, M.C. Global control of histone modification by the anaphase-promoting complex. Mol. Cell. Biol. 2003, 23, 9136–9149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Gruber, J.J.; Litzenburger, U.M.; Zhou, Y.; Miao, Y.R.; LaGory, E.L.; Li, A.M.; Hu, Z.; Yip, M.; Hart, L.S.; et al. Acetate supplementation restores chromatin accessibility and promotes tumor cell differentiation under hypoxia. Cell Death Dis. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, J.D.; Lawrence, R.T.; Healy, M.E.; Dominy, J.E.; Liao, J.A.; Breen, D.S.; Byrne, F.L.; Kenwood, B.M.; Lackner, C.; Okutsu, S.; et al. Genetic inhibition of hepatic acetyl-CoA carboxylase activity increases liver fat and alters global protein acetylation. Mol. Metab. 2014, 3, 419–431. [Google Scholar] [CrossRef]
- Rios Garcia, M.; Steinbauer, B.; Srivastava, K.; Singhal, M.; Mattijssen, F.; Maida, A.; Christian, S.; Hess-Stumpp, H.; Augustin, H.G.; Muller-Decker, K.; et al. Acetyl-CoA Carboxylase 1-Dependent Protein Acetylation Controls Breast Cancer Metastasis and Recurrence. Cell Metab. 2017, 26, 842–855.e845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soliman, M.L.; Rosenberger, T.A. Acetate supplementation increases brain histone acetylation and inhibits histone deacetylase activity and expression. Mol. Cell. Biochem. 2011, 352, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kanda, T.; Urai, H.; Kurokochi, A.; Kitahama, R.; Shigaki, S.; Ono, T.; Yukioka, H.; Hasegawa, K.; Tokuyama, H.; et al. NNMT activation can contribute to the development of fatty liver disease by modulating the NAD+ metabolism. Sci. Rep. 2018, 8, 8637. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; van Haren, M.J.; Buijs, N.; Innocenti, P.; Zhang, Y.; Sartini, D.; Campagna, R.; Emanuelli, M.; Parsons, R.B.; Jespers, W.; et al. Potent Inhibition of Nicotinamide N-Methyltransferase by Alkene-Linked Bisubstrate Mimics Bearing Electron Deficient Aromatics. J. Med. Chem. 2021, 64, 12938–12963. [Google Scholar] [CrossRef]
- Campagna, R.; Pozzi, V.; Sartini, D.; Salvolini, E.; Brisigotti, V.; Molinelli, E.; Campanati, A.; Offidani, A.; Emanuelli, M. Beyond Nicotinamide Metabolism: Potential Role of Nicotinamide N-Methyltransferase as a Biomarker in Skin Cancers. Cancers 2021, 13, 4943. [Google Scholar] [CrossRef]
- Wang, W.; Yang, C.; Wang, T.; Deng, H. Complex roles of nicotinamide N-methyltransferase in cancer progression. Cell Death Dis. 2022, 13, 267. [Google Scholar] [CrossRef]
- Nguyen, T.T.M.; Nguyen, T.H.; Kim, H.S.; Dao, T.T.P.; Moon, Y.; Seo, M.; Kang, S.; Mai, V.H.; An, Y.J.; Jung, C.R.; et al. GPX8 regulates clear cell renal cell carcinoma tumorigenesis through promoting lipogenesis by NNMT. J. Exp. Clin. Cancer Res. 2023, 42, 42. [Google Scholar] [CrossRef]
- Tomida, M.; Ohtake, H.; Yokota, T.; Kobayashi, Y.; Kurosumi, M. Stat3 up-regulates expression of nicotinamide N-methyltransferase in human cancer cells. J. Cancer Res. Clin. Oncol. 2008, 134, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Roessler, M.; Rollinger, W.; Palme, S.; Hagmann, M.L.; Berndt, P.; Engel, A.M.; Schneidinger, B.; Pfeffer, M.; Andres, H.; Karl, J.; et al. Identification of nicotinamide N-methyltransferase as a novel serum tumor marker for colorectal cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 6550–6557. [Google Scholar] [CrossRef] [Green Version]
- Kassem, H.; Sangar, V.; Cowan, R.; Clarke, N.; Margison, G.P. A potential role of heat shock proteins and nicotinamide N-methyl transferase in predicting response to radiation in bladder cancer. Int. J. Cancer 2002, 101, 454–460. [Google Scholar] [CrossRef]
- Thomas, M.G.; Saldanha, M.; Mistry, R.J.; Dexter, D.T.; Ramsden, D.B.; Parsons, R.B. Nicotinamide N-methyltransferase expression in SH-SY5Y neuroblastoma and N27 mesencephalic neurones induces changes in cell morphology via ephrin-B2 and Akt signalling. Cell Death Dis. 2013, 4, e669. [Google Scholar] [CrossRef] [Green Version]
- Mistry, R.J.; Klamt, F.; Ramsden, D.B.; Parsons, R.B. Nicotinamide N-methyltransferase expression in SH-SY5Y human neuroblastoma cells decreases oxidative stress. J. Biochem. Mol. Toxicol. 2020, 34, e22439. [Google Scholar] [CrossRef]
- Sartini, D.; Morganti, S.; Guidi, E.; Rubini, C.; Zizzi, A.; Giuliante, R.; Pozzi, V.; Emanuelli, M. Nicotinamide N-methyltransferase in non-small cell lung cancer: Promising results for targeted anti-cancer therapy. Cell Biochem. Biophys. 2013, 67, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xie, X.Y.; Yang, S.W.; Wang, J.; He, C. Nicotinamide N-methyltransferase protein expression in renal cell cancer. J. Zhejiang Univ. Sci. B 2010, 11, 136–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Z.; Mentch, S.J.; Gao, X.; Nichenametla, S.N.; Locasale, J.W. Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width. Nat. Commun. 2018, 9, 1955. [Google Scholar] [CrossRef]
- Sperber, H.; Mathieu, J.; Wang, Y.; Ferreccio, A.; Hesson, J.; Xu, Z.; Fischer, K.A.; Devi, A.; Detraux, D.; Gu, H.; et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 2015, 17, 1523–1535. [Google Scholar] [CrossRef]
- Cantoni, G.L. Biological methylation: Selected aspects. Annu. Rev. Biochem. 1975, 44, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Caudill, M.A.; Wang, J.C.; Melnyk, S.; Pogribny, I.P.; Jernigan, S.; Collins, M.D.; Santos-Guzman, J.; Swendseid, M.E.; Cogger, E.A.; James, S.J. Intracellular S-Adenosylhomocysteine Concentrations Predict Global DNA Hypomethylation in Tissues of Methyl-Deficient Cystathionine β-Synthase Heterozygous Mice. J. Nutr. 2001, 131, 2811–2818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibani, S.; Melnyk, S.; Pogribny, I.P.; Wang, W.; Hiou-Tim, F.; Deng, L.; Trasler, J.; James, S.J.; Rozen, R. Studies of methionine cycle intermediates (SAM, SAH), DNA methylation and the impact of folate deficiency on tumor numbers in Min mice. Carcinogenesis 2002, 23, 61–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S.-i.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Radak, Z.; Koltai, E.; Taylor, A.W.; Higuchi, M.; Kumagai, S.; Ohno, H.; Goto, S.; Boldogh, I. Redox-regulating sirtuins in aging, caloric restriction, and exercise. Free Radic. Biol. Med. 2013, 58, 87–97. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [Green Version]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Guarente, L. Sirtuins and ageing—New findings. EMBO Rep. 2013, 14, 750. [Google Scholar] [CrossRef] [Green Version]
- Pirinen, E.; Lo Sasso, G.; Auwerx, J. Mitochondrial sirtuins and metabolic homeostasis. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 759–770. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, C.; Satterstrom, F.K.; Haigis, M.C.; Mostoslavsky, R. From sirtuin biology to human diseases: An update. J. Biol. Chem. 2012, 287, 42444–42452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rongvaux, A.; Andris, F.; Van Gool, F.; Leo, O. Reconstructing eukaryotic NAD metabolism. Bioessays 2003, 25, 683–690. [Google Scholar] [CrossRef] [PubMed]
- German, N.J.; Haigis, M.C. Sirtuins and the Metabolic Hurdles in Cancer. Curr. Biol. 2015, 25, R569–R583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalabothula, N.; Al-jumaily, T.; Eteleeb, A.M.; Flight, R.M.; Xiaorong, S.; Moseley, H.; Rouchka, E.C.; Fondufe-Mittendorf, Y.N. Genome-Wide Profiling of PARP1 Reveals an Interplay with Gene Regulatory Regions and DNA Methylation. PLoS ONE 2015, 10, e0135410. [Google Scholar] [CrossRef] [PubMed]
- Ummarino, S.; Hausman, C.; Di Ruscio, A. The PARP Way to Epigenetic Changes. Genes 2021, 12, 446. [Google Scholar] [CrossRef] [PubMed]
- Caiafa, P.; Guastafierro, T.; Zampieri, M. Epigenetics: Poly(ADP-ribosyl)ation of PARP-1 regulates genomic methylation patterns. FASEB J. 2009, 23, 672–678. [Google Scholar] [CrossRef] [Green Version]
- Lobo, J.; Constâncio, V.; Guimarães-Teixeira, C.; Leite-Silva, P.; Miranda-Gonçalves, V.; Sequeira, J.P.; Pistoni, L.; Guimarães, R.; Cantante, M.; Braga, I.; et al. Promoter methylation of DNA homologous recombination genes is predictive of the responsiveness to PARP inhibitor treatment in testicular germ cell tumors. Mol. Oncol. 2021, 15, 846–865. [Google Scholar] [CrossRef]
- Guastafierro, T.; Cecchinelli, B.; Zampieri, M.; Reale, A.; Riggio, G.; Sthandier, O.; Zupi, G.; Calabrese, L.; Caiafa, P. CCCTC-binding Factor Activates PARP-1 Affecting DNA Methylation Machinery. J. Biol. Chem. 2008, 283, 21873–21880. [Google Scholar] [CrossRef] [Green Version]
- Ummarino, S.; Hausman, C.; Gaggi, G.; Rinaldi, L.; Bassal, M.A.; Zhang, Y.; Seelam, A.J.; Kobayashi, I.S.; Borchiellini, M.; Ebralidze, A.K.; et al. NAD Modulates DNA Methylation and Cell Differentiation. Cells 2021, 10, 2986. [Google Scholar] [CrossRef]
- Kim, M.Y.; Zhang, T.; Kraus, W.L. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev. 2005, 19, 1951–1967. [Google Scholar] [CrossRef] [Green Version]
- Kraus, W.L. Transcriptional control by PARP-1: Chromatin modulation, enhancer-binding, coregulation, and insulation. Curr. Opin. Cell Biol. 2008, 20, 294–302. [Google Scholar] [CrossRef] [Green Version]
- Kraus, W.L.; Lis, J.T. PARP goes transcription. Cell 2003, 113, 677–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnakumar, R.; Gamble, M.J.; Frizzell, K.M.; Berrocal, J.G.; Kininis, M.; Kraus, W.L. Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science 2008, 319, 819–821. [Google Scholar] [CrossRef] [PubMed]
- Ame, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef]
- Mentch, S.J.; Mehrmohamadi, M.; Huang, L.; Liu, X.; Gupta, D.; Mattocks, D.; Gomez Padilla, P.; Ables, G.; Bamman, M.M.; Thalacker-Mercer, A.E.; et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015, 22, 861–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mentch, S.J.; Locasale, J.W. One-carbon metabolism and epigenetics: Understanding the specificity. Ann. N. Y. Acad. Sci. 2016, 1363, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Moore, K.E.; Gozani, O. An unexpected journey: Lysine methylation across the proteome. Biochim. Biophys. Acta 2014, 1839, 1395–1403. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, A.K.; Cheng, X. Dynamics of histone lysine methylation: Structures of methyl writers and erasers. Prog. Drug Res. 2011, 67, 107–124. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [Green Version]
- Hino, S.; Sakamoto, A.; Nagaoka, K.; Anan, K.; Wang, Y.; Mimasu, S.; Umehara, T.; Yokoyama, S.; Kosai, K.; Nakao, M. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat. Commun. 2012, 3, 758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martignago, D.; Bernardini, B.; Polticelli, F.; Salvi, D.; Cona, A.; Angelini, R.; Tavladoraki, P. The Four FAD-Dependent Histone Demethylases of Arabidopsis Are Differently Involved in the Control of Flowering Time. Front. Plant Sci. 2019, 10, 669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Colibus, L.; Mattevi, A. New frontiers in structural flavoenzymology. Curr. Opin. Struct. Biol. 2006, 16, 722–728. [Google Scholar] [CrossRef] [PubMed]
- van Berkel, W.J.; Kamerbeek, N.M.; Fraaije, M.W. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 2006, 124, 670–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutter, J.; Winge, D.R.; Schiffman, J.D. Succinate dehydrogenase—Assembly, regulation and role in human disease. Mitochondrion 2010, 10, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Giancaspero, T.A.; Wait, R.; Boles, E.; Barile, M. Succinate dehydrogenase flavoprotein subunit expression in Saccharomyces cerevisiae--involvement of the mitochondrial FAD transporter, Flx1p. FEBS J. 2008, 275, 1103–1117. [Google Scholar] [CrossRef]
- Joosten, V.; van Berkel, W.J. Flavoenzymes. Curr. Opin. Chem. Biol. 2007, 11, 195–202. [Google Scholar] [CrossRef]
- Forneris, F.; Binda, C.; Vanoni, M.A.; Mattevi, A.; Battaglioli, E. Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett. 2005, 579, 2203–2207. [Google Scholar] [CrossRef] [Green Version]
- Opel, M.; Lando, D.; Bonilla, C.; Trewick, S.C.; Boukaba, A.; Walfridsson, J.; Cauwood, J.; Werler, P.J.; Carr, A.M.; Kouzarides, T.; et al. Genome-wide studies of histone demethylation catalysed by the fission yeast homologues of mammalian LSD1. PLoS ONE 2007, 2, e386. [Google Scholar] [CrossRef]
- Huang, J.; Sengupta, R.; Espejo, A.B.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007, 449, 105–108. [Google Scholar] [CrossRef]
- Hausinger, R.P. FeII/alpha-ketoglutarate-dependent hydroxylases and related enzymes. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 21–68. [Google Scholar] [CrossRef]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.T.; Manjeri, G.R.; Rodenburg, R.J.; Smeitink, J.A.; Notebaart, R.A.; Huynen, M.; Willems, P.H.; Koopman, W.J. Skeletal Muscle Mitochondria of Ndufs4 Mice Display Normal Maximal Pyruvate Oxidation and Atp Production. Biochim. Biophys. Acta 2015, 1847, 526–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, B.W.; Finley, L.W.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murugan, A.K.; Alzahrani, A.S. Isocitrate Dehydrogenase IDH1 and IDH2 Mutations in Human Cancer: Prognostic Implications for Gliomas. Br. J. Biomed. Sci. 2022, 79, 10208. [Google Scholar] [CrossRef]
- Yang, H.; Ye, D.; Guan, K.L.; Xiong, Y. IDH1 and IDH2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef] [Green Version]
- Oizel, K.; Gratas, C.; Nadaradjane, A.; Oliver, L.; Vallette, F.M.; Pecqueur, C. D-2-Hydroxyglutarate does not mimic all the IDH mutation effects, in particular the reduced etoposide-triggered apoptosis mediated by an alteration in mitochondrial NADH. Cell Death Dis. 2015, 6, e1704. [Google Scholar] [CrossRef] [Green Version]
- Parker, S.J.; Metallo, C.M. Metabolic consequences of oncogenic IDH mutations. Pharmacol. Ther. 2015, 152, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Reitman, Z.J.; Duncan, C.G.; Poteet, E.; Winters, A.; Yan, L.-J.; Gooden, D.M.; Spasojevic, I.; Boros, L.G.; Yang, S.-H.; Yan, H. Cancer-associated Isocitrate Dehydrogenase 1 (IDH1) R132H Mutation and d-2-Hydroxyglutarate Stimulate Glutamine Metabolism under Hypoxia. J. Biol. Chem. 2014, 289, 23318–23328. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, H.; Futakuchi-Tsuchida, A.; Takahashi, M.; Ishikawa, T.; Tsuji, M.; Ando, O. IDH1 and IDH2 have critical roles in 2-hydroxyglutarate production in D-2-hydroxyglutarate dehydrogenase depleted cells. Biochem. Biophys. Res. Commun. 2012, 423, 553–556. [Google Scholar] [CrossRef]
- Du, X.; Hu, H. The Roles of 2-Hydroxyglutarate. Front. Cell Dev. Biol. 2021, 9, 651317. [Google Scholar] [CrossRef] [PubMed]
- Carbonneau, M.; Gagné, L.M.; Lalonde, M.E.; Germain, M.A.; Motorina, A.; Guiot, M.C.; Secco, B.; Vincent, E.E.; Tumber, A.; Hulea, L.; et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 2016, 7, 12700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.; Yim, S.; Park, H. The cancer driver genes IDH1/2, JARID1C/KDM5C, and UTX/KDM6A: Crosstalk between histone demethylation and hypoxic reprogramming in cancer metabolism. Exp. Mol. Med. 2019, 51, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, S.; Lemonnier, F.; Mak, T.W. Roles of IDH1/2 and TET2 mutations in myeloid disorders. Int. J. Hematol. 2016, 103, 627–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vos, D.; van Overveld, W. Decitabine: A historical review of the development of an epigenetic drug. Ann. Hematol. 2005, 84 (Suppl. S1), 3–8. [Google Scholar] [CrossRef]
- Momparler, R.L. Pharmacology of 5-Aza-2’-deoxycytidine (decitabine). Semin. Hematol. 2005, 42, S9–S16. [Google Scholar] [CrossRef]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef]
- Derissen, E.J.; Beijnen, J.H.; Schellens, J.H. Concise drug review: Azacitidine and decitabine. Oncologist 2013, 18, 619–624. [Google Scholar] [CrossRef] [Green Version]
- Sorm, F.; Piskala, A.; Cihak, A.; Vesely, J. 5-Azacytidine, a new, highly effective cancerostatic. Experientia 1964, 20, 202–203. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S.; Vali, M.; Kunjithapatham, R.; Buijs, M.; Syed, L.H.; Rao, P.P.; Ota, S.; Kwak, B.K.; Loffroy, R.; Geschwind, J.F. 3-bromopyruvate: A new targeted antiglycolytic agent and a promise for cancer therapy. Curr. Pharm. Biotechnol. 2010, 11, 510–517. [Google Scholar] [CrossRef]
- Robinson, M.M.; McBryant, S.J.; Tsukamoto, T.; Rojas, C.; Ferraris, D.V.; Hamilton, S.K.; Hansen, J.C.; Curthoys, N.P. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J. 2007, 406, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, S.; Pramanik, S.; Williams, L.J.; Hodges, H.R.; Hudgens, C.W.; Fischer, G.M.; Luo, C.K.; Knighton, B.; Tan, L.; Lorenzi, P.L.; et al. The Glutaminase Inhibitor CB-839 (Telaglenastat) Enhances the Antimelanoma Activity of T-Cell-Mediated Immunotherapies. Mol. Cancer. Ther. 2021, 20, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Elhammali, A.; Ippolito, J.E.; Collins, L.; Crowley, J.; Marasa, J.; Piwnica-Worms, D. A high-throughput fluorimetric assay for 2-hydroxyglutarate identifies Zaprinast as a glutaminase inhibitor. Cancer Discov. 2014, 4, 828–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, N.E.; Tryndyak, V.P.; Pogribna, M.; Beland, F.A.; Pogribny, I.P. Modifying metabolically sensitive histone marks by inhibiting glutamine metabolism affects gene expression and alters cancer cell phenotype. Epigenetics 2012, 7, 1413–1420. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Meng, G.; Zheng, M.; Zhang, Y.; Chen, A.; Wu, J.; Wei, J. The Glutaminase-1 Inhibitor 968 Enhances Dihydroartemisinin-Mediated Antitumor Efficacy in Hepatocellular Carcinoma Cells. PLoS ONE 2016, 11, e0166423. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Sheng, X.; Clark, L.H.; Zhang, L.; Guo, H.; Jones, H.M.; Willson, A.K.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. Glutaminase inhibitor compound 968 inhibits cell proliferation and sensitizes paclitaxel in ovarian cancer. Am. J. Transl. Res. 2016, 8, 4265–4277. [Google Scholar]
- Emberley, E.; Pan, A.; Chen, J.; Dang, R.; Gross, M.; Huang, T.; Li, W.; MacKinnon, A.; Singh, D.; Sotirovska, N.; et al. The glutaminase inhibitor telaglenastat enhances the antitumor activity of signal transduction inhibitors everolimus and cabozantinib in models of renal cell carcinoma. PLoS ONE 2021, 16, e0259241. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef]
- Turner, N.D.; Lupton, J.R. Dietary fiber. Adv. Nutr. 2011, 2, 151–152. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wang, J.; He, T.; Becker, S.; Zhang, G.; Li, D.; Ma, X. Butyrate: A Double-Edged Sword for Health? Adv. Nutr. 2018, 9, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Geng, H.W.; Yin, F.Y.; Zhang, Z.F.; Gong, X.; Yang, Y. Butyrate Suppresses Glucose Metabolism of Colorectal Cancer Cells via GPR109a-AKT Signaling Pathway and Enhances Chemotherapy. Front. Mol. Biosci. 2021, 8, 634874. [Google Scholar] [CrossRef] [PubMed]
- Luu, M.; Riester, Z.; Baldrich, A.; Reichardt, N.; Yuille, S.; Busetti, A.; Klein, M.; Wempe, A.; Leister, H.; Raifer, H.; et al. Microbial short-chain fatty acids modulate CD8(+) T cell responses and improve adoptive immunotherapy for cancer. Nat. Commun. 2021, 12, 4077. [Google Scholar] [CrossRef] [PubMed]
- Bubna, A.K. Vorinostat-An Overview. Indian J. Dermatol. 2015, 60, 419. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M. Cancer biology: Mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Br. J. Cancer 2006, 95, S2–S6. [Google Scholar] [CrossRef]
- Valdez, B.C.; Brammer, J.E.; Li, Y.; Murray, D.; Liu, Y.; Hosing, C.; Nieto, Y.; Champlin, R.E.; Andersson, B.S. Romidepsin targets multiple survival signaling pathways in malignant T cells. Blood Cancer J. 2015, 5, e357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, C.; Rahman, F.; Piekarz, R.; Peer, C.; Frye, R.; Robey, R.W.; Gardner, E.R.; Figg, W.D.; Bates, S.E. Romidepsin: A new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev. Anticancer Ther. 2010, 10, 997–1008. [Google Scholar] [CrossRef]
- Pajak, B.; Siwiak, E.; Soltyka, M.; Priebe, A.; Zielinski, R.; Fokt, I.; Ziemniak, M.; Jaskiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2019, 21, 234. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Gueron, M. The inhibition of bovine heart hexokinase by 2-deoxy-D-glucose-6-phosphate: Characterization by 31P NMR and metabolic implications. Biochimie 1992, 74, 867–873. [Google Scholar] [CrossRef]
- Caravella, J.A.; Lin, J.; Diebold, R.B.; Campbell, A.M.; Ericsson, A.; Gustafson, G.; Wang, Z.; Castro, J.; Clarke, A.; Gotur, D.; et al. Structure-Based Design and Identification of FT-2102 (Olutasidenib), a Potent Mutant-Selective IDH1 Inhibitor. J. Med. Chem. 2020, 63, 1612–1623. [Google Scholar] [CrossRef]
- Stein, E.M.; Konopleva, M.; Gilmour, R.; Szpurka, A.M.; Hill, E.; Ward, R.; Kantarjian, H.M.; Dinardo, C.D. A Phase 1 Study of LY3410738, a First-in-Class Covalent Inhibitor of Mutant IDH in Advanced Myeloid Malignancies (Trial in Progress). Blood 2020, 136, 26. [Google Scholar] [CrossRef]
- Natsume, A.; Arakawa, Y.; Narita, Y.; Sugiyama, K.; Hata, N.; Muragaki, Y.; Shinojima, N.; Kumabe, T.; Saito, R.; Motomura, K.; et al. The first-in-human phase I study of a brain-penetrant mutant IDH1 inhibitor DS-1001 in patients with recurrent or progressive IDH1-mutant gliomas. Neuro. Oncol. 2023, 25, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Penas-Prado, M.; Peters, K.B.; Burris, H.A., 3rd; Maher, E.A.; Janku, F.; Cote, G.M.; de la Fuente, M.I.; Clarke, J.L.; Ellingson, B.M.; et al. Vorasidenib, a Dual Inhibitor of Mutant IDH1/2, in Recurrent or Progressive Glioma; Results of a First-in-Human Phase I Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 4491–4499. [Google Scholar] [CrossRef] [PubMed]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuser, M.; Palmisiano, N.; Mantzaris, I.; Mims, A.; DiNardo, C.; Silverman, L.R.; Wang, E.S.; Fiedler, W.; Baldus, C.; Schwind, S.; et al. Safety and efficacy of BAY1436032 in IDH1-mutant AML: Phase I study results. Leukemia 2020, 34, 2903–2913. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Levell, J.R.; Liu, G.; Caferro, T.; Sutton, J.; Shafer, C.M.; Costales, A.; Manning, J.R.; Zhao, Q.; Sendzik, M.; et al. Discovery and Evaluation of Clinical Candidate IDH305, a Brain Penetrant Mutant IDH1 Inhibitor. ACS Med. Chem. Lett. 2017, 8, 1116–1121. [Google Scholar] [CrossRef]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef] [Green Version]
- Okoye-Okafor, U.C.; Bartholdy, B.; Cartier, J.; Gao, E.N.; Pietrak, B.; Rendina, A.R.; Rominger, C.; Quinn, C.; Smallwood, A.; Wiggall, K.J.; et al. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat. Chem. Biol. 2015, 11, 878–886. [Google Scholar] [CrossRef]
- Davis, M.; Pragani, R.; Popovici-Muller, J.; Gross, S.; Thorne, N.; Salituro, F.; Fantin, V.; Straley, K.; Su, M.; Dang, L.; et al. ML309: A potent inhibitor of R132H mutant IDH1 capable of reducing 2-hydroxyglutarate production in U87 MG glioblastoma cells. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Kim, H.J.; Choi, B.Y.; Keum, Y.S. Identification of a new selective chemical inhibitor of mutant isocitrate dehydrogenase-1. J. Cancer Prev. 2015, 20, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.; Majeti, R. Optimizing Next-Generation AML Therapy: Activity of Mutant IDH2 Inhibitor AG-221 in Preclinical Models. Cancer Discov. 2017, 7, 459–461. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yang, J.; Sun, X.; Wang, Z.; Cheng, X.; Lu, W.; Cai, X.; Hu, C.; Shen, X.; Cao, P. Allosteric inhibitor remotely modulates the conformation of the orthestric pockets in mutant IDH2/R140Q. Sci. Rep. 2017, 7, 16458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, T.A.; Ahnert, J.R.; Piha-Paul, S.A.; Fu, S.; Janku, F.; Karp, D.D.; Naing, A.; Dumbrava, E.E.I.; Pant, S.; Subbiah, V.; et al. Phase I trial of IACS-010759 (IACS), a potent, selective inhibitor of complex I of the mitochondrial electron transport chain, in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2019, 37, 3014. [Google Scholar] [CrossRef]
- Tsuji, A.; Akao, T.; Masuya, T.; Murai, M.; Miyoshi, H. IACS-010759, a potent inhibitor of glycolysis-deficient hypoxic tumor cells, inhibits mitochondrial respiratory complex I through a unique mechanism. J. Biol. Chem. 2020, 295, 7481–7491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, T.; Saitoh, H.; Inoue, A.; Kobayashi, M.; Okitsu, Y.; Katsuoka, Y.; Fukuhara, N.; Onishi, Y.; Ishizawa, K.; Ichinohasama, R.; et al. 3-Deazaneplanocin A (DZNep), an inhibitor of S-adenosylmethionine-dependent methyltransferase, promotes erythroid differentiation. J. Biol. Chem. 2014, 289, 8121–8134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Kim, J.H.; Kim, S.C.; Yi, Y.S.; Yang, W.S.; Yang, Y.; Kim, H.G.; Lee, J.Y.; Kim, K.H.; Yoo, B.C.; et al. Adenosine dialdehyde suppresses MMP-9-mediated invasion of cancer cells by blocking the Ras/Raf-1/ERK/AP-1 signaling pathway. Biochem. Pharmacol. 2013, 86, 1285–1300. [Google Scholar] [CrossRef] [PubMed]
- Syed-Abdul, M.M.; Parks, E.J.; Gaballah, A.H.; Bingham, K.; Hammoud, G.M.; Kemble, G.; Buckley, D.; McCulloch, W.; Manrique-Acevedo, C. Fatty Acid Synthase Inhibitor TVB-2640 Reduces Hepatic de Novo Lipogenesis in Males With Metabolic Abnormalities. Hepatology 2020, 72, 103–118. [Google Scholar] [CrossRef]
- Falchook, G.; Infante, J.; Arkenau, H.T.; Patel, M.R.; Dean, E.; Borazanci, E.; Brenner, A.; Cook, N.; Lopez, J.; Pant, S.; et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine 2021, 34, 100797. [Google Scholar] [CrossRef]




















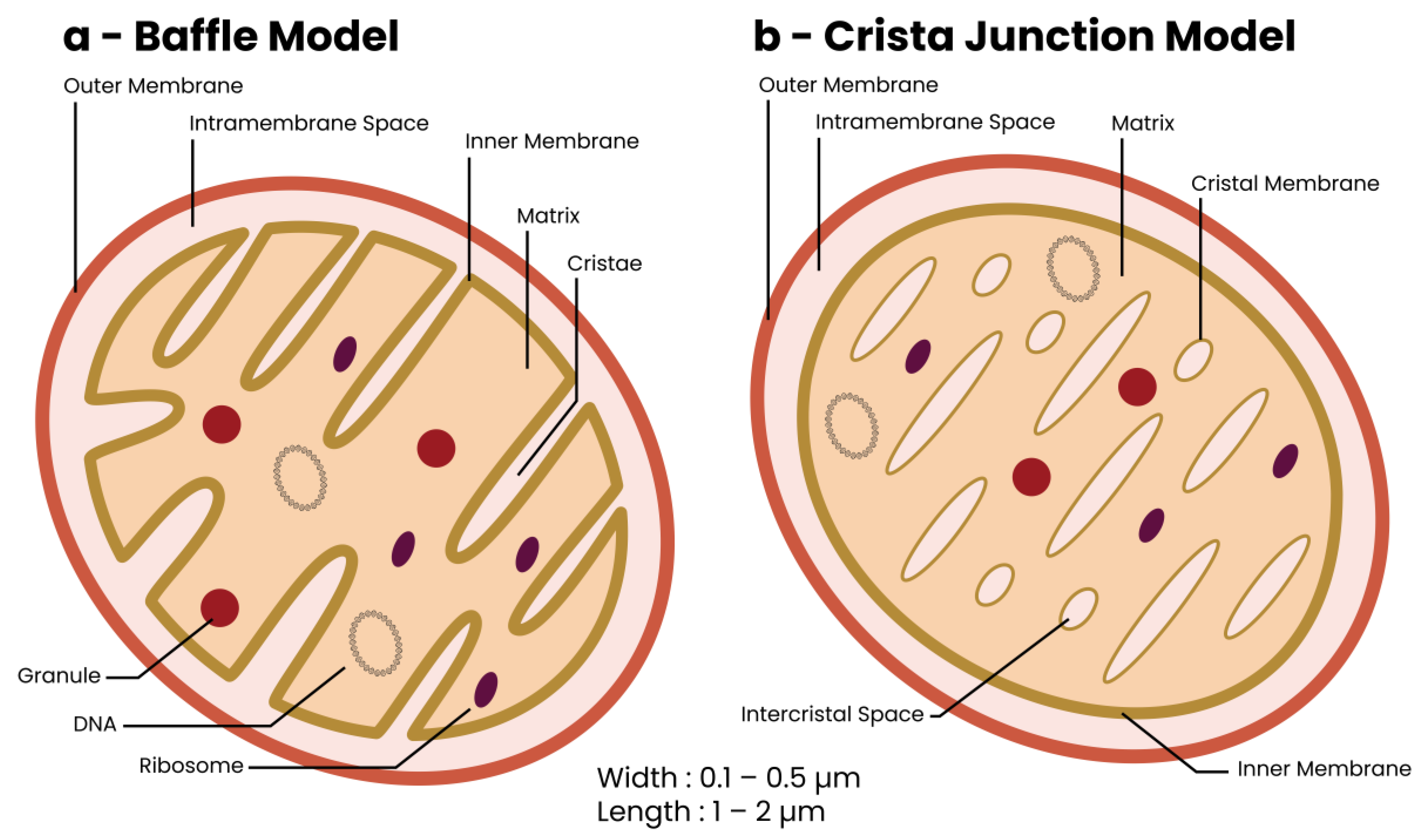{kind=link}
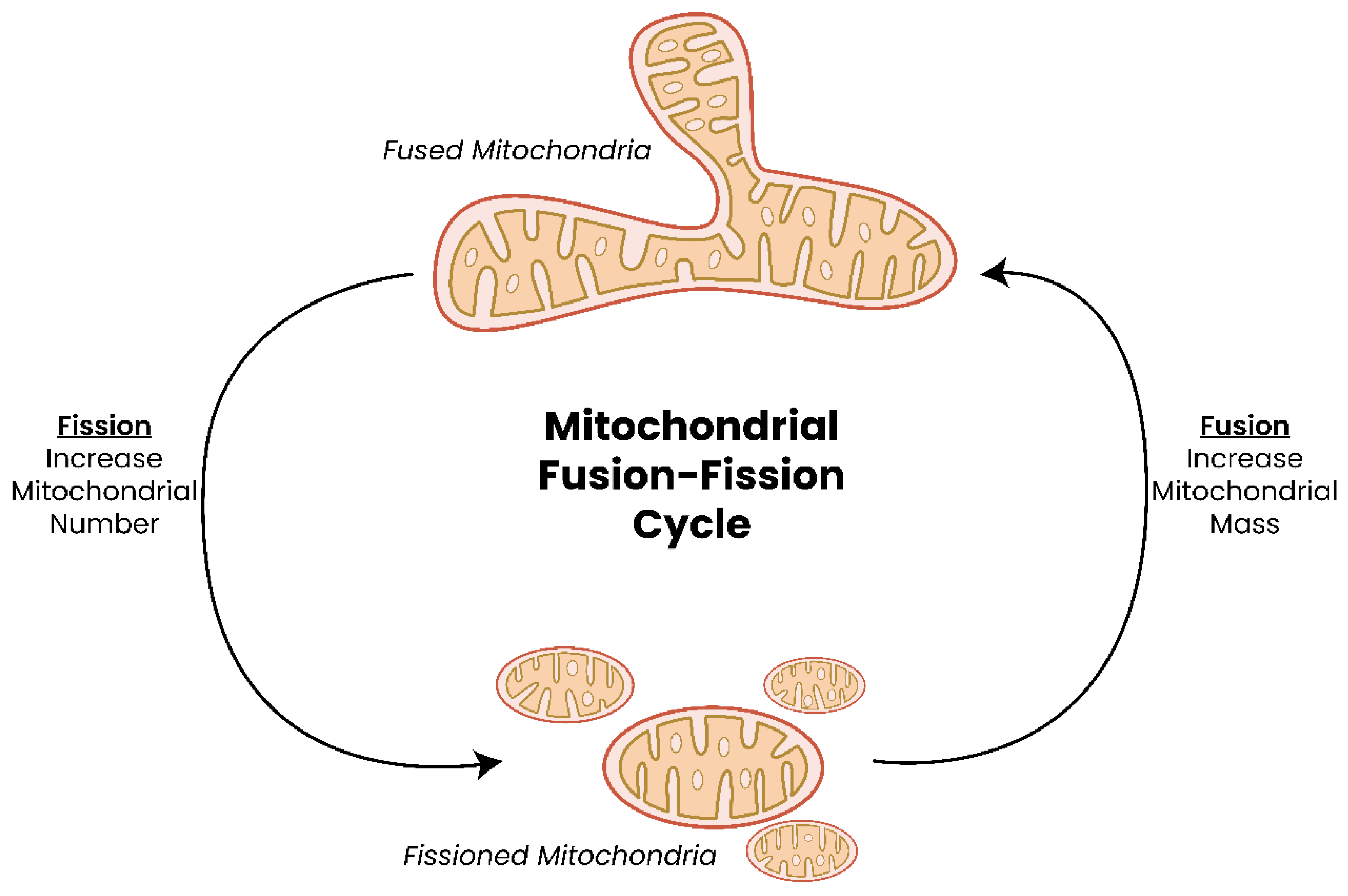{kind=link}
{kind=link}
{kind=link}
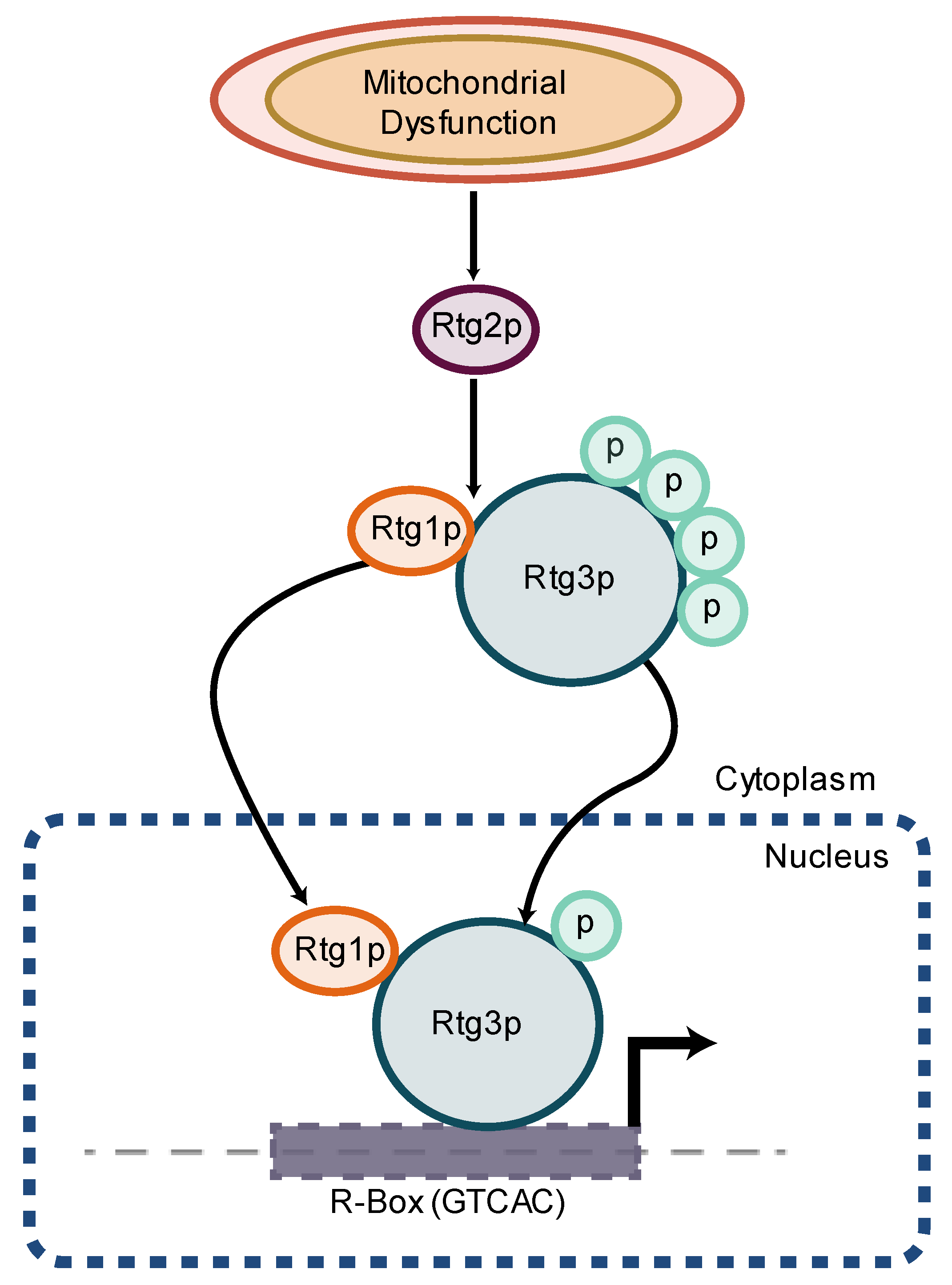{kind=link}
{kind=link}
{kind=link}
{kind=link}
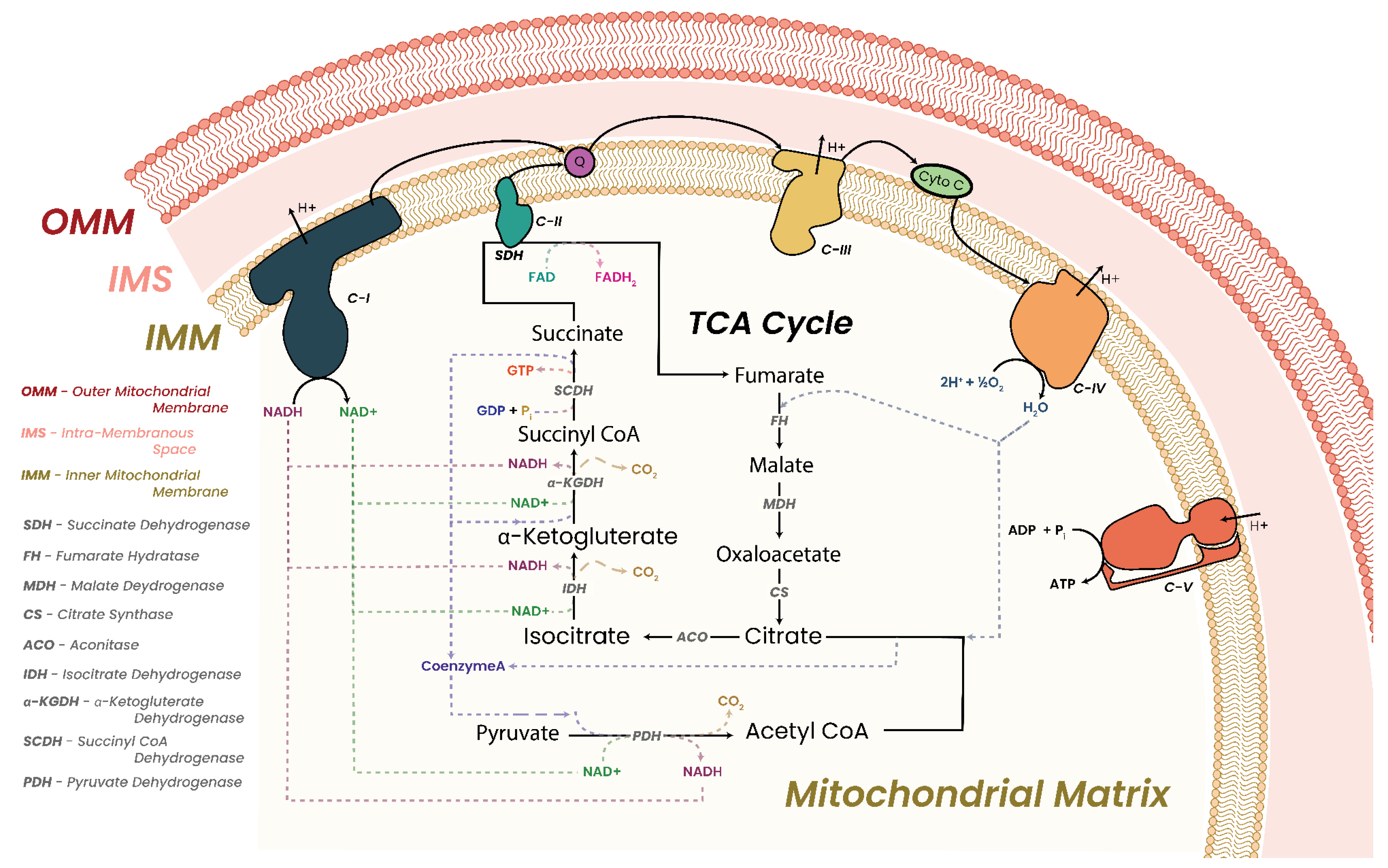{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | Somatic Mutation Frequency (%) |
|---|---|
| Skin | 16–75 |
| Head and neck | 49–78 |
| Thyroid | 23–100 |
| Breast | 30–93 |
| Lung | 43–79 |
| Esophageal | 5–55 |
| Gastric | 18–81 |
| Colorectal | 16–70 |
| Pancreatic | 16–92 |
| Liver | 40–68 |
| Renal | 27–79 |
| Urinary Bladder | 64–100 |
| Prostate | 19–88 |
| Ovarian | 20–80 |
| Endometrial | 9–63 |
| Cervical | 38–90 |
| Nervous system | <35 |
| Hematological | 30–50 |
| Connective tissue | <70 |
| Cancer (Reference) | C-I Gene Mutations Identified |
|---|---|
| Acute Myeloid Leukemia [31] | NDUFA12—3′ UTR—somatic NDUFA13—p.S57P—somatic NDUFAF2—intronic e2-16687—somatic NDUFS4—intronic e3-5101—somatic —5′ UTR—somatic NDUFS7—intronic e2+243—somatic NDUFV2—intronic e2-1653—somatic MT-ND1—p.Y43H—somatic —p.E204—somatic —p.E204—somatic MT-ND2—p.V43A—somatic MT-ND4—p.I8T—somatic —p.T350—somatic —p.V234—somatic MT-ND5—p.P242fs—somatic —p.L260P—somatic —p.S345P—somatic —p.S476P—somatic —p.N452S—somatic |
| Acute Lymphoid Leukemia [324] | MT-ND4—p.L-P variant—somatic |
| Chronic Lymphoid Leukemia [324] | MT-ND1—p.F-S variant—somatic |
| Thyroid Cancer [321] | MT-ND2—p.S-F variant—somatic —p.I-V variant—somatic —frameshift—somatic MT-ND4—p.S-F variant—somatic —p.E-K variant—somatic MT-ND5—p.L-K variant—somatic —p.S-F variant—somatic —p.I-V variant—somatic —p.D-G variant—somatic —p.A-G variant—somatic —p.S-M variant—somatic —p.I-V variant—somatic MT-ND6—p.V-A variant—somatic —p.W-R variant—somatic |
| Oncocytomas [303] | MT-ND1—p.G120X—somatic —p.G244X—somatic MT-ND4—p.374X—somatic MT-ND5—p.540X—somatic MT-ND6—p.87X—somatic |
| Modification | Writers | Readers | Erasers | Genes Mutated in Cancer |
|---|---|---|---|---|
| DNA Methylation | DNA Methyltransferases (DNMTs) | Methyl-CpG-Binding Domain Proteins (MBDs) | Ten–Eleven Translocation Proteins (TETs) | DNMT3A DNMT3B TET1 TET2 MBD4 IDH1/2 |
| Histone Acetylation | Histone Acetyltransferases (HATs) including GNAT, p300/CBP, MYST Protein Families | Bromodomain and Extra-Terminal Proteins (BETs) | Histone Deacetylases (HDACs) | CBP EP300 KAT6A KAT6B BRD1 BRD2 BRD3 BRD4 TRIM33 |
| Histone Methylation | Histone Methyltransferase (HMTs)/ Histone Lysine Methyltransferase (KMTs) | PHD Finger (PHF); Chromodomain-Containing (CHD); Malignant Brain Tumor (MBT); Tudor Domain; PWWP Domain | Histone Demethylases (HDMs)/ Histone Lysine Demethylases (KDMs) including LSD1 and Jumonji-C Proteins (JHDM/JmjC) | EZH2 MLL2 KDM3A KDM4C KDM5A KDM5C KDM6A MSD1/KMT3B SETD2/KMT3A EHMT1 |
| Histone Phosphorylation | Protein Tyrosine Kinases (PTKs) | 14-3-3 Proteins | Protein Tyrosine Phosphatases (PTPs) | PTPN1 PTPN11 PTPN13 PTPRB PTPRC PTPRD |
| Modification | Function | Reference(s) |
|---|---|---|
| DNA Methylation | ||
| CpG Islands, CpG Shores | Inversely associated with transcription | [490,492,521,538,539] |
| Gene Bodies | Positively associated with transcription | [495,529,540] |
| Intergenic | Positively associated with transcription | [541] |
| Enhancers | Inversely associated with transcription | [495,529,540] |
| Histone Modifications | ||
| H3K4me1 | Active enhancers and promoters; Poised enhancers and promoters when in conjunction with H3K4me3 + H3K27me3 | [529,532,540] |
| H3K4me2 | Active promoters when in conjunction with H3K4me3; Poised marker when alone | [535,542,543,544] [545,546,547] |
| H3K4me3 | Active promoters and transcription | [535,548,549,550] |
| H3K9ac | Active promoters | [529,532,548] |
| H3K9me1 | Active promoters | [529] |
| H3K9me2 | Silenced promoters and transcription | [551,552] |
| H3K9me3 | Silenced promoters and transcription | [553,554] |
| H3K27ac | Active enhancers and promoters | [506,555] |
| H3K27me1 | Active transcription | [556,557,558] |
| H3K27me2 | Active transcription | [558] |
| H3K27me3 | Silenced promoters and transcription | [559,560] |
| H3K36me3 | Active gene bodies | [529,532] |
| H3K79me2 | Active transcriptional elongation | [561] |
| H3R2me2 | Counter-correlates with H3K4me3 at promoters; Enriched in gene bodies | [562] |
| H3R8me2 | Silenced promoters | [563] |
| H3R17me2 | Active transcription | [564] |
| H3R42me2 | Active transcription | [565] |
| H4K20me1 | Active enhancers and promoters | [566] |
| Variant Histones | ||
| H2A.X | DNA repair site | [567] |
| H2A.Z | Active promoters and DNA repair sites | [568,569] |
| H3.3 | Active transcription | [570] |
| Non-coding RNA | ||
| miRNA | Repression of gene expression | [571,572,573,574] |
| lncRNA | Gene expression regulation | [575,576,577] |
| Drug | Target | Mode of Action | Epigenetic Consequence | Treats | References |
|---|---|---|---|---|---|
| 5-Aza-2′-deoxycytidine (Dacogen/Inqovi) | DNMTs | Integration into DNA to block DNMTs | Induces hypomethylation | AML and MDS | [701,702,703,704] |
| 5-azacytidine (Onureg/Vidaza) | DNMTs | Integration into DNA to block DNMTs | Induces hypomethylation | AML, MDS, CMML | [703,704,705] |
| 3-Bromopyruvate (BrPA) | Glyceraldehyde 3-phosphate dehydrogenase | Suppress production of ACoA resulting in depletion | Reduced histone acetylation | Prostate, breast, hepatic cancers | [706] |
| Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl) ethyl sulfide (BPTES) | Glutaminase | Suppress production of ACoA resulting in depletion Suppress 2-HG levels | Reduced histone acetylation | Breast cancer | [707] |
| CB-839 (Telaglenastat) | Glutaminase | Suppress production of ACoA resulting in depletion Suppress 2-HG levels | Reduced histone acetylation | Multiple cancers | [708,709,710] |
| Compound 968 (Glutaminase C-IN-1) | Glutaminase | Suppress production of ACoA resulting in depletion Suppress 2-HG levels | Reduced histone acetylation Reduced H3K4me3 methylation | Breast cancer | [711,712] |
| Zaprinast | Glutaminase | Suppress production of ACoA resulting in depletion Suppress 2-HG levels Reduces H3K27me2/me3 | IDH1 mutant cancers | AML, glioblastoma | [709,713] |
| Butyrate | HDACs | Reduces glucose update | Restores histone acetylation balance | T-cell lymphoma, colorectal cancer | [714,715,716,717,718] |
| Vorinostat (Zolinza) | HDACs | Reduces glucose update | Restores histone acetylation balance | T-cell lymphoma | [719,720] |
| Romidepsin | HDACs | Reduces glucose update | Restores histone acetylation balance | T-cell lymphoma | [721,722] |
| 2-Deoxyglucose (2-DG) | Hexokinases | Depletes ACoA stores | Reduced histone acetylation | Multiple cancers | [723,724] |
| FT-2102 (Olutasidenib) | Mutant IDH1 | Suppress 2-HG production | Enables proper TET function | AML, glioma, MDS | [725] |
| LY3410738 | Mutant IDH1 | Suppress 2-HG production | Enables proper TET function | AML and MDS | [726] |
| DS-1001b | Mutant IDH1 | Suppress 2-HG production | Enables proper TET function | Glioma | [727] |
| AG-881 (Vorasidenib) | Mutant IDH1 and IDH2 | Suppress 2-HG production | Enables proper TET function | AML, glioma, MDS, chondrosarcoma | [728] |
| AG-120 (Ivosidenib) | Mutant IDH1 | Suppress 2-HG production | Enables proper TET function | AML, glioma, MDS, chondrosarcoma | [729] |
| BAY1436032 | Mutant IDH1 | Suppress 2-HG production | Enables proper TET function | AML | [730] |
| IDH305 | Mutant IDH1 | Suppress 2-HG production Reduces H3K9me3 and H3K27me3 methylation | Enables proper TET function | Glioma, AML, MDS | [731] |
| AGI-5198 | Mutant IDH1 | Suppress 2-HG production Reduces H3K9me3 and H3K27me3 methylation | Enables proper TET function | AML, glioma, MDS, chondrosarcoma | [732] |
| GSK321 | Mutant IDH1 | Suppress 2-HG production | Enables proper TET function | AML | [372,733] |
| GSK864 (Derivative of CSK321) | Mutant IDH1 | Suppress 2-HG production | Enables proper TET function | AML | [733] |
| ML309 | Mutant IDH1 | Suppress 2-HG production | Enables proper TET function | AML, glioblastoma | [734] |
| 2-(3-Trifluoromethylphenyl) isothiazol-3(2H)-one | Mutant IDH1 | Suppress 2-HG production | Enables proper TET function | Glioma | [735] |
| AG-221 (Enasidenib) | Mutant IDH2 | Suppress 2-HG production | Enables proper TET function | AML, glioma, T-cell lymphomas, chondrosarcoma, cholangiocarcinoma | [736,737] |
| AGI-6780 | Mutant IDH2 | Suppress 2-HG production | Enables proper TET function | AML | [738] |
| CPI-613 (Devimistat) | TCA Cycle Intermediates | Pyruvate dehydrogenase alpha-ketoglutarate dehydrogenase | Blocks enzymatic activity | Burkitt’s lymphoma, MDS, T-cell lymphoma | Company press releases |
| IACS-010759 | NADH dehydrogenase (C-I) | Quinone-site inhibitor | Blocks C-I function and electron transfer | Multiple cancers | [739,740] |
| DZNep (3-deazaneeplanocin A) | SAH hydrolase | Increases SAH:SAM ratio Degrades EZH2 | Inhibits H3K27me3 and H4K20me3 | Multiple cancers | [741] |
| Adenosine dialdehyde | SAH hydrolase | Increases SAH:SAM ratio | Inhibits DNA and histone methylation Downregulates MMP-9 and inhibits Ras/Raf-1/ERK/AP-1 | Multiple cancers | [742] |
| N-methylnicotinamide | NNMTT | Increases SAM levels | Impairs methylation | AML, multiple cancers | NCT02746081, NCT03127735 |
| TVB-2640 | Fatty acid synthase | Blocks fatty acid processing | Solid tumors, lung, ovarian, breast | [743,744] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bassal, M.A. The Interplay between Dysregulated Metabolism and Epigenetics in Cancer. Biomolecules 2023, 13, 944. https://doi.org/10.3390/biom13060944
Bassal MA. The Interplay between Dysregulated Metabolism and Epigenetics in Cancer. Biomolecules. 2023; 13(6):944. https://doi.org/10.3390/biom13060944
Chicago/Turabian StyleBassal, Mahmoud Adel. 2023. "The Interplay between Dysregulated Metabolism and Epigenetics in Cancer" Biomolecules 13, no. 6: 944. https://doi.org/10.3390/biom13060944
APA StyleBassal, M. A. (2023). The Interplay between Dysregulated Metabolism and Epigenetics in Cancer. Biomolecules, 13(6), 944. https://doi.org/10.3390/biom13060944